
31996L0073[1]
Az Európai Parlament és a Tanács 96/73/EK irányelve (1996. december 16.) a kétkomponensű textilszálkeverékek mennyiségi elemzésének egyes módszereiről
AZ EURÓPAI PARLAMENT ÉS A TANÁCS 96/73/EK IRÁNYELVE
(1996. december 16.)
a kétkomponensű textilszálkeverékek mennyiségi elemzésének egyes módszereiről
AZ EURÓPAI PARLAMENT ÉS AZ EURÓPAI UNIÓ TANÁCSA,
tekintettel az Európai Közösséget létrehozó szerződésre és különösen annak 100a. cikkére,
tekintettel a Bizottság javaslatára ( 1 ),
tekintettel a Gazdasági és Szociális Bizottság véleményére ( 2 ),
a Szerződés 189b. cikkében megállapított eljárásnak megfelelően ( 3 ),
mivel a kétkomponensű textilszálkeverékek mennyiségi elemzésének egyes módszereire vonatkozó tagállami jogszabályok közelítéséről szóló, 1972. július 17-i 72/276/EGK tanácsi irányelvet ( 4 ) gyakran és jelentősen módosították, mivel ezért az átláthatóság és az ésszerűség érdekében szükséges az említett irányelv egységes szerkezetbe foglalása;
mivel a textiltermékek elnevezéséről szóló, 1996. december 16-i 96/74/EK európai parlamenti és tanácsi ( 5 ) irányelv előírja, hogy a címkén tüntessék fel a textiltermék szálösszetételét, és elemzés útján ellenőrizzék, hogy a címkén feltüntetett jelölések megfelelnek-e a textiltermék tényleges összetételének;
mivel a textiltermékek szálösszetételének meghatározása érdekében szükség van a tagállamok által alkalmazott hivatalos vizsgálati módszerek egységesítésére mind a minta előkezelésének, mind mennyiségi elemzésének a tekintetében;
mivel a 96/74/EK irányelvnek megfelelően a tagállamok által a termékek szálösszetételének meghatározására alkalmazandó mintavételi és elemzési módszerekről külön irányelvek rendelkeznek majd; mivel ennek megfelelően ezen irányelv II. melléklete 15 egységes elemzési módszert határoz meg, amelyek alkalmasak a piacon lévő, kétkomponensű szálkeverékekből álló textiltermékek túlnyomó többségének elemzésére;
mivel a technikai fejlődés az egyes irányelvekben a textiltermékek elemzésére meghatározott műszaki előírások gyakori kiegészítését teszi szükségessé; mivel az e célt szolgáló intézkedések végrehajtásának megkönnyítése érdekében a textiltermékek elnevezésére és címkézésére vonatkozó irányelvekkel foglalkozó bizottságon belül olyan eljárást kell megállapítani, amely a Bizottság és a tagállamok között szoros együttműködést hoz létre;
mivel az olyan kétkomponensű textilszálkeverékek esetében, amelyekre nem vonatkozik közösségi szinten egységesített elemzési módszer, a vizsgálatokért felelős laboratórium a rendelkezésére álló bármely érvényes módszerrel meghatározhatja az ilyen szálkeverékek összetételét, a vizsgálati jelentésben megjelölve a kapott eredményeket, valamint - amennyiben ismert - az alkalmazott módszer pontosságát;
mivel az irányelv rendelkezései összhangban vannak a textiltermékek elnevezésére és címkézésre vonatkozó irányelvekkel foglalkozó bizottság véleményével;
mivel ezen irányelv nem érinti a tagállamoknak azt a kötelezettségét, hogy a III. melléklet B. részében meghatározott irányelveket határidőre bevezessék,
ELFOGADTA EZT AZ IRÁNYELVET:
1. cikk
Ez az irányelv a kétkomponensű textilszálkeverékek mennyiségi elemzéséhez alkalmazott egyes módszerekkel foglalkozik, beleértve a vizsgálati minták és próbadarabok előkészítését is.
2. cikk
A "vizsgálati minta" kifejezés az elemzés céljainak megfelelő méretű mintát jelenti, amelyet laboratóriumi nyers mintából vettek, ez utóbbit pedig az elemzésre szánt árucikk szövethengeréből szabták ki.
A "próbadarab" a vizsgálati mintának egy része, amely egyedi vizsgálati eredményt szolgáltat.
3. cikk
A tagállamok megteszik a szükséges lépéseket annak biztosítására, hogy - a 96/74/EK irányelvvel összhangban - az I. és II. mellékletben található, bizonyos kétkomponensű textilszálkeverékek mennyiségi elemzéséhez alkalmazott módszerekre vonatkozó előírásokat, ideértve a vizsgálati minták és próbadarabok előkészítésére vonatkozó szabályokat is, a piacon megjelenő textiltermékek összetételének meghatározásánál alkalmazott összes hivatalos vizsgálati eljárás során betartsák.
4. cikk
Azok a kétkomponensű textilszálkeverékek vizsgálatáért felelős laboratóriumok, amelyekre nem vonatkozik a közösségi szinten egységesített elemzési módszer, a rendelkezésükre álló bármely érvényes módszerrel meghatározhatják az ilyen szálkeverékek összetételét, a vizsgálati jelentésben pedig jelezniük kell a kapott eredményeket, valamint - amennyiben ismert - az alkalmazott módszer pontossági fokát.
5. cikk
A Bizottság a II. mellékletben megadott mennyiségi elemzési módszereket hozzáigazítja a technikai fejlődéshez. Az ezen irányelv nem alapvető fontosságú elemeinek módosítására irányuló ilyen intézkedéseket a 6. cikk (2) bekezdésében említett, ellenőrzéssel történő szabályozási bizottsági eljárással összhangban kell elfogadni.
6. cikk
(1) A Bizottságot a textiltermékek elnevezésére és címkézésére vonatkozó irányelvekkel foglalkozó bizottság segíti.
(2) Az e bekezdésre történő hivatkozáskor az 1999/468/EK határozat 5a. cikkének (1)-(4) bekezdését és 7. cikkét kell alkalmazni, 8. cikkének rendelkezéseire is figyelemmel.
7. cikk
A tagállamok közlik a Bizottsággal nemzeti joguknak azokat a főbb rendelkezéseit, amelyeket az ezen irányelv által szabályozott területen fogadnak el.
8. cikk
A III. melléklet A. részében felsorolt irányelvek hatályon kívül helyezése e rendelkezéssel megtörténik, anélkül, hogy ez érintené a III. melléklet B. részében részletezett irányelvek határidőre való bevezetésének a tagállamokra vonatkozó kötelezettségét.
A hatályon kívül helyezett irányelvekre való hivatkozásokat a továbbiakban erre az irányelvre való hivatkozásként kell kezelni, és a IV. mellékletben megadott megfelelési táblázattal összhangban kell értelmezni.
9. cikk
Ennek az irányelvnek a tagállamok a címzettjei.
Ez az irányelv az Európai Közösségek Hivatalos Lapjában való kihirdetését követő 20. napon lép hatályba.
I. MELLÉKLET
VIZSGÁLATI MINTÁK ÉS PRÓBADARABOK ELŐKÉSZÍTÉSE TEXTILTERMÉKEK SZÁLÖSSZETÉTELÉNEK MEGHATÁROZÁSA CÉLJÁBÓL
1. ALKALMAZÁSI KÖR
Ez a melléklet a mennyiségi elemzéshez szükséges előkezelésnek megfelelő nagyságú (vagyis a 100 gramm tömeget meg nem haladó) laboratóriumi vizsgálati minták laboratóriumi nyersmintából történő kinyerésének, valamint a nem szálas anyagok eltávolítása érdekében előkezelésnek alávetett laboratóriumi vizsgálati mintákból próbadarabok kiválasztásának az eljárásait határozza meg ( 6 ).
2. MEGHATÁROZÁSOK
2.1. Vizsgálandó tétel - Az anyagnak az a mennyisége, amelynek minősítésére egy sor vizsgálati eredmény alapján kerül sor. Ez állhat például egy szövetszállítmányban található összes anyagból; egy lánchengeren szőtt összes szövetből, egy fonálszállítmányból, egy vagy több nyersrost-bálából.
2.2. Laboratóriumi nyers minta - A vizsgálandó tételből vett, a laboratórium számára rendelkezésre álló és az egész tétel szempontjából reprezentatívnak tekintett rész. A laboratóriumi nyers minta méretének és jellegének olyannak kell lennie, hogy megfelelően magában foglalja a vizsgálandó tételben található ingadozásokat és megkönnyítse a minta kezelését a laboratóriumi körülmények között ( 7 ).
2.3. Laboratóriumi vizsgálati minta - A laboratóriumi nyersmintának az a része, amelyet előkezelésnek vetettek alá a nem szálas anyagok eltávolítása érdekében, és amelyből a próbadarabok származnak. A laboratóriumi vizsgálati minta méretének és jellegének olyannak kell lennie, hogy megfelelően magában foglalja a laboratóriumi nyersmintában található ingadozásokat ( 8 ).
2.4. Próbadarab - Az anyagnak az a része, amely a laboratóriumi vizsgálati mintából származik és elegendő ahhoz, hogy egyedi vizsgálati eredményt adjon.
3. VIZSGÁLATI ALAPELV
A laboratóriumi vizsgálati mintát úgy kell kiválasztani, hogy az a laboratóriumi nyers minta szempontjából reprezentatív legyen.
A próbadarabokat úgy kell kiválasztani, hogy azok mindegyike a laboratóriumi vizsgálati minta szempontjából reprezentatív legyen.
4. MINTAVÉTEL LAZA SZÁLAKBÓL
4.1. Rendezetlen szálak - A laboratóriumi vizsgálati mintát a laboratóriumi nyersmintából találomra kiszakított pászmákból nyerjük. Laboratóriumi kártológép segítségével ( 9 ) az egész laboratóriumi vizsgálati mintát alaposan keverjük el. A kártyafátyolt, illetve keveréket a laza szálakkal és a keveréshez használt készülékre tapadt szálakkal egyetemben vessük alá előkezelésnek. Ezt követően az egyes mennyiségeknek megfelelő arányban válasszunk ki próbadarabokat a kártyafátyolból, illetve keverékből, a laza szálakból és a berendezéshez tapadt szálakból.
Amennyiben a kártolt kártyafátyol az előkezelést követően ép marad, a 4.2. pontban leírtak szerint válasszuk ki a próbadarabokat. Ha az előkezelés következtében a kártolt kártyafátyol sérül, a próbadarabokat úgy válasszuk ki, hogy legalább 16 kisebb, megfelelő és megközelítőleg azonos nagyságú pászmából találomra tépünk ki anyagot, majd ezeket állítjuk össze.
4.2. Rendezett szálak (kártolt kártyafátylak, fátyolszalagok, előfont anyagok) - A laboratóriumi nyers minta találomra kiválasztott részeiből vágjunk legalább 10 darab, egyenként kb. 1 gramm tömegű keresztmetszetet. Az ily módon nyert laboratóriumi vizsgálati mintákon végezzük el az előkezelést. Egymás mellé fektetve állítsuk újra össze a metszeteket és emeljük ki a próbadarabot úgy, hogy keresztirányú vágással mind a tíz hosszból vágjunk ki egy-egy darabot.
5. MINTAVÉTEL FONÁLBÓL
5.1. Motringban és csévén kiszerelt fonalak - A laboratóriumi nyers minta összes kiszerelési egységéből vegyünk mintát.
Folyamatosan húzzunk le minden motringról elegendő, azonos hosszúságú fonalat úgy, hogy azonos számú fordulattal ( 10 ), motringokat készítünk egy motollával vagy egy hasonló eszközzel. Egyesítsük egyetlen motringgá vagy kábellé a lecsévélt fonalakat, ügyelve arra, hogy minden motringról azonos hosszúságú fonal kerüljön az így létrehozott vizsgálati mintába.
Végezzük el a laboratóriumi vizsgálati minta előkezelését.
Vegyünk ki próbadarabokat a laboratóriumi vizsgálati mintából úgy, hogy azonos hosszúságú fonalakból álló kötegeket metszünk ki a motringból, illetve kábelből, ügyelve arra, hogy a fonálköteg a minta összes szálát tartalmazza.
Ha a fonál finomságának egysége t és a laboratóriumi nyersmintából kiválasztott fonálkiszerelési egységek száma n, akkor egy 10 g-os vizsgálati mintához minden egyes kiszerelési egységből
cm hosszúságú darabot kell lehúzni.
Ha az érték nt nagyságú, azaz értéke meghaladja a 2 000-et, csévéljünk egy nagyobb tömegű motringot, és azt két helyen vágjuk át, hogy megfelelő tömegű kábelt kapjunk. Bármely, kábel formájában kialakított minta végeit jól kössük el az előkezelés előtt és a próbadarabokat a lekötött végektől megfelelő távolságra vegyük ki.
5.2. Láncfonalak - A laboratóriumi vizsgálati minta vétele úgy történik, hogy a lánchenger végéről egy 20 centiméternél nem rövidebb rész levágunk, amely a láncban található összes fonalat tartalmazza, kivéve a szövetszegély fonalait, amelyeket eltávolítunk. A fonálköteget egyik végéhez közeli ponton megkötözzük. Ha a minta túl nagy ahhoz, hogy egyszerre lehessen előkezelni, osszuk két vagy több részre, mindegyik részt kössük össze az előkezeléshez, majd a külön-külön előkezelt részeket ismét egyesítsük. A laboratóriumi vizsgálati minta összekötött végétől távolabb eső vég felől vágjunk ki egy megfelelő hosszúságú próbadarabot, úgy, hogy a kötegben található összes láncfonál benne legyen. N számú, t finomságú fonálból álló lánc esetén az 1 g tömegű próbadarab hossza [Kép #1] cm lesz.
Kép #1
6. MINTAVÉTEL SZÖVETBŐL
6.1. A szövetet reprezentáló, egyetlen vágatot tartalmazó laboratóriumi nyers minta anyagából
- vágjunk ki keresztben egy csíkot az egyik sarkától a másikig, és a szövetszéleket távolítsuk el. Ez a csík lesz a laboratóriumi vizsgálati minta. Ahhoz, hogy g tömegű laboratóriumi vizsgálati mintát nyerjünk, a csík területének
-nek kell lennie, ahol G a szövet g/m
2
-ben megadott tömege.
- Vessük alá a laboratóriumi vizsgálati mintát előkezelésnek, majd a csíkot vágjuk fel keresztben négy egyforma hosszúságú csíkra és helyezzük a csíkokat egymás tetejére.
- A próbadarabot az így rétegzett anyag bármely részéből vehetjük úgy, hogy az összes réteg keresztülmetszésével kapott próbadarabok minden rétegből azonos hosszúságú részt tartalmazzanak.
- Ha az anyag szövött mintázatú, a laboratóriumi vizsgálati minta szélességét úgy vegyük föl, hogy a láncfonállal párhuzamosan mért irányban legalább egyszer ismétlődjön a szövött mintázat. Ha e feltétel teljesülése által a laboratóriumi vizsgálati minta túl nagy lesz ahhoz, hogy egyben kezeljük, vágjuk fel egyforma részekre, végezzük el az előkezelést, majd a próbadarabok kivétele előtt a darabokat helyezzük egymásra, ügyelve arra, hogy a szövött mintázat azonos részei ne essenek egymás alá.
6.2. Több vágatot tartalmazó laboratóriumi nyers minta anyagából
- minden kivágott darabot kezeljünk a 6.1. pontban leírtak szerint, és mindegyik eredményt külön adjuk meg.
7. MINTAVÉTEL KISZERELT ÉS KÉSZTERMÉKEKBŐL
A laboratóriumi nyers minta rendszerint egy teljes késztermék vagy kiszerelési egység, illetve ezek egy reprezentatív része.
Ahol szükséges, határozzuk meg, hogy a termék különböző részeinek hány százaléka nem azonos szálösszetételű, hogy ellenőrizhessük, megfelel-e a textiltermékek elnevezéséről szóló, 1996. december 16-i 96/74/EK európai parlamenti és tanácsi irányelv 9. cikkében foglaltaknak.
Vegyünk egy reprezentatív laboratóriumi vizsgálati mintát a késztermék, illetve kiszerelt termék azon részéből, amelynek összetételét a címkén fel kell tüntetni. Ha a terméken több címke is szerepel, olyan laboratóriumi vizsgálati mintákat készítsünk, amelyek mindegyike egy adott címkére nézve reprezentatív.
Ha a termék, amelynek összetételét meghatározzuk, nem egységes, a termék minden egyes részéből szükség lehet laboratóriumi vizsgálati mintavételre, illetve a különböző részek és a szóban forgó teljes termék közötti arány meghatározására.
Ezután számítsuk ki a százalékokat, figyelembe véve a mintázott részeknek az egészhez viszonyított arányait.
Végezzük el a laboratóriumi vizsgálati minták előkezelését.
Ezt követően válasszunk ki olyan próbadarabokat, amelyek az előkezelt laboratóriumi vizsgálati mintát reprezentálják.
II. MELLÉKLET
EGYES KÉTKOMPONENSŰ TEXTILSZÁLKEVERÉKEK MENNYISÉGI ELEMZÉSÉNEK MÓDSZEREI
1. ÁLTALÁNOS RÉSZ
Bevezetés
A textilszálkeverékek mennyiségi elemzésének módszerei alapvetően két fő eljáráson alapulnak, a szálak kézi, illetve kémiai úton történő szétválasztásán.
Amennyiben lehetséges, a kézi szétválasztás módszerét alkalmazzuk, mert általában pontosabb eredményt ad a kémiai módszernél. Minden olyan textília esetében alkalmazható, amelynek szálkomponensei nem alkotnak közvetlen keveréket, így például a több alkotóelemből összeállított fonalakban, ahol mindegyik alkotóelem csak egy szálféleségből készült, illetve olyan kelméknél, ahol a láncfonál eltér a vetülékfonál anyagától, vagy olyan felfejthető kötött kelméknél, amelyek különböző szálféleségekből készültek.
A mennyiségi kémiai elemzési módszerek általában az egyes összetevők eltérő oldhatóságán alapulnak. Az egyik komponens kioldását követően az oldhatatlan visszamaradt anyagot lemérjük és a kioldódott komponens arányát a kiindulási tömeg veszteségéből számítjuk ki. A melléklet első része a mellékletben felsorolt valamennyi szálkeveréknek az e módszerrel elvégzett elemzésére vonatkozóan általános tájékoztatást ad, tekintet nélkül a szálkeverékek összetételére. Ezt a részt együtt kell alkalmazni a melléklet további pontjaival, amelyek meghatározott szálkeverékekre vonatkozó eljárás részleteit tartalmazzák. Némely esetben az elemzés nem az eltérő oldhatóság elvén alapszik; ilyen esetekben a megfelelő részben teljes körű leírás szerepel.
A szálkeverékekben a feldolgozás során és kisebb mértékben a késztermékben is találhatóak akár természetes úton előforduló, akár a feldolgozás megkönnyítése érdekében hozzáadott, nem szálas anyagok, például zsírok, viaszok, kikészítőszerek vagy vízoldható anyagok. A nem szálas anyagokat az elemzés előtt el kell távolítani. Ezért megadunk egy módszert az olajok, zsírok, viaszok és vízoldható anyagok eltávolítására is.
Ezen túlmenően a textiltermékek gyantát vagy egyéb olyan anyagot tartalmazhatnak, amelyet különleges tulajdonságok kialakítása érdekében adtak a szálakhoz. Az ilyen anyagok, kivételes esetben akár a színezőanyagok is, megzavarhatják a reagenseknek az oldható komponensre kifejtett hatását, és/vagy a reagens teljesen vagy részlegesen eltávolíthatja azokat. Az ilyen típusú hozzáadott anyag hibák forrása lehet, ezért a minta elemzését megelőzően el kell távolítani ezeket. Ha az ilyen jellegű hozzáadott anyag eltávolítása nem lehetséges, akkor az e mellékletben ismertetett vegyi mennyiségi elemzési módszerek nem alkalmazhatók.
A színezett szálakban található színezőanyag a szálak szerves részének tekintendő, ezért eltávolítására nem kerül sor.
Az elemzés során a száraz tömeget vesszük alapul és ismertetjük a száraz tömeg meghatározásának eljárását is.
Az eredményt úgy kapjuk meg, ha az egyes szálak száraz tömegére a textiltermékek elnevezéséről szóló, 1996. december 16-i 96/74/EK európai parlamenti és tanácsi irányelvnek a II. mellékletében felsorolt, a száraztömegre számított egyezményes járulékanyag-hányadot alkalmazzuk.
Mielőtt bármelyik elemzéshez hozzálátnánk, a keverékben jelenlévő összes szálféleség azonosítását el kell végezni. Egyes módszereknél előfordulhat, hogy a keverék oldható komponensének eltávolítására szolgáló reagensben az oldhatatlan komponens is részben feloldódik. A reagenseket lehetőleg úgy kell megválasztani, hogy csak csekély vagy semmilyen hatásuk ne legyen az oldhatatlan komponensre. Ha ismeretes, hogy az elemzés során tömegveszteség várható, az eredményeket ennek megfelelően kell korrigálni; az erre vonatkozó korrekciós tényezők adottak. A tényezőket több laboratóriumban határozták meg úgy, hogy az előkezeléssel megtisztított szálakat az adott elemzési módszerben használt reagenssel kezelték. Ezek a korrekciós tényezők csak az ép szálakra vonatkoznak, amennyiben a szálak a feldolgozás alatt vagy még azt megelőzően károsodtak, más korrekciós tényezőkre lehet szükség. A megadott eljárások egyedi meghatározásokra vonatkoznak. Kézi és kémiai szétválasztás esetében egyaránt legalább két meghatározást kell elvégezni külön próbadarabon. Amennyiben technikailag lehetséges, megerősítés céljából alternatív eljárást is célszerű alkalmazni, amikor is azt az összetevőt oldjuk ki hamarabb, amely az első módszerben visszamaradt anyagként szerepelt.
I. A TEXTILTERMÉKEK SZÁLKEVERÉKEINEK MENNYISÉGI KÉMIAI ELEMZÉSI MÓDSZERÉRE VONATKOZÓ ÁLTALÁNOS ISMERTETŐ
A textilszálkeverékek kémiai úton történő mennyiségi elemzéséhez megadott valamennyi módszerre vonatkozó közös információ.
I. 1. Cél és alkalmazási kör
Minden megadott módszer alkalmazási köre meghatározza, mely szálak esetében lehet a módszert alkalmazni.
I. 2. Alapelv
Egy textilszálkeverék összetevőinek beazonosítását követően a nem szálas anyagot a megfelelő előkezeléssel eltávolítjuk, majd a két összetevő egyikének eltávolítására kerül sor, rendszerint az eltérő oldhatóság alapján ( 11 ). Az oldhatatlan maradékot lemérjük és a kioldott komponens arányát a tömegveszteségből kiszámítjuk. Azon esetek kivételével, ahol ez technikai nehézségekbe ütközik, ajánlatos a nagyobb arányban szereplő összetevőt kioldani, és a kisebb arányban jelen lévő szálat maradékként kinyerni.
I. 3. Eszközök és reagensek
I. 3.1. Eszközök
I. 3.1.1. Szűrőtégelyek és olyan méretű mérőlombikok, amelyekre ráférnek a szűrőtégelyek, vagy bármely más berendezés, amely azonos eredményt ad.
I. 3.1.2. Szívópalack
I. 3.1.3. Exszikkátor, amely nedvességindikátorral megfestett szilikagélt tartalmaz.
I. 3.1.4. Szellőztethető szárítószekrény a próbadarabok szárítására 105° ± 3 °C.
I. 3.1.5. 0,0002 g pontosságú analitikai mérleg.
I. 3.1.6. Soxhlet extrakciós készülék, vagy más, azonos eredményt adó eszköz.
I. 3.2. Reagensek
I. 3.2.1. Újradesztillált petroléter, forráspont 40-60 °C közötti.
I. 3.2.2. A többi reagenst az egyes módszerek ide vonatkozó pontjai adják meg. Az összes reagensnek vegytisztának kell lennie.
I. 3.2.3. Desztillált vagy ioncserélt víz.
I. 3.2.4. Aceton.
I. 3.2.5. Foszforsav.
I. 3.2.6. Karbamid.
I. 3.2.7. Nátrium-bikarbonát.
I. 4. Kondicionáló és vizsgálati légtér
Miután szárazanyag-tartalom meghatározására kerül sor, szükségtelen a próbadarabokat kondicionálni vagy az elemzést klimatizált légtérben végezni.
I. 5. A laboratóriumi vizsgálati minta
Olyan laboratóriumi vizsgálati mintát készítsünk, amely reprezentálja a laboratóriumi nyers mintát, és előállítható belőle az összes szükséges, legalább 1 g tömegű próbadarab.
I. 6. A laboratóriumi vizsgálati minta előkezelése ( 12 )
Ha valamely anyagot a (textiltermékek elnevezéséről szóló, 1996. december 16-i 96/74/EK európai parlamenti és tanácsi irányelv 12. cikke (3) bekezdésében leírt) százalékos számításokhoz nem szabad figyelembe venni, azt először el kell távolítani egy olyan módszerrel, amely egyik szálkomponensre sincs hatással.
Ennek érdekében a vízzel és petroléterrel eltávolítható nem szálas anyagok kivonása úgy történik, hogy a légszáraz mintát Soxhlet extrahálókészülékben egy órán keresztül petroléteres kivonásnak vetjük alá, óránként legalább 6 ciklus sebességgel. A petrolétert engedjük elpárologni a mintából, majd közvetlen kezeléssel extrahálunk, ami abból áll, hogy a mintát szobahőmérsékleten egy órán keresztül vízben áztatjuk, majd ezt követően az áztatást egy további órán keresztül megismételjük 65 ± 5 °C hőmérsékletű vízben, a folyadékot időnként felkeverve. A folyadék és a minta aránya 100:1 legyen. A felesleges vizet kipréseléssel, leszívatással vagy centrifugálással távolítsuk el a mintából, majd hagyjuk, hogy levegőn megszáradjon.
Elasztolefint, vagy elasztolefint és más szálas anyagot (gyapjú, állati szőr, selyem, pamut, len, valódi kender, juta, manilakender, alfa, kókuszrost, seprűzanót, hócsalán, szizál, cupro, modálszál, fehérje, viszkóz, akril, poliamid vagy nejlon, poliészter és elaszto-multiészter) tartalmazó textilszálkeverékek esetében az imént ismertetett eljárást kissé módosítani kell, azaz a petrolétert acetonnal kell felváltani.
Az elasztolefint és acetátot tartalmazó textilszálkeverékek esetében az alábbi eljárást kell előkezelésként alkalmazni. Extraháljuk a mintát 80 °C-on 10 percig 25 g/l 50 %-os foszforsavat és 50 g/l karbamidot tartalmazó oldattal. A folyadék és a minta aránya 100:1 legyen. A mintát mossuk ki vízben, majd távolítsuk el a vizet és mossuk le 0,1 %-os nátrium-bikarbonát oldattal, majd végezetül gondosan mossuk le vízzel.
Ha a nem szálas anyag petroléterrel és vízzel nem vonható ki, a fentebb leírt vizes eljárást célszerű olyan megfelelő módszerrel helyettesíteni, amely a szál összetevőit nem változtatja meg számottevő mértékben. Némely fehérítetlen, természetes növényi rostot tartalmazó szál esetében (például juta vagy kókuszrost) megjegyzendő, hogy a petroléterrel és vízzel végzett normál előkezelés nem távolítja el az összes nem szálas anyagot; ennek ellenére nem alkalmazunk további előkezelést, hacsak a minta nem tartalmaz vízben és petroléterben egyaránt oldhatatlan appretálószereket.
Az elemzésről készült jelentésnek részletesen ki kell térnie az alkalmazott előkezelési módszerekre.
I. 7. Vizsgálati eljárás
I. 7.1. Általános útmutató
I. 7.1.1. Szárítás
Az összes szárítási eljárást legalább négy, de legfeljebb 16 óra hosszat kell végezni 105 ± 3 °C-on szellőztetett szárítószekrényben, amelynek ajtaját végig zárva tartjuk. Ha a szárítás időtartama nem haladja meg a 14 órát, a próbadarabot a tömegállandóság végett meg kell mérni. A tömeg akkor tekinthető állandónak, ha további 60 perces szárítást követően a tömegváltozás kisebb, mint 0,05 %.
A szárítási, hűtési, valamint mérési eljárások során igyekezzünk elkerülni a szűrőtégelyek, mérőedények, próbadarabok vagy visszamaradt anyag puszta kézzel történő érintését.
A próbadarabokat mérőedényben szárítsuk, úgy, hogy a fedelet az edény mellé helyezzük. A szárítást követően a szárítószekrényből való kivételt megelőzően fedjük be a mérőedényt, és gyorsan helyezzük át az exszikkátorba.
A szűrőtégelyt mérőedényben szárítsuk, úgy, hogy közben a fedelet az edény mellé helyezzük. A szárítást követően fedjük be a mérőedényt, és gyorsan helyezzük át az exszikkátorba.
Ha nem szűrőtégelyt használtunk, a szárítószekrényben végzett szárítási folyamatot úgy kell kialakítani, hogy a szálak száraz tömegét veszteség nélkül meg lehessen határozni.
I. 7.1.2. Hűtés
Az exszikkátort a mérleg mellé helyezve minden hűtési műveletet addig folytatunk, amíg a mérőedény teljesen le nem hűlt, és ez két óránál nem lehet rövidebb.
I. 7.1.3. Tömegmérés
A hűtés után a mérőedényt az exszikkátorból való eltávolítást követő két percen belül le kell mérni. A mérési pontosság 0,0002 g legyen.
I. 7.2. Eljárás
Az előkezelt laboratóriumi vizsgálati mintából legalább 1 g tömegű próbadarabot készítünk. Vágjunk a szövetből, illetve fonalból körülbelül 10 mm hosszú darabokat, minél jobban bontsuk szét őket. A próbadarabokat mérőedényben megszárítjuk, az exszikkátorban visszahűtjük, majd megmérjük. A próbadarabot a vonatkozó uniós módszer megfelelő szakaszában meghatározott üvegedénybe tesszük át, a mérőedényt azonnal megmérjük és különbségképzéssel meghatározzuk a próbadarab tömegét. Az alkalmazott módszer megfelelő pontjában leírtaknak megfelelően befejezzük a vizsgálatokat. A visszamaradt anyagot mikroszkóp alatt megvizsgáljuk, hogy meggyőződjünk róla, a kezelés valóban teljes mértékben eltávolította a fonalból az oldható szálat.
I. 8. Eredmények kiszámítása és megadása
Az oldhatatlan komponens tömegét a keverékben lévő szálak teljes tömegének százalékában fejezzük ki. Az oldható komponens százalékát különbségszámítással határozzuk meg. Az eredményeket tiszta, száraz tömegre adjuk meg és a) a egyezményes járulékanyag-hányaddal, valamint b) az előkezelés és az elemzés során előforduló tömegveszteség figyelembevételéhez szükséges korrekciós tényezővel pontosítjuk. A számításokat az I.8.2. pontban megadott képletnek megfelelően kell elvégezni.
I. 8.1. Az oldhatatlan komponens kiszámításának módja tiszta, száraz tömeg százalékában kifejezve, az előkezelés során bekövetkezett tömegveszteség figyelembevétele nélkül.
ahol
P1 tiszta, száraz oldhatatlan komponens százalékban kifejezve
m a próbadarab száraz tömege az előkezelést követően
r a visszamaradt anyag száraz tömege
d az elemzés során a reagensben elveszett oldhatatlan komponens tömegveszteségére vonatkozó korrekciós tényező. A "d" alkalmazandó értékét az egyes módszerek megfelelő szövegrészénél található meg.
A "d" ezen értékeit természetesen normál értékeknek kell tekinteni, amelyek a vegyileg sértetlen szálak esetében alkalmazhatók.
I. 8.2. Az oldhatatlan komponens százalékarányának kiszámítási módja tiszta, száraz tömeg százalékában kifejezve és az egyezményes tényezőkkel, szükség esetén az előkezelés során bekövetkezett tömegveszteség figyelembevételére alkalmazott korrekciós tényezőkkel kiegészítve.
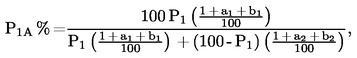
ahol
P1A az oldhatatlan komponens százaléka, az egyezményes járulékanyag-hányaddal és az előkezelés során bekövetkezett tömegveszteség értékével korrigálva
P1 a száraz, tiszta oldhatatlan komponens százaléka az I.8.1. pontban feltüntetett képlettel számolva
a1 az oldhatatlan komponens egyezményes járulékanyag-hányada (lásd a textiltermékek elnevezésekről szóló irányelv II. mellékletét)
a2 az oldható komponens egyezményes járulékanyag-hányada (lásd a textiltermékek elnevezésekről szóló irányelv II. mellékletét)
b1 az oldhatatlan komponens százalékos tömegvesztesége az előkezelés során
b2 az oldható komponens százalékos tömegvesztesége az előkezelés során
A második komponens százaléka (P2A%) = 100 - P1A%.
Ahol különleges előkezelést alkalmaztunk, a b1 és b2 értékeit lehetőség szerint úgy kell meghatározni, hogy az elemzés során alkalmazott előkezelésnek mindkét összetevőt alávetjük. Tiszta szálnak azt tekintjük, amely mentes a nem szálas anyagoktól (akár természeténél fogva, akár a gyártási eljárás következtében) kivéve azokat, amelyeket rendes körülmények között abban az állapotban (fehérítetlen vagy fehérített), amelyben az elemzett anyagban megtalálható.
Amennyiben nem áll rendelkezésre az elemzésre szánt anyag gyártási eljárása során alkalmazott tiszta, elkülönített szálkomponens, a b1 és b2 azon átlagos értékeit kell használni, amelyeket az elemzés alatt álló keverékben találhatóhoz hasonló tiszta szálakkal végzett elemzések során nyertek.
Ha a petroléterrel és vízzel végzett szabályos előkezelést alkalmazzuk, a b1 és b2 korrekciós tényezőket rendszerint nem kell figyelembe venni, kivéve a fehérítés nélküli pamut, len és kender esetét, ahol az előkezelés következtében előállott veszteséget egyezményesen 4& 14;%-nak vesszük, valamint a polipropilén esetét, amelynél ez az érték 1 %.
Más szálak esetében a számításoknál egyezményes alapon nem vesszük figyelembe az előkezelés során bekövetkezett veszteségeket.
II. A KÉZI ELVÁLASZTÁSSAL VÉGZETT MENNYISÉGI ELEMZÉS MÓDSZERE
II. 1. Alkalmazási kör
Ezt a módszert alkalmazzuk a textilszálak valamennyi típusánál, ha azok nem képeznek intim keveréket, vagyis kézzel szétválaszthatók.
II. 2. Alapelv
A textiltermék komponenseinek azonosítását követően a megfelelő előkezeléssel a nem szálas anyagokat eltávolítjuk, majd a szálakat kézzel elválasztjuk egymástól, megszárítjuk és megmérjük avégett, hogy kiszámíthassuk az egyes szálak százalékos arányát a keverékben.
II. 3. Eszközök
II. 3.1. Mérőedények vagy más, azonos eredményt adó eszközök.
II. 3.2. Exszikkátor, amely nedveségindikátorral megfestett szilikagélt tartalmaz.
II. 3.3. Szellőztethető szárítószekrény a próbadarabok szárítására 105 ± 3 °C-on.
II. 3.4. 0,0002 g pontosságú analitikai mérleg.
II. 3.5. Soxhlet extrakciós készülék, vagy más, azonos eredményt adó készülék.
II. 3.6. Tű.
II. 3.7. Sodratszámláló készülék vagy más hasonló eszköz.
II. 4. Reagensek
II. 4.1. Újradesztillált petroléter, forráspont 40-60 °C között.
II. 4.2. Desztillált vagy ioncserélt víz.
II. 5. Kondicionálás és vizsgálati légtér
Lásd I.4.
II. 6. Laboratóriumi vizsgálati minta
Lásd I.5.
II. 7. A laboratóriumi vizsgálati minta előkezelése
Lásd I.6.
II. 8. Eljárás
II. 8.1. Fonál elemzése
Az előkezelt laboratóriumi vizsgálati mintából válasszunk ki egy legalább 1 g tömegű próbadarabot. Nagyon finom fonalak esetében az elemzést legalább 30& ;m hosszban kell elvégezni, függetlenül annak tömegétől.
A fonalat alkalmas hosszúságú darabokra vágjuk fel, és a szálakat tű segítségével, illetve szükség esetén sodratszámlálóval válasszuk szét egymástól. Az így nyert száltípusokat előre lemért (kitárázott) mérőedényekbe helyezzük és 105 ± 3 °C-on tömegállandóságig szárítjuk az I.7.1., valamint I.7.2. pontban leírtak szerint.
II. 8.2. Szövet elemzése
Az előkezelt laboratóriumi vizsgálati mintából a szélektől jókora távolságban válasszunk ki egy legalább 1 g tömegű próbadarabot, amelyet a kirojtosodás elkerülése érdekében a szélek mentén gondosan levágunk, és amely a lánc vagy vetülékfonalakkal párhuzamosan, vagy kötött anyag esetén a szemsorok és szemoszlopok vonalában fut. Válasszuk szét a különböző szálféleségeket, gyűjtsük össze előre lemért mérőedényekben és folytassuk az eljárást a II.8.1. pontban leírtaknak megfelelően.
II. 9. Eredmények kiszámítása és megadása
Az egyes szálkomponensek tömegét a keverék teljes tömegének százalékában fejezzük ki. Az eredményeket tiszta, száraz tömegre adjuk meg és a) az egyezményes járulékanyag-hányaddal, valamint b) az előkezelés során előforduló tömegveszteség figyelembevételéhez szükséges korrekciós tényezővel pontosítjuk.
II. 9.1. A tiszta, száraz szálak tömegszázalékának kiszámítása az előkezelés során fellépő tömegveszteség figyelembevétele nélkül:
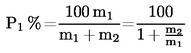
,
ahol
P1 a tiszta, száraz első komponens százaléka
m1 a tiszta, száraz első komponens tömege
m2 a tiszta, száraz második komponens tömege.
II. 9.2. Az egyes összetevők száraz tömegéhez, egyezményes járulékanyag-hányaddal, valamint szükség esetén az előkezelés során bekövetkezett tömegveszteségre tekintettel alkalmazandó korrekciós tényezőkkel helyesbített százalékarányának kiszámításához lásd az I.8.2. pontban leírtakat.
III. 1. A módszerek pontossága
Az egyes módszereknél megadott pontosság értéke a megismételhetőségre vonatkozik.
A megismételhetőség a megbízhatóságra utal, vagyis arra, hogy a különböző laboratóriumok személyzete által, illetve eltérő időpontokban elvégzett vizsgálatok eredményeként kapott kísérleti értékek mennyire szoros egyezést mutatnak, ha ugyanazt a módszert alkalmazzák és azonos, állandó keverékből származó próbadarabokból nyernek egyedi eredményeket.
A megismételhetőség fokát az eredményeknek 95& 14; %-os konfidenciaszinten megadott konfidenciahatárok fejezik ki.
Ez alatt azt értjük, hogy a módszer azonos és állandó szálkeverékre történő szabályos és helyes alkalmazása esetében a különböző laboratóriumokban végzett elemzések sorozatában két eredmény eltérése 100 esetből csak 5-ben haladja meg a konfidenciahatárt.
III. 2. Vizsgálati jelentés
III. 2.1. Közöljük, hogy az elemzést ennek a módszernek az alapján végeztük el.
III. 2.2. Adjuk meg az alkalmazott különleges előkezelés részleteit (lásd I.6.).
III. 2.3. Adjuk meg az egyedi eredményeket és a számtani középértéket, mindkettőt 1 tizedesjegy pontossággal.
2. ÖSSZEFOGLALÓ TÁBLÁZAT
| Módszer | Alkalmazási kör (1) | Reagens | |
| Oldható komponens | Oldhatatlan komponens | ||
| 1. | acetát | bizonyos más szálak | aceton |
| 2. | egyes fehérjeszálak | bizonyos más szálak | hipoklorit |
| 3. | viszkóz, réz-oxid vagy meghatározott típusú modálszálak | bizonyos más szálak | hangyasav és cink-klorid |
| 4. | poliamid vagy nejlon | bizonyos más szálak | hangyasav, 80 tömeg% |
| 5. | acetát | bizonyos más szálak | benzilalkohol |
| 6. | triacetát vagy polilaktid | bizonyos más szálak | diklór-metán |
| 7. | egyes cellulózszálak | bizonyos más szálak | kénsav, 75 tömeg% |
| 8. | akril, egyes modakrilok vagy polikloridok | bizonyos más szálak | dimetil-formamid |
| 9. | egyes polikloridok | bizonyos más szálak | széndiszulfid/aceton, 55,5/44,5 térfogat% |
| 10. | acetát | bizonyos más szálak | jégecet |
| 11. | selyem, poliamid vagy nejlon | bizonyos más szálak | kénsav, 75 tömeg% |
| 12. | juta | egyes állati eredetű szálak | nitrogéntartalmat mérő módszer |
| 13. | polipropilén | bizonyos más szálak | xilol |
| 14. | bizonyos szálak | bizonyos más szálak | koncentrált kénsavas módszer |
| 15. | polikloridok, egyes modakrilok, egyes elasztánok, acetátok, triacetátok | bizonyos más szálak | ciklohexanon |
| 16. | melamin | bizonyos más szálak | forró hangyasav, 90 tömeg% |
| (1) A szálak részletes felsorolása az egyes módszereknél található. | |||
1. MÓDSZER - ACETÁT ÉS BIZONYOS MÁS SZÁLAK
(Acetonos módszer)
1. ALKALMAZÁSI KÖR
Ez a módszer a nem szálas anyagok eltávolítását követően a következő összetételű, kétkomponensű textilszálkeverékek esetében alkalmazható:
1. acetát (19),
valamint
2. gyapjú (1), állati szőr (2 és 3), selyem (4), pamut (5), len (7), valódi kender (8), juta (9), manilakender (10), alfafű (11), kókuszrost (12), seprűzanót (13), rami (14), szizál (15), rézoxid-szál (21), modálszál (22), fehérje (23), viszkóz (25), akril (26), poliamid vagy nejlon (30), poliészter (35), polipropilén (37), elaszto-multiészter (46), elasztolefin (47), melamin (48) és polipropilén/poliamid kétkomponensű szál (49).
Ez a módszer semmilyen körülmények között nem alkalmazható olyan acetátszálak esetében, amelyek felületét deacetilezték.
2. ALAPELV
A keverék ismert száraz tömegéből acetonnal kioldjuk az acetátszálakat. A visszamaradt anyagot összegyűjtjük, kimossuk, megszárítjuk és megmérjük; tömegét szükség esetén korrigálva a keverék száraz tömegének százalékában fejezzük ki. Az acetát száraz tömegének százalékát a két adat különbsége adja.
3. ESZKÖZÖK ÉS REAGENSEK (az általános útmutatóban meghatározottakon kívül)
3.1. Eszközök
Legalább 200 ml űrtartalmú, becsiszolt üvegdugóval ellátott Erlenmeyer-lombikok.
3.2. Reagens
Aceton.
4. VIZSGÁLATI ELJÁRÁS
Kövessük az általános útmutatóban leírtakat, és a következőképpen járjunk el:
A legalább 200 ml űrtartalmú, becsiszolt üvegdugóval ellátott Erlenmeyer-lombikban lévő próbadarab minden grammjára 100 ml acetont öntünk, az Erlenmeyer-lombikot összerázzuk és 30 percig szobahőmérsékleten állni hagyjuk, majd a folyadékot az előzetesen lemért szűrőtégelyen keresztül dekantáljuk.
A kezelést még kétszer megismételjük (összesen három extrakciót végezve), de csupán 15 perces időszakokra, úgy, hogy ezzel az acetonos kezelés teljes időtartama egy óra legyen. A visszamaradt anyagot szűrőtégelyre visszük át. A szűrőtégelyben található szálmaradékot acetonnal lemossuk és leszívással ürítjük. Ezután ismét megtöltjük a szűrőtégelyt acetonnal és engedjük, hogy magától leszivárogjon.
Végül a szűrőtégelyt leszívással ürítjük, majd megszárítjuk, lehűtjük, és a szálmaradékkal együtt megmérjük.
5. AZ EREDMÉNYEK KISZÁMÍTÁSA ÉS KIFEJEZÉSE
Az eredményeket az általános útmutatóban meghatározottak szerint számítsuk ki. A "d" értéke 1,00, kivéve a melamin szálak esetében, amelyeknél "d" = 1,01.
6. PONTOSSÁG
Textilanyagok homogén keveréke esetében az e módszerrel nyerhető eredmények konfidenciahatára 95& 14;%-os konfidenciaszint esetén legfeljebb ± 1.
2. MÓDSZER - BIZONYOS FEHÉRJESZÁLAK ÉS BIZONYOS MÁS SZÁLAK
(Hipokloritos módszer)
1. ALKALMAZÁSI KÖR
Ez a módszer a nem szálas anyagok eltávolítását követően a következő összetételű, kétkomponensű textilszálkeverékek esetében alkalmazható:
1. bizonyos fehérjeszálak, nevezetesen gyapjú (1), állati szőrök (2 és 3), selyem (4), fehérje (23),
valamint
2. pamut (5), rézoxid-szál (21), viszkóz (25), akril (26), polikloridok (27), poliamid vagy nejlon (30), poliészter (35), polipropilén (37), elasztán (43), üvegszál (44), elaszto-multiészter (46), elasztolefin (47), melamin (48) és polipropilén/poliamid kétkomponensű szál (49).
Ha különböző fehérjeszálak vannak jelen, akkor a módszerrel ezek összes tömegét határozzuk meg és nem egyedi mennyiségüket.
2. ALAPELV
Az ismert száraz tömegű keverékből hipoklorit oldattal kioldjuk a fehérjeszálakat. A visszamaradt anyagot összegyűjtjük, kimossuk, megszárítjuk és megmérjük, tömegét pedig, szükség esetén korrigálva, a keverék száraz tömegének százalékában adjuk meg. A fehérje száraz tömegének százalékát a két adat különbsége adja.
A hipoklorit oldat elkészítéséhez vagy lítium-hipoklorit, vagy nátrium-hipoklorit használható.
A lítium-hipoklorit akkor ajánlható, ha kevés elemzést igénylő esetről van szó, illetve ha az elemzéseket nagyobb időközönként végezzük. Ennek az az oka, hogy a szilárd formájú lítium-hipokloritban - ellentétben a nátrium-hipoklorittal - a hipokrit százalékos aránya lényegileg állandó. Ha a hipoklorit százalékos aránya ismert, a hipoklorit tartalmat nem szükséges jodometriásan ellenőrizni minden egyes elemzés esetében, mert a konstans, lemért lítium-hipokloritot lehet alkalmazni.
3. ESZKÖZÖK ÉS REAGENSEK (az általános útmutatóban meghatározottakon kívül)
3.1. Eszközök
i. 250 ml-es, becsiszolt üvegdugóval ellátott Erlenmeyer-lombik;
ii. 20 (± 2) °C-ra állítható termosztát.
3.2. Reagensek
i. Hipoklorit reagens
a) Lítium hipoklorit oldat
Ez egy frissen elkészített 35 (± 2) g/l aktív klórt tartalmazó oldatból (kb. 1 M) áll, amelyhez 5 (± 0,5) g/l előzetesen feloldott nátrium-hidroxidot adunk. Az elkészítéshez 35 % aktív klórt tartalmazó 100 gramm (vagy 115 gramm 30 % aktív klórtartalmú anyagot) lítium-hipokloritot megközelítőleg 700 ml desztillált vízben kell feloldani, ehhez hozzáadni 5 gramm körülbelül 200 ml desztillált vízben feloldott nátrium-hidroxidot, és az egészet desztillált vízzel 1 literre kiegészíteni. A frissen elkészített oldat jodometriás ellenőrzése nem szükséges.
b) Nátrium-hipoklorit oldat
Ez egy frissen elkészített 35 (± 2) g/l aktív klórt tartalmazó oldatból (kb. 1 M) áll, amelyhez 5 (± 0,5) g/l korábban feloldott nátrium hidroxidot adunk.
Az oldat aktív klórtartalmát minden elemzés előtt jodometriásan ellenőrizni kell.
ii. Ecetsav, hígított oldat
5 ml jégecetet vízzel hígítsunk fel 1 literre.
4. VIZSGÁLATI ELJÁRÁS
Kövessük az általános útmutatóban leírtakat, és a következőképpen járjunk el: a körülbelül 1 gramm tömegű mintát megközelítőleg 100 ml hipoklorit oldattal (lítium vagy nátrium-hipoklorit) helyezzük egy 250 ml-es lombikba és alaposan rázzuk fel, hogy a próbadarab átnedvesedjen.
Ezt követően 40 percig melegítsük a lombikot 20 °C-on állandó vagy legalábbis rendszeres időközönként végzett rázogatás mellett. Miután a gyapjú kioldódása hőtermeléssel jár, a módszerben keletkező reakcióhőt el kell oszlatni és ki kell űzni. Ellenkező esetben jelentős mértékű hiba állhat elő a nem oldható szálak kezdődő kioldódása miatt.
A 40 perc elteltével a lombik tartalmát szűrjük le egy lemért üveg szűrőtégelyen, és a visszamaradt szálakat tegyük szintén át a tégelybe úgy, hogy a lombikot egy kevés hipoklorit reagenssel kiöblítjük. A tégelyt leszívással ürítsük le, ezt követően a szálmaradékot mossuk át vízzel, híg ecetsavval, végül ismét vízzel, miközben a tégelyt minden egyes folyadék-hozzáadás után leszívatjuk. A leszívást addig nem kell elkezdeni, amíg a mosófolyadék magától el nem távozott.
Végül szívassuk le a tégelyt, a visszamaradt anyaggal együtt szárítsuk meg, hűtsük le és mérjük meg.
5. AZ EREDMÉNYEK KISZÁMÍTÁSA ÉS KIFEJEZÉSE
Az eredményeket az általános útmutatóban meghatározottak szerint számítsuk ki. A "d" értéke 1,00, kivéve a pamut, viszkóz, modál és melamin szálak esetében, amelyeknél "d" = 1,01, valamint a fehérítés nélküli pamutnál, amelynél "d" = 1,03.
6. PONTOSSÁG
Textilanyagok homogén keveréke esetében az ezen módszerrel nyerhető eredmények konfidenciahatára 95 14%-os konfidenciaszint esetén legfeljebb ± 1.
3. MÓDSZER - VISZKÓZ, RÉZOXID-SZÁL VAGY BIZONYOS MODÁLSZÁLTÍPUSOK ÉS BIZONYOS MÁS SZÁLAK
(Hangyasavas és cink-kloridos módszer)
1. ALKALMAZÁSI KÖR
Ez a módszer a nem szálas anyagok eltávolítását követően a következő összetételű, kétkomponensű textilszálkeverékek esetében alkalmazható:
1. viszkóz (25) vagy cupro (21), beleértve a modál szálak bizonyos típusait is (22),
valamint
2. pamut (5), polipropilén (37), elasztolefin (47) és melamin (48).
Ha modálszál is jelen van, előzetes vizsgálatra van szükség annak megállapítására, hogy az oldódik-e a reagensben.
Ez a módszer nem alkalmas olyan keverékek vizsgálatára, amelyekben a pamutot erős kémiai roncsoló hatás érte, illetve ahol a viszkóz vagy rézoxid-szál részlegesen oldhatóvá vált bizonyos teljes mértékben el nem távolítható színezékek vagy kikészítőanyagok jelenléte következtében.
2. ALAPELV
A viszkóz, cupro vagy modál szálakat az ismert száraz tömegű keverékből hangyasavval vagy cink-kloriddal oldjuk ki. A visszamaradt anyagot összegyűjtjük, szárítjuk és mérjük, szükség esetén korrigált tömegértékét pedig a keverék száraz tömegének százalékában fejezzük ki. A száraz viszkóz, cupro, illetve modál szálak százalékát a különbségképzéssel határozzuk meg.
3. ESZKÖZÖK ÉS REAGENSEK (az általános útmutatóban meghatározottakon kívül)
3.1. Eszközök
i. legalább 200 ml űrtartalmú, becsiszolt üvegdugóval ellátott Erlenmeyer-lombikok;
ii. az Erlenmeyer-lombik 40 (± 2) °C-on tartására alkalmas készülék.
3.2. Reagensek
i. Az oldathoz feloldunk 20 gramm olvasztott vízmentes cink-kloridot és 68 g vízmentes hangyasavat, majd vízzel 100 g-ra egészítjük ki (vagyis 80 tömegegység 85 tömeg%-os hangyasavhoz adunk 20 tömegegység vízmentes ömlesztett cink-kloridot).
Megjegyzés:
Legyünk figyelemmel az I.3.2.2. pontra, amely azt szabályozza, hogy a felhasznált összes reagensnek vegytisztának kell lennie, emellett létfontosságú, hogy csak olvasztott vízmentes cink-kloridot használjunk.
ii. Ammónium hidroxid oldat: 20 ml tömény ammónium hidroxidot (melynek fajlagos súlya 0,880 g/ml) vízzel hígítsunk fel 1 literre.
4. VIZSGÁLATI ELJÁRÁS
Kövessük az általános útmutatóban leírtakat, és a következőképpen járjunk el: a próbadarabot azonnal helyezzük el a 40 °C-ra előmelegített Erlenmeyer-lombikban. A próbadarab minden grammjához adjunk hozzá 100 ml-t a hangyasav és cink-klorid oldatából, amelyet szintén 40 °C-ra melegítettünk fel. Zárjuk le a lombikot és rázzuk fel erősen. Két és fél órán keresztül óránként felrázva tartsuk a lombikot és tartalmát állandó, 40 °C-os hőmérsékleten. Szűrjük le a lombik tartalmát egy lemért szűrőtégelyen és a reagens segítségével a lombikban maradt esetleges szálmaradványokat tegyük át a szűrőtégelyre. A reagens 20 milliliterével öblítsük el.
Vízzel alaposan mossuk ki a szűrőtégelyt és a visszamaradt anyagokat 40 °C-on. A szálmaradékot öblítsük ki kb. 100 ml hideg ammóniaoldat segítségével (3.2. ii. pont), és biztosítsuk, hogy a visszamaradt anyagot ez az oldat 10 percen keresztül teljesen ellepje ( 13 ); majd alaposan öblítsük ki hideg vízzel.
Amíg a mosófolyadék önmagáról el nem folyt, a leszívást ne kezdjük meg. Végül szívassuk le a tégelyt, szárítsuk meg, hűtsük le és mérjük meg a szálmaradékkal együtt.
5. AZ EREDMÉNYEK KISZÁMÍTÁSA ÉS KIFEJEZÉSE
Az eredményeket az általános útmutatóban meghatározottak szerint számítsuk ki. A "d" értéke 1,00, kivéve a pamut esetében, amelynél "d" = 1,02, valamint a melamin esetében, amelynél "d"= 1,01.
6. PONTOSSÁG
Textilanyagok homogén keveréke esetében az e módszerrel nyerhető eredmények konfidenciahatára 95 14%-os konfidenciaszint esetén legfeljebb ± 2.
4. MÓDSZER - POLIAMID VAGY NEJLON ÉS BIZONYOS MÁS SZÁLAK
(80 tömeg%-os hangyasavat alkalmazó módszer)
1. ALKALMAZÁSI KÖR
Ez a módszer a nem szálas anyagok eltávolítását követően a következő összetételű, kétkomponensű textilszálkeverékek esetében alkalmazható:
1. poliamid vagy nejlon, (30),
valamint
2. gyapjú (1), állati szőr (2 és 3), pamut (5), rézoxid-szál (21), modál (22), viszkóz (25), akril (26), poliklorid (27), poliészter (35), polipropilén (37), üvegszál (44), elaszto-multiészter (46), elasztolefin (47) és melamin (48).
Mint azt fentebb említettük, ezt a módszert gyapjúkeverékekre is lehet alkalmazni, de ahol a gyapjútartalom meghaladja a 25 %-ot, a 2. módszert kell alkalmazni (a gyapjút lúgos nátrium-hipoklorit oldattal kell kioldani).
2. ALAPELV
A poliamid szálakat a ismert száraz tömegű keverékből hangyasavval oldjuk ki. A visszamaradt anyagot összegyűjtjük, szárítjuk és mérjük, szükség esetén korrigált tömegértékét pedig a keverék száraz tömegének százalékában fejezzük ki. A száraz poliamid- vagy nejlonszál százalékát különbségképzéssel számoljuk ki.
3. ESZKÖZÖK ÉS REAGENSEK (az általános útmutatóban meghatározottakon kívül)
3.1. Eszközök
Legalább 200 ml űrtartalmú, becsiszolt üvegdugóval ellátott Erlenmeyer-lombikok.
3.2. Reagensek
i. Hangyasav (80 tömeg%-os, relatív sűrűsége 20 °C-on: 1,186 g/cm3). 880 ml 90 tömeg%-os hangyasavat (relatív sűrűsége 20 °C-on: 1,204 g/cm3) vízzel hígítsunk fel 1 literre. Alternatív lehetőségként 780 ml 98-100 tömeg %-os hangyasavat (relatív sűrűsége 20 °C-on: 1,220 g/cm3) egészítsünk ki vízzel 1 literre.
A hangyasav töménysége 77 és 83 tömeg% között nem kritikus.
ii. Híg ammóniaoldat: tömény ammóniaoldatból 80 ml-t (relatív sűrűsége 20 °C-on: 0,880 g/cm3) egészítsünk ki vízzel 1 literre.
4. VIZSGÁLATI ELJÁRÁS
Kövessük az általános útmutatóban leírtakat, és a következőképpen járjunk el: a legalább 200 ml űrtartalmú lombikba helyezett minta minden grammjához adjunk 100 ml hangyasavat. Tegyük rá az üvegdugót és alaposan rázzuk fel, hogy a minta átnedvesedjen. Ezt követően tartsuk a lombikot 15 percig szobahőmérsékleten, rendszeres időközönként felrázva. A lombik tartalmát ürítsük ki leszívással egy lemért üveg szűrőtégelyen, és a visszamaradó szálakat a tégelyen mossuk át egymást követően hangyasavval, forró vízzel, híg ammóniaoldattal, végezetül pedig hideg vízzel, miközben a tégelyt minden újabb anyag hozzáadása után leszívással kiürítjük. A leszívást addig nem kezdjük el, amíg a mosófolyadék magától el nem folyik. Végül szívassuk le a tégelyt, a szárítsuk meg, hűtsük le és mérjük meg a szálmaradékkal együtt.
5. AZ EREDMÉNYEK KISZÁMÍTÁSA ÉS KIFEJEZÉSE
Az eredményeket az általános útmutatóban meghatározottak szerint számítsuk ki. A "d" értéke 1,00, kivéve a melamin szálak esetében, amelyeknél "d" = 1,01.
6. PONTOSSÁG
Textilanyagok homogén keveréke esetében az e módszerrel nyerhető eredmények konfidenciahatára 95& 14;%-os konfidenciaszinten legfeljebb ± 1.
5. MÓDSZER - ACETÁT ÉS BIZONYOS MÁS SZÁLAK
(Benzil-alkoholos módszer)
1. ALKALMAZÁSI KÖR
Ez a módszer a nem szálas anyagok eltávolítását követően a következő összetételű, kétkomponensű textilszálkeverékek esetében alkalmazható:
1. acetát (19),
valamint
2. triacetát (24), polipropilén (37), elasztolefin (47), melamin (48) és polipropilén/poliamid kétkomponensű szál (49).
2. VIZSGÁLATI ALAPELV
Az acetát szálat ismert száraz tömegű keverékéből 52 ± 2 °C-on benzil-alkohollal oldjuk ki.
A visszamaradt anyagot összegyűjtjük, szárítjuk és mérjük, tömegét pedig, szükség esetén korrigálva, a keverék száraz tömegének százalékában fejezzük ki. A száraz acetát szál százalékát különbségképzéssel számoljuk ki.
3. ESZKÖZÖK ÉS REAGENSEK (az általános útmutatóban meghatározottakon kívül)
3.1. Eszközök
i. Legalább 200 ml űrtartalmú, becsiszolt üvegdugóval ellátott Erlenmeyer-lombik.
ii. . Mechanikus rázógép.
iii. Termosztát vagy egyéb készülék, amely a lombikot 52 ± 2 °C-on tudja tartani.
3.2. Reagensek
i. Benzil-alkohol,
ii. Etanol.
4. VIZSGÁLATI ELJÁRÁS
Kövessük az általános útmutatóban leírtakat, és a következőképpen járjunk el:
Az Erlenmeyer-lombikban lévő próbadarab minden grammjához adjunk 100 ml benzil-alkoholt. Helyezzük az üvegdugót a lombik nyílásába, majd rögzítsük a lombikot a rázógépen úgy, hogy az belemerüljön az 52 ± 2 °C-on tartott vízfürdőbe, majd ezen a hőmérsékleten rázassuk mintegy 20 percen keresztül.
(Mechanikus rázógép alkalmazása helyett a lombikban lévő anyagot heves kézzel történő rázogatással is keverhetjük).
A lemért szűrőtégelyen keresztül dekantáljuk a folyadékot. Öntsünk még egy további adag benzil-alkoholt a lombikba, és az előbbiek szerint rázzuk 20 percig 52 ± 2 °C-on.
A szűrőtégelyen keresztül dekantáljuk a folyadékot. Ismételjük meg a folyamatot harmadszor is.
Végül öntsük a folyadékot és a visszamaradt anyagot az üveg szűrőtégelybe; majd mossuk át a visszamaradó szálakat a lombikból a tégelyre még egy adag 52 ± 2 °C-os benzil-alkohollal. A tégelyt alaposan ürítsük ki.
A szálakat tegyük át egy lombikba, etanollal öblítsük ki, és miután kézzel felráztuk, dekantáljuk a tégelybe.
Ismételjük meg ezt az öblítőműveletet további két vagy három alkalommal. A visszamaradt anyagot vigyük át a tégelybe, és azt ürítsük ki alaposan. A tégelyt a szálmaradékokkal együtt szárítsuk meg, hűtsük le és mérjük meg.
5. AZ EREDMÉNYEK KISZÁMÍTÁSA ÉS KIFEJEZÉSE
Az eredményeket az általános útmutatóban meghatározottak szerint számítsuk ki. A "d" értéke 1,00, kivéve a melamin szálak esetében, amelyeknél "d" = 1,01.
6. PONTOSSÁG
Textilanyagok homogén keveréke esetében az e módszerrel nyerhető eredmények konfidenciahatára 95 14%-os konfidenciaszinten legfeljebb ± 1.
6. MÓDSZER - TRIACETÁTOK VAGY POLILAKTID ÉS BIZONYOS MÁS SZÁLAK
(Diklór-metános módszer)
1. ALKALMAZÁSI KÖR
Ez a módszer a nem szálas anyagok eltávolítását követően a következő összetételű, kétkomponensű textilszálkeverékek esetében alkalmazható:
1. triacetát (24) vagy polilaktid (33a),
valamint
2. gyapjú (1), állati szőr (2 és 3), selyem (4), pamut (5), rézoxid-szál (21), modálszál (22), viszkóz (25), akril (26), poliamid vagy nejlon (30), poliészter (35), polipropilén (37), üvegszál (44), elaszto-multiészter (46), elasztolefin (47), melamin (48) és polipropilén/poliamid kétkomponensű szál (49).
Megjegyzés:
Az olyan triacetátszálak, amelyeket részleges hidrolízishez vezető kikészítésnek vetettek alá, már nem teljes mértékben oldódnak ebben a reagensben. Ilyen esetekben a módszer nem alkalmazható.
2. VIZSGÁLATI ALAPELV
A triacetát vagy polilaktid szálakat a keverék ismert száraz tömegéből diklór-metánnal oldjuk ki. A visszamaradt anyagot összegyűjtjük, kimossuk, megszárítjuk és megmérjük; tömegét - szükség esetén korrigálva - a keverék száraz tömegének százalékában fejezzük ki. A száraz triacetát vagy polilaktid százalékát a két adat különbsége adja.
3. ESZKÖZÖK ÉS REAGENSEK (az általános útmutatóban meghatározottakon kívül)
3.1. Eszközök
Legalább 200 ml űrtartalmú, becsiszolt üvegdugóval ellátott Erlenmeyer-lombik.
3.2. Reagens
Diklór-metán.
4. VIZSGÁLATI ELJÁRÁS
Kövessük az általános útmutatóban leírtakat, és a következőképpen járjunk el:
A 200 ml-es, üvegdugóval ellátott Erlenmeyer-lombikban található próbadarab minden grammjához adjunk 100 ml diklór-metánt, helyezzük rá az üvegdugót, tízpercenként rázzuk fel a lombik tartalmát, hogy a próbadarab átnedvesedjen, majd hagyjuk állni szobahőmérsékleten 30 percig, miközben rendszeres időközönként felrázzuk. A folyadékot a lemért szűrőtégely segítségével dekantáljuk. Öntsünk a szálmaradékot tartalmazó lombikba 60 ml diklór-metánt, kézzel rázzuk fel és a lombik tartalmát szűrjük át a szűrőtégelyen. A visszamaradt szálakat vigyük át a szűrőtégelyre úgy, hogy a lombikot kis mennyiségű diklór-metánnal kiöblítjük. Ürítsük ki a szűrőtégelyből a felesleges folyadékot leszívatással, töltsük fel a szűrőtégelyt diklór-metánnal és hagyjuk, hogy magától lefolyjon.
Végezetül ismét szívassuk le a felesleges folyadékot, majd a visszamaradt anyagot kezeljük forró vízzel az összes oldószer eltávolítása céljából, a tégelyt szívassuk le, szárítsuk meg és a szálmaradékot, hűtsük vissza és mérjük le.
5. AZ EREDMÉNYEK KISZÁMÍTÁSA ÉS KIFEJEZÉSE
Az eredményeket az általános útmutatóban meghatározottak szerint számítsuk ki. A "d" értéke 1,00, kivéve a poliészter, elaszto-multiészter, elasztolefin és melamin szálak esetében, amelyeknél a "d" értéke 1,01.
6. PONTOSSÁG
Textilanyagok homogén keveréke esetében az e módszerrel nyerhető eredmények konfidenciahatára 95& 14;%-os konfidenciaszinten legfeljebb ± 1.
7. MÓDSZER - BIZONYOS CELLULÓZSZÁLAK ÉS BIZONYOS MÁS SZÁLAK
(75 tömeg%-os kénsavas módszer)
1. ALKALMAZÁSI KÖR
Ez a módszer a nem szálas anyagok eltávolítását követően a következő összetételű, kétkomponensű textilszálkeverékek esetében alkalmazható:
1. pamut (5), len (7), valódi kender (8), rami (14), cupro (21), modál szál (22), viszkóz (25),
valamint
2. poliészter (35), polipropilén (37), elaszto-multiészter (46), elasztolefin (47) és polipropilén/poliamid kétkomponensű szál (49).
2. VIZSGÁLATI ALAPELV
A cellulózszálakat a keverék ismert száraz tömegű mennyiségéből 75 % m/m kénsavval oldjuk ki. A visszamaradt anyagot összegyűjtjük, szárítjuk és mérjük, tömegét pedig a keverék száraz tömegének százalékában fejezzük ki. A száraz cellulóz rostszázalékát a különbségből számoljuk ki.
3. ESZKÖZÖK ÉS REAGENSEK (az általános útmutatóban meghatározottakon kívül)
3.1. Eszközök
i. . Legalább 500 ml űrtartalmú, üvegdugóval ellátott Erlenmeyer-lombik.
i. Termosztát vagy egyéb olyan eszköz, amely a lombikot 50 ± 5 °C-on tudja tartani.
3.2. Reagensek
i. . Kénsav, 75 ± 2 % m/m
Készítésénél óvatosan járjunk el, folyamatos hűtés mellett 700 ml kénsavat adjunk (sűrűsége 20 °C-on: 1,84) 350 ml desztillált vízhez. Miután szobahőmérsékletre visszahűtöttük, egészítsük ki az oldatot vízzel 1 literre.
i. Híg ammóniaoldat:
80 ml tömény ammóniaoldatot (sűrűsége 20 °C-on: 0,880) vízzel hígítsunk fel 1 literre.
4. VIZSGÁLATI ELJÁRÁS
Kövessük az általános útmutatóban leírtakat, és a következőképpen járjunk el:
A legalább 500 ml-es, üvegdugóval ellátott Erlenmeyer-lombikban található próbadarab minden grammjához adjunk 200 ml 75 14%-os kénsavat, helyezzük rá az üvegdugót, rázzuk fel a lombik tartalmát, hogy a minta átitatódjon. Hagyjuk állni 50 ± 5 °C-on egy óra hosszat, miközben kb. tízpercenként rendszeresen rázogatjuk. A palack tartalmát a lemért szűrőtégely segítségével leszívással szűrjük le. A visszamaradt szálakat vigyük át a szűrőtégelyre úgy, hogy a lombikot kis mennyiségű 75 %-os kénsavval kiöblítjük. Szívassuk le a szűrőtégelyt a felesleges folyadék eltávolítására, töltsük fel a szűrőtégelyt kénsavval. Ne alkalmazzunk leszívást, amíg gravitációs úton magától le nem csöpögött.
A visszamaradt anyagot mossuk ki egymás után többször előbb hideg vízzel, kétszer a híg ammóniaoldattal, majd alaposan hideg vízzel, és a tégelyt minden alkalommal alaposan csöpögtessük le. Ne alkalmazzunk leszívást addig, amíg mindegyik mosófolyadék gravitációs úton teljesen le nem csöpögött. Végezetül ismét szívjuk meg a tégelyt a felesleges folyadék eltávolítása érdekében, szárítsuk meg a tégelyt és a visszamaradt anyagot, hűtsük vissza és mérjük le.
5. AZ EREDMÉNYEK KISZÁMÍTÁSA ÉS KIFEJEZÉSE
Az eredményeket az általános útmutatóban meghatározottak szerint számítsuk ki. A "d" értéke 1,00, kivéve a polipropilén/poliamid kétkomponensű szál esetében, amelynél "d" = 1,01.
6. PONTOSSÁG
Textilanyagok homogén keveréke esetében az e módszerrel nyerhető eredmények konfidenciahatára 95& 14;%-os konfidenciaszinten legfeljebb ± 1.
8. MÓDSZER - AKRILOK, EGYES MODAKRILOK VAGY KLÓRSZÁLAK ÉS BIZONYOS EGYÉB SZÁLAK
(Dimetil-formamidos módszer)
1. ALKALMAZÁSI KÖR
Ez a módszer a nem szálas anyagok eltávolítását követően a következő összetételű, kétkomponensű textilszálkeverékek esetében alkalmazható:
1. akrilok (26), egyes modakrilok (29), vagy egyes klórszálak (27) ( 14 ),
valamint
2. gyapjú (1), állati szőr (2 és 3), selyem (4), pamut (5), rézoxid-szál (21), modálszál (22), viszkóz (25), poliamid vagy nejlon (30), poliészter (35), polipropilén (37), elaszto-multiészter (46), elasztolefin (47), melamin (48) és polipropilén/poliamid kétkomponensű szál (49).
A módszer egyaránt alkalmas előzetesen metallizált festékkel festett akrilok és egyes modakrilok esetében, kivétel a krómos utánkezeléssel színezett modakrilok.
2. VIZSGÁLATI ALAPELV
Az akril, modakril vagy klórszál műszálakat a keverék ismert száraz tömegű mennyiségéből forráspontig hevített vízfürdőben dimetil-formamiddal oldjuk ki. A visszamaradt anyagot összegyűjtjük, szárítjuk és mérjük. Szükség esetén korrigált tömegértékét pedig a keverék száraz tömegének százalékában fejezzük ki, a száraz akril, modakril vagy klórszál százalékát pedig a különbségből számoljuk ki.
3. ESZKÖZÖK ÉS REAGENSEK (az általános útmutatóban meghatározottakon kívül)
3.1. Eszközök
i. . Legalább 200 ml űrtartalmú, üvegdugóval ellátott Erlenmeyer-lombik.
i. Forráspontig hevített vízfürdő.
3.2. Reagens
Dimetil-formamid (forráspont 153 ± 1 °C) 0,1 14%-nál kisebb víztartalommal.
Ez a reagens mérgező, ezért vegyifülke használata ajánlott.
4. VIZSGÁLATI ELJÁRÁS
Kövessük az általános útmutatóban leírtakat, és a következőképpen járjunk el:
A 200 ml-es, üvegdugóval ellátott Erlenmeyer-lombikban található próbadarab minden grammjához adjunk 80 ml dimetil-formamidot, amelyet előzetesen forráspontig hevített vízfürdőben előmelegítettünk, helyezzük rá az üvegdugót, rázzuk fel a lombik tartalmát a minta átitatása érdekében, és a forráspontig hevített vízfürdőben melegítsük 1 óra hosszat. A palackot és tartalmát ez alatt az idő alatt öt alkalommal kézzel gyengéden rázzuk fel.
A folyadékot a lemért szűrőtégely segítségével dekantáljuk, a szálakat hagyjuk a lombikban. Öntsünk a visszamaradt anyagot tartalmazó lombikba 60 ml dimetil-formamidot és melegítsük további 30 percig. A palack tartalmát ez alatt az idő alatt két alkalommal kézzel enyhén rázzuk fel.
A lombik tartalmát leszívással szűrjük át a szűrőtégelyen.
A visszamaradt szálakat vigyük át a szűrőtégelyre úgy, hogy a lombikot kis mennyiségű dimetil-formamiddal kiöblítjük. A felesleges folyadék eltávolítása érdekében szívassuk le a szűrőtégelyt. A visszamaradt anyagot mossuk ki körülbelül 1 liter 70-80 oC-os meleg vízzel, miközben a tégelyt minden alkalommal töltsük tele. A tégelyt minden alkalommal gyengéden szívassuk le, de csak azután, hogy mindegyik mosófolyadék gravitációs úton teljesen lecsöpögött. Ha a mosófolyadék túl lassan csöpög le, enyhe leszívást lehet alkalmazni.
Végezetül szárítsuk meg a tégelyt és a visszamaradt anyagot, hűtsük vissza és mérjük le.
5. AZ EREDMÉNYEK KISZÁMÍTÁSA ÉS KIFEJEZÉSE
Az eredményeket az általános útmutatóban meghatározottak szerint számítsuk ki. A "d" értéke 1,00, kivéve a gyapjú, pamut, rézoxid, modál, poliészter, elaszto-multiészter és melamin szálak esetében, amelyeknél a "d" értéke 1,01.
6. PONTOSSÁG
Textilanyagok homogén keveréke esetében az e módszerrel nyerhető eredmények konfidenciahatára 95 14%-os konfidenciaszinten legfeljebb ± 1.
9. MÓDSZER - EGYES KLÓRSZÁLAK ÉS BIZONYOS EGYÉB SZÁLAK
(Széndiszulfid és aceton 55,5/44,5 arányú keverékét alkalmazó módszer)
1. ALKALMAZÁSI KÖR
Ez a módszer a nem szálas anyagok eltávolítását követően a következő összetételű, kétkomponensű textilszálkeverékek esetében alkalmazható:
1. egyes klórszálak (27), nevezetesen egyes polivinil-klorid szálak, utóklórozással vagy anélkül ( 15 ),
valamint
2. gyapjú (1), állati szőr (2 és 3), selyem (4), pamut (5), rézoxid-szál (21), modálszál (22), viszkóz (25), akril (26), poliamid vagy nejlon (30), poliészter (35), polipropilén (37), üvegszál (44), elaszto-multiészter (46), melamin (48) és polipropilén/poliamid kétkomponensű szál (49).
Ha a keverék gyapjú- vagy selyemtartalma meghaladja a 25 %-ot, a 2. módszert kell alkalmazni.
Ha a keverék poliamid- vagy nejlontartalma meghaladja a 25 %-ot, a 4. módszert kell alkalmazni.
2. VIZSGÁLATI ALAPELV
A klórszálakat a keverék ismert száraz tömegű mennyiségéből széndiszulfid és aceton állandó forrpontú elegyével oldjuk ki. A visszamaradt anyagot összegyűjtjük, szárítjuk és mérjük; szükség esetén korrigált tömegértékét pedig a keverék száraz tömegének százalékában fejezzük ki. A száraz polivinil-klorid műszál százalékát a különbségből számoljuk ki.
3. ESZKÖZÖK ÉS REAGENSEK (az általános útmutatóban meghatározottakon kívül)
3.1. Eszközök
i. Legalább 200 ml űrtartalmú, üvegdugóval ellátott Erlenmeyer-lombik.
ii. Mechanikus rázógép.
3.2. Reagensek
i. Széndiszulfid és aceton állandó forrpontú elegye (55,5 % térfogatszázalék széndiszulfid és 44,5 % aceton). Miután a reagens mérgező, vegyifülke használata feltétlenül ajánlott.
ii. Etanol (92 % térfogatszázalékos) vagy metanol.
4. VIZSGÁLATI ELJÁRÁS
Kövessük az általános útmutatóban leírtakat, és a következőképpen járjunk el:
A 200 ml-es, üvegdugóval ellátott Erlenmeyer-lombikban található próbadarab minden grammjához adjunk 100 ml-t az állandó forráspontú elegyből. Biztonságosan zárjuk le a lombikot és szobahőmérsékleten 20 percen keresztül rázassuk intenzíven mechanikus rázógépen vagy manuálisan. A felül úszó folyadékot a lemért szűrőtégelyen keresztül dekantáljuk.
Ismételjük meg a folyamatot 100 ml friss reagenssel. Ezt a műveletsort egészen addig kell folytatni, amíg az óraüvegre cseppentett extrakciós folyadék elpárolgása után az üvegen semmiféle polimer-lerakódás nem marad. A visszamaradt anyagot további reagens hozzáadásával vigyük át a szűrőtégelyre, és a tégelyt, illetve a visszamaradt anyagot 20 ml alkohol, majd három alkalommal víz felhasználásával öblítsük ki. Mielőtt leszívásos szárítást alkalmaznánk, engedjük, hogy a mosófolyadék gravitációs úton csöpögjön le. Szárítsuk meg a tégelyt és a visszamaradt anyagot, hűtsük vissza és mérjük le.
Megjegyzés:
Bizonyos magas klórszáltartalmú keverékeknél a szárítási folyamat során jelentős zsugorodás következhet be, aminek következtében az oldat a klórszálat lassan oldja fel. Ennek ellenére a klórszál végül teljesen feloldódik az oldószerben.
5. AZ EREDMÉNYEK KISZÁMÍTÁSA ÉS KIFEJEZÉSE
Az eredményeket az általános útmutatóban meghatározottak szerint számítsuk ki. A "d" értéke 1,00, kivéve a melamin szálak esetében, amelyeknél "d" = 1,01.
Calculate the results as described in the general instructions. The value of ‘d' is 1,00, except for melamine, for which ‘d' = 1,01.
6. PONTOSSÁG
Textilanyagok homogén keveréke esetében az e módszerrel nyerhető eredmények konfidenciahatára 95 14%-os konfidenciaszinten legfeljebb ± 1.
10. MÓDSZER - ACETÁT ÉS BIZONYOS MÁS SZÁLAK
(Jégecetes módszer)
1. ALKALMAZÁSI KÖR
Ez a módszer a nem szálas anyagok eltávolítását követően a következő összetételű, kétkomponensű textilszálkeverékek esetében alkalmazható:
1. acetát (19),
valamint
2. bizonyos polikloridok (27), nevezetesen polivinil-klorid szálak, utóklórozással vagy anélkül, polipropilén (37), elasztolefin (47), melamin (48) és polipropilén/poliamid kétkomponensű szál (49).
2. VIZSGÁLATI ALAPELV
Az acetát műszálakat a keverék ismert száraz tömegű mennyiségéből jégecettel oldjuk ki. A visszamaradt anyagot összegyűjtjük, szárítjuk és mérjük, korrigált tömegértékét pedig a keverék száraz tömegének százalékában fejezzük ki. A száraz acetát rostszázalékát a különbségből számoljuk ki.
3. ESZKÖZÖK ÉS REAGENSEK (az általános útmutatóban meghatározottakon kívül)
3.1. Eszközök
i. . Legalább 200 ml űrtartalmú, üvegdugóval ellátott Erlenmeyer-lombik.
i. Mechanikus rázógép.
3.2. Reagens
Jégecet (99 % felett). Ezt a reagenst fokozott körültekintéssel kell kezelni, mert erősen maró hatású.
4. VIZSGÁLATI ELJÁRÁS
Kövessük az általános útmutatóban leírtakat, és a következőképpen járjunk el:
A 200 ml-es, üvegdugóval ellátott Erlenmeyer-lombikban található próbadarab minden grammjához adjunk 100 ml jégecetet. Biztonságosan zárjuk le a lombikot és szobahőmérsékleten 20 percen keresztül intenzíven rázassuk mechanikus rázógépen vagy manuálisan. A felül úszó folyadékot a lemért szűrőtégelyen keresztül dekantáljuk. Ismételjük meg a műveletet további két alkalommal, mindannyiszor 100 ml friss reagenst használjunk, és összesen három extrahálást végezzünk. Vigyük át a visszamaradt anyagot a szűrőtégelyre, leszívással szárítsuk meg, távolítsuk el a folyadékot, és öblítsük ki a tégelyt, illetve a visszamaradt anyagot 50 ml jégecet, majd három alkalommal víz felhasználásával. Mielőtt leszívásos szárítást alkalmaznánk, engedjük, hogy a mosófolyadék gravitációs úton csöpögjön le. Szárítsuk meg a tégelyt és a visszamaradt anyagot, hűtsük vissza és mérjük le.
5. EREDMÉNYEK KISZÁMÍTÁSA ÉS MEGADÁSA
Az eredményeket az általános útmutatóban meghatározottak szerint számítsuk ki. A "d" értéke 1,00.
6. PONTOSSÁG
Textilanyagok homogén keveréke esetében az e módszerrel nyerhető eredmények konfidenciahatára 95 14%-os konfidenciaszinten legfeljebb ± 1.
11. módszer
SELYEM VAGY POLIAMID ÉS BIZONYOS MÁS SZÁLAK
(75 tömeg%-os kénsavas módszer)
1. ALKALMAZÁSI KÖR
Ez a módszer a nem szálas anyagok eltávolítását követően a következő összetételű, kétkomponensű textilszálkeverékek esetében alkalmazható:
1. selyem (4) vagy poliamid vagy nejlon (30)
valamint
2. gyapjú (1), állati szőr (2 és 3), polipropilén (37), elasztolefin (47), melamin (48) és polipropilén/poliamid kétkomponensű szál (49).
2. VIZSGÁLATI ALAPELV
A selyem-, poliamid- vagy nejlonszálakat a keverék ismert száraz tömegű mennyiségéből 75 tömegszázalékos kénsavval oldjuk ki.
A visszamaradt anyagot összegyűjtjük, kimossuk, megszárítjuk és megmérjük; tömegét - szükség esetén korrigálva - a keverék száraz tömegének százalékában fejezzük ki. A különbség alapján megkapjuk a száraz selyem, poliamid vagy nejlon százalékát.
3. ESZKÖZÖK ÉS REAGENSEK (az általános útmutatóban meghatározottakon kívül)
3.1. Eszközök
Legalább 200 ml űrtartalmú, üvegdugóval ellátott Erlenmeyer-lombik.
3.2. Reagensek
i. Kénsav (75 ± 2 % m/m)
Készítésénél körültekintően járjunk el, folyamatos hűtés mellett 700 ml kénsavat adjunk (sűrűsége 20 °C-on: 1,84) 350 ml desztillált vízhez.
Miután szobahőmérsékletre visszahűtöttük, egészítsük ki az oldatot vízzel 1 literre.
ii. Kénsav, hígító oldat: 100 ml kénsavat (sűrűsége 20 °C-on: 1,84) fokozatosan adjunk hozzá 1 900 ml desztillált vízhez.
iii. Híg ammóniaoldat: hígítsunk fel 200 ml tömény ammóniát (sűrűsége 20 °C-on: 0,880) 1 000 ml vízzel.
4. VIZSGÁLATI ELJÁRÁS
Kövessük az általános útmutatóban leírtakat, és a következőképpen járjunk el:
A legalább 200 ml-es, becsiszolt üvegdugóval ellátott Erlenmeyer-lombikban található próbadarab minden grammjához adjunk 100 ml 75 tömegszázalékos kénsavat, és zárjuk le az üvegdugóval. Erősen rázzuk fel, és hagyjuk szobahőmérsékleten állni 30 percig. Rázzuk fel ismét, és hagyjuk állni 30 percig. Végezetül még egyszer rázzuk fel, és szűrjük le a lombik tartalmát a lemért szűrőtégelyen. A 75 tömegszázalékos kénsav reagenssel mossuk ki a visszamaradt szálakat a lombikból. A szűrőn visszamaradt anyagot előbb 50 ml híg kénsavoldattal, majd 50 ml vízzel, végül 50 ml híg ammóniaoldattal mossuk ki. Leszívás előtt minden alkalommal engedjük, hogy a szálak a folyadékkal legalább 10 percig érintkezzenek. Végezetül vízzel öblítsük ki, de hagyjuk, hogy a szálak a vízzel körülbelül 30 percen át érintkezzenek. Szívassuk le a tégelyt, szárítsuk meg a visszamaradt anyaggal együtt, hűtsük vissza és mérjük le.
A poliamid és a polipropilén/poliamid szál kétkomponensű keverékei esetében a szálaknak a lemért szűrőtégelyen történő leszűrése után és a leírt mosási eljárás alkalmazása előtt kétszer mossuk át a szűrőtégelyen visszamaradó szálakat, mindannyiszor 50 ml 75 %-os kénsav reagenssel.
5. AZ EREDMÉNYEK KISZÁMÍTÁSA ÉS KIFEJEZÉSE
Az eredményeket az általános útmutatóban meghatározottak szerint számítsuk ki. A "d" értéke 1,00, kivéve a gyapjú esetében, amelynél "d" = 0,985, továbbá a polipropilén/poliamid kétkomponensű szál esetében, amelynél "d" = 1,005, valamint a melamin esetében, amelynél "d" = 1,01.
6. PONTOSSÁG
Textilanyagok homogén keveréke esetében az e módszerrel nyerhető eredmények konfidenciahatára 95 %-os konfidenciaszinten legfeljebb ± 1, kivéve a poliamid és a polipropilén/poliamid szál kétkomponensű keverékeit, amelyek esetében az eredmények konfidenciahatára legfeljebb ± 2.
12. módszer
JUTA ÉS BIZONYOS ÁLLATI SZÁLAK
(Nitrogéntartalom meghatározásán alapuló módszer)
1. ALKALMAZÁSI KÖR
Ez a módszer a nem szálas anyagok eltávolítását követően a következő összetételű, kétkomponensű textilszálkeverékek esetében alkalmazható:
1. juta (9)
és
2. egyes állati szálak.
Az állati eredetű komponens vagy csak szőr (2 és 3), vagy gyapjú (1), vagy e kettő bármilyen arányú keveréke. A módszer nem alkalmazható olyan textilipari keverékek esetében, amelyek nitrogén alapú nem rostszerű anyagot is tartalmaznak (festékek, kikészítő anyagok stb.).
2. VIZSGÁLATI ALAPELV
Meghatározzuk a keverék nitrogéntartalmát, majd ennek, valamint a két összetevő feltételezett vagy ismert nitrogéntartalma alapján kiszámítjuk az egyes komponensek részarányát.
3. ESZKÖZÖK ÉS REAGENSEK (az általános útmutatóban meghatározottakon kívül)
3.1. Eszközök
i. 200-300 ml űrtartalmú Kjeldahl feltáró lombik.
ii. Gőz befecskendezéssel működő Kjeldahl desztilláló készülék.
iii. 0,05 ml pontosságú titráló készülék.
3.2. Reagensek
i. Toluol.
ii. Metanol.
iii. Kénsav, relatív sűrűsége 20 °C-on: 1,84 ( 16 ).
iv. Kálium-szulfát (16) .
v. Szelén-dioxid (16) .
vi. Nátrium-hidroxid oldat (400 g/liter). Négyszáz gramm nátrium-hidroxidot oldjunk fel 400-500 ml vízben és egészítsük ki vízzel 1 literre.
vii. Vegyes indikátor. Oldjunk fel 0,1 g metilvöröset 95 ml etanolban és 5 ml vízben, majd keverjük el 475 ml etanolban és 25 ml vízben feloldott 0,5 g brómkrezol zölddel.
viii. Bórsavoldat. Oldjunk fel 20 g bórsavat 1 liter vízben.
ix. Kénsav, 0,02N (normál térfogat szerinti oldat).
4. A PRÓBADARAB ELŐKEZELÉSE
Az alábbiakban ismertetett előkezelési eljárás az általános útmutatóban leírtak helyett alkalmazandó:
A légszáraz mintát 1 térfogat toluol és 3 térfogat metanol keverékével Soxhlet készülékben négy órán keresztül óránként legalább 5 alkalommal vessük alá extrakciós eljárásnak. Engedjük, hogy az oldószer a levegőn elpárologjon a mintából, és az utolsó nyomokat 105 ± 3 °C-ra állított szárítószekrényben távolítsuk el. Ezután a mintát vízben (50 ml a minta minden grammjára) visszafolyó hűtés mellett forralva 30 percig extraháljuk. Szűrjük le, vigyük vissza a mintát a lombikba és ismételjük meg az eljárást azonos mennyiségű vízzel még egyszer. Szűrjük le, a felesleges vizet préseléssel, leszívással vagy centrifugálással távolítsuk el, majd engedjük, hogy a minta ismét felvegye légszáraz állapotát.
Megjegyzés:
A toluol és a metanol mérgező hatásáról nem szabad elfeledkezni, ezért használatuk során fokozott elővigyázatossággal kell eljárni.
5. VIZSGÁLATI ELJÁRÁS
5.1. Általános útmutató
A próbadarab kiválasztásánál, szárításánál és lemérésénél kövessük az általános útmutatóban leírtakat.
5.2. Részletes eljárás
Tegyük a próbadarabot egy Kjeldahl feltáró lombikba. A lombikban elhelyezett, legalább egy gramm súlyú próbadarabhoz a következő sorrendben adjunk 2,5 g kálium-szulfátot, 0,1-0,2 g szelén-dioxidot és 10 ml kénsavat (relatív sűrűsége 1,84). Melegítsük a lombikot, először csak enyhén, amíg az összes rost el nem roncsolódik, majd erőteljesebben, amíg az oldat tiszta és majdnem színtelen lesz. Melegítsük további 15 percen át. Engedjük a lombikot lehűlni, tartalmát óvatosan hígítsuk fel 10-20 ml vízzel, hűtsük le, a teljes tartalmát tegyük át egy 200 ml-es kalibrált lombikba (mérőlombik), és a feltáró oldat elkészítéséhez öntsük fel vízzel a jelzésig.
Körülbelül 20 ml bórsavas oldatot helyezzünk egy 100 ml-es Erlenmeyer-lombikba és a lombikot rakjuk a Kjeldahl-féle desztilláló készülék kondenzátora alá úgy, hogy a kifolyócső éppen a bórsavoldat felszíne alá merüljön. Pontosan 10 ml feltáró oldatot tegyünk bele a desztilláló lombikba, a tölcsérbe adagoljunk legalább 5 ml nátrium-hidroxid oldatot, kissé emeljük meg a dugót és hagyjuk, hogy a nátrium-hidroxid oldat lassan lecsorogjon az edénybe. Ha a feltáró oldat és a nátrium-hidroxidos oldat külön fázisban marad mint két, egymástól elkülönült réteg, enyhe rázogatással keverjük el őket. Enyhén melegítsük a lombikot és helyezzük a gőzfejlesztőből származó gőzbe. Gyűjtsünk össze mintegy 20 ml desztillátumot, engedjük le az Erlenmeyer-lombikot úgy, hogy a kondenzátor kifolyócsöve mintegy 20 mm-rel a folyadék felszíne alatt legyen és további egy percen át folytassuk a desztillálást. A desztilláló kifolyócsövének hegyét mossuk le vízzel és a mosófolyadékot fogjuk fel az Erlenmeyer-lombikba. Vegyük el az Erlenmeyer-lombikot, a helyére tegyünk egy másik Erlenmeyer-lombikot, amely körülbelül 10 ml bórsavas oldatot tartalmaz és gyűjtsünk bele körülbelül 10 ml desztillátumot.
A két desztillátumot a vegyes indikátor alkalmazásával külön-külön titráljuk meg 0,02N kénsavval. Jegyezzük fel a két desztillátum teljes fogyását. Ha a második desztillátum fogyása meghaladja a 0,2 ml-t, ismételjük meg a vizsgálatot és indítsuk el újból a desztillációs folyamatot, ehhez ismét adagoljunk feltáró folyadékot.
Végezzünk el egy vakpróbát is, vagyis folytassuk le a feltárást és a desztillálást csak a reagensekkel.
6. EREDMÉNYEK KISZÁMÍTÁSA ÉS MEGADÁSA
6.1. Számítsuk ki a százalékos nitrogéntartalmat a minta szárazanyag-tartalmára vonatkoztatva a következők szerint:
ahol
A = a nitrogén százalékos aránya a száraz, tiszta minta anyagában,
V = a meghatározáskor felhasznált normál kénsavoldat teljes térfogata milliliterben megadva,
b = a vakpróbánál felhasznált normál kénsavoldat teljes térfogata milliliterben megadva,
N = a normál kénsavoldat normalitása,
W = a próbadarab száraz tömege grammokban kifejezve.
6.2. A juta esetében 0,22 %, az állati szálakra pedig 16,2 % nitrogéntartalmat feltételezve és mindkét százalékos arányt a szál száraz tömegének százalékában kifejezve a keverék összetételét a következőképpen lehet kiszámítani:
,
ahol
PA% = az állati szálak százalékos aránya a tiszta, száraz mintában.
7. PONTOSSÁG
Textilanyagok homogén keveréke esetében az e módszerrel nyerhető eredmények konfidenciahatára 95 14%-os konfidenciaszinten legfeljebb ± 1.
13. MÓDSZER - POLIPROPILÉN SZÁLAK ÉS BIZONYOS EGYÉB SZÁLAK
(Xilolos módszer)
1. ALKALMAZÁSI KÖR
Ez a módszer a nem szálas anyagok eltávolítását követően a következő összetételű, kétkomponensű textilszálkeverékek esetében alkalmazható:
1. polipropilén szál (36)
valamint
2. gyapjú (1), állati szőr (2 és 3), selyem (4), pamut (5), acetát (19), rézoxid-szál (21), modál (22), triacetát (24), viszkóz (25), akril (26), poliamid vagy nejlon (30), poliészter (35), üvegszál (44), elaszto-multiészter (46) és melamin (48).
2. VIZSGÁLATI ALAPELV
A polipropilén műszálakat a keverék ismert száraz tömegű mennyiségéből forrásban lévő xilollal oldjuk ki. A visszamaradt anyagot összegyűjtjük, szárítjuk és mérjük, a korrigált tömeg értékét pedig szükség esetén a keverék száraz tömegének százalékában fejezzük ki. A száraz polipropilén műszál százalékát a különbségből számoljuk ki.
3. ESZKÖZÖK ÉS REAGENSEK (az általános útmutatóban meghatározottakon kívül)
3.1. Eszközök
i. Legalább 200 ml űrtartalmú, üvegdugóval ellátott Erlenmeyer-lombik.
ii. Visszafolyós hűtő (magas forrpontú folyadékok számára is megfelelő), amely illeszthető az Erlenmeyer-lombikhoz (i.)
3.2. Reagens
137 és 142 °C között desztillált xilol.
Megjegyzés:
Ez a reagens igen gyúlékony és gőzei mérgezőek. Használata során fokozott elővigyázatosság szükséges.
4. VIZSGÁLATI ELJÁRÁS
Kövessük az általános útmutatóban leírtakat, majd a következőképpen járjunk el:
Adjunk a (3.1. i.) pont szerinti Erlenmeyer-lombikban található próbadarabhoz a minta minden grammjára számítva 100 ml xilolt (3.2.). Csatlakoztassuk a visszafolyós hűtőt (3.1. ii.), forraljuk fel a lombik tartalmát és tartsuk három percen át forráspont közelében. A folyadékot a lemért szűrőtégelyen keresztül (lásd az 1. megjegyzést) azonnal dekantáljuk. Ismételjük meg a műveletet további két alkalommal, mindannyiszor adjunk az anyaghoz 50 ml oldószert.
Mossuk ki a lombikban visszamaradt anyagot egymás után 30 ml forrásban lévő xilollal (kétszer), majd 75 ml petroléterrel (lásd az általános útmutató I.3.2.1. pontját) (kétszer). A második petroléteres öblítést követően szűrjük le a lombik tartalmát a szűrőtégelyen keresztül, a visszamaradt szálakat kis mennyiségű petroléter alkalmazásával vigyük át a tégelyre és hagyjuk, hogy az oldószer elpárologjon. Szárítsuk meg a tégelyt és a visszamaradt anyagot, hűtsük vissza és mérjük le.
Megjegyzés:
1. A szűrőtégelyt, amelyen keresztül a xilol dekantálását akarjuk végezni, elő kell melegíteni.
2. A forrásban lévő xilollal végzett kezelést követően győződjünk meg róla, hogy a maradványanyagokat tartalmazó Erlenmeyer-lombik kellően lehűlt-e, mielőtt a petrolétert hozzáöntenénk.
3. A laboránst érintő tűz- és mérgezésveszély csökkentése érdekében azonos eredményt adó, a megfelelő eljárást alkalmazó meleg extraháló berendezés is használható ( 17 ).
5. AZ EREDMÉNYEK KISZÁMÍTÁSA ÉS KIFEJEZÉSE
Az eredményeket az általános útmutatóban meghatározottak szerint számítsuk ki. A "d" értéke 1,00, kivéve a melamin szálak esetében, amelyeknél "d" = 1,01.
6. PONTOSSÁG
Textilanyagok homogén keveréke esetében az e módszerrel nyerhető eredmények konfidenciahatára 95& 14;%-os konfidenciaszinten legfeljebb ± 1.
14. MÓDSZER - BIZONYOS SZÁLAK ÉS BIZONYOS MÁS SZÁLAK
(Tömény kénsavas módszer)
1. ALKALMAZÁSI KÖR
Ez a módszer a nem szálas anyagok eltávolítását követően a következő összetételű, kétkomponensű textilszálkeverékek esetében alkalmazható:
1. pamut (5), acetát (19), rézoxid-szál (21), modál (22), triacetát (24), viszkóz (25), egyes akrilok (26), egyes modakrilok (29), poliamid vagy nejlon (30), poliészter (35) és elaszto-multiészter (46);
valamint
2. polikloridok (27) vinilklorid-homopolimerre alapozva, utóklórozással vagy anélkül, polipropilén (37), elasztolefin (47), melamin (48) és polipropilén/poliamid kétkomponensű szál (49).
A szóban forgó modakrilok tömény kénsavba mártva átlátszó oldatot adnak (a kénsav relatív sűrűsége 20 °C-on 1,84).
Ezt a módszert a 8. és 9. módszer helyett lehet alkalmazni.
2. ALAPELV
A keverék nem poliklorid, nem polipropilén, nem elasztolefin, nem melamin és nem polipropilén/poliamid kétkomponensű szál összetevőjét (vagyis az 1.1. pontban említett szálakat) tömény kénsavval (relatív sűrűsége 20 °C-on 1,84) kioldjuk az ismert száraz tömegű keverékből. A polikloridból, polipropilénből, elasztolefinből, melaminból vagy polipropilén/poliamid kétkomponensű szálból álló visszamaradt anyagot összegyűjtjük, kimossuk, megszárítjuk és lemérjük, tömegét szükség esetén a megfelelő módon korrigálva a keverék száraz tömegére vonatkoztatott százalékos értékben fejezzük ki. A különbség alapján megkapjuk a második összetevő százalékos arányát.
3. ESZKÖZÖK ÉS REAGENSEK (az általános útmutatóban meghatározottakon kívül)
3.1. Eszközök
i. Legalább 200 ml űrtartalmú, üvegdugóval ellátott Erlenmeyer-lombik.
ii. Lapos végű üvegbot.
3.2. Reagensek
i. Tömény kénsav, (relatív sűrűsége 20 °C-on 1,84).
ii. Kénsav, megközelítőleg 50& 14;%-os (m/m) vizes oldat.
Készítésénél körültekintően járjunk el, állandó hűtés mellett adjunk 400 ml kénsavat (relatív sűrűség 20 °C-on 1,84) 500 ml desztillált vagy ioncserélt vízhez. Szobahőmérsékletre hűtés után az oldatot vízzel egy literre egészítjük ki.
iii. Híg ammóniaoldat.
60 ml tömény ammóniaoldatot (relatív sűrűsége 20 °C-on 0,880) desztillált vízzel 1 literre hígítsunk fel.
4. VIZSGÁLATI ELJÁRÁS
Kövessük az általános útmutatóban leírtakat, majd a következőképpen járjunk el:
Az Erlenmeyer-lombikban található próbadarab (3.1. i..) minden grammjához adjunk 100 ml kénsavat (3.2. i.).
Hagyjuk a lombik tartalmát szobahőmérsékleten állni 10 percen keresztül, és ez alatt az idő alatt egy üvegbot segítségével alkalmanként keverjük meg a próbadarabot. Ha szövött vagy kötött anyagot kezelünk, szorítsuk be a mintát az Erlenmeyer-lombik fala és az üvegbot közé és gyakoroljunk rá enyhe nyomást, hogy elválasszuk a kénsav által kioldott anyagot a többitől.
Dekantáljuk a folyadékot egy lemért szűrőtégelyen. Ismételten adjunk a lombik tartalmához 100 ml friss kénsavat (3.2. i.) és ismételjük meg a műveletet. A lombik tartalmát és - az üvegbot segítségével - a rostos maradványokat vigyük át a szűrőtégelyre. Szükség esetén öntsünk még egy kevés tömény kénsavat (3.2. i.) a lombikba, annak érdekében, hogy a lombik falához esetleg hozzátapadó szálakat el tudjuk távolítani. Szívjuk le a szűrőtégelyt, távolítsuk el a szűrletet a szűrőlombik eltávolításával vagy kiürítésével, ezt követően mossuk ki a tégelyben maradt anyagot 50& 14;%-os kénsavoldattal (3.2. ii.), desztillált vagy ioncserélt vízzel (az útmutató I.3.2.3. pontja, ammóniaoldat (3.2. iii.), majd végezetül alaposan mossuk át desztillált vagy ioncserélt vízzel, a tégelyt pedig minden alkalommal csöpögtessük le. (A mosás alatt ne alkalmazzunk leszívást, először hagyjuk a folyadékot gravitációs úton lecsöpögni.)
Szárítsuk meg a tégelyt és a visszamaradt anyagot hűtsük vissza és mérjük le.
5. AZ EREDMÉNYEK KISZÁMÍTÁSA ÉS KIFEJEZÉSE
Az eredményeket az általános útmutatóban meghatározottak szerint számítsuk ki. A "d" értéke 1,00, kivéve a melamin és a polipropilén/poliamid kétkomponensű szál esetében, amelyeknél "d" = 1,01.
6. PONTOSSÁG
Textilanyagok homogén keveréke esetében az e módszerrel nyerhető eredmények konfidenciahatára 95 14%-os konfidenciaszinten legfeljebb ± 1.
15. MÓDSZER - KLÓRSZÁLAK, BIZONYOS MODAKRILOK, EGYES ELASZTÁNOK, ACETÁTOK, TRIACETÁTOK ÉS BIZONYOS EGYÉB SZÁLAK
(Ciklohexanonos módszer)
1. ALKALMAZÁSI KÖR
Ez a módszer a nem szálas anyagok eltávolítását követően a következő összetételű, kétkomponensű textilszálkeverékek esetében alkalmazható:
1. acetát (19), triacetát (24), poliklorid (27), bizonyos modakril szálak (29), bizonyos elasztánok (43);
valamint
2. gyapjú (1), állati szőr (2 és 3), selyem (4), pamut (5), rézoxid-szál (21), modál (22), viszkóz (25), poliamid vagy nejlon (30), akril (26), üvegszál (44) és melamin (48).
Ha modakrilok vagy elasztánok is jelen vannak, először előzetes vizsgálattal meg kell állapítani, hogy a szál teljesen oldódik-e a reagensben.
A polikloridokat tartalmazó keverékeket a 9. vagy a 14. módszerrel lehet elemezni.
2. VIZSGÁLATI ALAPELV
Az acetát és triacetát műszálak, a klórszálak, bizonyos modakrilok és elasztánok forrásponthoz közeli hőmérsékleten a ciklohexán hatására kioldódnak az ismert száraz tömegű anyagból. A visszamaradt anyagot összegyűjtjük, kimossuk, megszárítjuk, lemérjük és tömegét szükség esetén a megfelelő módon korrigálva a keverék száraz tömegére vonatkoztatott százalékos értékben fejezzük ki. A klórszál, modakril, elasztán, acetát és triacetát százalékos értékét különbségszámítással kapjuk meg.
3. ESZKÖZÖK ÉS REAGENSEK (az általános útmutatóban meghatározottakon kívül)
3.1. Eszközök
i. Meleg extraháló készülék, amely a 4. részben leírt vizsgálati eljáráshoz alkalmas. (Lásd az ábrát: ez a Melliand Textilberichte 56 (1975) 643-645. oldalán leírt készülék egy bizonyos változata).
ii. A próbadarabot tartó szűrőtégely.
iii. Elválasztó lemez (1-es fokú porozitás).
iv. Visszafolyós hűtő, amely a desztillációs lombikhoz illeszthető.
v. Melegítő eszköz.
3.2. Reagensek
i. Ciklohexanon, forráspontja 156 °C.
ii. Etilalkohol, 50 térfogatszázalékos.
Megjegyzés: A ciklohexanon gyúlékony és mérgező anyag. Használata során kellő elővigyázatossággal járjunk el.
4. VIZSGÁLATI ELJÁRÁS
Kövessük az általános útmutatóban leírtakat, és a következőképpen járjunk el:
A desztilláló lombikba öntsünk az anyag minden grammjához 100 ml ciklohexanont, helyezzük bele az extrakciós készüléket, amelyre előzetesen ráhelyeztük a próbadarabot és az enyhén lejtő elválasztó lemezt tartalmazó szűrőtégelyt. Illesszük a helyére a visszafolyós hűtőt. Hevítsük forrásig és folytassuk a kivonást 60 percen keresztül óránként legalább 12 ciklus gyakorisággal. A kivonást és hűtést követően távolítsuk el az extraháló készüléket, vegyük ki belőle a szűrőtégelyt és távolítsuk el a elválasztó lemezt. Háromszor-négyszer mossuk ki a szűrőtégelyt körülbelül 60 oC-ra melegített 50& 14;%-os etilalkohollal, majd ezt követően 1 liter 60 °C-os vízzel.
A mosási műveletek között vagy alatt leszívást ne alkalmazzunk. Hagyjuk a folyadékot a gravitációs hatás folytán lecsöpögni, és csak ezután alkalmazzuk a leszívást.
Végül szárítsuk meg a szűrőtégelyt és a visszamaradt anyagot, hűtsük le és mérjük meg.
5. AZ EREDMÉNYEK KISZÁMÍTÁSA ÉS KIFEJEZÉSE
| selyem és melamin | 1,01 |
| akril | 0,98. |
6. PONTOSSÁG
Textilanyagok homogén keveréke esetében az e módszerrel nyerhető eredmények konfidenciahatára 95 14%-os konfidenciaszinten legfeljebb ± 1.
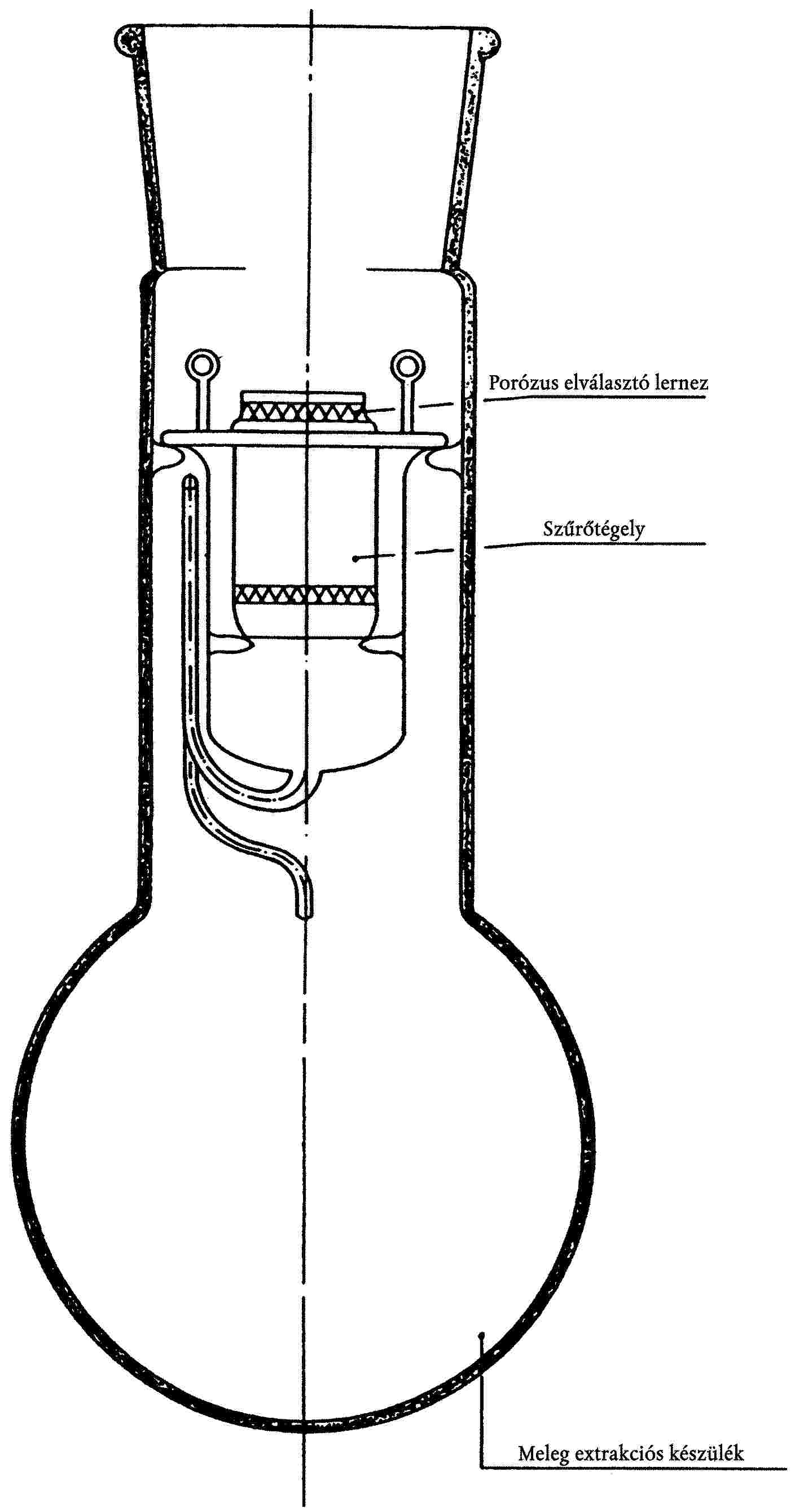
A 15. módszer 3.1. i. pontjában hivatkozott ábra
16. MÓDSZER - MELAMIN ÉS BIZONYOS MÁS SZÁLAK
(Forró hangyasavas módszer)
1. ALKALMAZÁSI KÖR
Ez a módszer a nem szálas anyagok eltávolítását követően a következő összetételű, kétkomponensű textilszálkeverékek esetében alkalmazható:
1. melamin (48);
valamint
2. pamut (5), aramid (31) és polipropilén (37).
2. ALAPELV
A melamint az ismert száraz tömegű keverékből (90 tömeg%-os) forró hangyasavval oldjuk ki.
A visszamaradt anyagot összegyűjtjük, kimossuk, megszárítjuk és megmérjük; tömegét szükség esetén a megfelelő módon korrigálva a keverék száraz tömegére vonatkoztatott százalékos értékben fejezzük ki. A különbség alapján megkapjuk a második összetevő százalékos arányát.
Megjegyzés: Szigorúan az ajánlott hőmérsékleti tartományon belül kell maradni, mivel a melamin oldhatósága nagymértékben függ a hőmérséklettől.
3. ESZKÖZÖK ÉS REAGENSEK (az általános útmutatóban meghatározottakon kívül)
3.1. Eszközök
i. Legalább 200 ml űrtartalmú, üvegdugóval ellátott Erlenmeyer-lombik.
ii. Rázó vízfürdő vagy egyéb olyan eszköz, amely rázni és 90 ± 2 °C-on tudja tartani a lombikot.
3.2. Reagensek
i. Hangyasav (90 tömeg%-os, relatív sűrűsége 20 °C-on: 1,204 g/ml). 890 ml 98-100 tömeg%-os hangyasavat (relatív sűrűsége 20 °C-on: 1,220 g/ml) egészítsünk ki vízzel 1 literre.
A forró hangyasav erősen maró hatású, ezért fokozott körültekintéssel kell vele bánni.
ii. Híg ammóniaoldat: tömény ammóniaoldatból 80 ml-t (relatív sűrűsége 20 °C-on: 0,880) egészítsünk ki vízzel 1 literre.
4. VIZSGÁLATI ELJÁRÁS
Kövessük az általános útmutatóban leírtakat, majd a következőképpen járjunk el:
A legalább 200 ml-es, üvegdugóval ellátott Erlenmeyer-lombikban található próbadarab minden grammjához adjunk 100 ml hangyasavat. Tegyük rá az üvegdugót és alaposan rázzuk fel, hogy a minta átnedvesedjen. Erőteljesen rázassuk a lombikot rázó vízfürdőben 90 ± 2 °C-on egy óra hosszat. Hagyjuk a lombikot szobahőmérsékletűre hűlni. Dekantáljuk a folyadékot egy lemért szűrőtégelyen. Öntsünk a szálmaradékot tartalmazó lombikba 50 ml hangyasavat, kézzel rázzuk fel, és a lombik tartalmát szűrjük át a szűrőtégelyen. A visszamaradt szálakat vigyük át a szűrőtégelyre úgy, hogy a lombikot kis mennyiségű hangyasavval kiöblítjük. A tégelyt leszívással ürítsük le, ezt követően a szálmaradékot mossuk hangyasavval, forró vízzel, híg ammóniaoldattal, végül hideg vízzel, miközben a tégelyt minden egyes folyadék-hozzáadás után leszívatjuk. Ne alkalmazzunk leszívást addig, amíg mindegyik mosófolyadék gravitációs úton teljesen le nem csöpögött. Végül szívassuk le a tégelyt, szárítsuk meg, hűtsük le és mérjük meg a szálmaradékkal együtt.
Megjegyzés: A hőmérséklet nagyon erősen befolyásolja a melamin oldhatóságát, ezért gondosan ellenőrizni kell.
5. AZ EREDMÉNYEK KISZÁMÍTÁSA ÉS KIFEJEZÉSE
Az eredményeket az általános útmutatóban meghatározottak szerint számítsuk ki. A "d" értéke pamut és aramid esetében 1,02.
6. PONTOSSÁG
Textilanyagok homogén keveréke esetében az e módszerrel nyerhető eredmények megbízhatósági tartománya 95 %-os megbízhatósági szinten nem nagyobb ± 2-nél.
III. MELLÉKLET
A. RÉSZ
HATÁLYON KÍVÜL HELYEZETT IRÁNYELVEK
(A 8. cikkben hivatkozott irányelvek)
- A Tanács 72/276/EGK irányelve (HL L 173., 1972.7.31., 1. o.) és annak későbbi módosításai:
-
- A Bizottság 79/76/EGK irányelve (HL L 17., 1979.1.24., 17. o.)
- A Tanács 81/75/EGK irányelve (HL L 57., 1981.3.4., 23. o.)
- A Bizottság 87/184/EGK irányelve (HL L 75., 1987.3.17., 21. o.)
B. RÉSZ
BEVEZETÉSI HATÁRIDŐK
| Irányelv | A bevezetés határideje |
| 72/276/EGK | 1974. január 18. |
| 79/76/EGK | 1979. június 28. |
| 81/75/EGK | 1982. február 27. |
| 87/184/EGK | 1988. szeptember 1. |
IV. MELLÉKLET
MEGFELELÉSI TÁBLÁZAT
| Ez az irányelv | A 72/276/EGK irányelv |
| 1. cikk | 1. cikk |
| 2. cikk | 2. cikk |
| 3. cikk | 3. cikk |
| 4. cikk | 4. cikk |
| 5. cikk | 5. cikk |
| 6. cikk | 6. cikk |
| 7. cikk | 7. cikk (2) bekezdése |
| 8. cikk | – |
| 9. cikk | 8. cikk |
| I. melléklet | I. melléklet |
| II. melléklet (1) bekezdése | II. melléklet (1) bekezdése |
| II. melléklet (2) bekezdése | II. melléklet (2) bekezdése |
| II. melléklet, 1. módszer | II. melléklet, 1. módszer |
| II. melléklet, 2. módszer | II. melléklet, 2. módszer |
| II. melléklet, 3. módszer | II. melléklet, 3. módszer |
| II. melléklet, 4. módszer | II. melléklet, 4. módszer |
| II. melléklet, 5. módszer | II. melléklet, 5. módszer |
| II. melléklet, 6. módszer | II. melléklet, 6. módszer |
| II. melléklet, 7. módszer | II. melléklet, 7. módszer |
| II. melléklet, 8. módszer | II. melléklet, 8. módszer |
| II. melléklet, 9. módszer | II. melléklet, 9. módszer |
| II. melléklet, 10. módszer | II. melléklet, 10. módszer |
| II. melléklet, 11. módszer | II. melléklet, 11. módszer |
| II. melléklet, 12. módszer | II. melléklet, 13. módszer |
| II. melléklet, 13. módszer | II. melléklet, 14. módszer |
| II. melléklet, 14. módszer | II. melléklet, 15. módszer |
| II. melléklet, 15. módszer | II. melléklet, 16. módszer |
| III. melléklet | –– |
| IV. melléklet | –– |
( 1 ) HL C 96., 1994.4.6., 20. o.
( 2 ) HL C 195., 1994.7.18., 20. o.
( 3 ) Az Európai Parlament 1995. február 15-i állásfoglalása (HL C 56., 1995.3.6., 53. o.), a Tanács 1996. február 26-i közös álláspontja (HL C 196., 1996.7.6., 20. o.), valamint az Európai Parlament 1996. június 18-i határozata (HL C 198., 1996.7.8., 25. o.), és a Tanács 1996. október 7-i határozata.
( 4 ) HL L 173., 1972.7.31., 1. o. A legutóbb a 87/184/EGK irányelvvel (HL L 75., 1987.3.17., 21. o.) módosított irányelv.
( 5 ) HL L 32., 1997.2.3., 38. o.
( 6 ) Bizonyos esetekben szükséges az egyes próbadarabok előkezelése.
( 7 ) Kiszerelt és késztermékek vonatkozásában lásd a 7. pontot.
( 8 ) Lásd az 1. pontot.
( 9 ) A laboratóriumi kártológépet szálkeverővel is lehet helyettesíteni, illetve a szálakat a "pászmák és selejtek" módszerével is lehet keverni.
( 10 ) Ha a kiszerelési egységek egy arra alkalmas motollára felvethetők, egyszerre több is csévélhető.
( 11 ) A 12. módszer kivétel. Ez ugyanis a két összetevő egyikében található egyik anyag meghatározásán alapszik.
( 12 ) Lásd az I.1. mellékletet.
( 13 ) Annak biztosítására, hogy a szálmaradék tízperces időtartamra az ammóniaoldatban maradjon, olyan csappal ellátott szűrőtégelyt lehet felszerelni, amellyel az ammóniaoldat áramlása szabályozható.
( 14 ) Az elemzés elvégzése előtt a modakrilok és klórszálak oldódását a reagensben ellenőrizni kell.
( 15 ) Az elemzés elvégzése előtt a polivinil-klorid műszálak oldódását a reagensben ellenőrizni kell.
( 16 ) Ezek a reagensek nem tartalmazhatnak nitrogént.
( 17 ) Lásd például a Melliand Textilberichte 56 (1975), 643-645. oldalain leírt berendezést.
Lábjegyzetek:
[1] A dokumentum eredetije megtekinthető CELEX: 31996L0073 - https://eur-lex.europa.eu/legal-content/HU/ALL/?uri=CELEX:31996L0073&locale=hu Utolsó elérhető, magyar nyelvű konszolidált változat CELEX: 01996L0073-20110819 - https://eur-lex.europa.eu/legal-content/HU/ALL/?uri=CELEX:01996L0073-20110819&locale=hu