
31980L1335[1]
A Bizottság első irányelve (1980. december 22.) a kozmetikai termékek összetételének ellenőrzéséhez szükséges vizsgálati módszerekre vonatkozó tagállami jogszabályok közelítéséről
A BIZOTTSÁG ELSŐ IRÁNYELVE
(1980. december 22.)
a kozmetikai termékek összetételének ellenőrzéséhez szükséges vizsgálati módszerekre vonatkozó tagállami jogszabályok közelítéséről
(80/1335/EGK)
AZ EURÓPAI KÖZÖSSÉGEK BIZOTTSÁGA,
tekintettel az Európai Gazdasági Közösséget létrehozó szerződésre,
tekintettel a 79/661/EGK irányelvvel ( 1 ) módosított, a kozmetikai termékekre vonatkozó tagállami jogszabályok közelítéséről szóló, 1976. július 27-i 76/768/EGK tanácsi irányelvre ( 2 ) és különösen annak 8. cikke (1) bekezdésére,
mivel a 76/768/EGK irányelv rendelkezik a kozmetikai termékek hivatalos vizsgálatáról azzal a céllal, hogy biztosítsa a kozmetikai termékek összetételére vonatkozó közösségi rendelkezések által előírt feltételek teljesülését;
mivel minden szükséges vizsgálati módszert a lehető leggyorsabban meg kell határozni; mivel a mintavételi és a laboratóriumi mintaelőkészítési módszerek rögzítése, a szabad nátrium- és kálium-hidroxid azonosítása és adagolása, az oxálsavnak és alkálisóinak hajápolási termékekben történő azonosítása és adagolása, a kloroform és a cink fogkrémekben történő adagolása, továbbá a fenolszulfonsav azonosítása és adagolása jelenti az első lépést ebben az irányban;
mivel az ezen irányelv által megállapított rendelkezések összhangban vannak a 76/768/EGK irányelvnek a műszaki fejlődéshez való hozzáigazításával foglalkozó bizottság véleményével,
ELFOGADTA EZT AZ IRÁNYELVET:
1. cikk
A tagállamok megtesznek minden szükséges intézkedést, hogy a kozmetikai termékek hivatalos vizsgálata során:
- a mintavétel,
- a vizsgálati minták laboratóriumi előkészítése,
- a szabad nátrium- és kálium-hidroxid azonosítása és mennyiségi meghatározása,
- az oxálsavnak és alkálisóinak hajápolási termékekben történő azonosítása és mennyiségi meghatározása,
- a kloroform fogkrémekben történő mennyiségi meghatározása,
- a cink mennyiségi meghatározása,
- a fenolszulfonsav azonosítása és mennyiségi meghatározása
a mellékletben leírt módszereknek megfelelően történjék.
2. cikk
A tagállamok hatályba léptetik azokat a törvényi, rendeleti és közigazgatási rendelkezéseket, amelyek szükségesek ahhoz, hogy ennek az irányelvnek legkésőbb 1982. december 31-ig megfeleljenek.
Erről haladéktalanul tájékoztatják a Bizottságot.
3. cikk
Ennek az irányelvnek a tagállamok a címzettjei.
MELLÉKLET
I. KOZMETIKAI TERMÉKEK MINTAVÉTELE
1. CÉL ÉS ALKALMAZÁSI TERÜLET
A kozmetikai termékek mintavételi eljárása a különböző laboratóriumokban történő vizsgálatuk céljából kerül leírásra.
2. FOGALOMMEGHATÁROZÁSOK
2.1. Elemi minta:
a értékesítésre szánt tételből vett mintaegység.
2.2. Összes minta:
az azonos tételszámmal rendelkező elemi minták összessége.
2.3. Laboratóriumi minta:
az összes mintának az egyes laboratóriumokban vizsgálandó reprezentatív része.
2.4. Vizsgálati minta:
a laboratóriumi mintából az egy elemzéshez szükséges reprezentatív adag.
2.5. Tárolóedény:
a terméket magában foglaló, vele folyamatosan, közvetlenül érintkező tárgy.
3. MINTAVÉTELI ELJÁRÁS
3.1. A kozmetikai termékeket eredeti csomagolásukban mintázzák, és ilyen formában juttatják el az analitikai laboratóriumba.
3.2. A nem kimért adagokban kereskedelmi forgalomba hozott, vagy a kiskereskedelemben az eredeti csomagolástól eltérő tárolóedénybe átcsomagolt kozmetikai termékek esetében különleges mintavételi szabályok kerülnek megállapításra.
3.3. A laboratóriumi minta elkészítéséhez szükséges elemi minták számát az analitikai módszer és az egyes laboratóriumok által elvégzendő vizsgálatok száma határozza meg.
4. A MINTA AZONOSÍTÁSA
4.1. A mintákat a mintavétel helyén lezárják, és a mintavétel helye szerinti tagállamban hatályban levő szabályoknak megfelelően azonosítással látják el.
4.2. Minden kivett elemi mintán a következő adatokat tartalmazó címkét helyeznek el:
- a kozmetikai termék neve,
- a mintavétel kelte, időpontja és helye,
- a mintavételért felelős személy neve,
- az ellenőrzést végző hatóság neve.
4.3. A mintavételről a mintavétel helye szerinti tagállamban hatályban levő szabályoknak megfelelő jegyzőkönyvet kell felvenni.
5. A MINTÁK TÁROLÁSA
5.1. Az elemi mintákat a gyártó címkén szereplő tárolási utasításainak megfelelően kell tárolni.
5.2. Ha nem határoznak meg más körülményeket, a laboratóriumi mintákat sötét helyen, 10 °C és 25 °C között tárolják.
5.3. Az elemi mintákat csak a vizsgálat kezdetekor szabad kinyitni.
II. A VIZSGÁLATI MINTÁK LABORATÓRIUMI ELŐKÉSZÍTÉSE
1. ÁLTALÁNOS RENDELKEZÉSEK
1.1. Ahol lehetséges, a vizsgálatot minden elemi mintán elvégzik, vagy - ha az elemi minta mennyisége túl kicsi - a szükséges legkisebb számú elemi mintát használják, és ezeket a vizsgálati minta kivétele előtt alaposan összekeverik.
1.2. A tartóedényt - ha az analitikai módszer úgy kívánja inert gáz alatt - kinyitják, és a lehető leggyorsabban kiveszik a szükséges számú vizsgálati mintát, ezután késlekedés nélkül lefolytatják a vizsgálatot. Ha a mintát meg kell őrizni, a tartályt ismét légmentesen le kell zárni inert gáz alatt.
1.3. A kozmetikai termékek háromféle halmazállapotban fordulhatnak elő: szilárd, félszilárd, folyékony. Előfordulhat, hogy az eredetileg homogén állapotú terméknél a különböző fázisok során szétválás tapasztalható. Ebben az esetben újra kell ezeket homogenizálni.
1.4. Amennyiben a kozmetikai termék különleges módon csomagolt, és emiatt nem lehet a fenti utasítások szerint eljárni, és megfelelő vizsgálati módszerek nem kerültek előírásra, elfogadható valamilyen egyedi eljárás alkalmazása, feltéve hogy az analízisről készült jegyzőkönyvben ennek leírása szerepel.
2. FOLYADÉKOK
2.1. Ezek a termékek olajos, alkoholos és vizes oldatok, kölnivizek, krémek vagy tejek formájában fordulhatnak elő, és flakonokba, palackokba, ampullákba vagy tubusokba csomagolják őket.
2.2. Vizsgálati minta kivétele:
- kinyitás előtt rázzuk össze erőteljesen a tárolóedényt,
- nyissuk ki a tárolóedényt,
- öntsünk néhány milliliter folyadékot a vizsgálatra szolgáló kémcsőbe, hogy a minta jellegzetességeinek szemrevételezésével kivegyük a vizsgálati mintát,
- zárjuk vissza a tárolóedényt, vagy
- vegyük ki a kívánt mennyiségű vizsgálati mintákat,
- gondosan zárjuk vissza a tartályt.
3. FÉLSZILÁRD TERMÉKEK
3.1. Ezek a termékek paszták, kenőcsök, sűrű emulziók és gélek formájában fordulhatnak elő, és tubusokba, műanyag palackokba vagy tégelyekbe csomagolják őket.
3.2. Vizsgálati minta kivétele, két eset lehetséges:
3.2.1. szűknyakú tartóedények esetében. Távolítsuk el a vizsgált termék legalább felső 1 cm-ét. Nyomjuk ki a vizsgálati mintát, majd azonnal zárjuk le a tartályt.
3.2.2. széles nyakú tartályok esetében: A felső réteget egyenletesen kaparjuk le és távolítsuk el. Vegyük ki a vizsgálati mintát és azonnal zárjuk vissza a tartályt.
4. SZILÁRD TERMÉKEK
4.1. Ezek a termékek laza porok, kompakt porok, stiftek formájában fordulhatnak elő, és különféle tartóedényekbe csomagolhatják őket.
4.2. Vizsgálati minta kivétele, két eset lehetséges:
4.2.1. laza porok esetén: a dugó kivétele vagy a tartóedény kinyitása előtt erőteljesen rázzuk össze a port. Nyissuk ki és vegyük ki a vizsgálati mintát.
4.2.2. tömörített por vagy stift esetén: egyenletesen kaparjuk le és távolítsuk el a felső réteget szilárd termékről, az így szabaddá váló részből vegyük ki a vizsgálati mintát.
5. TÚLNYOMÁSOS CSOMAGOLÁSBAN FORGALMAZOTT TERMÉKEK (aeroszolos flakonok)
5.1. Ezek definícióját az 1975. május 20-i 75/324/EGK tanácsi irányelv ( 3 ) 2. cikke tartalmazza.
5.2. Vizsgálati minta:
Az aeroszolos flakon erőteljes felrázását követően a flakon tartalmának reprezentatív mennyiségét, megfelelő összekötő elem segítségével (lásd például az 1. ábrát: különleges esetekben az analitikai módszer más összekötő elem használatát teheti szükségessé), egy aeroszol szeleppel ellátott, de bemerülő csövet nem tartalmazó műanyag bevonatú, áttetsző üvegpalackba (4. ábra) juttatjuk. Az átvitel során a flakont szeleppel lefelé kell tartani. Az átvitel után tartalma jól látható, és az alábbi négy eset valamelyikének megfelelően viselkedik:
5.2.1. A flakon tartalma homogén oldat, amely közvetlenül vizsgálható.
5.2.2. A flakon két folyadékfázisú aeroszol terméket tartalmaz. Az alsó fázisnak egy második továbbító flakonba történő leválasztását követően mindkét fázis vizsgálata elvégezhető. Az első flakont ilyenkor szeleppel lefelé kell tartani. Az esetek többségében ilyenkor gyakran az alsó a vizes fázis, és nem tartalmaz hajtógázt (pl. bután/víz receptúra).
5.2.3. A flakon szuszpenzióban port tartalmaz. A folyadékfázis a por eltávolítását követően vizsgálható.
5.2.4. Hab vagy krém. Először mérjünk be pontosan a mintagyűjtő palackba 5-10 g 2-metoxi-etanolt. Ez az anyag a gázmentesítés során megakadályozza a habképződést, és ezután folyadékveszteség nélkül elvégezhető a hajtógázok kihajtása.
5.3. Segédeszközök
Az összekötő elem (1. ábra) dúralumíniumból vagy sárgarézből készül. Úgy van kialakítva, hogy egy polietilén adapteren keresztül különböző szeleprendszerekhez csatlakoztatható. A bemutatott eszköz példaként szolgál; más összekötő elemek is használhatók (lásd a 2. és 3. ábrát).
A mintagyűjtő palack (4. ábra) kívül átlátszó, műanyag védőréteggel bevont opálüvegből készül. 50-100 ml minta befogadására alkalmas. A palack egy bemerülő cső nélküli aeroszol szeleppel van ellátva.
5.4. Eljárás
Megfelelő mennyiségű vizsgálati minta befogadása érdekében a mintagyűjtő palackot légteleníteni kell. Ehhez az összekötő elemen keresztül vezessünk be körülbelül 10 ml diklór-difluor-metánt vagy butánt (a vizsgálandó aeroszol terméktől függően), majd a mintagyűjtő palackot szeleppel fölfelé tartva teljesen gázmentesítsük a folyadékfázis eltűnéséig. Vegyük le az összekötő elemet és mérjük le a mintagyűjtő palackot ("a" gramm). Erőteljesen rázzuk föl az aeroszolos flakont, amelyből a mintavétel történik. Csatlakoztassuk az összekötő elemet a mintául szolgáló aeroszolos flakonon levő szelephez (szelep fölfelé néz), illesszük a mintagyűjtő palackot (nyakkal lefelé) az összekötő elemhez, majd nyomjuk meg. Töltsük föl a mintagyűjtő palackot körülbelül kétharmad részig. Ha az átvitel a nyomás kiegyenlítődése miatt a kívánt szint elérése előtt megszűnik, a mintagyűjtő palack lehűtésével újra megindítható a folyamat. Vegyük le az összekötő elemet, mérjük meg a feltöltött palackot ("b" gramm), és számítsuk ki az átvitt minta mennyiségét (m1) (m1 = b - a).
Az így kapott minta felhasználható:
1. a szokásos kémiai vizsgálat elvégzésére;
2. az illékony komponensek gázkromatográfiás analízisére.
5.4.1. Kémiai vizsgálat
A mintagyűjtő palackot szeleppel fölfelé tartva a következőképpen folytassuk a vizsgálatot:
- gázmentesítés. Ha a gázmentesítés habképződéssel jár, olyan mintagyűjtő palackot használjunk, amelybe az átvitelt megelőzően az összekötő elemen keresztül egy fecskendő segítségével pontosan lemért mennyiségű (5-10 g) 2-metoxi-etanolt adagoltunk be.
- vigyázva, hogy anyagveszteség ne történjék, teljesen távolítsuk el az illékony alkotókat 40 °C-os vízfürdőn rázatva a mintát. Vegyük le az összekötő elemet.
- ismét mérjük le a mintagyűjtő palackot ("c" gramm) a maradék, mennyiségének meghatározásához (m2) (m2 = c - a).
-
(NB:A maradék tömegének számítása során vonjuk le a bemért 2-metoxi-etanol tömegét.)
- a szelep levételével nyissuk ki a mintagyűjtő palackot,
- oldjuk fel a mintát hiánytalanul ismert mennyiségű, megfelelő oldószerben,
- a minta egy részéből végezzük el a kívánt vizsgálatot.
- A számítási képletek:
-
és
,
ahol
m1 = az aeroszol mennyisége a mintagyűjtő palackban;
m2 = a maradék mennyisége 40 °C-on történő rázatás után;
r = a vizsgált anyag százalékos aránya m2-ben (a megfelelő módszerrel meghatározva);
R = a vizsgált anyag százalékos aránya az aeroszol mintában;
Q = a vizsgált anyag összes tömege az aeroszolos flakonban;
P = a kiindulási aeroszolos flakon nettó tömege (elemi minta).
5.4.2. Az illékony komponensek gázkromatográfiás vizsgálata
5.4.2.1. Alapelv
Gázkromatográfiás fecskendővel vegyünk ki megfelelő mennyiségű anyagot a mintagyűjtő palackból. Ezután a fecskendőben található anyagot fecskendezzük be a gázkromatográfiás készülékbe.
5.4.2.2. Segédeszközök
25 és 50 μl-es, A2 sorozatba tartozó "precíziós mintavételre" megjelölésű vagy ezzel egyenértékű gázkromatográfiás fecskendő (5. ábra). A fecskendő a tű felőli végénél egy tolattyúval van ellátva. A fecskendő csatlakoztatása a mintagyűjtő palackhoz a palack felőli oldalon egy összekötő elemen, a fecskendőnél pedig egy (8 mm hosszú, 2,5 mm átmérőjű) polietilén csövön keresztül történik.
5.4.2.3. Eljárás
Miután a mintagyűjtő palackba átjutott a szükséges mennyiségű aeroszol termék, a fecskendő kúpos végét csatlakoztassuk az 5.4.2.2. pontban leírt módon a mintagyűjtő palackhoz. Nyissuk ki a szelepet és szívjuk fel a szükséges mennyiségű folyadékot. Távolítsuk el a gázbuborékokat a dugattyú többszöri ki-be mozgatásával (szükség esetén hűtsük le a fecskendőt). Amikor a fecskendőbe a szükséges mennyiségű buborékmentes oldat bekerült, zárjuk a szelepet és vegyük le a fecskendőt a mintagyűjtő palackról. Illesszük a fecskendőre a tűt és helyezzük be a gázkromatográf injektorába, nyissuk ki a szelepet és fecskendezzük be a mintát.
5.4.2.4. Belső standard
Ha a vizsgálathoz belső standard szükséges, azt a mintagyűjtő palackba kell beadagolni (összekötő elemen keresztül egy közönséges üvegfecskendő segítségével).
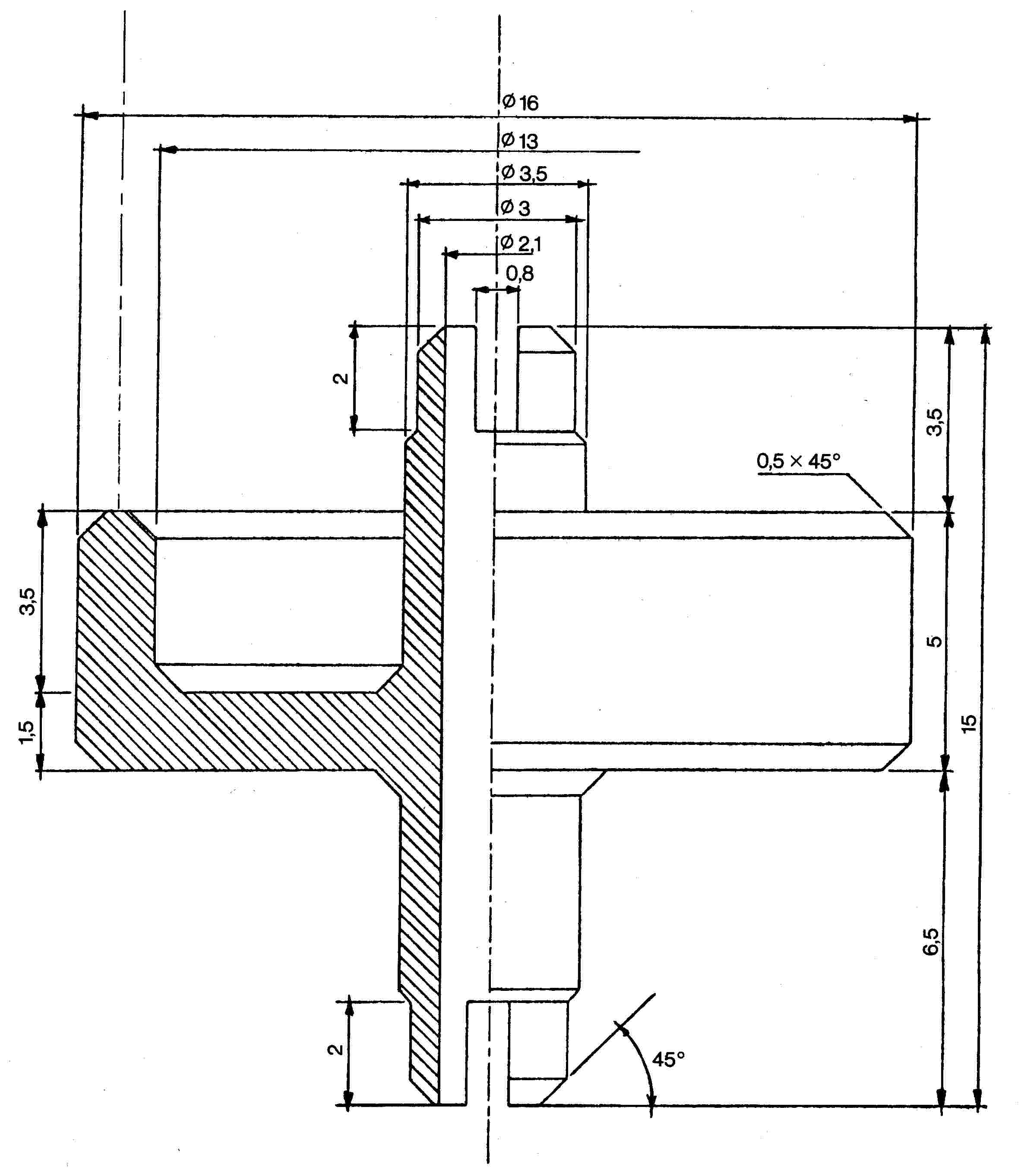
1. ábra
P1 összekötő elem
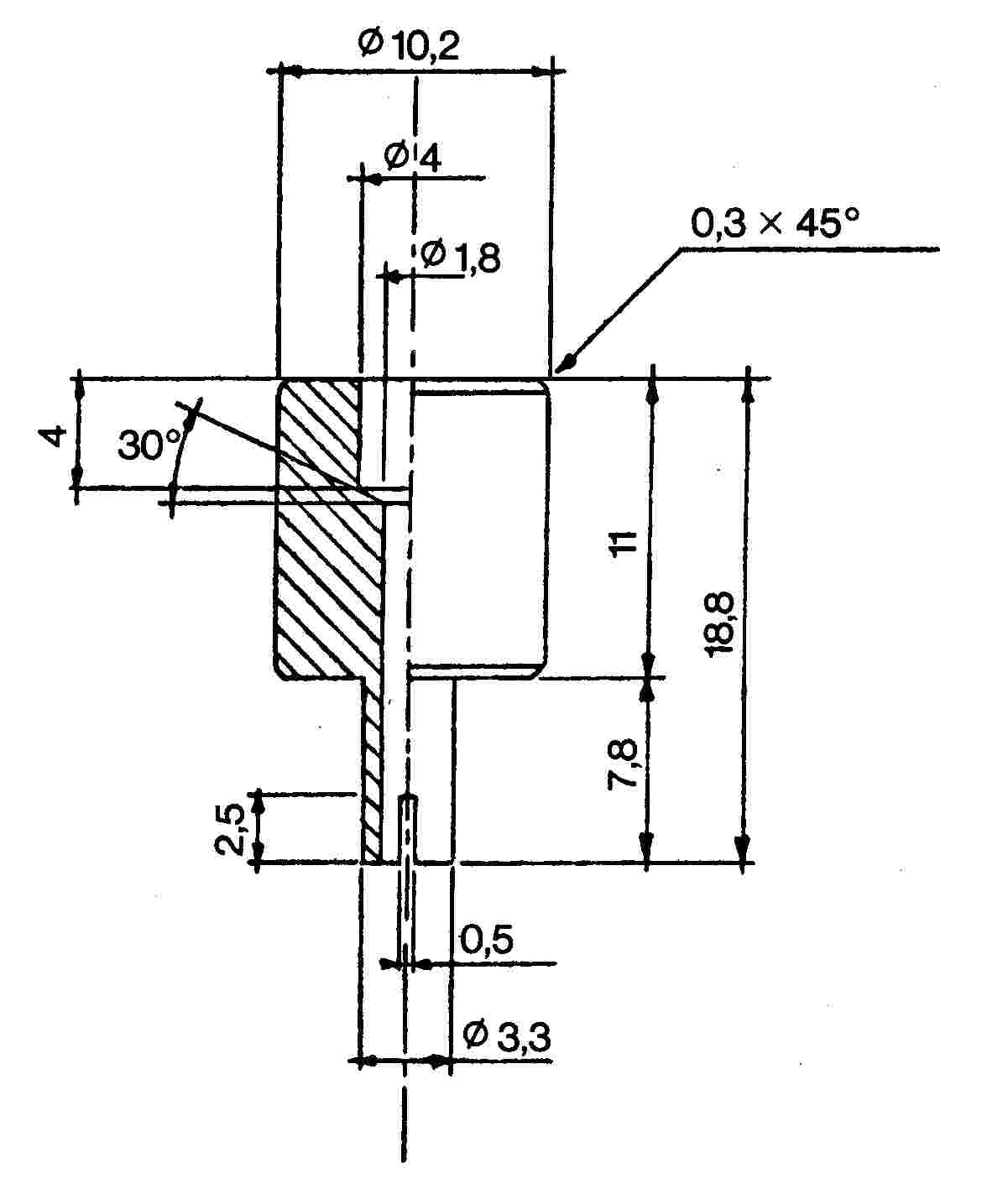
2. ábra
M2 összekötő elem
Külső és belső menetes szelepek közötti átvitelre
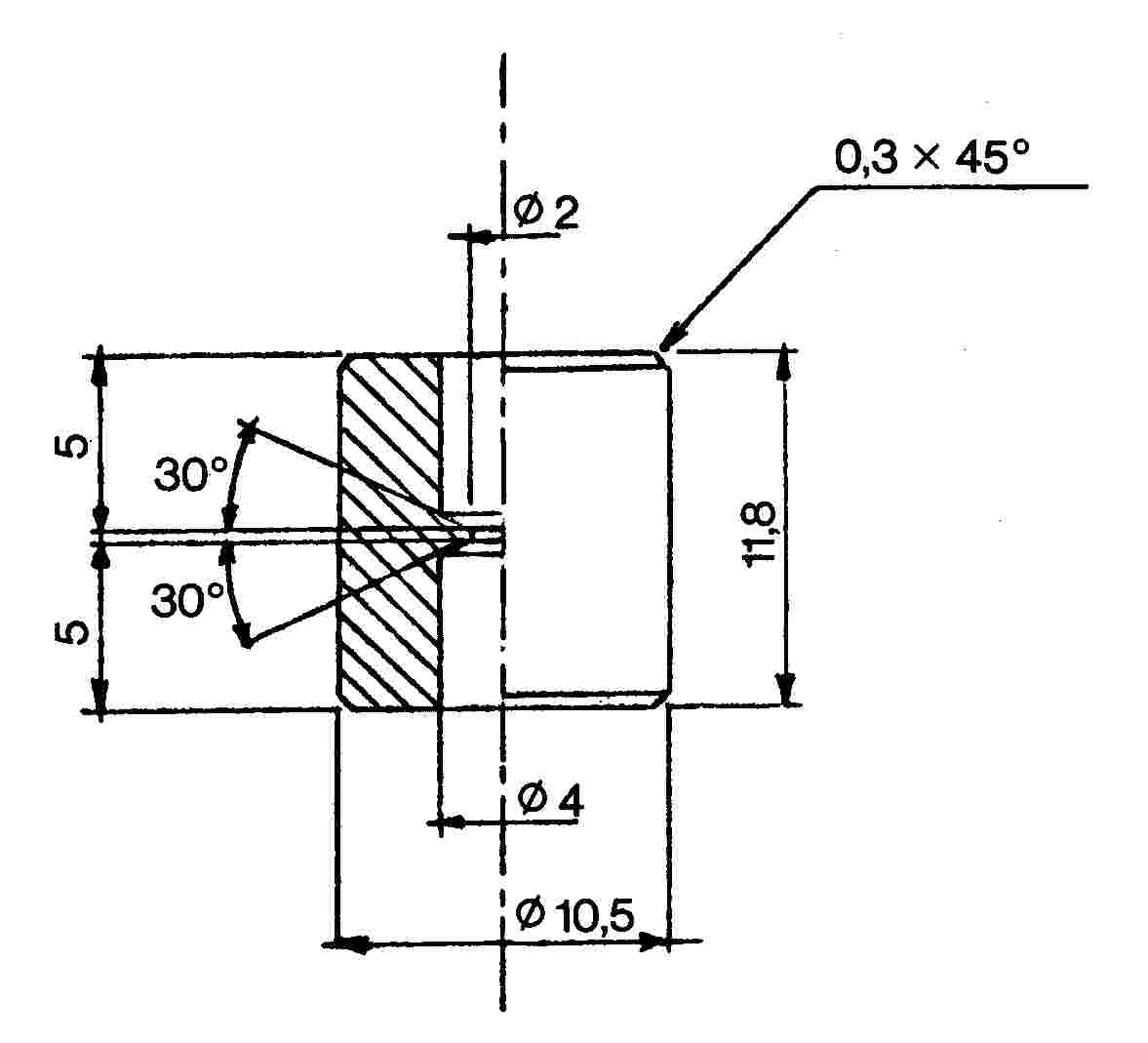
3. ábra
M1 összekötő elem
két külső menetes szelep közötti átvitelre

4. ábra
Mintagyűjtő palack
űrtartalom 50-100 ml
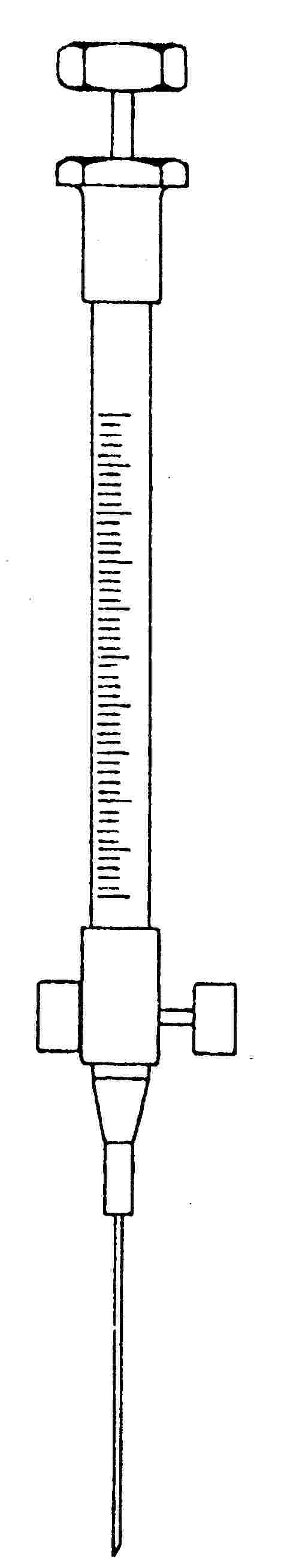
5. ábra
Gáznyomásos fecskendő
III. SZABAD NÁTRIUM- ÉS KÁLIUM-HIDROXID MENNYISÉGI MEGHATÁROZÁSA ÉS AZONOSÍTÁSA
1. CÉL ÉS ALKALMAZÁSI TERÜLET
Ez a módszer a jelentős mennyiségű szabad nátrium és/vagy kálium-hidroxidot tartalmazó kozmetikai termékek azonosítására és a hajegyenesítő, valamint körömágybőr-eltávolító készítményekben az ilyen szabad nátrium- és/vagy kálium-hidroxid-tartalom meghatározására szolgáló eljárást írja le.
2. FOGALOMMEGHATÁROZÁS
A szabad nátrium- és kálium-hidroxid tartalom a termék semlegesítéséhez szükséges savmérő oldat térfogata meghatározott körülmények között. Az elért eredmény szabad nátrium-hidroxid formájában kerül megadásra.
3. ALAPELV
A mintát vízben feloldjuk vagy diszpergáljuk, és savmérő oldattal titráljuk. A sav hozzáadása közben folyamatosan regisztráljuk a pH-értékét: tiszta nátrium- vagy kálium-hidroxid oldat esetében a végpontot a regisztrált pH-érték változási sebességének maximuma egyértelműen meghatározza.
Az egyszerű titrálási görbét a következő anyagok jelenléte torzíthatja:
a) ammónia és más gyenge szerves bázisok, amelyek titrálási görbéje meglehetősen lapos. Az ammónia eltávolítása szobahőmérsékleten csökkentett nyomáson történik.
b) gyenge savak sói, amelyek titrálási görbéje több inflexiós pontot is tartalmazhat. Ilyenkor, több inflexiós pont esetén a görbének csupán az első inflexiós pontig tartó első szakasza felel meg a szabad nátrium- és kálium-hidroxidból származó hidroxil ionok semlegesítésének.
Létezik egy alternatív eljárás alkoholos oldatban történő titrálása esetére akkor, ha gyenge szervetlen savak sói túlzott mértékben zavarják a meghatározást.
Elméletileg ugyan fennáll a lehetősége, hogy más erős bázisok oldata, például lítium-hidroxid vagy kvaterner ammónium-hidroxid okozza a magas pH-t, ezek jelenléte azonban ilyen típusú kozmetikai termékekben erősen valószínűtlen.
4. AZONOSÍTÁS
4.1. Reagensek
4.1.1. Lúgos standard pufferoldat, 25 °C-on pH = 9,18: 0,05 M nátrium-tetraborát-dekahidrát.
4.2. Eszközök
4.2.1. Szokásos laboratóriumi üvegeszközök
4.2.2. pH-mérő
4.2.3. Üvegelektród
4.2.4. Kalomel referenciaelektród
4.3. Eljárás
A standard pufferoldat segítségével végezzük el az elektródok kalibrációját.
Készítsük el a vizsgálandó termék 10 %-os vizes oldatát vagy diszperzióját, és szűrjük meg. Mérjük meg a pH-t. Ha a pH 12 vagy azt meghaladó érték, mennyiségi meghatározást kell végezni.
5. MENNYISÉGI MEGHATÁROZÁS
5.1. Titrálás vizes közegben
5.1.1. Reagens
5.1.1.1. 0,1 N sósav mérőoldat
5.1.2. Eszközök
5.1.2.1. Szokásos laboratóriumi üvegeszközök
5.1.2.2. pH-mérő, lehetőleg regisztrálóval
5.1.2.3. Üvegelektród
5.1.2.4. Kalomel referenciaelektród
5.1.3. Eljárás
Pontosan mérjünk be 0,5-1,0 g vizsgálati mintát egy 150 ml-es főzőpohárba. Amennyiben a mintában ammónia van, dobjunk néhány üveggyöngyöt a főzőpohárba, helyezzük a főzőpoharat vákuum deszikkátorba, és vízsugárszivattyúval vákuumozzuk, amíg az ammóniaszag meg nem szűnik (kb. három óra).
Adjunk hozzá 100 ml vizet, oldjuk fel vagy diszpergáljuk a maradékot, és titráljuk (5.1.1.1.) 0,1 N sósavoldattal, regisztrálva a pH változását (5.1.2.2.).
5.1.4. Számítás
Állapítsuk meg a titrálási görbék inflexiós pontjait. Az első inflexiós pontnál, amikor az 7 pH alatt található, a mintában nincs nátrium- és kálium-hidroxid.
Ha a görbén két vagy több inflexiós pont figyelhető meg, csak az elsőt kell figyelembe venni.
Jegyezzük föl a titrálószer térfogatát az első inflexiós pontban.
Legyen "V" a titrálószer térfogata ml-ben,
"M" a vizsgálati minta tömege grammban.
A minta nátrium- és/vagy kálium-hidroxid-tartalmát % (m/m) nátrium-hidroxidban kifejezve az alábbi képlettel számítjuk ki:
.
Előfordulhat, hogy noha a jelek szerint a mintában jelentős mennyiségű nátrium- és/vagy kálium-hidroxid van jelen, a titrálási görbén mégsem fedezhető fel határozott inflexiós pont. Ilyen esetben a mennyiségi meghatározást izopropanolban kell megismételni.
5.2. Titrálás izopropanolban
5.2.1. Reagensek
5.2.1.1. Izopropanol
5.2.1.2. 1,0 N sósav vizes mérőoldata
5.2.1.3. Az izopropanolos 0,1 N sósavoldat közvetlenül felhasználás előtt készítendő az 1,0 N vizes sósavoldat izopropanollal történő hígításával.
5.2.2. Eszközök
5.2.2.1. Szokásos laboratóriumi üvegeszközök
5.2.2.2. pH-mérő, lehetőleg regisztrálóval
5.2.2.3. Üvegelektród
5.2.2.4. Kalomel referenciaelektród
5.2.3. Eljárás
Pontosan mérjünk be 0,5-1,0 g vizsgálati mintát egy 150 ml-es főzőpohárba. Amennyiben a mintában ammónia van, dobjunk néhány üveggyöngyöt a főzőpohárba, helyezzük a főzőpoharat vákuum deszikkátorba, és vízsugárszivattyúval vákuumozzuk, amíg az ammóniaszag meg nem szűnik (kb. három óra).
Adjunk hozzá 100 ml izopropanolt, oldjuk fel vagy diszpergáljuk a vákuumozás maradékát, és titráljuk (5.2.1.3.) 0,1 N sósavas izopropanollal, regisztrálva a látszólagos pH változását (5.2.2.2.).
5.2.4. Számítás
Az 5.1.4. pont szerint. Az első inflexiós pont helye 9 körüli látszólagos pH-nál van.
5.3. Megismételhetőség ( 4 )
Nátrium-hidroxidban kifejezve 5 % (m/m) körüli nátrium- vagy kálium-hidroxid-tartalom esetén, az azonos mintán, párhuzamosan végzett két mennyiségi meghatározás eredményei közötti különbség abszolút értéke nem haladhatja meg a 0,25 %-ot.
IV. OXÁLSAVNAK ÉS ALKÁLISÓINAK MENNYISÉGI MEGHATÁROZÁSA ÉS AZONOSÍTÁSA HAJÁPOLÁSI TERMÉKEKBEN
1. CÉL ÉS ALKALMAZÁSI TERÜLET
Az alábbiakban leírt módszer alkalmas oxálsavnak és alkálisóinak hajápolási termékben történő mennyiségi meghatározására és azonosítására. A módszer körülbelül 5 % oxálsavat vagy ezzel azonos mennyiségű alkáli-oxalátot tartalmazó színtelen, vizes/alkoholos oldatok és bőrlemosó-folyadékok esetében alkalmazható.
2. FOGALOMMEGHATÁROZÁS
A minta oxálsav- és/vagy alkáli-oxalát-tartalmát ennek az eljárásnak megfelelően adjuk meg, és a szabad oxálsav tömegszázalékában (m/m) fejezzük ki.
3. ALAPELV
Az esetleg jelenlevő anionos felületaktív anyagok p-toluidin hidro-kloroddal történő eltávolítását követően az oxálsavat és/vagy az oxalátokat kalcium-oxalát formájában kicsapatjuk, majd az oldatot szűrjük. A csapadékot kénsavban feloldjuk és kálium-permanganáttal titráljuk.
4. REAGENSEK
Valamennyi reagensnek analitikai tisztaságúnak kell lennie.
4.1. 5 % (m/m) ammónium-acetát-oldat
4.2. 10 % (m/m) kalcium-klorid-oldat
4.3. 95 %-os (V/V) etanol
4.4. szén-tetraklorid
4.5. dietil-éter
4.6. 6,8 % (m/m) p-toluidin-dihidroklorid-oldat
4.7. 0,1 N kálium-permanganát-oldat
4.8. 20 % (m/m) kénsav
4.9. 10 % (m/m) sósav
4.10. nátrium-acetát-trihidrát
4.11. jégecet
4.12. (1:1) kénsav
4.13. telített bárium-hidroxid-oldat
5. ESZKÖZÖK
5.1. 500 ml-es választótölcsér
5.2. 50 ml-es és 600 ml-es főzőpohár
5.3. G-4 üvegszűrő
5.4. 25 ml-es és 100 ml-es mérőhenger
5.5. 10 ml-es pipetta
5.6. 500 ml-es szívópalack
5.7. Vízsugárszivattyú
5.8. 0-100°C közötti beosztású hőmérő
5.9. Fűthető mágneses keverő
5.10. Teflonbevonatú mágneses keverőpálcák
5.11. 25 ml-es büretta
5.12. 250 ml-es Erlenmeyer-lombik
6. ELJÁRÁS
6.1. Mérjünk be 6-7 g mintát egy 50 ml-es főzőpohárba, állítsuk be pH-ját 3-ra hígított sósavval (4.9.), és mossuk be 100 ml desztillált vízzel egy választótölcsérbe. Ezután adjunk hozzá 25 ml etanolt (4.3.), 25 ml p-toluidin-dihidroklorid (4.6.) oldatot és 25-30 ml széntetrakloridot (4.4.), majd erőteljesen rázzuk össze a keveréket.
6.2. A fázisok szétválása után távolítsuk el az alsó (szerves) fázist, ismételjük meg az extrakciót a 6.1. pontban leírt reagensekkel és újra távolítsuk el a szerves fázist.
6.3. Mossuk a vizes oldatot egy 600 ml-es főzőpohárba, és az oldat forralásával távolítsuk el a visszamaradt szén-tetrakloridot.
6.4. Adjunk hozzá 50 ml ammónium-acetát-oldatot (4.1.), forraljuk az (5.9.) oldatot, és keverjünk be 10 ml forró kalcium-klorid-oldatot (4.2.) a forrásban levő oldatba; hagyjuk kiülepedni a csapadékot.
6.5. Ellenőrizzük, hogy teljes-e a csapadék leválása néhány csepp kalcium-klorid-oldat (4.2.) hozzáadásával, hagyjuk lehűlni szobahőmérsékletre, majd keverjünk bele 200 ml etanolt (4.3.); (5.10.), hagyjuk állni 30 percet.
6.6. Szűrjük a folyadékot (5.3.) üvegszűrő tégelyen, vigyük át a csapadékot kismennyiségű forró vízzel (50-60 °C) a szűrőtégelybe, és mossuk a csapadékot hideg vízzel.
6.7. Mossuk a csapadékot ötször kismennyiségű etanollal (4.3.), majd ötször kismennyiségű dietil-éterrel (4.5.), és oldjuk fel 50 ml forró kénsavban (4.8.) úgy, hogy utóbbit vákuummal átszívatjuk a szűrőtégelyen.
6.8. Vigyük át az oldatot veszteség nélkül egy (5.12.) Erlenmeyer-lombikba, majd titráljuk kálium-permanganát-oldattal (4.7.) halvány rózsaszín szín megjelenéséig.
7. SZÁMÍTÁS
A minta tömegszázalékban kifejezett oxálsavtartalmát az alábbi képlettel számítjuk:
,
ahol:
A 0,1 N kálium-permanganát fogyása a 6.8. pont szerint mérve;
E a (6.1.) vizsgálati minta mennyisége grammban;
4,50179 az oxálsav átszámítási faktora.
8. MEGISMÉTELHETŐSÉG ( 5 )
5 % (m/m) körüli oxálsavtartalom esetén az azonos mintán, párhuzamosan végzett két mennyiségi meghatározás eredményei közötti különbség abszolút értékben nem haladhatja meg a meg a 0,15 %-ot.
9. AZONOSÍTÁS
9.1. Alapelv
Az oxálsavat és/vagy az oxalátokat kalcium-oxalát formájában kicsapatjuk, és kénsavban feloldjuk. Az oldathoz kevés kálium-permanganát oldatot adunk, amely széndioxid képződése közben elszíntelenedik. A keletkező széndioxidot bárium-hidroxid oldaton átengedve fehér (tejszerű) bárium-karbonát csapadék képződik.
9.2. Eljárás
9.2.1. A vizsgálandó minta egy részét kezeljük a 6.1-6.3. pontokban leírtaknak megfelelően; ez a műveletsor eltávolítja az esetleg jelenlevő detergenseket.
9.2.2. Adjunk egy spatulahegynyi (4.10.) nátrium-acetátot a 9.2.1. pontnak megfelelően kapott oldat 10 ml-éhez, és savasítsuk az oldatot néhány csepp (4.11.) jégecettel.
9.2.3. Adjunk hozzá 10 %-os kalcium-klorid-oldatot (4.2.), és szűrjük le. Oldjuk fel a kalcium-oxalát-csapadékot 2 ml (1:1) (4.12.) kénsavban.
9.2.4. Vigyük át az oldatot egy kémcsőbe, és adjunk hozzá cseppenként körülbelül 0,5 ml 0,1 N kálium-permanganát-oldatot (4.7.). Oxalát jelenlétében az oldat színe előbb fokozatosan halványul, majd gyorsan elszíntelenedik.
9.2.5. A kálium-permanganát hozzáadását követően azonnal helyezzünk egy megfelelő méretű dugós üvegcsövet a kémcső fölé, enyhén melegítsük föl a tartalmát, és gyűjtsük össze a képződött széndioxidot a telített bárium-hidroxid-oldatba (4.13.). A három-öt perc után megjelenő tejszerű bárium-karbonát-felhő oxálsav jelenlétére utal.
V. KLOROFORM MEGHATÁROZÁSA FOGKRÉMBEN
1. CÉL ÉS ALKALMAZÁSI TERÜLET
Ez a módszer a fogkrém kloroformtartalmának gázkromatográfiás meghatározását írja le. A módszer legfeljebb 5 % kloroformtartalom meghatározására alkalmas.
2. FOGALOMMEGHATÁROZÁS
A módszerrel meghatározott kloroformtartalmat a termék tömegszázalékában adjuk meg.
3. ALAPELV
A fogkrémet olyan dimetil-formamid-metanol elegyben szuszpendáljuk, amihez belső standardként ismert mennyiségű acetonitrilt adunk. Centrifugálás után a folyadékfázisból kivett mintát gázkromatografáljuk, és kiszámítjuk annak kloroformtartalmát.
4. REAGENSEK
Minden reagensnek analitikai tisztaságúnak kell lennie.
4.1. 80-100 mesh szemcseméretű Porapak Q, szirén-divinil-benzil 101 vagy ezekkel egyenértékű töltet
4.2. Acetonitril
4.3. Kloroform
4.4. Dimetil-formamid
4.5. Metanol
4.6. Belső standard oldat
Pipettázzunk 5 ml dimetil-formamidot (4.4.) egy 50 ml-es mérőlombikba, és adjunk hozzá 300 mg (M mg) pontosan mért acetonitrilt. Töltsük föl a jelig dimetil-formamiddal és keverjük össze.
4.7. A relatív válaszjel faktor meghatározására szolgáló oldat. Pipettázzunk pontosan 5 ml belső standard oldatot (4.6.) egy 10 ml-es mérőlombikba, és adjunk hozzá 300 mg (M mg) pontosan mért kloroformot. Töltsük föl a jelig dimetil-formamiddal és keverjük össze.
5. ESZKÖZÖK
5.1. Analitikai mérleg
5.2. Gázkromatográf lángionizációs detektorral felszerelve
5.3. 5-10 ml-es, 0,1 ml beosztású mikrofecskendők
5.4. 1, 4 és 5 ml-es hasas pipetta
5.5. 10 és 50 ml-es mérőlombik
5.6. Körülbelül 20 ml-es, menetes kupakkal rendelkező, Sovirel France No. 20 típusú vagy ezzel egyenértékű próbacsövek. A menetes kupak egyik oldalán teflonbevonatú belső tömítőlappal rendelkezik.
5.7. Centrifuga.
6. ELJÁRÁS
6.1. Gázkromatográfiás körülmények
6.1.1. Oszlop anyaga: üveg,
Hossza: 150 cm
Belső átmérő: 4 mm
Külső átmérő: 6 mm.
6.1.2. Töltsük meg a oszlopot Porapak Q, sztirén-divinil-bentén 101 vagy ezekkel egyenértékű (4.1.) 80-100 mesh szemcseméretű töltettel.
6.1.3. Lángionizációs detektor: úgy állítsuk be az érzékenységet, hogy a 4.7. pontban ismertetett oldat 3 μl-ét befecskendezve az acetonitril csúcs magassága a teljes fokbeosztásnak körülbelül háromnegyedénél legyen.
6.1.4. Gázok
Vivőgáz: nitrogén, áramlási sebesség 65 ml/perc.
Kisegítő gáz: állítsuk be a lángionizációs detektorhoz áramló gázok térfogatáramát úgy, hogy a levegő vagy oxigén térfogatárama a hidrogénének 5-10-szerese legyen.
6.1.5. Hőmérsékletek
| Injektor blokk | 210 °C |
| Detektor blokk | 210 °C |
| Oszlop | 175 °C |
6.1.6. Papírdiagram sebessége: kb. 100 cm/óra
6.2. Mintaelőkészítés
Olyan tubusból vegyük a vizsgálati mintát, amely még nem volt kinyitva. Nyomjuk ki a tubus tartalmának egyharmadát, ezután tegyük vissza a kupakot, gondosan keverjük össze a tubus tartalmát, majd vegyük ki a vizsgálati mintát.
6.3. Mennyiségi meghatározás
6.3.1. Egy csavaros kupakkal ellátott (5.6.) csőbe mérjünk ki 10 mg pontossággal 6-7 grammot (M0 g) a 6.2. pontnak megfelelően előkészített fogkrémből, majd adjunk hozzá három kis üveggyöngyöt.
6.3.2. Pipettázzunk pontosan 5 ml belső standard oldatot (4.6.), 4 ml dimetil-formamidot (4.4.) és 1 ml metanolt (4.5.)a kémcsőbe, zárjuk le, és keverjük össze a tartalmát.
6.3.3. Mechanikus rázógépen rázassuk fél órán keresztül, majd 15 percig centrifugáljuk a lezárt csövet olyan fordulatszámon, hogy a fázisok teljesen szétváljanak.
Megjegyzés:Időnként előfordul, hogy a folyadékfázis a centrifugálást követően is zavaros. Némi javulás érhető el, ha 1-2 gramm nátrium-kloridot adunk a folyadékfázishoz, hagyjuk leülepedni, majd centrifugáljuk.
6.3.4. Fecskendezzünk 3 μl-t ebből a (6.3.3.) oldatból a 6.1. pontban leírt körülmények között. Ismételjük meg a műveletet. A fenti körülmények között tájékoztató jelleggel az alábbi retenciós idők adhatók meg:
| metanol | körülbelül egy perc |
| acetonitril | körülbelül 2,5 perc |
| kloroform | körülbelül hat perc |
| dimetil-formamid | > 15 perc |
6.3.5. A relatív válaszjel faktor meghatározása Fecskendezzük be a 4.7. pont szerinti oldat 3 μl-ét a faktor meghatározása céljából. Ismételjük meg a műveletet. A relatív válaszjel faktort naponta határozzuk meg.
7. SZÁMÍTÁSOK
7.1. A relatív válaszjel számítása
7.1.1. Mérjük le az acetonitril és kloroform csúcsok magasságát és a magasság felénél ezek szélességét, majd számítsuk ki a két csúcs területét a következő képlet szerint: magasság szorozva a magasság felénél mért szélességgel.
7.1.2. Határozzuk meg az acetonitril és kloroform csúcsok területét a 6.3.5. szakasznak megfelelően felvett kromatogramokon, és számítsuk ki a relatív válaszjelet, fs, a következő képlet segítségével:
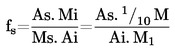
,
ahol:
fs = a kloroform relatív válaszjel faktora;
As = a kloroform csúcs területe (6.3.5)
Ai = az acetonitril csúcs területe (6.3.5);
Ms = a kloroform mennyisége a 6.3.5. szakaszban említett 10 ml oldatban, mg-ban (= M1)
Mi = az acetonitril mennyisége a 6.3.5. szakaszban említett 10 ml oldatban, mg-ban (= 1/10 M).
Számítsuk ki a kapott értékek átlagát.
7.2. A kloroformtartalom számítása
7.2.1. A 6.3.4. szakaszban leírt eljárásban kapott kromatogramokon a 7.1.1. szakasznak megfelelően számítsuk ki a kloroform és acetonitril csúcsok területét.
7.2.2. Számítsuk ki a fogkrém kloroformtartalmát a következő képlettel:
,
ahol
% X = fogkrém kloroformtartalma tömegszázalékban kifejezve;
As = a kloroform csúcs területe (6.3.4.);
Ai = az acetonitril csúcs területe (6.3.4.);
Msx = a 6.3.1. szakaszban említett minta tömege mg-ban (1000 M0);
Mi = az acetonitril mennyisége a 6.3.2. pontnak megfelelően kapott oldat 10 ml-ében, mg-ban (= 1/10 M).
Számítsuk ki a kapott értékek átlagát, és az eredményt adjuk meg 0,1 % pontossággal.
8. MEGISMÉTELHETŐSÉG ( 6 )
3 % (m/m) körüli kloroformtartalom esetén az azonos mintán, párhuzamosan végzett két mennyiségi meghatározás eredményei közötti különbség abszolút értéke nem haladhatja meg a 0,3 %-ot.
VI. CINK MEGHATÁROZÁSA
1. CÉL ÉS ALKALMAZÁSI TERÜLET
Ez a módszer a cink klorid, szulfát vagy 4-hidroxi-benzol-szulfonát formájában vagy ezeknek a cinksóknak a kozmetikumokban asszociátumaként előforduló meghatározására alkalmas..
2. FOGALOMMEGHATÁROZÁS
A minta cinktartalmát bis-(2-metil-8-kinolil-oxid) só formájában gravimetrikusan határozzuk meg, és az eredményt a minta tömegére vonatkoztatva tömegszázalékban adjuk meg.
3. ALAPELV
Az oldatban jelenlevő cinket savas közegben kicsapatjuk cink-bis-(2-metil-8-kinolil-oxid) formájában. Szűrés után a csapadékot szárítjuk és megmérjük.
4. REAGENSEK
Valamennyi reagensnek analitikai tisztaságúnak kell lennie.
4.1. 25 %-os (m/m) tömény ammónia oldat; [Kép #1]
Kép #1
4.2. Jégecet
4.3. Ammónium-acetát
4.4. 2-metil-kinolin-8-ol
4.5. 6 %-os (m/V) ammóniaoldat
Tegyünk 240 g tömény ammóniaoldatot (4.1.) egy 1000 ml-es mérőlombikba, töltsük föl a jelig desztillált vízzel és keverjük össze.
4.6. 0,2 M ammónium-acetát-oldat
Oldjunk fel 15,4 g ammónium-acetátot (4.3.) desztillált vízben, töltsük jelig egy 1000 ml-es mérőlombikban, és keverjük össze.
4.7. 2-metil-kinolin-8-ol-oldat
Oldjunk fel 5 g 2-metil-kinolin-8-ol-t 12 ml jégecetben, és desztillált vízzel mossuk egy 100 ml-es mérőlombikba. Töltsük föl a jelig desztillált vízzel, és keverjük össze.
5. ESZKÖZÖK
5.1. 100 ml-es és 1000 ml-es mérőlombik
5.2. 400 ml-es főzőpohár
5.3. 50 ml-es és 150 ml-es mérőhenger
5.4. 10 ml-es mérőpipetta
5.5. G-4 üvegszűrő tégely
5.6. 500 ml-es szívópalack
5.7. Vízsugárszivattyú
5.8. 0-100 °C közötti beosztású hőmérő
5.9. Deszikkátor megfelelő szárítószerrel és páratartalom-jelzővel, pl. szilikagél vagy ezzel egyenértékű
5.10. 150 ± 2 °C-ra szabályozható hőmérsékletű szárítószekrény
5.11. pH-mérő
5.12. Főzőlap.
5.13. Szűrőpapír, 4. sz. Whatman-papír vagy azzal egyenértékű.
6. ELJÁRÁS
6.1. Egy 400 ml-es főzőpohárba mérjünk be 5-10 g (M gramm) vizsgálandó mintát úgy, hogy a cinktartalom 50-100 mg között legyen, adjunk hozzá 50 ml desztillált vizet, és keverjük össze.
6.1.1. Szűrjük le, ha szükséges vákuum-szivattyú segítségével, és a szűrletet fogjuk fel.
6.1.2. Az extrakciós lépést még 50 ml desztillált vízzel ismételjük meg. Szűrjük le és a szűrleteket öntsük össze.
6.2. A ( 6.1.2.) oldatban levő cink minden 10 mg-jára számítva adjunk hozzá 2 ml 2-metil-kinolin-8-ol-oldatot (4.7.), és keverjük össze.
6.3. Hígítsuk a keveréket 150 ml desztillált vízzel, melegítsük 60 °C-ig (5.12.), és folyamatos keverés közben adjunk hozzá 45 ml 0,2 M ammónium-acetát-oldatot (4.6.).
6.4. A 6 %-os ammóniaoldattal (4.5.) folyamatos keverés mellett állítsuk be az oldat pH-ját 5,7-5,9 közé; az oldat pH-ját a pH-mérővel mérjük.
6.5. Hagyjuk az oldatot állni 30 percig. Szűrjük vízsugárszivattyú segítségével egy előzőleg kiszárított (150 °C) és lemért G4-es szűrőn, majd mossuk a csapadékot 150 ml 95 °C-os desztillált vízzel.
6.6. Helyezzük a tégelyt 150 °C-ra szabályozott hőmérsékletű szárítószekrénybe, és szárítsuk egy órán át.
6.7. Vegyük ki a tégelyt a szárítószekrényből, tegyük (5.9.) deszikkátorba és amikor elérte a szobahőmérsékletet, mérjük meg a tömegét (M1 gramm).
7. SZÁMÍTÁS
Számítsuk ki a minta százalékos (% m/m) cinktartalmát a következő képlet segítségével:
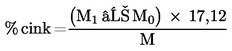
,
ahol:
M = a 6.1. szakasznak megfelelően vett minta tömege grammban;
M0 = az üres, kiszárított szűrőtégely tömege grammban (6.5.);
M1 = a csapadékot tartalmazó szűrőtégely tömege grammban (6.7.);
8. MEGISMÉTELHETŐSÉG ( 7 )
1 % (m/m) körüli cinktartalom esetén az azonos mintán, párhuzamosan végzett két mennyiségi meghatározás eredményei közötti különbség abszolút értéke nem haladhatja meg a 0,1 %-ot.
VII. 4-HIDROXI-BENZOL-SZULFONSAV MENNYISÉGI MEGHATÁROZÁSA ÉS AZONOSÍTÁSA
1. CÉL ÉS ALKALMAZÁSI TERÜLET
A módszer 4-hidroxi-benzol-szulfonsav meghatározására és azonosítására alkalmas kozmetikai termékekben, például aeroszolokban és arcvizekben.
2. FOGALOMMEGHATÁROZÁS
Ezzel a módszerrel meghatározott 4-hidroxi-benzol-szulfonsav-tartalmat vízmentes cink-4-hidroxi-benzol-szulfonát sóként fejezzük ki, és a termék tömegszázalékában adjuk meg.
3. ALAPELV
A vizsgálati mintát csökkentett nyomás mellett betöményítjük, feloldjuk vízben, és kloroformos extrakcióval tisztítjuk. A 4-hidroxi-benzol-szulfonsav-tartalom meghatározása a szűrt vizes oldatból kivett aliquot jodometrikus titrálásával történik.
4. REAGENSEK
Valamennyi reagensnek analitikai tisztaságúnak kell lennie.
4.1. 36 %-os (m/m) tömény sósav; [Kép #2]
Kép #2
4.2. Kloroform
4.3. Bután-1-ol
4.4. Jégecet
4.5. Kálium-jodid
4.6. Kálium-bromid
4.7. Nátrium-karbonát
4.8. Szulfanilsav
4.9. Nátrium-nitrit
4.10. 0,1 N kálium-bromát
4.11. 0,1 N nátrium-tioszulfát-oldat
4.12. 1 % (m/V) vizes keményítőoldat
4.13. 2 % (m/V) nátrium-karbonát vizes oldat
4.14. 4,5 % (m/V) nátrium-nitrit vizes oldat
4.15. 0,05 % (m/V) ditizon kloroformos oldata
4.16. Előhívó szer: bután-1-ol/jégecet/víz (4:1:5 térfogatarányban); választótölcsérben történő összekeverést követően távolítsuk el az alsó fázist.
4.17. Pauly reagens
Oldjunk fel 4,5 g szulfanilsavat (4.8.) 45 ml tömény sósavban (4.1.) melegítés közben, és hígítsuk az oldatot vízzel 500 ml-re. Az oldat 10 ml-ét hűtsük le jeges vízzel egy edényben, és keverés közben adjunk hozzá 10 ml hideg nátrium-nitrit-oldatot (4.14.). Hagyjuk állni az oldatot 15 percig 0 °C-on (e hőmérsékleten az oldat 1-3 napig eltartható), és közvetlenül a lefújása előtt (7.5.) adjunk hozzá 20 ml nátrium-karbonát-oldatot (4.13.).
4.18. Készgyártmány vékonyréteg-kromatográfiás cellulóz lapok: méret 20 x 20 cm, az adszorbens réteg vastagsága 0,25 mm.
5. ESZKÖZÖK
5.1. 100 ml-es gömblombik csiszolt üvegdugóval
5.2. 100 ml-es választótölcsér
5.3. 250 ml-es Erlenmeyer-lombik csiszolt dugóval
5.4. 25 ml-es büretta
5.5. 1, 2 és 10 ml-es hasas pipetta
5.6. 5 ml-es mérőpipetta
5.7. 10 μl-es, 0,1 μl beosztású fecskendő
5.8. 0-100°C közötti beosztású hőmérő
5.9. Fűthető vízfürdő
5.10. Jól szellőző, 80 °C-ra termosztálható szárítószekrény
5.11. Vékonyréteg-kromatográfia szokásos eszközei
6. MINTAELŐKÉSZÍTÉS
A hidroxi-benzol-szulfonsav aeroszolokban történő azonosítására és mennyiségi meghatározására szolgáló, alábbiakban leírt módszerben az aeroszolos dobozból légköri nyomáson elpárolgó oldószerek és a hajtógázok felszabadulása után kapott maradékot használjuk.
7. AZONOSÍTÁS
7.1. A mikrofecskendővel (5.7.) vigyünk fel 5-5 μl-t a maradékból (6) vagy a mintából a vékonyréteg-kromatográfiás lap (4.18.) alsó szélétől 1 cm távolságra levő alapvonalra, összesen hat helyre.
7.2. Helyezzük a lapot az előhívó szert tartalmazó előhívó kádba (4.16.), és addig végezzük az előhívást, amíg az oldószerfront a kiindulástól számított 15 cm távolságra nem jut.
7.3. Vegyük ki a lapot a kádból, és szárítsuk 80 °C-on, amíg az ecetsavszag meg nem szűnik. Fújjuk le a lapot nátrium-karbonát-oldattal (4.13.), és levegőn szárítsuk meg.
7.4. Fedjük le a lap egyik felét egy üveglappal, és a fedetlen részt fújjuk le 0,05 %-os ditizonoldattal (4.15.). Bíborvörösbe hajló piros foltok megjelenése a kromatogramon cinkionok jelenlétére utal.
7.5. Ezt követően fedjük le a lap eddig fedetlen részét egy üveglappal, és fújjuk le a másik felét a Pauly reagenssel (4.17.). A kromatogramon 0,26 Rf értéknél látható sárgásbarna foltok 4-hidroxi-benzol-szulfonsav jelenlétére, míg a 0,45 Rf értéknél látható sárga foltok 3-hidroxi-benzol-szulfonsav jelenlétére utalnak.
8. MENNYISÉGI MEGHATÁROZÁS
8.1. Mérjünk be 10 g mintát vagy maradékot (6) egy 100 ml-es gömblombikba, helyezzük 40 °C-os vízfürdőbe, és rotadeszten pároljuk majdnem teljesen szárazra vákuum alatt.
8.2. Pipettázzunk 10,0 ml vizet (V1) a lombikba, és melegítés közben oldjuk fel a bepárlási maradékot (8.1.).
8.3. Vigyük át az oldatot veszteség nélkül egy választótölcsérbe (5.2.), és vonjuk ki a vizes oldatot kétszer 20 ml kloroformmal (4.2.). Az kivonás után dobjuk el a kloroformos fázist.
8.4. Szűrjük le az oldatot redős szűrőn. A várt hidroxi-benzol-szulfonsav-tartalomtól függően pipettázzuk a szűrlet 1,0-2,0 ml-ét (V2) egy 250 ml-es Erlenmeyer-lombikba (5.3.), majd hígítsuk 75 ml-re vízzel.
8.5. Adjunk hozzá 2,5 ml 36 %-os sósavat (4.1.) és 2,5 g kálium-bromidot (4.6.), keverjük össze, és melegítsük vízfürdőn 50 °C-os hőmérsékletre az oldatot.
8.6. Egy bürettából adjunk hozzá annyi 0,1 N kálium-bromátot (4.10.), hogy a változatlanul 50 °C-os oldat színe sárgára változzon.
8.7. Adjunk hozzá még 3,0 ml kálium-bromát-oldatot (4.10.), zárjuk a lombikot a dugóval, és melegítsük további 10 percen át 50 °C-os vízfürdőn.
Amennyiben 10 perc múlva az oldat elveszíti a színét, adjunk hozzá még 2,0 ml kálium-bromát-oldatot (4.10.), zárjuk a lombikot a dugóval, és melegítsük további 10 percen át 50 °C-os vízfürdőn. Jegyezzük le a hozzáadott kálium-bromát-oldat összes mennyiségét (a).
8.8. Hűtsük le az oldatot szobahőmérsékletűre, adjunk hozzá 2 g kálium-jodidot (4.5.), és keverjük össze.
8.9. Titráljuk a keletkező jódot 0,1 N nátrium-tioszulfát-oldattal (4.11.). A titrálás vége felé indikátorként adjunk az oldathoz néhány csepp keményítőoldatot (4.12.). Jegyezzük fel a nátrium-tioszulfát-oldat fogyását (b).
9. SZÁMÍTÁS
A minta vagy a maradék (6) cink-hidroxi-benzol-szulfonsav-tartalmát tömegszázalékban (% m/m) a következő képlettel számítjuk:
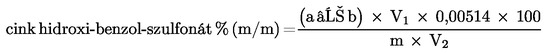
,
ahol:
a = a hozzáadott 0,1 N kálium-bromát-oldat összes mennyisége milliliterben (8.7.);
b = a visszatitráláshoz fogyott 0,1 N nátrium-tioszulfát-oldat mennyisége milliliterben (8.9.);
m = a vizsgált termék vagy maradék mennyisége milligrammban (8.1.);
V1 = a 8.2. pontnak megfelelően kapott oldat térfogata milliliterben;
V2 = a vizsgálathoz felhasznált oldott bepárlási maradék térfogata (8.4.) milliliterben.
Megjegyzés:Aeroszolok esetében a maradék (6) %-ban (m/m) kifejezett mérési eredményét át kell számítani az eredeti termék mennyiségre. Az átszámítással kapcsolatban lásd az aeroszolok mintavételi szabályainak leírását.
10. MEGISMÉTELHETŐSÉG ( 8 )
5 % (m/m) körüli cink-hidroxi-benzol-szulfonát-tartalom esetén az azonos mintán, párhuzamosan végzett két mennyiségi meghatározás eredményei közötti különbség abszolút értékben nem haladhatja meg a 0,5 %-ot.
11. AZ EREDMÉNYEK ÉRTELMEZÉSE
A kozmetikai termékekre vonatkozó irányelv szerint a cink-4-hidroxi-benzol-szulfonát legnagyobb megengedett koncentrációja arclemosókban és dezodorokban 6 % (m/m). Ez a megfogalmazás azt jelenti, hogy a hidroxi-benzol-szulfonsav-tartalom mellett a cinktartalmat is meg kell határozni. A számított cink-hidroxi-benzol-szulfonát tartalmát (9) 0,1588-kal megszorozva megkapjuk azt a minimális cinktartalmat, ami biztosan jelen van a termékben annak hidroxi-benzol-szulfonsav-tartalma miatt. A gravimetrikusan meghatározott cinktartalom azonban ennél nagyobb is lehet, mert a kozmetikai termékek cink-kloridot és cink-szulfátot is tartalmazhatnak (lásd az ide vonatkozó rendeleteket).
( 1 ) HL 192., 1979.7.31., 35. o.
( 2 ) HL 262., 1976.9.27., 169. o.
( 3 ) HL L 147., 1975.6.9., 40. o.
( 4 ) ISO 5725 szabvány szerint.
( 5 ) ISO 5725 szabvány szerint.
( 6 ) ISO 5725 szabvány szerint.
( 7 ) ISO 5725.
( 8 ) ISO 5725 szabvány szerint.
Lábjegyzetek:
[1] A dokumentum eredetije megtekinthető CELEX: 31980L1335 - https://eur-lex.europa.eu/legal-content/HU/ALL/?uri=CELEX:31980L1335&locale=hu Utolsó elérhető, magyar nyelvű konszolidált változat CELEX: 01980L1335-19870213 - https://eur-lex.europa.eu/legal-content/HU/ALL/?uri=CELEX:01980L1335-19870213&locale=hu