
32002D0657[1]
A Bizottság határozata (2002. augusztus 14.) a 96/23/EK tanácsi irányelvnek az analitikai módszerek elvégzése és az eredmények értelmezése tekintetében történő végrehajtásáról (az értesítés a C(2002) 3044. számú dokumentummal történt)
A BIZOTTSÁG HATÁROZATA
(2002. augusztus 14.)
a 96/23/EK tanácsi irányelvnek az analitikai módszerek elvégzése és az eredmények értelmezése tekintetében történő végrehajtásáról
(az értesítés a C(2002) 3044. számú dokumentummal történt)
(EGT vonatkozású szöveg)
(2002/657/EK)
2. TELJESÍTMÉNYKRITÉRIUMOK ÉS EGYÉB KÖVETELMÉNYEK ANALITIKAI MÓDSZEREKHEZ
Az alábbiakban szereplőktől eltérő analitikai módszerek vagy ezek kombinációi csak szűrési vagy megerősítési célokra alkalmazhatók akkor, ha bizonyítható, hogy megfelelnek az e határozatban megállapított követelményeknek.
2.1. ÁLTALÁNOS KÖVETELMÉNYEK
2.1.1. A minták kezelése
A minták nyerése, kezelése és feldolgozása úgy történjen, hogy az anyag kimutatásának esélye a lehető legnagyobb legyen. A mintakezelési eljárásoknak meg kell akadályozniuk az analitek véletlen beszennyeződésének vagy elveszésének a lehetőségét.
2.1.2. Vizsgálatok végzése
2.1.2.1. Visszanyerés
A minták elemzése során a visszanyerést minden egyes mintatételnél meg kell határozni, amennyiben fix visszanyerési korrekciós tényezőt használnak. Akkor használható fix visszanyerési korrekciós tényező, ha a visszanyerés bizonyos határértékek között van. Egyéb esetben az adott tételre kapott visszanyerési tényező használandó, hacsak nem a mintában levő analit konkrét visszanyerési tényezőjét kell alkalmazni, amely esetben a minta analittartalmának kvantitatív meghatározásához a standard hozzáadási eljárás (3.5. pont) vagy egy belső standard használandó.
2.1.2.2. Specificitás
Egy módszernek képesnek kell lennie arra, hogy a kísérleti viszonyok mellett különbséget tegyen az analit és az egyéb anyagok között. Ennek lehetséges mértékét illetően becslést kell adni. A leírt mérési technika alkalmazása során az anyagok vonatkozásában előrelátható kölcsönhatások (pl. a kérdéses maradvány homológjai, analógjai, bomlástermékei) felszámolására stratégiákat kell alkalmazni. A mátrix komponenseinek tulajdonítható esetleges zavaró hatás azonosítása elsődleges fontosságú.
2.2. SZŰRÉSI MÓDSZEREK
Szűrési célokra - a 96/23/EK irányelvvel összhangban - csak azok az analitikai technikák használhatók, amelyek esetében dokumentált és visszakereshető módon bizonyítható az, hogy megtörtént a validálás és hogy a hamis megfelelt ráta (β-hiba) 5 %-nál kisebb a kérdéses szinten. Gyaníthatóan nem-megfelelt eredmény esetén az ilyen eredményt megerősítő módszerrel kell megerősíteni.
2.3. ELLENŐRZÉSI MÓDSZEREK SZERVES MARADVÁNYOKHOZ ÉS SZENNYEZŐ ANYAGOKHOZ
A szerves maradványok vagy szennyező anyagok megerősítő módszerei az analit kémiai szerkezetére vonatkozóan szolgálnak információkkal. Ebből következően a csupán kromatográfiás elemzésen alapuló - spektrometriás kimutatás nélküli - módszerek önmagukban nem alkalmasak arra, hogy megerősítő módszerek legyenek. Ha azonban egy adott technika nem rendelkezik a szükséges specificitással, akkor azt az analitikai eljárások tisztítást, kromatográfiás elválasztás(oka)t és a spektrometriás kimutatást magukban foglaló kombinációi segítségével kell biztosítani.
Az alábbi módszerek vagy módszerkombinációk a feltüntetett anyagcsoportokat illetően alkalmasnak kell tekinteni a szerves maradványok vagy szennyező anyagok azonosítására:
1. táblázat
Szerves maradékanyagokhoz vagy szennyező anyagokhoz alkalmas ellenőrzési módszerek
| Mérési technika | Anyagok 96/23/EK 1. melléklet | Korlátozások |
| LC vagy GC, tömeg-spektrometriás kimutatással | A. és B. csoport | Csak ha on-line vagy off-line kromatográfiás elválasztást követ Csak ha teljes letapogatású technika, vagy a teljes tömegspektrum rögzítésére nem képes technikák esetén legalább 3 (B. csoport) vagy 4 (A. csoport) azonosítási pontszám kerül alkalmazásra |
| LC vagy GC, IR spektrometriás kimutatással | A. és B. csoport | Az IR spektrometria esetén meghatározott abszorpciós követelményeknek kell teljesülniük |
| LC teljes letapogatású DAD | B. csoport | Az UV spektrometria esetén meghatározott abszorpciós követelményeknek kell teljesülniük |
| LC fluoreszcencia | B. csoport | Csak a natív fluoreszcenciát mutató molekulák, valamint a transzformálás vagy derivatizálás után fluoreszcenciát mutató molekulák esetén |
| 2-D TLC teljes letapogatású UV/VIS | B. csoport | Kétdimenziós HPTLC és együttes kromatográfia kötelező |
| GC elektronbefogásos kimutatás | B. csoport | Csak két eltérő polaritású kolonna alkalmazása esetén |
| LC-immunogram | B. csoport | Csak legalább két különböző kromatográfiás rendszer vagy egy második, önálló kimutatási módszer alkalmazása esetén |
| LC-UV/VIS (egyetlen hullámhossz) | B. csoport | Csak legalább két különböző kromatográfiás rendszer vagy egy második, önálló kimutatási módszer alkalmazása esetén |
2.3.1. Általános teljesítménykritériumok és követelmények
A megerősítő módszerek az analit kémiai szerkezetére vonatkozóan szolgálnak információkkal. Amikor egynél több vegyület ugyanazt a választ adja, akkor a módszer nem képes azok között különbséget tenni. A csupán kromatográfiás elemzésen alapuló - spektrometriás kimutatás nélküli - módszerek önmagukban nem alkalmasak arra, hogy ellenőrzési módszerek legyenek.
Amennyiben az a módszer része, úgy egy megfelelő belső standardot kell a vizsgálati részhez hozzáadni az extrakciós eljárás kezdetén. Rendelkezésre állástól függően vagy az analit stabil, izotóppal megjelölt formái - amelyek tömeg-spektrometriás kimutatásra különösen alkalmasak - vagy az analittel szerkezeti rokonságot mutató vegyületek alkalmazandók erre a célra.
Amikor nem használható megfelelő belső standard, akkor az analit azonosításának ellenőrzése együttes kromatográfiával történik. Ilyen esetben csak egy csúcs jelentkezik, mivel a megnövelt csúcsmagasság (terület) a hozzáadott analit mennyiségének felel meg. Gázkromatográfia (GC) vagy folyadék-kromatográfia (LC) esetén a csúcs szélessége a maximális magasság felénél az eredeti szélesség 90-110 %-os tartományába esik, a retenciós idők pedig 5 %-os tűrésen belül azonosak. Vékonyréteg-kromatográfiás (TLC) módszerek esetén csak az analitnek tulajdonított folt erősödik fel; nem jelenik meg új folt és a nem változik meg a külső megjelenés.
Az analit ismert mennyiségeit tartalmazó referencia- vagy megerősített anyagot - az engedélyezett határérték vagy a döntési határérték értékén vagy annak közelében (nem-megfelelt kontrollminta) -, valamint a megfelelt kontrollanyagokat és reagens-vakokat lehetőség szerint az elemzés tárgyát képező vizsgálati minták tételeivel egy időben kell a teljes eljáráson végigvinni. Az extraktumok a következő sorrendben kerülnek injektálásra az analitikai műszerbe: reagens-vak, megfelelt kontrollminta, megerősítendő minta/minták, megfelelt kontrollminta újra, végül pedig a nem-megfelelt kontrollminta. Az ettől a sorrendtől való bármely eltérést indokolni kell.
2.3.2. További teljesítmény-kritériumok és egyéb követelmények a kvantitatív analitikai módszerekhez
2.3.2.1. A kvantitatív módszerek egzaktsága
Valamely hiteles anyagminta ismételt elemzései esetén a kísérleti úton meghatározott és visszanyerésre korrigált közepes tömeghányadnak a hitelesített értéktől való eltérésére vonatkozó referenciatartományai a következők:
2. táblázat
A kvantitatív módszerek minimális egzaktsága
| Tömeghányad | Tartomány |
| ≤ 1 μg/kg | – 50 % és + 20 % között |
| > 1 μg/kg – 10 μg/kg | – 30 % és + 10 % között |
| ≥ 10 μg/kg | – 20 % és + 10 % között |
Ilyen CRM-ek hiányában az is elfogadható, hogy a mérések egzaktságának a kiértékelése ismert analitmennyiség(ek) vak mátrixhoz való hozzáadásainak a visszanyerésén keresztül történjen. A közepes visszanyeréssel korrigált adatok csak akkor fogadhatók el, ha a 2. táblázatban szereplő tartományokba esnek.
2.3.2.2. A kvantitatív módszerek precizitása
Egy referencia- vagy megerősített anyag - reprodukálhatósági körülmények közötti - ismételt elemzésére vonatkozó laborközi variációs koefficiens (CV) nem haladhatja meg a Horwitz-egyenlettel kalkulált szintet. Az egyenlet a következő:
ahol C a 10 hatványaként (kitevőjeként) kifejezett tömeghányadot jelenti (pl. 1 mg/g = 10-3). Példák a 3. táblázatban szerepelnek.
3. táblázat
A kvantitatív módszerek reprodukálhatósági CV példái különböző analit-tömeghányad tartományokban
| Tömeghányad | Reprodukálhatósági CV ( %) |
| 1 μg/kg | (*1) |
| 10 μg/kg | (*1) |
| 100 μg/kg | 23 |
| 1 000 μg/kg (1 mg/kg) | 16 |
| (*1) 100 μg/kg alatti tömeghányadok esetén a Horwitz-egyenlet alkalmazása elfogadhatatlanul magas értékeket eredményez. Ezért a 100 μg/kg alatti koncentrációk CV-i a lehető legkisebb értéken tartandók. | |
A reprodukálhatósági körülmények között elvégzett elemzéseknél a laboratóriumon belüli CV rendszerint a fenti értékek fele és kétharmada közé esik. A laboratóriumon belüli reprodukálhatósági körülmények között elvégzett elemzéseknél a laboratóriumon belüli CV nem lehet nagyobb, mint a reprodukálhatósági CV.
Megállapított engedélyezett határértékkel rendelkező anyagok esetében a módszerrel elért laboratóriumon belüli reprodukálhatóság nem lehet nagyobb, mint a hozzá tartozó reprodukálhatósági CV 0,5 × engedélyezett határérték koncentráció mellett.
2.3.3. Teljesítménykritériumok és egyéb követelmények tömeg-spektrometriás kimutatáshoz
A tömeg-spektrometriás módszerek megerősítő módszerekként való alkalmazása csak vagy on-line vagy off-line kromatográfiás elválasztást követően jöhet szóba.
2.3.3.1. Kromatográfiás elválasztás
A GC-MS eljárásoknál a gázkromatográfiás elválasztást kapilláris kolonnák alkalmazásával kell elvégezni. Az LC-MS eljárásoknál a kromatográfiás elválasztást megfelelő LC kolonnák alkalmazásával kell elvégezni. A vizsgálat tárgyát képező analit elfogadható minimum retenciós ideje minden esetben kétszerese a kolonna üres térfogatához tartozó retenciós időnek. A vizsgálati részben levő analit retenciós ideje (vagy relatív retenciós ideje) - egy meghatározott retenciós időablakon belül - megfelel a kalibráló standard hasonló értékének. A retenciós időablaknak összemérhetőnek kell lennie a kromatográfiás rendszer felbontóképességével. Az analit kromatográfiás retenciós idejének a belső standard retenciós idejéhez viszonyított aránya (azaz az analit relatív retenciós ideje) GC esetén ± 0,5 % és LC esetén ± 2,5 % határon belül egyenlő a kalibráló oldatéval.
2.3.3.2. Tömeg-spektrometriás kimutatás
A tömeg-spektrometriás kimutatást MS technikák - például teljes tömegspektrumok regisztrálása (teljes letapogatások) vagy kiválasztott ion monitoring (SIM) - valamint MS-MSn technikák - például kiválasztott reakció monitoring (SRM) - vagy megfelelő ionizálási módokkal kombinált egyéb alkalmas MS vagy MS-MSn technikák alkalmazásával kell elvégezni. Nagy felbontású tömeg-spektrometria (HRMS) esetén a felbontásnak a teljes tömegtartományra vonatkozóan rendszerint 10 000 -nél nagyobbnak kell lennie 10 %-os mélypont mellett.
Teljes letapogatás: Amikor a tömeg-spektrometriás meghatározás teljes letapogatású spektrumok regisztrálásával történik, akkor az összes mért diagnosztikai ionnak (a molekulaion, a molekulaion jellemző adduktjai, jellemző töredékionok és izotópionok) 10 %-nál nagyobb relatív intenzitással kell jelen lennie a kalibráló standard referenciaspektrumában.
SIM: Amikor a tömeg-spektrometriás meghatározás fragmentográfiával történik, akkor a molekulaion lehetőség szerint a kiválasztott diagnosztikai ionok egyike legyen (a molekulaion, a molekulaion jellemző adduktjai, jellemző töredékionok és azok összes izotópionja). A kiválasztott diagnosztikai ionok nem származhatnak a molekulának kizárólag ugyanabból a részéből. A jel-zaj hányados minden egyes diagnosztikai ion esetében 3:1.
Teljes letapogatás és SIM: A kimutatott ionok relatív intenzitásai - a legintenzívebb ion vagy átmenet intenzitásának százalékos értékeként kifejezve - meg kell, hogy feleljenek a kalibráló standard - akár kalibráló standard oldatokból, akár preparált mintákból származó - relatív intenzitásainak összehasonlítható koncentrációk mellett, ugyanazon körülmények között, az alábbi tűréseken belül:
4. táblázat
Relatív ionintenzitásokhoz engedélyezett maximum tűrések tömeg-spektrometriás technikák alkalmazása esetén
| Relatív intenzitás (alapcsúcs %-a) | EI-GC-MS (relatív) | CI-GC-MS, GC-MSn LC-MS, LC-MSn |
| > 50 % | ± 10 % | ± 20 % |
| > 20 % és 50 % között | ± 15 % | ± 25 % |
| > 10 % és 20 % között | ± 20 % | ± 30 % |
| ≤ 10 % | ± 50 % | ± 50 % |
A tömeg-spektrometriás adatok értelmezése: A diagnosztikai ionok és/vagy prekurzor/termék ionpárok relatív intenzitásait a spektrumok összehasonlítása vagy az egyes tömegnyomok jeleinek integrálása útján kell azonosítani. Háttérkorrekció esetén azt az egész tételen keresztül egységesen kell alkalmazni (lásd a 2.3.1. pont 4. bekezdését) és egyértelműen jelezni kell.
Teljes letapogatás: Amikor teljes letapogatású spektrumok egyetlen tömeg-spektrometriában kerülnek regisztrálásra, akkor minimum négy ionnak kell az alapcsúcs legalább 10 %-ának megfelelő relatív intenzitással jelen lennie. A molekulaion akkor értendő ide, ha az legalább 10 %-os relatív intenzitással van jelen a referencia spektrumban. Legalább négy ionnak kell a relatív ionintenzitásokra engedélyezett maximum tűréshatárokon belül lennie (5. táblázat). Számítógéppel segített könyvtári kutatás is alkalmazható. Ilyen esetben a vizsgálati mintákból származó tömeg-spektrometriás adatoknak a kalibráló oldat ugyanezen adataival való összehasonlítása meg kell, hogy haladjon egy kritikus megfeleltetési tényezőt. Ezt a tényezőt a validálási folyamat során kell meghatározni minden egyes analitre vonatkozóan, mégpedig az alábbi kritériumoknak megfelelő spektrumok alapján. A mintamátrix és a detektorteljesítmény által a spektrumokban előidézett variabilitást ellenőrizni kell.
SIM: Amikor nem teljes letapogatású technikákkal mérnek tömegfragmentumokat, akkor az adatok értelmezéséhez azonosítási pontszámok rendszerét kell alkalmazni. A 96/23/EK irányelv I. mellékletének A. csoportjában felsorolt anyagok megerősítő vizsgálatához minimum 4 azonosítási pontszámra van szükség. A 96/23/EK irányelv I. mellékletének B. csoportjában felsorolt anyagok megerősítő vizsgálatához minimum 3 azonosítási pontszámra van szükség. Az alábbi táblázat azt mutatja, hogy az egyes tömeg-spektrometriás alaptechnikák mennyi azonosítási pontszámot kaphatnak. Az ellenőrzéshez szükséges azonosítási pontszámokhoz és az azonosítási pontszámok kiszámítandó összegéhez azonban:
a) legalább egy ionarányt kell megmérni, és
b) valamennyi vonatkozó mért ionaránynak teljesítenie kell a fenti kritériumokat, és
c) a minimális számú azonosítási pontszám megszerzése érdekében maximum három különálló technika kombinálható.
5. táblázat
A tömegfragmentum-osztályok és a kapható azonosítási pontszámok közötti kapcsolat
| MS technika | Iononként kapható azonosítási pontszámok |
| Kis felbontású tömeg-spektrometria (LR) | 1,0 |
| LR-MSn prekurzor ion | 1,0 |
| LR-MSn átmeneti termékek | 1,5 |
| HRMS | 2,0 |
| HR-MSn prekurzor ion | 2,0 |
| HR-MSn átmeneti termékek | 2,5 |
| Lábjegyzetek: (1) Minden ion csak egyszer vehető számításba. (2) Az elektronütközéses ionizációt alkalmazó GC-MS és a kémiai ionizációt alkalmazó GC-MS két külön technikának tekintendő. (3) Az azonosítási pontszámok számának növelése érdekében csak akkor alkalmazhatók különböző analitek, ha a származékokra különböző reakciókémiák jellemzők. (4) A 96/23/EK irányelv I. mellékletének A. csoportjában felsorolt anyagok esetében, ha az analitikai eljárás során a következő technikák valamelyike kerül alkalmazásra: teljes letapogatású diódasoros spektrofotometriával (DAD) párosított HPLC; fluoreszcenciás detektálással párosított HPLC; immunogrammal párosított HPLC; spektrometriás detektálással párosított kétdimenziós TLC, akkor maximum egy azonosítási pontszám adható, feltéve, hogy az említett technikákra vonatkozó kritériumok teljesülnek. (5) Az átmeneti termékek közé tartoznak mind a leányelemek, mind az unokaelemek. | |
6. táblázat
Példák az egyes technikák és azok kombinálása esetén kapható azonosítási pontszámokra (n = egész)
| Technika/technikák | Ionok száma | Azonosítási pontszámok |
| GC-MS (EI vagy CI) | N | n |
| GC-MS (EI és CI) | 2 (EI) + 2 (CI) | 4 |
| GC-MS (EI vagy CI) 2 származék | 2 („A” származék) + 2 („B” származék) | 4 |
| LC-MS | N | n |
| GC-MS-MS | 1 prekurzor és 2 leányelem | 4 |
| LC-MS-MS | 1 prekurzor és 2 leányelem | 4 |
| GC-MS-MS | 2 prekurzor ion, egyenként 1 leányelemmel | 5 |
| LC-MS-MS | 2 prekurzor ion, egyenként 1 leányelemmel | 5 |
| LC-MS-MS-MS | 1 prekurzor, 1 leányelem és 2 unokaelem | 5,5 |
| HRMS | N | 2 n |
| GC-MS és LC-MS | 2 + 2 | 4 |
| GC-MS és HRMS | 2 + 1 | 4 |
2.3.4. Teljesítménykritériumok és egyéb követelmények infravörös detektálással párosított kromatográfiához
Megfelelő csúcsok: A megfelelő csúcsok valamely kalibráló standard infravörös spektrumának abszorpciós maximumait jelentik, az alábbi követelmények teljesülése mellett.
2.3.4.1. Infravörös detektálás
Abszorpciós maximum: Ennek a 4 000 és 500 cm-1 közötti hullámhossztartományban kell jelentkeznie.
Abszorpciós intenzitás: Ez nem lehet kisebb, mint
a) egy 40-es értékű moláris abszorbancia a csúcs alappontjához képest; vagy
b) a 4 000 és 500 cm-1 közötti tartományban jelentkező legintenzívebb csúcs abszorbanciájának 12,5 %-át kitevő relatív abszorbancia
amennyiben mindkettőt nulla abszorbanciához képest mérik, illetve a 4 000 és 500 cm-1 közötti tartományban jelentkező legintenzívebb csúcs abszorbanciájának 5 %-a, amennyiben mindkettőt a csúcs alappontjához képest mérik.
Megjegyzés:
Bár elméleti szempontból az a) szerinti megfelelő csúcsok a kedvezőbbek, a b) szerintiek gyakorlati meghatározása könnyebb.
Az analit infravörös spektrumában jelentkező azon csúcsok száma kerül meghatározásra, amelyek frekvenciája ± 1 cm-1 határon belül tartozik egy a kalibráló standard spektrumában jelentkező megfelelő csúcshoz.
2.3.4.2. Az infravörös tartomány adatainak értelmezése
Abszorpciónak az analit-spektrum mindazon régióiban kell lennie, amelyek a kalibráló standard referenciaspektrumának valamely megfelelő csúcsához tartoznak. A kalibráló standard infravörös spektrumában minimum hat megfelelő csúcsnak kell lennie. Ha a megfelelő csúcsok száma hatnál kevesebb (7), akkor a szóban forgó spektrum nem alkalmazható referenciaspektrumként. A "találati aránynak" - vagyis az analit infravörös spektrumában található megfelelő csúcsok százalékos arányának - legalább 50-nek kell lennie. Ahol egy megfelelő csúcsot illetően nincs pontos illeszkedés, ott az analitspektrum vonatkozó régiójának egy illeszkedő csúcs meglétével kell konzisztenciát mutatnia. Az eljárás a mintaspektrum csak azon abszorpciós csúcsaira érvényes, amelyek intenzitása legalább háromszorosa a két csúcs közötti zajnak.
2.3.5. Teljesítménykritériumok és egyéb követelmények LC és más kimutatási technikák segítségével történő analit-meghatározáshoz
2.3.5.1. Kromatográfiás elválasztás
Belső standardot kell használni, amennyiben egy erre a célra alkalmas anyag áll rendelkezésre. Ez lehetőség szerint valamilyen, az analitéhoz hasonló retenciós idővel rendelkező rokon standard legyen. Az analit annál a retenciós időnél eluálódik, amely - ugyanazon kísérleti körülmények mellett - a hozzá tartozó kalibráló standardra jellemző. Egy analit elfogadható minimum retenciós ideje kétszerese a kolonna üres térfogatához tartozó retenciós időnek. Az analit retenciós idejének a belső standard retenciós idejéhez viszonyított aránya (azaz az analit relatív retenciós ideje) ± 2,5 % határon belül egyenlő a megfelelő mátrixban levő kalibráló standardéval.
2.3.5.2. Teljes letapogatású UV/VIS kimutatás
Az LC módszerekre vonatkozó teljesítménykritériumoknak teljesülniük kell.
Az analit-spektrum abszorpciós maximumainak ugyanazokon a hullámhosszokon kell jelentkezniük, mint a kalibráló standardéinak, a kimutatási rendszer felbontásától függő határon belül. Diódasoros detektálás esetén ez rendszerint ± 2 nm. Az analit 220 nm feletti spektruma - a két spektrum azon részeit illetően, amelyeknek legalább 10 % a relatív abszorbanciája - nem mutathat szemmel észlelhető különbséget a kalibráló standard spektrumához képest. Ez a kritérium akkor teljesül, amikor - először - ugyanazok a maximumok jelentkeznek és - másodszor - amikor a két spektrum között egy ponton sem figyelhető meg nagyobb különbség, mint a kalibráló standard abszorbanciájának 10 %-a. Számítógéppel segített könyvtári kutatás és megfeleltetés alkalmazása esetén a vizsgálati mintákból származó spektrometriás adatoknak a kalibráló oldat ugyanezen adataival való összehasonlítása meg kell hogy haladjon egy kritikus megfeleltetési tényezőt. Ezt a tényezőt a validálási folyamat során kell meghatározni minden egyes analitre vonatkozóan, mégpedig a fenti kritériumoknak megfelelő spektrumok alapján. A mintamátrix és a detektorteljesítmény által a spektrumokban előidézett variabilitást ellenőrizni kell.
2.3.5.3. Teljesítménykritériumok fluorimetriás detektáláshoz
Az LC módszerekre vonatkozó teljesítménykritériumoknak teljesülniük kell.
Ez csak a natív fluoreszcenciát mutató molekulákra, valamint a transzformálás vagy derivatizálás után fluoreszcenciát mutató molekulákra érvényes. A gerjesztési és emissziós hullámhosszoknak a kromatográfiás körülményekkel kombinált megválasztását úgy kell végezni, hogy a vakminta extraktumokban minimális legyen a zavaró komponensek előfordulása.
A kromatogram legközelebbi csúcsmaximumának a kijelölt analit-csúcstól legalább egy teljes csúcsszélességgel kell elkülönülnie, az analit-csúcs maximum magasságának 10 %-ánál.
2.3.5. 4 Teljesítménykritériumok az analit LC immunogrammal történő meghatározásához
Az LC immunogram önmagában nem alkalmas arra, hogy ellenőrzési módszerként használják.
Az LC módszerekre vonatkozó kritériumoknak teljesülniük kell.
Az előre meghatározott minőségellenőrzési paramétereknek (pl. nem specifikus kötődés, a kontrollminták relatív kötődése, a vak abszorbancia-értéke) a vizsgálat validálása során kapott határokon belül kell lenniük.
Az immunogramnak legalább öt frakcióból kell állnia.
Mindegyik frakciónak a csúcsszélesség felénél kisebbnek kell lennie.
A maximális mennyiségű analitet tartalmazó frakciónak a gyanús minta, a nem-megfelelt kontrollminta és a standard esetében ugyanannak kell lennie.
2.3.5.5. Analit meghatározása UV/VIS detektálás (egyetlen hullámhossz) melletti LC segítségével
Az UV/VIS detektálás (egyetlen hullámhossz) melletti LC önmagában nem alkalmas arra, hogy megerősítő vizsgálati módszerként használják.
A kromatogram legközelebbi csúcsmaximumának a kijelölt analit-csúcstól legalább egy teljes csúcsszélességgel kell elkülönülnie, az analit-csúcs maximum magasságának 10 %-ánál.
2.3.6. Teljesítménykritériumok és egyéb követelmények teljes letapogatású UV/VIS spektrometriás detektálással párosított 2-D TLC útján történő analit-meghatározáshoz
Kétdimenziós HPTLC és együttes kromatográfia kötelező.
Az analit RF értékeinek ± 5 % határon belül meg kell egyezniük a standardok RF értékeivel.
Külső megjelenésre az analitnek nem szabad különböznie a mintától.
Az ugyanolyan színű foltok esetében a legközelebbi folt középpontjának legalább a foltátmérők összegének felével kell elkülönülnie az analit foltjának a középpontjától.
Az analit spektruma nem mutathat szemmel észrevehető különbséget a kalibráló standard spektrumához képest, amint az a teljes letapogatású UV/VIS detektálásnál is szerepel.
Számítógéppel segített könyvtári kutatás és megfeleltetés alkalmazása esetén a vizsgálati mintákból származó spektrometriás adatoknak a kalibráló oldat ugyanezen adataival való összehasonlítása meg kell, hogy haladjon egy kritikus megfeleltetési tényezőt. Ezt a tényezőt a validálási folyamat során kell meghatározni minden egyes analitre vonatkozóan, mégpedig a fenti kritériumoknak megfelelő spektrumok alapján. A mintamátrix és a detektorteljesítmény által a spektrumokban előidézett variabilitást ellenőrizni kell.
2.3.7. Teljesítménykritériumok és egyéb követelmények elektronbefogásos kimutatással (ECD) kombinált GC útján történő analit-meghatározáshoz
Belső standardot kell használni, amennyiben egy erre a célra alkalmas anyag áll rendelkezésre. Ez lehetőség szerint valamilyen, az analitéhoz hasonló retenciós idővel rendelkező rokon anyag legyen. Az analit annál a retenciós időnél eluálódik, amely - ugyanazon kísérleti körülmények mellett - a hozzá tartozó kalibráló standardra jellemző. Egy analit elfogadható minimum retenciós ideje kétszerese a kolonna üres térfogatához tartozó retenciós időnek. Az analit retenciós idejének a belső standard retenciós idejéhez viszonyított aránya (azaz az analit relatív retenciós ideje) ± 0,5 % határon belül egyenlő a megfelelő mátrixban levő kalibráló standardéval. A kromatogram legközelebbi csúcsmaximumának a kijelölt analit-csúcstól legalább egy teljes csúcsszélességgel kell elkülönülnie, az analit-csúcs maximum magasságának 10 %-ánál. További információk igénye esetén együttes kromatográfia alkalmazható.
2.4 MEGERŐSÍTŐ VIZSGÁLATI MÓDSZEREK ELEMEKHEZ
A kémiai elemekre vonatkozó megerősítő elemzési módszereknek az egyértelmű azonosításra, valamint az adott kémiai elem szempontjából egyedinek számító fizikai-kémiai tulajdonságok (pl. a kibocsátott vagy elnyelt sugárzás elemre jellemző hullámhossza, atomtömeg) útján történő helyes és pontos számszerűsítésére kell alapulnia a kérdéses szinten.
A kémiai elemek azonosításához az alábbi módszerek, illetve módszerkombinációk tekintettek alkalmasnak:
7. táblázat
Alkalmas megerősítő módszerek kémiai elemekhez
| Technika | Mért paraméter |
| Differenciál impulzus anódos stripping voltametria | Elektromos jel |
| Atomabszorpciós spektrometria | |
| Láng | Abszorpciós hullámhossz |
| Hidridképződés | Abszorpciós hullámhossz |
| Hideggőz | Abszorpciós hullámhossz |
| Elektrotermális atomizálás (grafitkályha) | Abszorpciós hullámhossz |
| Atomemissziós spektrometria | |
| Induktív csatolású plazma | Emissziós hullámhossz |
| Tömeg-spektrometria | |
| Induktív csatolású plazma | Tömeg-töltet arány |
2.4.1. Általános teljesítménykritériumok és egyéb követelmények megerősítő módszerekhez
Az analit ismert mennyiségeit tartalmazó referencia- vagy megerősített anyagot - az engedélyezett maximális határérték vagy a döntési határérték értékén vagy annak közelében (nem-megfelelt kontrollminta) -, valamint a megfelelt kontrollanyagokat és reagens-vakokat lehetőség szerint az elemzés tárgyát képező vizsgálati minták tételeivel egy időben kell a teljes eljáráson végigvinni. Az extraktumok a következő sorrendben kerülnek injektálásra az analitikai műszerbe: reagens-vak, megfelelt kontrollminta, megerősítendő minta/minták, megfelelt kontrollminta újra, végül pedig a nem-megfelelt kontrollminta. Az ettől való bármely eltérést indokolni kell.
Az analit-meghatározás előtti oldatnyerés érdekében a legtöbb analitikai módszernél rendszerint a szerves mátrix teljes feltárására van szükség. Ehhez mikrohullámos mineralizációs eljárások alkalmazhatók, amelyekkel minimálisra csökkenthető a kérdéses analitek elveszésének és/vagy szennyeződésének a kockázata. Szennyezéstől mentesített, jó minőségű teflonedényeket kell használni. Más nedves vagy száraz feltárási módszerek alkalmazása esetén dokumentált bizonyítékoknak kell rendelkezésre állniuk az elveszés vagy szennyeződés lehetőségének kizárása érdekében. A feltárás alternatívájaként bizonyos körülmények között elválasztási eljárások (pl. extrakció) is választhatók az analiteknek a mátrixkomponensektől való elválasztásához és/vagy az analitek koncentrálásához, az analitikai berendezésbe való bevezethetőségük érdekében.
A kalibrálást illetően - legyen az külső vagy a standard hozzáadás módszerén alapuló - figyelni kell arra, hogy az elemzéshez meghatározott munkatartomány túllépése ne következzék be. Külső kalibrálás esetén a mintaoldat összetételének a lehető legjobban megfelelő oldatban kell kalibráló standardokat készíteni. Háttérkorrekciót is kell alkalmazni, amennyiben azt konkrét analitikai körülmények megkövetelik.
2.4.2. További teljesítménykritériumok és egyéb követelmények a kvantitatív analitikai módszerekhez
2.4.2.1. A kvantitatív módszerek egzaktsága
Valamely hiteles anyagminta elemekre vonatkozó ismételt elemzései esetén a kísérleti úton meghatározott közepes tartalomnak a hitelesített értéktől való eltérése nem eshet kívül a ± 10 %-os határon. Ilyen CRM-ek hiányában az is elfogadható, hogy a mérések egzaktságának a kiértékelése ismert elemmennyiség(ek) ismeretlen mintákhoz való hozzáadásainak a visszanyerésén keresztül történjen. Nem szabad figyelmen kívül hagyni azt a tényt, hogy az analittól eltérően a hozzáadott elem kémiailag nem kötődik a valódi mátrixban, és így az ezzel a módszerrel kapott eredmények érvényessége kisebb, mint a CRM-ek használatával kapottaké. A visszanyerési adatok csak akkor elfogadhatók, ha a célzott érték ± 10 %-án belül vannak.
2.4.2.2. A kvantitatív módszerek precizitása
Egy minta - laboratóriumon belüli reprodukálhatósági körülmények közötti - ismételt elemzése esetén az átlagérték laboratóriumon belüli variációs koefficiense (CV) nem haladhatja meg az alábbi értékeket:
8. táblázat
A kvantitatív módszerek CV-i különböző elem-tömeghányad tartományokban
| Tömeghányad | CV ( %) |
| ≥ 10 μg/kg és 100 μg/kg között | 20 |
| > 100 μg/kg és 1 000 μg/kg között | 15 |
| ≥ 1 000 μg/kg | 10 |
2.4.3. Konkrét követelmények a differenciál impulzus anódos stripping voltametriához (DPASV)
A DPASV meghatározások előtt rendkívül fontos a minták szervesanyag-tartalmának tökéletes roncsolása. A voltamogramok nem mutathatnak szerves anyag jelenlétére utaló széles jeleket. A mátrix szervetlen komponensei befolyásolhatják a DPASV csúcsmagasságait. A számszerűsítést ezért a standard hozzáadások módszerével kell végezni. A módszerhez tartozékként mellékelni kell egy mintaoldat tipikus voltamogram-mintáit.
2.4.4. Konkrét követelmények az atomabszorpciós spektrometriához (AAS)
Mivel ez alapvetően egyelemes technika, ezért szükségessé teszi a kísérleti beállításoknak a számszerűsítendő elemtől függő optimalizálását. Lehetőség szerint az eredményeket kvalitatív és kvantitatív ellenőrzésnek kell alávetni alternatív abszorpciós vonalak (ideális esetben két különböző vonal) segítségével. A kalibráló standardokat olyan oldatmátrixban kell elkészíteni, amely a lehető legjobban megfelel a minta mérőoldaténak (pl. savkoncentráció vagy modifikáló-összetétel). A vakértékek minimalizálása érdekében valamennyi reagensnek a lehető legnagyobb tisztaságúnak kell lennie. A minta elpárologtatásához és/vagy atomizálásához választott módszertől függően különböző AAS típusok léteznek.
2.4.4.1. Konkrét követelmények a lángos AAS-hez
A műszerbeállításokat minden egyes elemre optimalizálni kell. Különösen a gázösszetételt és az áramlási sebességeket kell ellenőrizni. A háttérabszorpció által okozott interferenciák elkerülése érdekében folytonos forráskorrektort kell használni. Ismeretlen mátrixok esetén ellenőrzéssel kell megállapítani azt, hogy szükség van-e háttérkorrekcióra.
2.4.4.2. Konkrét követelmények a grafitkályhás AAS-hez
A grafitkályhával történő ultranyom szintű munkavégzés során a laboratóriumban bekövetkező szennyeződés gyakran befolyásolja a pontosságot. Ezért a minta és a standard kezeléséhez nagytisztaságú reagenseket, ionmentesített vizet és inert műanyagárukat kell használni. A műszerbeállításokat minden egyes elemre optimalizálni kell. Különösen az előkezelési és atomizálási körülményeket (hőmérséklet, időtartam), valamint a mátrixmódosítást kell ellenőrizni.
Az izotermikus atomizálási körülmények (pl. keresztfűtéses grafitcső integrált Lvov platformmal) (8) közötti munka csökkenti a mátrix hatását az analit atomizálását illetően. Mátrix modifikálással és Zeeman-féle háttérkorrekcióval (9) kombinálva, a vizes standard oldatok mérésén alapuló kalibrációs görbével történő számszerűsítés engedélyezett.
2.4.5. Konkrét követelmények a hidrides atomabszorpciós spektrometriához
Az arzén, bizmut, germánium, ólom, antimon, szelén, ón és tellúr tartalmú szerves vegyületek nagyfokú stabilitást mutathatnak, és az összes elemtartalomra vonatkozó korrekt eredmények nyerése érdekében oxidatív lebontást igényelhetnek. Ezért mikrohullámos feltárás vagy erős oxidatív körülmények közötti nagynyomású hamvasztás ajánlott. Az elemek teljes és reprodukálható módon történő hidriddé alakítására kell legjobban odafigyelni.
A sósavoldatban NaBH4 jelenlétében történő arzén-hidrid képződés az arzén oxidációs állapotától függ (As III: gyors képződés, As V: hosszabb képződési időszak). Az As V áramlásos rendszerű injektálási technikával történő meghatározása esetén az érzékenység-csökkenés - amit a rendszer rövid reakcióideje eredményez - elkerülése érdekében az As V az oxidatív lebontást követően As III-ra redukálandó. Erre a célra káliumjodid/aszkorbinsav vagy cisztein alkalmas. A vakok, kalibráló oldatok és mintaoldatok ugyanígy kezelendők. Batch rendszer alkalmazása esetén a pontosság befolyásolása nélkül van lehetőség mindkét arzénfajta meghatározására. Az arzén (V) hidrid elhúzódó képződése miatt a kalibrálást csúcsterület-integrálással kell elvégezni. A műszerbeállításokat optimalizálni kell. A hidridet az atomizálóhoz szállító gázáram különösen fontos és ellenőrizendő.
2.4.6. Konkrét követelmények a hideggőzös atomabszorpciós spektrometriához
Hideggőz kizárólag a higany esetében használatos. Az elemi higany párolgása és adszorpciós vesztesége miatt a teljes elemzés során különös gonddal kell eljárni. A reagensek okozta vagy a környezetből származó szennyeződést gondosan el kell kerülni.
A higanytartalmú szerves vegyületek az összes higanytartalomra vonatkozó korrekt eredmények nyerése érdekében oxidatív lebontást igényelnek. A lebontáshoz mikrohullámos feltáráson vagy nagynyomású hamvasztáson alapuló zárt rendszereket kell alkalmazni. A higannyal kapcsolatba került berendezések tisztításánál különös gonddal kell eljárni.
Az áramlásos rendszerű injektálási technikával való munkavégzés előnyös. Alacsonyabb döntési határértékeknél ajánlott az elemi higany arany/platina adszorbensen történő megkötése, ezt követően pedig a termikus deszorpció. Az adszorbens vagy a cella nedvességgel való érintkezése zavarja a mérést és kerülendő.
2.4.7. Konkrét követelmények az induktív csatolású plazmás atomemissziós spektrometriához (ICP-AES)
Az induktív csatolású plazmás atomemissziós spektrometria (10) sokelemes módszer, amely különböző elemek egyidejű mérését teszi lehetővé. Az ICP-AES alkalmazásához a mintákat előbb fel kell tárni a szerves mátrixok lebontása érdekében. Ehhez mikrohullámos feltáráson vagy nagynyomású hamvasztáson alapuló zárt rendszereket kell alkalmazni. Az érdemi ICP-AES elemzésnél a műszer kalibrálása és az elem, illetve hullámhossz kiválasztása lényeges szerepet játszik. A műszer kalibrálásához - lineáris kalibráló görbék esetén - rendszerint csak négy koncentráció kalibráló oldatait kell megmérni, mert az ICP-AES kalibráló görbék általában négy-hat koncentráció-nagyságrenden keresztül lineárisak. Az ICP-AES rendszer kalibrálását normál esetben egy olyan sokelemes standarddal kell végezni, amelyet a mérőoldatéval megegyező savkoncentrációt tartalmazó oldatban kell elkészíteni. A lineáris görbénél ellenőrizni kell az elemkoncentrációkat.
Az analitek emissziójának mérésére szolgáló hullámhosszok kiválasztása megfelel a meghatározandó elemek koncentrációinak. Ha az analit-koncentráció egy emissziós vonal munkatartományán kívül esik, akkor egy másik emissziós vonal alkalmazandó. Először a legérzékenyebb (interferencia nélküli) emissziós vonal, majd egy kevésbé érzékeny vonal választandó ki. A kimutatási határérték értékén vagy annak közelében történő munkavégzés esetén rendszerint az adott analitre legérzékenyebb vonal jelenti a legjobb választást. Az ICP-AES tekintetében a spektrális és a háttér-interferenciák okozzák a legnagyobb nehézségeket. A lehetséges interferenciák közé tartozik pl. az egyszerű háttéreltolódás, ferde háttéreltolódás, direkt spektrális átfedés és összetett háttéreltolódás. Ezen interferenciák mindegyikének megvan a maga oka és orvoslása. A mátrixoktól függően interferencia-korrekciót és optimalizálást kell végezni a működési paraméterek vonatkozásában. Bizonyos interferenciákat a mátrix hígításával vagy adaptálásával el lehet kerülni. Az elemzett vizsgálati minták minden egyes tétele esetében az ismert analit-mennyiség(ek)et tartalmazó referencia- vagy megerősített anyagot, valamint a vakanyagot ugyanúgy kell kezelni, mint a vizsgálati mintákat. Valamilyen elcsúszás vizsgálata esetén szükség van a standard ellenőrzésére, pl. minden 10 minta után. Az összes reagensnek és a plazmagáznak a lehető legnagyobb tisztaságúnak kell lennie.
2.4.8. Konkrét követelmények az induktív csatolású tömeg-spektrometriához (ICP-MS) (11)
Az átlagos atomtömegű nyomelemek (például króm, réz és nikkel) meghatározásakor más izobár és sokatomos ionok erős interferenciát produkálhatnak. Ez csak úgy kerülhető meg, ha legalább 7 000 -8 000 -szeres felbontóképesség áll rendelkezésre. Az MS technikákkal összefüggő nehézségek közé kell sorolni a műszerelcsúszást, a mátrixhatásokat és a molekulaion-interferenciát (m/z < 80). A műszerelcsúszás és a mátrixhatások korrigálásához a meghatározandó elemekével megegyező tömegtartományt lefedő többszörös belső standardizálásra van szükség.
Az ICP-MS mérések előtt a minták szervesanyag-tartalmának teljes lebontása szükséges. Csakúgy, mint az AAS esetében, a zárt rendszerben történő lebontást követően, az illékony elemeket (pl. jód) stabil oxidációs állapotba kell helyezni. A legsúlyosabb interferenciát az argon (plazmagáz), hidrogén, szén, nitrogén és oxigén (bomlási savak, plazmagáz szennyeződések és légköri gázzárványok) molekulaion kombinációi és a mintamátrix okozzák. Az interferenciák elkerüléséhez teljes lebontásra, háttérmérésekre, valamint az alkalmanként alacsonyabb előfordulással (gyengébb kimutatási határérték) társuló analitikai tömegek és a bomlási savak (pl. salétromsav) megfelelő kiválasztására van szükség.
A meghatározandó elemeknél az interferenciákat a konkrét analitikai tömegek megfelelő kiválasztásával kell elkerülni, ideértve az izotóparányok ellenőrzését is. A Fano-tényezőkkel kapcsolatos műszerválaszt belső standardok használatával kell ellenőrizni minden egyes mérésnél.
3. VALIDÁLÁS
A validálásnak azt kell bizonyítania, hogy az analitikai módszer megfelel az adott teljesítményjellemzőkre vonatkozó kritériumoknak.
Az eltérő kontrollcélok eltérő módszercsoportokat igényelnek. Az alábbi táblázat azt mutatja, hogy az egyes módszertípusoknál milyen teljesítményjellemzőket kell igazolni.
9. táblázat
Az analitikai módszerek osztályozása a meghatározandó teljesítményjellemzők szerint
| Kimutatási határérték CCβ | Döntési határérték CCα | Egzaktság/visszanyerés | Precizitás | Szelektivitás/specificitás | Alkalmazha-tóság/zavartűrés/stabilitás | ||
| Kvalitatív módszerek | S | + | – | – | – | + | + |
| C | + | + | – | – | + | + | |
| Kvantitatív módszerek | S | + | – | – | + | + | + |
| C | + | + | + | + | + | + | |
| S = szűrési módszerek; C = megerősítő vizsgálati módszerek; + = meghatározás kötelező. | |||||||
3.1. VALIDÁLÁSI ELJÁRÁSOK
Ez a fejezet az analitikai módszerek validálási eljárásaira hoz példákat és/vagy referenciákat. Egyéb megközelítések is használhatók annak bizonyítására, hogy az analitikai módszer megfelel a teljesítményjellemzőkre vonatkozó kritériumoknak, feltéve, hogy az ilyen megközelítések ugyanolyan szintű és minőségű információkat biztosítanak.
Validálás végzése történhet a Codex Alimentarius, az ISO vagy az IUPAC (12) által előírt laborközi vizsgálatok valamelyikének a lefolytatásával vagy olyan alternatív módszerek szerint is, mint az egylaboros vizsgálatok vagy a házon belüli validálás (13) (14). Ez a rész a valamilyen moduláris megközelítést alkalmazó egylaboros vizsgálatokat (házon belüli validálást) tárgyalja. Ez a megközelítés a következőkből áll:
1. az alkalmazott validálási modelltől független általános teljesítményjellemzők halmaza; és
2. konkrétabb, a 10. táblázatban leírt modellfüggő eljárások.
10. táblázat
Modellfüggetlen és modellfüggő teljesítményparaméterek
| Validálás | ||
| Modellfüggetlen teljesítményparaméterek | Modellfüggő teljesítményparaméterek | |
| Általános teljesítményjellemzők (3.1.1.) | Hagyományos validálási megközelítés (3.1.2.) | Házon belüli validálási megközelítés (3.1.3.) |
| Specificitás | Visszanyerés | Visszanyerés |
| Egzaktság | Ismételhetőség | Ismételhetőség |
| Zavartűrés: kisebb változások | Laboratóriumon belüli reprodukálhatóság | Laboratóriumon belüli reprodukálhatóság |
| Stabilitás | Reprodukálhatóság | Reprodukálhatóság |
| Döntési határérték (CCα) | Döntési határérték (CCα) | |
| Kimutatási képesség (CCβ) | Kimutatási képesség (CCβ) | |
| Kalibráló görbék | Kalibrációs görbe | |
| Zavartűrés: nagyobb változások | Zavartűrés | |
3.1.1. Modellfüggetlen teljesítményjellemzők
A kiválasztott validálási megközelítéstől függetlenül az alábbi teljesítményjellemzőket kell meghatározni. A munkamennyiség minimalizálása érdekében gondosan megtervezett és statisztikailag megalapozott megközelítés alkalmazható a különböző paraméterek meghatározása céljából elvégzett kísérletek kombinálásához.
3.1.1.1. Specificitás
Az analitikai módszereknél fontos szempont az analit és a vele szoros rokonságot mutató anyagok (izomerek, metabolitok, bomlástermékek, endogén anyagok, mátrixkomponensek stb.) közötti különbségtétel képessége. Az interferenciák ellenőrzéséhez két megközelítésre van szükség.
Ezért egymással potenciálisan interferáló anyagokat kell kiválasztani és a vonatkozó vakmintákat elemezni kell az esetleges interferenciák kimutatása és az interferenciák hatásának becslése céljából:
- válasszon ki egy sor kémiailag rokon vegyületet (metabolitok, származékok stb.) vagy a kérdéses vegyülettel valószínűleg együtt előforduló olyan egyéb anyagokat, amelyek jelen lehetnek a mintákban,
- elemezzen megfelelő számú reprezentatív vakmintát (n = 20) és keressen interferenciákat (jelek, csúcsok, ionnyomok) a kérdéses tartományban, ahol a célzott analit eluálása várható,
- emellett a reprezentatív vakmintákat a vonatkozó koncentrációnál olyan anyagokkal kell erősíteni, amelyek várhatóan interferálnak az analit azonosításával és/vagy mennyiségi meghatározásával,
- az elemzést követően vizsgálja meg, hogy:
- a jelenlét vezethet-e hamis azonításhoz,
- a célzott analit azonosítását akadályozza-e egy vagy több interferencia jelenléte, vagy
- észlelhető mértékű-e a mennyiségi meghatározás befolyásolása.
3.1.1.2. Egzaktság
Ez a pont az egzaktság (a pontosság egyik eleme) meghatározását írja le. Az egzaktság csak hiteles anyagminta (CRM) segítségével állapítható meg. CRM használandó minden olyan esetben, amikor rendelkezésre áll. A részletes eljárást az ISO 5725-4 (5) tartalmazza. Az alábbiakban egy példát mutatunk be:
- végezze el a CRM hat ismétlésének elemzését a módszerre vonatkozó vizsgálati instrukciók szerint,
- határozza meg az egyes ismétlések mintáiban található analit koncentrációját,
- számítsa ki ezekre a koncentrációkra az átlagot, a szórást és a variációs koefficienst (%),
- számítsa ki az egzaktságot úgy, hogy a kimutatott átlagkoncentrációt elosztja a (koncentrációként mért) hitelesített értékkel és megszorozza 100-zal, hogy az eredmény százalékos formájú legyen.
Egzaktság (%) = visszanyeréssel korrigált kimutatott átlagkoncentráció × 100/hitelesített érték.
Ha nem áll rendelkezésre CRM, a visszanyerést az alábbi 4.1.2.1. pontban leírt módon lehet meghatározni.
3.1.1.3. Alkalmazhatóság/zavartűrés (kisebb változások)
Ezek a vizsgálatok a laboratórium által szándékosan végrehajtott kisebb ésszerű változtatásokra és a változtatások következményeinek a megfigyelésére irányulnak.
A vizsgálatokat megelőzően ki kell választani a minta előkezelésének, tisztításának és elemzésének azon faktorait, amelyek befolyásolhatják a mérési eredményeket. Ilyen faktor lehet az elemzést végző személy, a reagensek, oldószerek, standardok és minta-extraktumok forrása és életkora, a melegítés üteme, a hőmérséklet, a pH-érték, valamint számos egyéb faktor, ami a laboratóriumban előfordulhat. Ezeket a faktorokat olyan nagyságrendben kell módosítani, ami a laboratóriumok között rendszerint észlelt eltéréseknek megfelel.
- Azonosítsa azokat a lehetséges faktorokat, amelyek befolyásolhatják az eredményeket.
- Hajtsa végre minden egyes faktor kismértékű módosítását.
- Youden megközelítését (15) (16) alkalmazva végezzen zavartűrés-próbát. (Ezen a ponton más jóváhagyott módszerek is alkalmazhatók. A Youden-megközelítéssel azonban az idő- és munkaszükséglet minimális szinten tartható). A Youden-megközelítés törtszerű faktoriális kialakítású. Az egyes faktorok közötti kölcsönhatások észlelését nem teszi lehetővé.
- Ha kiderül, hogy valamelyik faktor szignifikánsan befolyásolja a mérési eredményeket, akkor további kísérletek alapján döntsön ennek a faktornak az elfogadhatósági határértékeiről.
- Az eredményeket szignifikánsan befolyásoló faktorokat egyértelműen azonosítani kell a módszer protokolljában.
Az alapelképzelés nem az elváltozások egyenkénti tanulmányozása, hanem több változtatás egyszerre történő bevezetése. Tegyük fel például, hogy A, B, C, D, E, F és G annak a hét különböző faktornak a nominális értékét jelenti, amelyek képesek az eredmények befolyásolására, ha nominális értékük enyhén megváltozik. Az alternatív értékeket jelöljük a fenti betűkhöz tartozó kisbetűkkel (a, b, c, d, e, f és g). Ebből összesen 27, vagyis 128 különböző kombináció adódik.
Ezek közül lehetséges egy nyolc kombinációból álló olyan részhalmazt kiemelni, amelyben a nagy- és a kisbetűk egyensúlyban vannak (11. táblázat). Nyolc meghatározást kell elvégezni, ami a kiválasztott faktorok (A-G) valamilyen kombinációjával történik. A meghatározások eredményeit a 11. táblázat mutatja (S-Z).
11. táblázat
Kísérleti minta zavartűrési vizsgálatokhoz (kisebb változások)
| Faktorérték, F | A meghatározások számának kombinációja | |||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |
| A/a | A | A | A | A | a | a | a | a |
| B/b | B | B | b | b | B | B | b | b |
| C/c | C | c | C | c | C | c | C | c |
| D/d | D | D | d | d | d | d | D | D |
| E/e | E | e | E | e | e | E | e | E |
| F/f | F | f | f | F | F | f | f | F |
| G/g | G | g | g | G | g | G | G | g |
| Megfigyelt eredmény, R | S | T | U | V | W | X | Y | Z |
| A számításokat lásd a zavartűrés-vizsgálat példáinál a 3.3. pontban. | ||||||||
3.1.1.4. Stabilitás
A megfigyelések szerint az elemzési eredmények szignifikáns deviációját okozhatja az, ha a tárolás vagy elemzés során nem megfelelő a mintában levő analit vagy mátrixkomponensek stabilitása. Emellett ellenőrizni kell az oldatban levő kalibráló standard stabilitását is. Az analit különböző tárolási körülmények közötti stabilitása rendszerint jól jellemzett. A tárolási körülmény figyelemmel kísérése részét képezi a szokásos laboratóriumi akkreditációs rendszernek. Az alábbi példák arra vonatkoznak, hogy miként határozható meg a stabilitás akkor, ha a tárolási körülmény nem ismert.
Az analit stabilitása oldatban:
- Készítsen az analit(ek)ből friss törzsoldatokat és a vizsgálati instrukciók szerint végezzen hígítást, hogy minden kiválasztott koncentrációból elegendő aliquot (pl. 40) álljon rendelkezésre (kb. a minimálisan megkövetelt teljesítményszint azon anyagoknál, amelyekre vonatkozóan még nincs kialakítva engedélyezett határérték, illetve kb. az engedélyezett határérték egyéb anyagoknál). Készítse el az analit mindkét oldatát, amelyek az erősítéshez és a végső elemzési oldatban kerülnek felhasználásra, illetve minden egyéb szükséges oldatot (pl. derivatizált standardok).
- A vizsgálati instrukcióknak megfelelően mérje meg az analit-tartalmat a frissen készített oldatban.
- Öntsön megfelelő mennyiségeket alkalmas tárolóedényekbe, címkézze fel és tárolja azokat a séma szerint:
12. táblázat
Séma az oldatban levő analit stabilitásának meghatározásához
| – 20 °C | + 4 °C | + 20 °C | |
| Sötét | 10 aliquot | 10 aliquot | 10 aliquot |
| Világos | 10 aliquot |
- A tárolási idő egy, két, három és négy hét, vagy szükség esetén még hosszabb is lehet, pl. amíg az azonosítás és/vagy mennyiségi meghatározás során nem mutatkoznak az első bomlási jelek. A maximális tárolási időt és az optimális tárolási körülményeket fel kell jegyezni.
- Az egyes aliquotokban levő analit(ek) koncentrációjának kiszámításához az elemzés időpontjában frissen készített analitoldat 100 %-nak tekintendő.
Ci = adott időpontbeli koncentráció
Cfriss = a friss oldat koncentrációja
Az analit stabilitása mátrixban
- Lehetőség szerint reális mintákat kell használni. Ha nem áll rendelkezésre reális anyag, akkor analittel erősített mátrix használandó.
- Ha rendelkezésre áll reális anyag, akkor az anyagban levő koncentrációt addig kell megállapítani, amíg az anyag friss. Egy, két, négy és 20 hét elteltével az anyagból további aliquotok vehetők, és meg kell határozni a koncentrációkat. A szövetet legalább mínusz 20 °C-os vagy szükség esetén ennél is alacsonyabb hőmérsékleten kell tárolni.
- Ha nem áll rendelkezésre reális anyag, akkor vegyen egy kis vakanyagot, majd homogenizálja azt. Ossza fel az anyagot öt aliquotra. Erősítse meg mindegyik aliquotot az analittel, amit lehetőség szerint kis mennyiségű vizes oldatban kell elkészíteni. Egy aliquot esetében azonnal végezze el az elemzést. A fennmaradó aliquotokat legalább mínusz 20 °C-os vagy szükség esetén ennél is alacsonyabb hőmérsékleten tárolja, majd azokat egyenként elemezze egy, két, négy és 20 hét elteltével.
3.1.1.5. Kalibráló görbék
Amikor számszerűsítés céljából kalibráló görbék kerülnek alkalmazásra:
- legalább öt szintet (a nullát is beleértve) kell használni a görbe felépítéséhez,
- le kell írni a görbe munkatartományát,
- le kell írni a görbe matematikai képletét és az adatok görbéhez illeszkedési jóságát,
- le kell írni a görbeparaméterek elfogadhatósági tartományait.
Amikor egy standard oldat alapján sorozatkalibrálásra van szükség, akkor jelezni kell a kalibráló görbe paramétereire vonatkozó elfogadható tartományokat, amelyek sorozatról sorozatra változhatnak.
3.1.2. Hagyományos validálási eljárások
A paraméterek hagyományos módszerekkel történő kiszámítása jó néhány egyedi kísérlet elvégzését igényli. Minden nagyobb változást illetően meg kell határozni minden egyes teljesítményjellemzőt (lásd a fenti Alkalmazhatóság/zavartűrés cím alatt). Sokanalites módszerek esetén több analit is elemezhető egy időben, feltéve, hogy előzetesen sikerül kizárni az esetleges interferenciákat. Több teljesítményjellemző határozható meg hasonló módon. A munkamennyiség minimalizálása érdekében tehát ajánlatos a kísérleteket minél inkább összevonni (pl. ismételhetőség és laboratóriumon belüli reprodukálhatóság a specificitással, a vakmintáknak a döntési határérték meghatározására szolgáló elemzésével és a specificitás vizsgálatával).
3.1.2.1. Visszanyerés
Ha nem áll rendelkezésre CRM, akkor a visszanyerést megerősített vakmátrixos kísérletekkel kell meghatározni, például az alábbi séma alkalmazása mellett:
- válassza ki egy vakanyag 18 aliquotját és hat-hat aliquot esetében végezzen a minimálisan megkövetelt teljesítményszint 1-szeresének, 1,5-szeresének és 2-szeresének, vagy az engedélyezett határérték 0,5-szeresének, 1-szeresének és 1,5-szeresének megfelelő erősítést,
- végezze el a minták elemzését és számítsa ki az egyes minták koncentrációját,
- az alábbi egyenlet segítségével számítsa ki az egyes minták visszanyerését,
- a szintenkénti hat eredményből számítsa ki az átlagos visszanyerést és CV-t,
- visszanyerés % = 100 × mért tartalom/erősített szint.
A visszanyerés meghatározásának ez a hagyományos módszere a 3.5. pontban leírt standard hozzáadás módszerének az egyik változata, amikor:
- a mintát elemzendő minta helyett vakmintának tekintik,
- úgy tekintik, hogy a kihozatal ( 1 ) és a visszanyerés ( 2 ) a két vizsgálati részre vonatkozóan hasonló,
- a vizsgálati mintáknak ugyanaz a tömegük, a vizsgálatirész-extraktumoknak pedig ugyanaz a térfogatuk,
- a kalibráló standardnak a második (preparált) vizsgálati részhez hozzáadott mennyisége ismert: xADD. (xADD = ρA.VA),
- x1 a vak mért értékét, x2 pedig a második (preparált) vizsgálati rész mért értékét jelenti,
- akkor, a visszanyerési % = 100 (x2 - x1)/xADD.
Amikor a fenti feltételek bármelyike nem (vagy vélhetően nem) teljesíthető, akkor a visszanyerés meghatározásának a teljes eljárását a 3.5. pontban leírt standard hozzáadás módszerével kell elvégezni.
3.1.2.2. Ismételhetőség
- Készítse el azonos mátrixok mintacsoportjait és ezeket úgy erősítse meg az analittel, hogy a kapott koncentrációk a minimálisan megkövetelt teljesítményszint 1-szeresének, 1,5-szeresének és 2-szeresének, vagy az engedélyezett határérték 0,5-szeresének, 1-szeresének és 1,5-szeresének feleljenek meg.
- Az elemzést minden szinten legalább hat ismétléssel kell végezni.
- Végezze el a minták elemzését.
- Számítsa ki az egyes mintákban kimutatott koncentrációt.
- Határozza meg a megerősített minták átlagkoncentrációját, szórását és variációs koefficiensét (%).
- Legalább két másik alkalommal ismételje meg a fenti lépéseket.
- Számítsa ki a megerősített mintákra vonatkozó teljes átlagkoncentrációkat és CV-ket.
3.1.2.3. Laboratóriumon belüli reprodukálhatóság
- Készítse el meghatározott vizsgálati anyag (azonos vagy különböző mátrixok) mintacsoportjait és ezeket úgy erősítse meg az analittel/analitekkel, hogy a kapott koncentrációk a minimálisan megkövetelt teljesítményszint 1-szeresének, 1,5-szeresének és 2-szeresének, vagy az engedélyezett határérték 0,5-szeresének, 1-szeresének és 1,5-szeresének feleljenek meg.
- Az elemzést minden szinten legalább hat ismétléssel kell végezni.
- Legalább két másik alkalommal ismételje meg a fenti lépéseket, lehetőség szerint más vizsgáló személyekkel és más környezeti körülmények között, pl. más reagensek, oldószerek stb., más szobahőmérsékletek, más műszerek stb. alkalmazásával.
- Végezze el a minták elemzését.
- Számítsa ki az egyes mintákban kimutatott koncentrációt.
- Határozza meg a megerősített minták átlagkoncentrációját, szórását és variációs koefficiensét (%).
3.1.2.4. Reprodukálhatóság
Amikor a reprodukálhatóságot ellenőrizni kell, a laboratóriumoknak az ISO 5725-2 (5) szerinti kollaboratív vizsgálatokban kell részt venniük.
3.1.2.5. Döntési határérték (CCα)
A döntési határértéket az azonosítás vagy az azonosítás plusz a számszerűsítés "Teljesítménykritériumok és egyéb követelmények analitikai módszerekhez" (2. rész) szerinti követelményei alapján kell meghatározni.
Azon anyagoknál, amelyekre vonatkozóan még nincs kialakítva engedélyezett határérték, a CCα meghatározása a következők szerint történhet:
- vagy az ISO 11843 (17) szerinti kalibráló görbe eljárással (ottani hivatkozása a nettó állapotváltozó kritikus értéke). Ebben az esetben vakanyagot kell használni, amely ekvidisztáns osztásokban van megerősítve a minimálisan megkövetelt teljesítményszint értékén és a fölött. Végezze el a minták elemzését. Az azonosítást követően ábrázolja grafikusan a jel görbéjét a hozzáadott koncentrációval szemben. Az y-metszékhez tartozó koncentráció plusz a metszék laboratóriumon belüli reprodukálhatósága szórásának 2,33-szorosa adja a döntési határértéket. Ez csak kvantitatív próbákra érvényes (α = 1 %),
- vagy mátrixonként legalább 20 vakanyag elemzésével annak érdekében, hogy kiszámítható legyen a jel/zaj viszony az analit várható időablakánál. Döntési határértékként a jel/zaj viszony háromszorosa használható. Ez kvantitatív és kvalitatív próbákra érvényes.
Megállapított engedélyezett határértékkel rendelkező anyagok esetében a CCα meghatározása a következők szerint történhet:
- vagy az ISO 11843 (17) szerinti kalibráló görbe eljárással (ottani hivatkozása a nettó állapotváltozó kritikus értéke). Ebben az esetben vakanyagot kell használni, amely ekvidisztáns osztásokban van megerősítve az engedélyezett határérték értéke körül. Végezze el a minták elemzését. Az azonosítást követően ábrázolja grafikusan a jel görbéjét a hozzáadott koncentrációval szemben. Az engedélyezett határértékhez tartozó koncentráció plusz a laboratóriumon belüli reprodukálhatóság szórásának 1,64-szorosa adja a döntési határértéket (α = 5 %),
- vagy az analittel/analitekkel az engedélyezett határérték értékén megerősített mátrixonként legalább 20 vakanyag elemzésével. Az engedélyezett határértékhez tartozó koncentráció plusz a hozzá tartozó szórás 1,64-szorosa adja a döntési határértéket (α = 5 %).
Lásd még az 5. cikket és a 3.2. pontot.
3.1.2.6. Kimutatási képesség (CCβ)
A kimutatási képességet a szűrés, az azonosítás vagy az azonosítás plusz a számszerűsítés előírás (lásd a 2. részt) szerinti követelményei alapján kell meghatározni.
Azon anyagoknál, amelyekre vonatkozóan még nincs kialakítva engedélyezett határérték, a CCβ meghatározása a következők szerint történhet:
- az ISO 11843 (17) szerinti kalibráló görbe eljárással (ottani hivatkozása a nettó állapotváltozó minimálisan kimutatható értéke). Ebben az esetben reprezentatív vakanyagot kell használni, amely ekvidisztáns osztásokban van megerősítve a minimálisan megkövetelt teljesítményszint értékén és az alatt. Végezze el a minták elemzését. Az azonosítást követően ábrázolja grafikusan a jel görbéjét a hozzáadott koncentrációval szemben. A döntési határértékhez tartozó koncentráció plusz a döntési határértéknél mért átlagtartalom laboratóriumon belüli reprodukálhatósága szórásának 1,64-szorosa adja a kimutatási képességet (β = 5 %),
- az analittel/analitekkel a döntési határérték értékén megerősített mátrixonként legalább 20 vakanyag elemzésével. Végezze el a minták elemzését és az analitek azonosítását. A döntési határérték értéke plusz a döntési határértéknél mért tartalom laboratóriumon belüli reprodukálhatósága szórásának 1,64-szorosa adja a kimutatási képességet (β = 5 %),
- amennyiben nem állnak rendelkezésre kvantitatív eredmények, a kimutatási képesség meghatározása történhet a döntési határérték értékén és afölött megerősített vakanyag vizsgálatával is. Ebben az esetben az a koncentrációszint jelenti a módszer kimutatási képességét, ahol csupán 5 % hamis megfelelt minta marad. Ehhez a meghatározáshoz ezért csak akkor biztosítható megbízható alap, ha legalább egy koncentrációszintre legalább 20 vizsgálat kerül elvégzésre.
Azon anyagoknál, amelyekre vonatkozóan már van kialakítva engedélyezett határérték, a CCβ meghatározása a következők szerint történhet:
- vagy az ISO 11843 (17) szerinti kalibráló görbe eljárással (ottani hivatkozása a nettó állapotváltozó minimálisan kimutatható értéke). Ebben az esetben reprezentatív vakanyagot kell használni, amely ekvidisztáns osztásokban van megerősítve az engedélyezett határérték értéke körül. Végezze el a minták elemzését és az analit(ek) azonosítását. Számítsa ki a döntési határértéknél mért átlagtartalom szórását. A döntési határérték értékéhez tartozó koncentráció plusz a laboratóriumon belüli reprodukálhatóság szórásának 1,64-szorosa adja a kimutatási képességet (β = 5 %),
- vagy az analittel/analitekkel a döntési határérték értékén megerősített mátrixonként legalább 20 vakanyag elemzésével. A döntési határérték értéke plusz a hozzá tartozó szórás 1,64-szorosa adja a kimutatási képességet (β = 5 %).
Lásd még a 3.2. részt.
3.1.2.7. Zavartűrés (nagyobb változások)
Az analitikai módszert különböző kísérleti körülmények között kell tesztelni, így például különböző fajok, különböző mátrixok vagy különböző mintavételi körülmények alkalmazásával. A végrehajtott változtatásoknak nagyoknak kell lenniük. E változtatások jelentőségét például a Youden-megközelítéssel (15) (16) lehet kiértékelni. Minden egyes teljesítményjellemzőt meg kell határozni mindazon nagy változásokra vonatkozóan, amelyek kimutatottan jelentős hatást gyakorolnak a próba teljesítményére.
3.1.3. Validálás alternatív modellek szerint
Alternatív validálási eljárások alkalmazása esetén az alapmodellt és stratégiát - a vonatkozó feltételekkel, feltételezésekkel és formulákkal együtt - rögzíteni kell a validálási protokollban, vagy legalább hivatkozni kell azok rendelkezésre állására. Az alábbiakban egy alternatív megközelítés példája szerepel. Amikor például a házon belüli validálási modell kerül alkalmazásra, akkor a teljesítményjellemzők meghatározása úgy történik, hogy ugyanaz a validálási eljárás lehetővé tegye a nagyobb változásokra vonatkozó validálás elvégzését is. Ehhez validálási kísérleti tervet kell összeállítani.
3.1.3.1. Kísérleti terv
A vizsgálat tárgyát képező különböző fajok és egyéb faktorok számától függően kísérleti tervet kell összeállítani. Így tehát a validálási eljárás legelső lépéseként fel kell mérni a jövőben a laboratóriumban elemzendő mintapopulációkat annak érdekében, hogy ki lehessen választani a legfontosabb fajokat és azokat a faktorokat, amelyek befolyásolhatják a mérési eredményeket. Ezt követően a koncentrációtartományt célhoz szabottan kell megválasztani a kérdéses szint szerint.
Példa:
- az analitikai módszer validálásával egy időben több analit vizsgálata is folyhat,
- a főfaktor két variációja került azonosításra (A és B). A főfaktorok képezik a faktorszintek kombinálásának az alapját. A főfaktorok között olyan faktorok szerepelhetnek, mint a faj vagy a mátrix. Ebben a példában a főfaktor variálására két szinten került sor, azaz két különböző faj (A faj és B faj) került vizsgálat alá. A főfaktorok variálása rendszerint kettőnél több szinten is lehetséges, ami csak az elvégzendő elemzések számát növeli,
- a kiválasztott faktorok két szinten variálandók (jelölésük + vagy -).
13. táblázat
Példák a validálási eljáráshoz fontosnak tartott faktorokra
| Az állat ivara | (1. faktor) |
| Fajta | (2. faktor) |
| Szállítási körülmények | (3. faktor) |
| Tárolási körülmények | (4. faktor) |
| A minta frissessége | (5. faktor) |
| Hizlalási körülmények | (6. faktor) |
| Különböző tapasztalatokkal rendelkező különböző vizsgáló személyek | (7. faktor) |
14. táblázat
Lehetséges kísérleti terv a fenti példához
| Faj | 1. faktor | 2. faktor | 3. faktor | 4. faktor | 5. faktor | 6. faktor | 7. faktor | Minta száma |
| A | + | + | + | + | – | + | – | 1 |
| A | + | + | – | – | + | – | – | 2 |
| A | + | – | + | – | – | – | + | 3 |
| A | + | – | – | + | + | + | + | 4 |
| A | – | + | + | – | + | + | + | 5 |
| A | – | + | – | + | – | – | + | 6 |
| A | – | – | + | + | + | – | – | 7 |
| A | – | – | – | – | – | + | – | 8 |
| B | + | + | + | + | + | – | + | 9 |
| B | + | + | – | – | – | + | + | 10 |
| B | + | – | + | – | + | + | – | 11 |
| B | + | – | – | + | – | – | – | 12 |
| B | – | + | + | – | – | – | – | 13 |
| B | – | + | – | + | + | + | – | 14 |
| B | – | – | + | + | – | + | + | 15 |
| B | – | – | – | – | + | – | + | 16 |
Mivel minden mintát (minden faktorszint-kombinációt) a kérdéses szint körüli négy különböző koncentrációval kell preparálni, továbbá minden szintre vonatkozóan egy vakmintát kell elemezni, így a teljes validálási kísérlet során 5 × 16 = 80 elemzést kell elvégezni.
Ebből a 80 mérési eredményből kiszámítható (13) (14):
Visszanyerés
- ismételhetőség/koncentrációszint (sir),
- laboratóriumon belüli reprodukálhatóság/koncentrációszint (sir),
- döntési határérték (CCα),
- kimutatási képesség (CCβ),
- teljesítmény-jelleggörbe (β-hiba arány a koncentrációhoz képest) (lásd 3.1.3.2.),
- zavartűrés nagyobb változásoknál; a zavartűrés kisebb változásoknál a 3.1.1.3. pont szerint határozható meg,
- 16 darab, mintához kapcsolt kalibráló görbe,
- egy darab átfogó kalibráló görbe,
- az átfogó kalibráló görbe predikciós intervalluma,
- mátrix indukálta deviációk (smat),
- üzemi ciklus indukálta deviációk (srun),
- az egyedi faktorok hatása a mérési eredményekre.
Ezek a teljesítményjellemzők lehetővé teszik a módszer teljesítményszintjének átfogó kiértékelését, mivel nem csupán az egyedi faktorok hatása képezi vizsgálat tárgyát, hanem e faktorok releváns kombinációi is. Ennek a kísérleti tervnek a segítségével az is eldönthető, hogy a kiválasztott faktorok közül egyet vagy többet ki kell-e zárni az átfogó kalibráló görbéből amiatt, mert szignifikánsan eltér a többi faktor szórásaitól.
3.1.3.2. Teljesítmény-jelleggörbe
A teljesítmény-jelleggörbe arra nézve szolgál információkkal, hogy a kiválasztott koncentrációtartományban milyen a módszer kimutatási képessége. A vizsgált módszer alkalmazása során a β-hiba kockázatára utal. A teljesítmény-jelleggörbe segítségével egy adott β-hibánál (pl. 5 %) kiszámíthatók a módszerosztályokra (szűrés, megerősítés) vagy a módszertípusokra (kvalitatív vagy kvantitatív) vonatkozó kimutatási képességek.
[Kép #1] ábra 1 Teljesítmény-jelleggörbe
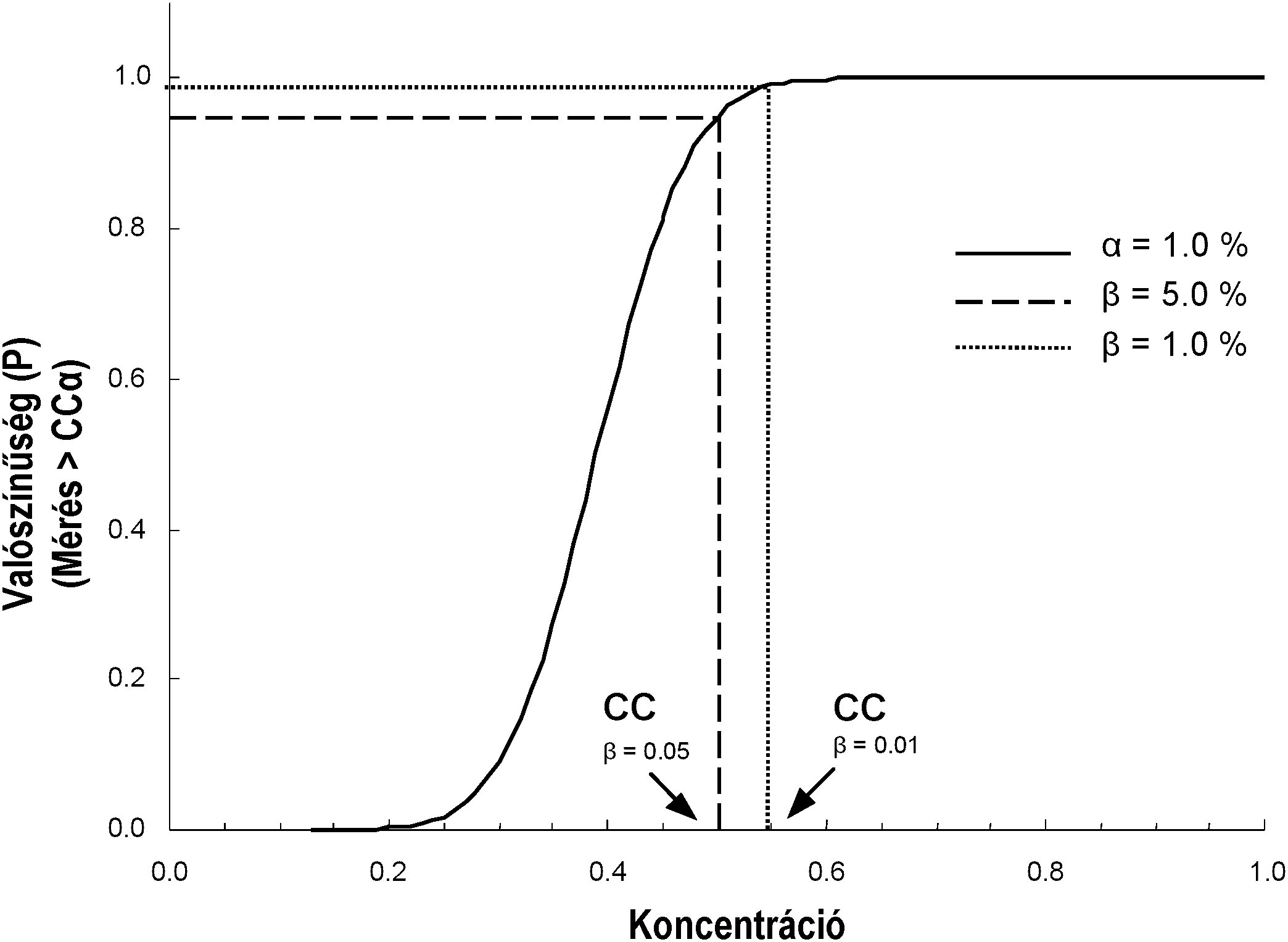
Kép #1
Az 1. ábra grafikus példán szemlélteti egy analitikai módszer kimutatási képességét (CCβ). Ez a konkrét módszer 5 %-os maradványkockázatot jelent arra nézve, hogy 0,5 μg/kg koncentrációnál hamis döntés születik. 0,55 μg/kg koncentrációnál a hamis megfelelt döntés meghozatalának kockázata 1 %-ra csökken.
3.1.3.3. Reprodukálhatóság
Egy módszer reprodukálhatóságának az egylaboros vizsgálatok (házon belüli validálás) koncepció útján történő meghatározásához az alkalmasság-vizsgálatokban való ismételt részvételre van szükség, az ISO 43-1 (3) és 43-2 (4) útmutatás szerint. A laboratóriumok maguk választhatják meg saját módszerüket, feltéve, hogy ezeket a módszereket rutinkörülmények között alkalmazzák. A laboratórium szórása a módszer reprodukálhatóságának a felmérésére használható.
3.2. KÜLÖNBÖZŐ ANALITIKAI HATÁRÉRTÉKEK GRAFIKUS MEGJELENÍTÉSE
[Kép #2] ábra 2 Anyagok, amelyekre vonatkozóan még nincs megállapítva engedélyezett határérték
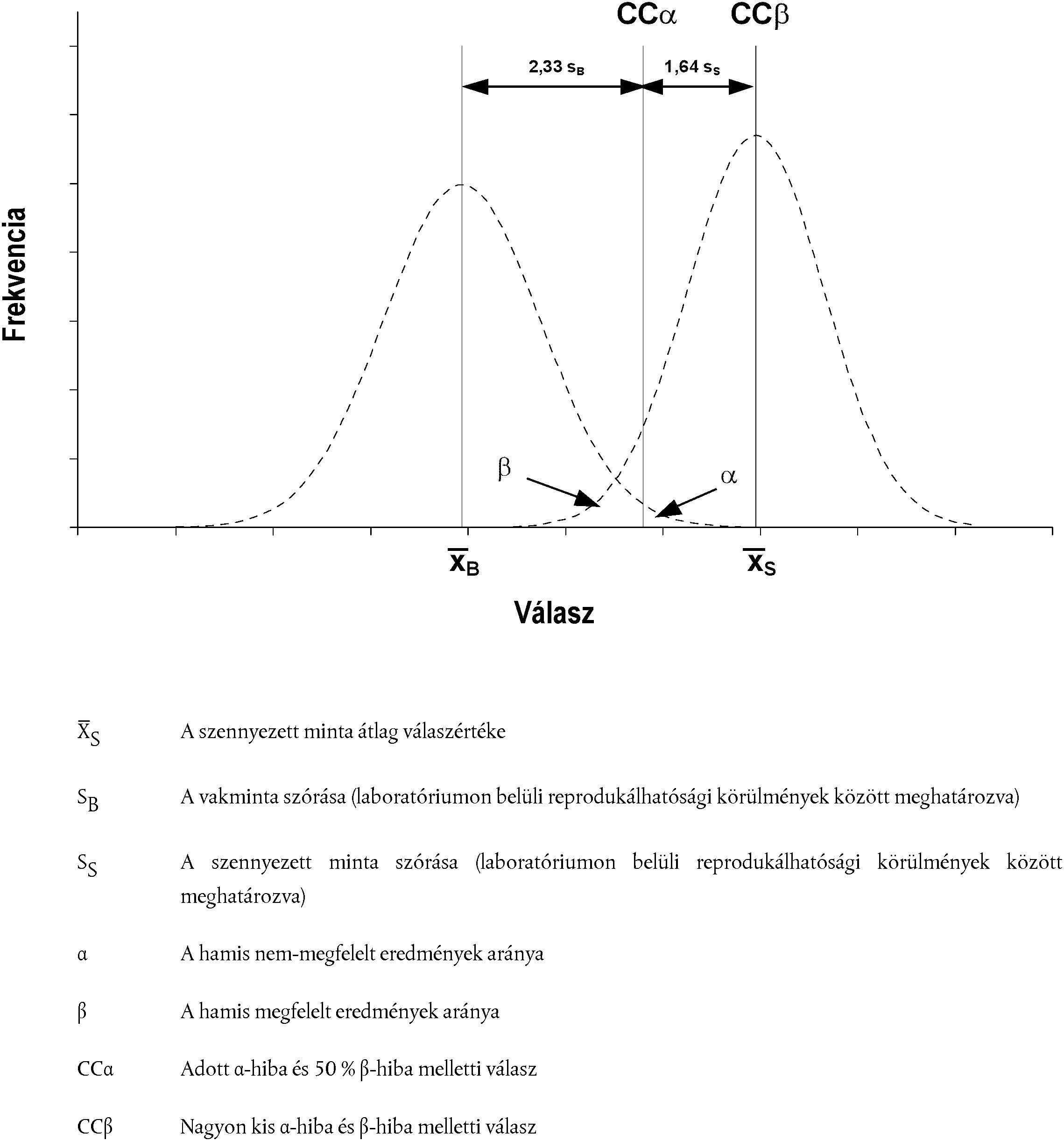
Kép #2
[Kép #3] ábra 3 Anyagok, amelyekre vonatkozóan már van megállapított engedélyezett határérték
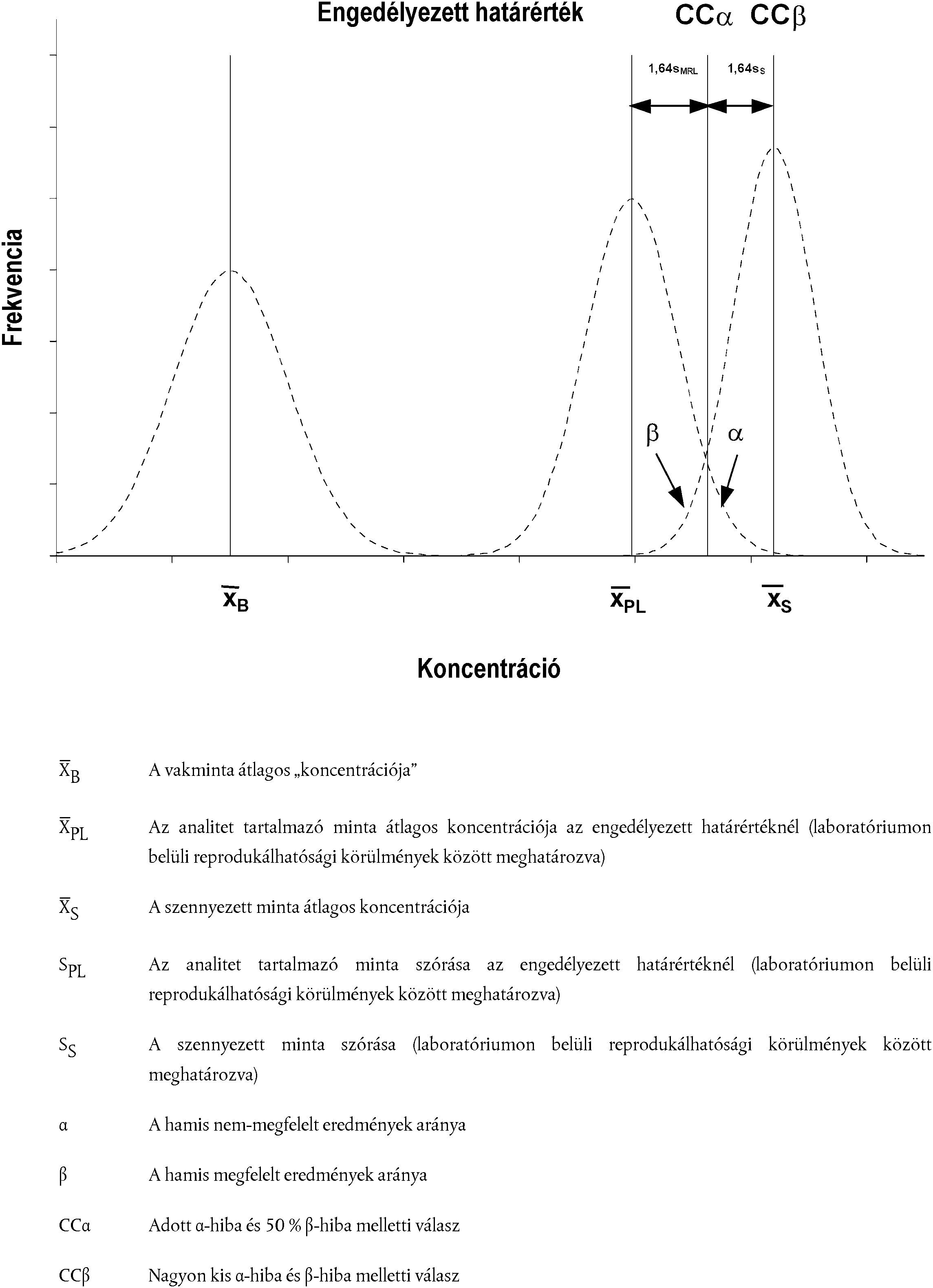
Kép #3
3.3. KISEBB VÁLTOZÁSOK ZAVARTŰRÉSÉNEK SZÁMÍTÁSI PÉLDÁJA A YOUDEN-MEGKÖZELÍTÉS SZERINT (16)
Az átlagok (A) összehasonlítása
| AA = Σ(Ai)/4 AB = Σ(Bi)/4 AC = Σ(Ci)/4 AD = Σ(Di)/4 AE = Σ(Ei)/4 AF = Σ(Ei)/4 AG = Σ(Gi)/4 Aa = Σ(ai)/4 Ab = Σ(bi)/4 Ac = Σ(ci)/4 Ad = Σ(di)/4 Ae = Σ(ei)/4 Af = Σ(fi)/4 Ag = Σ(gi)/4 | Hasonlítsa össze a nagybetűk átlagait (AA – AG) a hozzájuk tartozó kisbetűk átlagaival (Aa – Ag). Ha egy faktornak hatása van, akkor a különbség szignifikánsan nagyobb lesz, mint a többi faktor különbsége. Egy igazán zavartűrő módszert nem befolyásolhatnak az egyes laboratóriumok között csaknem bizonyosan előforduló változások. Ha nincs kiemelkedő különbség, akkor a véletlenszerű hibát legreálisabban a hét különbség adja meg. |
| Különbségek (Di) | Különbségnégyzetek (Di 2) |
| Da = A – a = Σ(Ai) – Σ(ai) Db = B – b = Σ(Bi) – Σ(bi) Dc = C – c = Σ(Ci) – Σ(ci) Dd = D – d = Σ(Di) – Σ(di) De = E – e = Σ(Ei) – Σ(ei) Df = F – f = Σ(Fi) – Σ(fi) Dg = G – g = Σ(Gi) – Σ(gi) | Da 2 = a érték Db 2 = b érték Dc 2 = c érték Dd 2 = d érték De 2 = e érték Df 2 = f érték Dg 2 = g érték |
Az átlagok (A) összehasonlítása
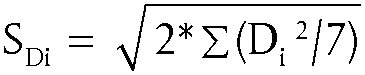
Ha az SDi szignifikánsan nagyobb, mint a laboratóriumon belüli reprodukálhatósági körülmények között végrehajtott módszer szórása (lásd fent), akkor elkerülhetetlenül adódik az a következtetés, hogy az összes faktor együttesen befolyással van az eredményre még akkor is, ha az egyes faktorok külön nem mutatnak szignifikáns befolyást, és hogy a módszer nem eléggé zavartűrő a választott módosításokkal szemben.
3.4. A HÁZON BELÜLI VALIDÁLÁS SZÁMÍTÁSI PÉLDÁJA
A "Validálás alternatív modellek szerint" c. részben (3.1.3.) leírt házon belüli validálási protokollra vonatkozó példák és számítások (13) (14).
3.5. A STANDARD HOZZÁADÁS MÓDSZER PÉLDÁI
Egy T analit-tartalmú vizsgálati mintát két (1. és 2.) vizsgálati részre osztunk, amelyek tömege m1 és m2. A 2. vizsgálati rész az analit VA térfogatú, ρA koncentrációjú oldatával van preparálva. A módszer extrakciós és tisztítási lépéseit követően a vizsgálati részekből két, V1 és V2 térfogatú extraktumot kapunk. Az analit visszanyeréseként rc értéket feltételezünk. Mindkét extraktumot b érzékenységű mérési módszerrel vizsgáljuk, és x1 és x2 analitikai választ kapunk.
Ha azt feltételezzük, hogy az analit rc és b értéke ugyanaz a natív és a preparált mintában, akkor a T tartalom a következők szerint számítható ki:
A módszerrel meghatározható az rc visszanyerés. Ezt követően - a fent leírt próbán kívül - az 1. vizsgálati rész extraktumának egy részét (V3 térfogat) az analit ismert ρB.VB mennyiségével preparáljuk és megvizsgáljuk. Az analitikai válasz x3, a visszanyerés pedig a következő:
Ezenkívül kiszámítható a b érzékenység is:
Az összes alkalmazási körülmény és részlet le van írva (18).
II. MELLÉKLET
Minimálisan megkövetelt teljesítményszintek
| Anyag és/vagy metabolit | Hordozóanyag | MRPL |
| Klóramfenikol | Hús | |
| Tojás | 0,3 μg/kg | |
| Tej | ||
| Vizelet | ||
| Akvakultúra-termékek | ||
| Méz | ||
| Medroxiprogeszteron-acetát | Sertésvese zsírja | 1 μg/kg |
| Nitrofurán metabolitok; | ||
| – furazolidon | Baromfihús | 1 μg/kg valamennyi esetében |
| – furaltadon | Akvakultúra-termékek | |
| – nitrofurantoin | ||
| – nitrofurazon | ||
| Malachitzöld és leuko-malachitzöld összesített mennyisége | Akvakultúra-termékek húsa | 2 μg/kg |
( 1 ) Kihozatal: a mintában található analitnak a végső extraktumban jelen levő tömeghányada.
( 2 ) Visszanyerés (itt): a mintához hozzáadott analitnak a végső extraktumban jelen levő tömeghányada. A dokumentum további részében azt feltételezzük, hogy a kihozatal és a visszanyerés egyenlő, és így csak a "visszanyerés" kifejezést használjuk.
Lábjegyzetek:
[1] A dokumentum eredetije megtekinthető CELEX: 32002D0657 - https://eur-lex.europa.eu/legal-content/HU/ALL/?uri=CELEX:32002D0657&locale=hu Utolsó elérhető, magyar nyelvű konszolidált változat CELEX: 02002D0657-20210610 - https://eur-lex.europa.eu/legal-content/HU/ALL/?uri=CELEX:02002D0657-20210610&locale=hu