
31972L0199[1]
A Bizottság harmadik irányelve (1972. április 27.) a takarmányok hatósági ellenőrzésére szolgáló közösségi analitikai módszerek meghatározásáról
A BIZOTTSÁG HARMADIK IRÁNYELVE
(1972. április 27.)
a takarmányok hatósági ellenőrzésére szolgáló közösségi analitikai módszerek meghatározásáról
(72/199/EGK)
AZ EURÓPAI KÖZÖSSÉGEK BIZOTTSÁGA,
tekintettel az Európai Gazdasági Közösséget létrehozó szerződésre,
tekintettel a takarmányok hatósági ellenőrzésére szolgáló közösségi mintavételi és analitikai módszerek bevezetéséről szóló, 1970. július 20-i tanácsi irányelvre ( 1 ) és különösen annak 2. cikkére,
mivel az említett irányelv előírja, hogy a takarmányok hatósági ellenőrzését, amelynek célja a takarmányok minőségére és összetételére vonatkozó törvényi, rendeleti és közigazgatási rendelkezésekből eredő követelmények betartásának ellenőrzése, közösségi mintavételi és analitikai módszerek alkalmazásával kell végrehajtani;
mivel az 1971. június 15-i 71/250/EGK ( 2 ) és az 1971. november 18-i 71/393/EGK bizottsági irányelv ( 3 ) már megállapított számos közösségi analitikai módszert; mivel az azóta eltelt időben, az adott területen történt fejlődésre való tekintettel egy harmadik módszersorozatot is el kell fogadni;
mivel az ebben az irányelvben előírt intézkedések összhangban vannak a Takarmányok Állandó Bizottsága véleményével,
ELFOGADTA EZT AZ IRÁNYELVET:
1. cikk
A tagállamok előírják, hogy a takarmányok hatósági ellenőrzésére szolgáló, azok keményítő-, nyersfehérje-, pepszinnel és sósavval oldható nyersfehérje-, szabad és összes gosszipol-szintjének, és pepszin-aktivitásának meghatározására irányuló analitikai vizsgálatokat ezen irányelv I. mellékletében leírt módszerek alkalmazásával kell végrehajtani.
2. cikk
A tagállamok előírják, hogy a takarmányok hatósági ellenőrzésére szolgáló analitikai vizsgálatokat, amelyek célja a -----a takarmányokban megtalálható -----tilozin és virginiamicin szintjének meghatározása, ezen irányelv II. mellékletében leírt módszerek alkalmazásával kell végrehajtani.
3. cikk
A tagállamok legkésőbb 1973. július 1-ig hatályba léptetik azokat a törvényi, rendeleti és közigazgatási rendelkezéseket, amelyek szükségesek ahhoz, hogy ennek az irányelvnek megfeleljenek. Erről haladéktalanul tájékoztatják a Bizottságot.
4. cikk
Ennek az irányelvnek a tagállamok a címzettjei.
I. MELLÉKLET
1. A KEMÉNYÍTŐ MEGHATÁROZÁSA - POLARIMETRIÁS MÓDSZER
1. Cél és alkalmazási terület
E módszer lehetővé teszi a keményítő és a nagy molekulasúlyú keményítő-bomlástermékek szintjének takarmányokban történő meghatározását a 86/174/EGK bizottsági irányelv és a 96/25/EK tanácsi irányelv betartásának ellenőrzése céljából.
2. Vizsgálati alapelv
A módszer két meghatározásból áll. Az első meghatározás során a mintát, annak forró állapotában híg sósavval kezeljük. Tisztítás és szűrés után az oldat optikai forgatóképességét polarimetriával mérjük.
A második meghatározás során a mintát 40 %-os etanollal extraháljuk. A szűrlet sósavval való savanyítása, tisztítása és szűrése után az optikai forgatóképességet ugyanúgy mérjük, mint az első meghatározás esetében.
A minta keményítőtartalmát úgy kapjuk meg, hogy a két mért érték különbségét megszorozzuk egy ismert együtthatóval.
3. Reagensek
3.1. 25 %-os (w/w) sósav, d: 1,126 g/ml.
3.2. 1,128 %-os (w/v) sósav.
A koncentrációt 0,1 mól/liter koncentrációjú nátrium-hidroxid oldat alkalmazásával végzett titrálással kell ellen- őrizni, 94 %-os (v/v) etanolban oldott 0,1 % (w/v) metilvörös jelenlétében. 10 ml = 30,94 ml 0,1 mól/liter koncentrációjú NaOH.
3.3. Carrez I. oldat: vízben oldjunk fel 21,9 g cink-acetátot, Zn(CH3COO)2·2H2O és 3 g jégecetet. Töltsük fel vízzel 100 ml-re.
3.4. Carrez II. oldat: vízben oldjunk fel 10,6 g kálium-hexaciano-ferrátot [K4(Fe(CN)6]·3H2O. Töltsük fel vízzel 100 ml-re.
3.5. 40 %-os (v/v) etanol, d: 0,948 g/ml 20 °C-on.
4. Eszközök
4.1. 250 ml-es Erlenmeyer-lombik szabványos üvegcsiszolattal és visszafolyós hűtővel.
4.2. Polariméter vagy szachariméter.
5. A vizsgálat módja
5.1. A minta előkészítése
Őröljük a mintát olyan finomságúra, hogy az teljes egészében áthaladjon egy 0,5 mm szemméretű, kerek szemű szitán.
5.2. A teljes optikai forgatóképesség (P vagy S) meghatározása (lásd a 7.1. megjegyzést)
Mérjünk ki a megőrölt mintából mg pontossággal 2,5 g-ot, és helyezzük azt egy 100 ml-es mérőlombikba. Adjunk hozzá 25 ml sósavat (3.2.), rázzuk össze a vizsgálandó minta egyenletes eloszlásának biztosítása céljából és adjunk hozzá további 25 ml sósavat (3.2.). Merítsük a lombikot forrásban levő vízfürdőbe, az első három percben erőteljesen és folyamatosan rázogassuk a csomóképződés megakadályozása céljából. A vízfürdőben elegendő víznek kell lennie ahhoz, hogy a fürdő a lombik behelyezésekor forrásponton maradjon. A lombikot rázogatás közben nem szabad kivenni a vízfürdőből. Pontosan 15 perc elteltével vegyük ki a lombikot a vízfürdőből, adjunk hozzá 30 ml hideg vizet és azonnal hűtsük le 20 °C-ra.
Adjunk hozzá 5 ml-t a Carrez I. oldatból (3.3.), és rázogassuk egy percen át. Ezt követően adjunk hozzá 5 ml-t a Carrez II. oldatból (3.4.), és ismét rázzuk egy percen át. Töltsük fel vízzel térfogatra, keverjük össze és szűrjük meg. Ha a szűrlet nem teljesen tiszta (ami ritkán fordul elő), az Carrez I. és II. oldat nagyobb mennyiségével (pl. 10 ml) ismételjük meg a meghatározást.
Egy 200 mm-es csőben a polariméterrel vagy a szachariméterrel mérjük meg az oldat optikai forgatóképességét.
5.3. A 40 %-os etanolban oldható anyagok optikai forgatóképességének (P′ vagy S′) meghatározása
Mérjünk ki a mintából mg pontossággal 5 g-ot, helyezzük egy 100 ml-es mérőlombikba, és adjunk hozzá mintegy 80 ml etanolt (3.5.) (lásd a 7.2 megjegyzést). Hagyjuk a lombikot állni szobahőmérsékleten 1 órán át; ezalatt hatszor erőteljesen rázzuk össze, hogy a vizsgálandó minta alaposan összekeveredjen az etanollal. Töltsük fel térfogatra etanollal (3.5.), keverjük össze és szűrjük meg. A szűrletből pipettázzunk 50 ml-t (= 2,5 g minta) egy 250Xml-es Erlenmeyer-lombikba, adjunk hozzá 2,1 ml sósavat (3.1.), és erősen rázzuk össze. Csatlakoztassunk visszafolyós hűtőt az Erlenmeyer-lombikhoz, és merítsük az utóbbit forrásban levő vízfürdőbe. Pontosan 15 perc elteltével vegyük ki az Erlenmeyer-lombikot a vízfürdőből, öntsük át annak tartalmát egy 100 ml-es mérőlombikba, öblítsük kis mennyiségű hideg vízzel, majd hűtsük le 20 °C-ra. Derítsük az Carrez I.oldat (3.3.) és a Carrez II. oldat (3.4.) alkalmazásával, vízzel töltsük fel térfogatra, keverjük össze, szűrjük meg, majd mérjük meg az optikai forgatóképességét az 5.2. pont második és harmadik bekezdésében ismertetett módon.
6. Az eredmények kiszámítása
A keményítőtartalmat ( %) a következőképpen számítjuk ki:
6.1. Mérés polariméterrel
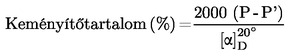
P = a teljes optikai forgatóképesség szögfokban
P′ = a 40 %-os (V/V) etanolban oldható anyagok optikai forgatóképessége szögfokban
= a tiszta keményítő fajlagos optikai forgatóképessége. Az együttható elfogadott számszerű értékei a következők:
+ 185,9° : rizskeményítő
+ 185,4° : burgonyakeményítő
+ 184,6° : kukoricakeményítő
+ 182,7° : búzakeményítő
+ 181,5° : árpakeményítő
+ 181,3° : zabkeményítő
+ 184,0° : összetett takarmányokban használt egyéb keményítőtípusok és keményítőkeverékek
6.2. Mérés szachariméterrel
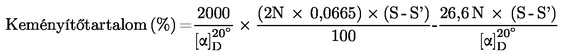
S = teljes optikai forgatóképesség szachariméter-fokban
S′ = a 40 %-os (V/V) etanolban oldható anyagok optikai forgatóképessége szachariméter-fokban
N
=
az a szacharózmennyiség (g) 100 ml vízben, amely 100 szachariméter-foknak megfelelő optikai forgatóképességet ad 200 mm-es csőben mérve
16,29 g a francia szachariméterek esetében
26,00 g a német szachariméterek esetében
20,00 g a vegyes szachariméterek esetében.
[Kép #1] = a tiszta keményítő fajlagos optikai forgatóképessége (lásd 6.1.).
Kép #1
6. 3 Ismételhetőség
Az ugyanazon a mintán végzett két párhuzamos meghatározás eredményének különbsége 40 %-nál alacsonyabb keményítőtartalom esetében, abszolút értékben nem lehet több, mint 0,4, a 40 %-os vagy annál nagyobb keményítőtartalmak esetében pedig, nem haladhatja meg az 1,1 %-os relatív értéket.
7. Észrevételek
7.1. Ha a minta több, mint 6 %-nyi karbonátot tartalmaz kalcium-karbonátra számítva, e karbonátot híg kénsav pontosan megfelelő mennyiségével történő kezeléssel el kell roncsolni a teljes optikai forgatóképesség meghatározását megelőzően.
7.2. Magas laktóztartalmú termékek - pl. tejsavópor vagy sovány tejpor - esetében 80 ml etanol (3.5.) hozzáadását követően, a következőképpen kell eljárni. Csatlakoztassunk visszafolyós hűtőt a lombikhoz, és merítsük az utóbbit 50 °C-os vízfürdőbe 30 percre. Hagyjuk lehűlni, majd folytassuk az analízist az 5.3. pontban leírt módon.
7.3. A következő takarmány-alapanyagokról ismert, hogy, a takarmányokban való nagy mennyiségű jelenlétük esetén, olyan interferenciákat idéznek elő a keményítőtartalom polarimetriás módszerrel való meghatározásakor, amelyek téves eredményekhez vezethetnek:
- (cukor)répakészítmények, pl. (cukor)répapép, (cukor)répamelasz, (cukor)répapép-melasz, (cukor)répaseprő, (répa)cukor,
- citruspép,
- lenmag; lenmagpogácsa; lenmagkivonat,
- repcemag; repcemagpogácsa; repcemagkivonat; repcemaghéj,
- napraforgómag; napraforgómag kivonat; részlegesen hántolt napraforgómag kivonat,
- koprapogácsa, koprakivonat,
- burgonyapép,
- szárított élesztő,
- inulinban gazdag termékek (pl. csicsókaszeletek és csicsókaliszt);
- töpörtyű.
2. A NYERSFEHÉRJE MEGHATÁROZÁSA
1. Cél és alkalmazási terület
E módszer lehetővé teszi a takarmányok nyersfehérje-tartalmának meghatározását a Kjeldahl-módszer szerint meghatározott nitrogéntartalom alapján.
2. Vizsgálati alapelv
A mintát katalizátor jelenlétében kénsavval roncsoljuk. A savas oldatot nátrium-hidroxid oldattal lúgosítjuk. Az ammóniát lepároljuk, és ismert mennyiségű kénsavban vesszük fel, amelynek feleslegét nátrium-hidroxid standard oldatával titráljuk.
3. Reagensek
3.1. Kálium-szulfát.
3.2. Katalizátor: réz(II)-oxid, CuO vagy réz(II)-szulfát-pentahidrát, CuSO4 · 5H2O
3.3. Granulált cink.
3.4. Kénsav, p20 = 1,84 g/ml.
3.5. Kénsav, c(½H2SO4) = 0,5 mol/l.
3.6. Kénsav, c(½H2SO4) = 0,1 mol/l.
3.7. Metilvörös indikátor; oldjunk fel 300 mg metilvöröst 100 ml etanolban, σ = 95-96 % (v/v)
3.8. Nátrium-hidroxid oldat (technikai minőségű is használható) σ = 40 g/100 ml (m/v: 40 %).
3.9. Nátrium-hidroxid oldat, c = 0,25 ml/l.
3.10. Nátrium-hidroxid oldat, c = 0,1 mol/l.
3. 11 Granulált horzsakő, sósavban mosott és meggyújtott.
3.12. Acetanilid (olvadáspont = 114 °C, N = 10,36 %)
3.13. Szacharóz (nitrogénmentes).
4. Eszközök
A Kjeldahl-eljárás szerinti roncsolás, desztillálás és titrálás elvégzésére alkalmas készülék.
5. A vizsgálat módja
5.1. Roncsolás
Mérjünk ki 0,001 g pontossággal 1 g mintát, és helyezzük be a roncsoló lombikjába. Adjunk hozzá 15 g kálium-szulfátot (3.1.), megfelelő mennyiségű katalizátort (3.2.) (0,3-0,4 g réz(II)-oxidot vagy 0,9-1,2 g réz(II)-szulfát-pentahidrátot), 25 ml kénsavat (3.4.) és néhány szemcse horzsakövet (3.11.), majd keverjük össze. Először mérsékelten hevítsük a lombikot, szükség esetén időnként keverjük meg annak tartalmát, egészen addig, amíg a massza karbonizálódik és a hab eltűnik; ezt követően erősebben hevítsük, amíg a folyadék erős forrásba nem jön. A hevítés akkor megfelelő, ha a forrásban levő sav a lombik falán lecsapódik. Akadályozzuk meg a lombik oldalának túlhevülését, és azt, hogy ahhoz szerves részecskék tapadjanak. Amikor az oldat feltisztult és világoszöld színűvé vált, folytassuk a forralást további két órán át, majd hagyjuk lehűlni az oldatot.
5.2. Lepárlás
Óvatosan adjunk hozzá annyi vizet, amennyi a szulfátok teljes feloldódásához elegendő. Engedjük lehűlni, majd adjunk hozzá néhány szemcsényi cinket (3.3.).
A lepárlókészülék gyűjtőlombikjába mérjünk be pontosan 25 ml kénsavat (3.5. vagy 3.6.), a feltételezett nitrogéntartalomtól függően. Adjunk hozzá néhány csepp metilvörös indikátort (3.7.).
Kapcsoljuk össze a Kjeldahl-lombikot a lepárlókészülék hűtőjével, és legalább 1 cm mélyen merítsük be a hűtő végét a gyűjtőlombikban lévő folyadékba (lásd a 8.3. észrevételt). Lassan, ammóniaveszteség nélkül öntsünk 100 ml nátrium-hidroxid oldatot (3.8.) a Kjeldahl-lombikba (lásd a 8.1. észrevételt).
Hevítsük a lombikot az ammónia lepárlódásáig.
5.3. Titrálás
A gyűjtőlombikban levő kénsavfelesleget a végpont eléréséig titráljuk nátrium-hidroxid oldattal (3.9. vagy 3.10.) az alkalmazott kénsav-koncentrációtól függően.
5.4. Vakpróba
A reagensek nitrogénmentességének igazolására végezzünk vakpróbát (roncsolás, lepárlás és titrálás) a minta helyett 1 g szacharózt (3.13.) használva.
6. Az eredmények kiszámítása
A nyersfehérje-tartalom kiszámítása a következő képlettel történik:
Ahol:
V0
=
a vakpróbában használt
NaOH (3.9. vagy 3.10.) térfogata (ml).
V1
=
a minta titrálásához használt
NaOH (3.9. vagy 3.10.) térfogata (ml).
c
=
a nátrium-hidroxid (3.9. vagy 3.10.)
koncentrációja (mol/l).
m = a minta tömege (g).
7. A módszer validálása
7.1. Ismételhetőség
Az ugyanazon a mintán végzett két párhuzamos meghatározás eredménye közötti különbség nem haladhatja meg:
a 0,2 %-ot abszolút értékben, 20 %-nál alacsonyabb nyersfehérje-tartalom esetében;
a magasabb értékhez viszonyított 1,0 %-ot, 20 % és 40 % közötti nyersfehérje-tartalom esetében;
a 0,4 %-ot abszolút értékben, a 40 %-nál magasabb nyersfehérje-tartalom esetében.
7.2. Pontosság
Végezzük el az analízist (roncsolás, lepárlás és titrálás) 1,5-2,0 g acetaniliden (3.12.) 1 g szacharóz (3.13.) jelenlétében; 1 g acetanilid 14,80 ml kénsavat (3.5.) fogyaszt. A visszanyerés mértékének legalább 99 %-osnak kell lennie.
8. Észrevételek
8.1. A készülék lehet kézi, félautomata vagy automata típusú. Ha az alkalmazott készülék esetében az emésztési és a lepárlási lépések között mintaátvitelre van szükség, ezt anyagveszteség nélkül kell végrehajtani. Ha a lepárlókészülék lombikja nincs felszerelve csepegtetőtölcsérrel, a nátrium-hidroxidot közvetlenül a lombik és a hűtő összekapcsolása előtt kell hozzáadni, a folyadékot lassan az edény belső oldala mentén folyatva.
8.2. Ha a roncsolt anyag megszilárdul, a fent megadottnál nagyobb mennyiségű kénsavval (3.4.) kezdjük újra a meghatározást.
8.3. Alacsony nitrogéntartalmú termékek esetén a gyűjtőlombikba mérendő kénsav (3.6.) térfogata szükség esetén 10 vagy 15 ml-re csökkenthető, és vízzel 25 ml-re feltölthető.
3. A PEPSZINBEN ÉS SÓSAVBAN OLDOTT NYERSFEHÉRJE MEGHATÁROZÁSA
1. Cél és alkalmazási terület
E módszer lehetővé teszi a pepszinnel és sósavval oldott nyersfehérje-frakció adott körülmények között történő meghatározását. Minden takarmánynál alkalmazható.
2. Vizsgálati alapelv
A mintát 40 °C-on, 48 órán át sósavas pepszinoldatban melegítjük. A szuszpenziót leszűrjük, és a szűrlet nitrogéntartalmát a nyersfehérje meghatározásánál ismertetett módszer szerint meghatározzuk.
3. Reagensek
3.1. Sósav, d: 1,125.
3.2. Sósav 0,075 N.
3.3. 2,0 E/mg pepszin; a pepszinaktivitás ezen melléklet 4. részében ismertetett módszeren belül kerül leírásra, és e módszer szerint kell azt megállapítani.
3.4. Sósavban (3.2) frissen oldott kb. 0,2 %-os (w/v) pepszin; aktivitása: 400 E/l.
3.5. Habzásgátló emulzió (pl. szilikon).
3.6. Mindazok a reagensek, amelyek a nyersfehérje-tartalom meghatározási módszerét ismertető rész 3. pontjában kerültek felsorolásra.
4. Eszközök
4.1. Vízfürdő vagy inkubátor, 40 °C ± 1 °C-ra beállítva.
4.2. Kjeldahl-módszerrel történő emésztésre és lepárlásra szolgáló készülék.
5. A vizsgálat módja
5.1. Oldatkészítés (lásd a 7.2 észrevételt).
Mérjünk ki 2 g mintát mg pontossággal, és helyezzük bele egy 500 ml-es mérőlombikba. Adjunk hozzá 450 ml, 40 °C-ra előmelegített sósavas pepszinoldatot (3.4), és rázzuk össze, hogy megelőzzük a csomóképződést. Ellenőrizzük, hogy a szuszpenzió pH-ja 1,7 alatt van. Tegyük a mérőlombikot a vízfürdőbe vagy az inkubátorba (4.1), és 48 órán át hagyjuk ott. Rázzuk össze 8, 24 és 32 óra elteltével. Negyvennyolc óra elteltével adjunk hozzá 15 ml sósavat (3.1), hűtsük le 20 °C-ra, vízzel töltsük fel térfogatra, és szűrjük át.
5.2. Emésztés
A szűrletből mérjünk ki 250 ml-t és helyezzük a lepárlókészülék lombikjába (4.2). Adjuk hozzá a nyersfehérje-tartalom meghatározási módszerét leíró 5.1 pont második mondatában feltüntetett, az emésztéshez szükséges reagenseket. Homogenizáljuk és forraljuk fel. Habképződés esetén adjunk hozzá néhány cseppet a habzásgátló emulzióból (3.5). Folytassuk a forralást, amíg a víz csaknem teljesen elpárolog. Csökkentsük a hőt, és óvatosan vonjuk el a maradék vizet is.
Amikor az oldat letisztul és színtelenné (illetve réz alapú katalizátor használata esetén világoszöld színűvé) válik, folytassuk a forralást további egy órán át. Ezután hagyjuk hűlni.
5.3. Lepárlás és titrálás
A műveletet a nyersfehérje-meghatározási módszer leírásának 5.2 és 5.3 pontjában leírtak szerint hajtsuk végre.
5.4. Vakpróba
Végezzünk vakpróbát a fent ismertetett eljárással, az analizálandó minta elhagyásával.
6. Az eredmények kiszámítása
Vonjuk ki a vakpróba során fogyott kénsav mennyiségét abból a kénsavmennyiségből, amely a minta feldolgozása során fogyott. 1 ml 0,1 N kénsav 1,4 mg nitrogénnek felel meg.
Szorozzuk meg a nitrogén mennyiségét a 6,25-os szorzóval. Az eredményt a mintára vonatkoztatva, százalékosan fejezzük ki.
Ismételhetőség
Az ugyanazon a mintán végzett két párhuzamos meghatározás eredménye közötti különbség nem haladhatja meg:
- 20 %-nál alacsonyabb nyersfehérje-tartalom esetében, abszolút értékben a 0,4-et,
- 20 %-nál nem alacsonyabb és 40 %-osnál nem magasabb nyersfehérje-tartalom esetében, relatív értékben a 2,0 %-ot,
- 40 %-ot meghaladó nyersfehérje-tartalom esetében, abszolút értékben a 0,8-et.
7. Észrevételek
7.1. Az e módszer során kapott eredményeknek nincs közvetlen összefüggésük az in vivo emészthetőséggel.
7.2. A 10 %-nál több olajat vagy zsírt tartalmazó termékek vizsgálata esetén a terméket először petroléterrel, extrahálás útján zsírtalanítani kell (B.P. 40-60 °C).
4. A PEPSZINAKTIVITÁS MEGHATÁROZÁSA
1. Cél és alkalmazási terület
Ez a módszer lehetővé teszi a pepszinben és sósavban oldott nyersfehérje meghatározásánál használt pepszin aktivitásának megállapítását.
2. Vizsgálati alapelv
Meghatározott körülmények között, sósavas közegben, hemoglobint pepszinnel kezelünk. A fehérje nem hidrolizált frakciója kicsapódik triklór-ecetsavban. A szűrlethez nátrium-hidroxidot és Folin-Ciocalteu reagenst adunk. Megmérjük az oldat optikai sűrűségét 750 nm-en, és egy kalibrációs görbéről leolvassuk a tirozin megfelelő mennyiségét.
Definíció: E módszer szerint egy egységnyi pepszin az az enzimmennyiség, amely a módszer feltételei mellett percenként annyi hidroxi-aril csoportot szabadít fel, amely Folin-Ciocalteu reagenssel festve ugyanolyan optikai sűrűséget mutat, mint egy μmol tirozin, ugyanezzel a módszerrel festve.
3. Reagensek
3.1. Sósav 0,2 N.
3.2. Sósav 0,06 N.
3.3. Sósav 0,025 N.
3.4. 5 %-os triklór-ecetsav-oldat (w/v).
3.5. Nátrium-hidroxid-oldat, 0,5 N.
3.6. Folin-Ciocalteu reagens. Tegyünk egy kétliteres, szabványos üvegcsiszolattal ellátott gömblombikba 100 g nátrium-wolframátot (Na2WO4 · 2H2O), 25 g nátrium-molibdenátot (Na2MoO4 · 2H2O) és 700 ml vizet. Adunk hozzá 50 ml foszforsavat (d: 1,71) és 100 ml tömény sósavat (d: 1,19), csatlakoztassunk a lombikhoz egy visszafolyós hűtőt, forraljuk fel, és hagyjuk finoman forrni az oldatot 10 órán át. Hagyjuk lehűlni, válasszuk le a visszafolyós hűtőt, és adjunk hozzá 175 g lítium-szulfátot (Li2SO4 · 2H2O), 50 ml vizet és 1 ml brómot. 15 percen át forraljuk, hogy a brómfelesleg elpárologjon.
Hagyjuk lehűlni, töltsük át az oldatot egy 1 literes mérőlombikba, töltsük fel vízzel térfogatra, homogenizáljuk és szűrjük át. Zöldes elszíneződés nem maradhat. Felhasználás előtt 1 rész reagenst hígítsunk fel 2 rész vízzel.
3.7. Hemoglobinoldat: mérjünk ki hemoglobint olyan mennyiségben (kb. 2 g Anson szerint meghatározott fehérje-szubsztrátum), amely 354 mg nitrogénnek ( 4 ) felel meg, és tegyük bele egy szabványos üvegcsiszolattal ellátott lombikba. Adjunk hozzá néhány ml sósavat (3.2), és csatlakoztassuk a lombikot egy vákuumszivattyúhoz, majd addig rázzuk, amíg a hemoglobin teljesen fel nem oldódik. Szüntessük meg a vákuumot, és folyamatos rázás közben töltsük fel 100 ml-re sósavval (3.2). Közvetlenül a felhasználás előtt készítsük el.
3.8. Standard tirozinoldat: oldjunk fel 181,2 mg tirozint sósavban (3.1), és töltsük fel 1 literre, ugyanazzal a savval (törzsoldat). Vegyünk belőle 20 ml-t, és hígítsuk fel 100 ml-re sósavval (3.1). Ezen oldat 1 ml-e 0,2 μmol tirozint tartalmaz.
4. Eszközök
4.1. Ultratermosztáttal 25 °C ± 0,1 °C-ra beállított vízfürdő.
4.2. Spektrofotométer.
4.3. Precíziós óra (pontosság: 1 másodperc).
4.4. pH-mérő.
5. A vizsgálat módja
5.1. Oldatkészítés (lásd a 7.1 megjegyzést)
Oldjunk fel 150 mg pepszint 100 ml sósavban (3.2). Pipettázzunk ebből az oldatból 2 ml-t egy 50 ml-es mérőlombikba, és töltsük fel térfogatra sósavval (3.3). Ellenőrizzük a pH-értéket, amely nem lehet több 1,6 ± 0,1-nél. Merítjük a lombikot vízfürdőbe (4.1).
5.2. Hidrolízis
Pipettázzunk 5,0 ml hemoglobinoldatot (3.7) egy kémcsőbe, hevítsük 25 °C-ra a vízfürdőben (4.1), adjunk hozzá 1,0 ml-t az 5.1 pont szerint elkészített pepszinből, majd kevergessük egyik végén megvastagított üvegbottal kb. tízszer, oda-vissza irányuló mozdulattal. Hagyjuk a kémcsövet a 25 °C-os vízfürdőben, a pepszinoldat hozzáadásától számított pontosan 10 percen keresztül (az időtartamot és a hőmérsékletet szigorúan be kell tartani). Ezután adjunk hozzá 10,0 ml triklór-ecetsav-oldatot (3.4), amelyet 25 °C-ra előmelegítettünk, homogenizáljuk és szűrjük át egy száraz szűrőn.
5.3. Az elszíneződés kialakulása és az optikai sűrűség mérése.
Pipettázzunk a szűrletből 5,0 ml-t egy 50 ml-es Erlenmeyer-lombikba, adjunk hozzá 10,0 ml nátrium-hidroxid-oldatot (3.5), és folyamatos rázás közben adjunk hozzá 3,0 ml hígított Folin-Ciocalteu reagenst (3.6). Öt-tíz perc múlva mérjük meg az oldat optikai sűrűségét spektrofotométerrel, 750 nm-en, víz ellenében, 1 cm-es küvettákban.
5.4. Vakpróba
Minden meghatározás esetén vakpróbát kell végezni az alábbiak szerint:
Pipettázzunk 5,0 ml hemoglobinoldatot (3.7) egy kémcsőbe, hevítsük 25 °C-ra a vízfürdőben (4.1), adjunk hozzá 25 °C-ra előmelegített, 10,0 ml triklór-ecetsavat (3.4), homogenizáljuk, majd adjunk hozzá az 5.1 pont szerint készített pepszinoldatból 1,0 ml-t. Keverjük össze üvegpálcával, és hagyjuk pontosan 10 percen át a 25 °C-os vízfürdőben (4.1). Homogenizáljuk, és szűrjük át egy száraz szűrőn. Kövessük az 5.3 pontban leírt eljárást.
5.5. Kalibrációs görbe
A standard tirozinoldatból (3.8) mérjünk be 1,0, 2,0, 3,0, 4,0 és 5,0 ml aliquot mennyiségeket, amelyek 0,2, 0,4, 0,6, 0,8 és 1,0 μmol tirozintartalomnak felelnek meg, egy-egy 50 ml-es Erlenmeyer-lombikba. A sorozatot egészítsük ki egy tirozinmentes referenciaoldattal. Töltsük fel sósavval (3.1) 5,0 ml-re. Adjunk hozzá 10,0 ml nátrium-hidroxid-oldatot (3.5), és folyamatos rázás közben adjunk hozzá 3,0 ml hígított Folin-Ciocalteu reagenst (3.6). Mérjük meg az optikai sűrűséget az 5.3 pont utolsó mondata szerint. Szerkesszük meg a kalibrációs görbét úgy, hogy az optikai sűrűséget a tirozinmennyiség függvényében ábrázoljuk.
6. Az eredmények kiszámítása
Olvassuk le a kalibrációs görbéről a μmol-ban kifejezett tirozin mennyiségét, amely megfelel a vakpróbával korrigált színes oldat optikai sűrűségének.
A tirozin mg-onkénti és percenkénti pepszinaktivitását, μmol-ban, 25 °C-on, az alábbi képlet segítségével számítjuk ki:
ahol:
a = a kalibrációs görbéről leolvasott tirozin mennyisége μmol-ban;
p = az 5.2 pont szerint hozzáadott pepszinmennyiség súlya mg-ban mérve.
7. Észrevételek
7.1. A feloldandó pepszin mennyisége akkora legyen, hogy a végső fotometriás mérés szerint 0,35 ± 0,035 optikai sűrűséget kapjunk.
7.2. Az e módszerrel nyert mg-onkénti két egység megfelel:
3,64 Anson milliegység/mg-nak (μmol tirozin/mg/perc 35,5 °C-on) vagy
36,400 kereskedelmi egység/g-nak (μmol tirozin/g 10 perc alatt 35,5 °C-on).
5. A SZABAD ÉS AZ ÖSSZES GOSSZIPOL MEGHATÁROZÁSA
1. Cél és alkalmazási terület
Ez a módszer lehetővé teszi a szabad gosszipol, az összes gosszipol és a kémiailag rokon anyagok mennyiségi meghatározását a gyapotmagban, a gyapotmaglisztben és a gyapotmag-pogácsában, valamint az ezen anyagokat tartalmazó összetett takarmányokban, amennyiben a gosszipolból több mint 20 ppm van jelen.
2. Vizsgálati alapelv
A gosszipolt 3-aminopropán-1-ol jelenlétében, vagy propán-2-ol és hexán keverékével (a szabad gosszipol meghatározása esetén) vagy dimetil-formamiddal (az összes gosszipol meghatározása esetén) extraháljuk. A gosszipol anilinnel gosszipol-dianilinné alakul, amelynek optikai sűrűségét 440 nm-nél mérjük.
3. Reagensek
3.1. Propán-2-ol-hexán keverék: keverjünk össze 60 térfogategységnyi propán-2-ol a.r.-t 40 térfogategységnyi n-hexánnal.
3.2. A-oldószer: tegyünk 1 literes mérőlombikba kb. 500 ml propán-2-ol-hexán keveréket (3.1), 2 ml 3-aminopropán-1-ol-t, 8 ml jégecetet, valamint 50 ml vizet. Töltsük fel térfogatra a propán-2-ol-hexán keverékkel (3.1). Ez a reagens egy hétig stabil.
3.3. B-oldószer: pipettázzunk 2 ml 3-aminopropán-1-ol-t és 10 ml jégecetet egy 100 ml-es mérőlombikba. Hűtsük le szobahőmérsékletre, és töltsük fel térfogatra N,N-dimetil-formamiddal. Ez a reagens egy hétig stabil.
3.4. Anilin a.r.: ha a vakpróba optikai sűrűsége meghaladja a 0,022-et, pároljuk le az anilint cinkpor fölött, és öntsük el a párlat első és utolsó 10-10 %-át. Lehűtve és barna, lezárt üveglombikban tárolva a reagens több hónapig eláll.
3.5. Gosszipol-A standard oldat: tegyünk 27,9 mg gosszipol-acetátot egy 250 ml-es mérőlombikba. Oldjuk fel, és töltsük fel térfogatra az A-oldószerrel (3.2). Pipettázzunk 50 ml-t ebből az oldatból egy 250 ml-es mérőlombikba, és töltsük fel térfogatra az A-oldószerrel. Ennek az oldatnak a gosszipol-koncentrációja 0,02 mg/ml. Használat előtt hagyjuk egy órán át szobahőmérsékleten állni.
3.6. Gosszipol-B standard oldat: tegyünk 27,9 mg gosszipol-acetátot egy 50 ml-es mérőlombikba. Oldjuk fel, és töltsük fel térfogatra a B-oldószerrel (3.3). Ennek az oldatnak a gosszipol-koncentrációja 0,5 mg/ml.
Ha fénytől védve tároljuk, a gosszipol-A és -B standard oldat 24 órán át stabil marad.
4. Eszközök
4.1. Keverőgép (kb. 35 fordulat/perc).
4.2. Spektrofotométer.
5. A vizsgált módja
5.1. A vizsgálati minta
A vizsgálati minta mennyisége a minta feltételezett gosszipoltartalmától függ. Jobb kisebb mennyiségű mintával dolgozni, és a szűrlet viszonylag nagyobb mennyiségű aliquot részét használni, hogy elegendő mennyiségű gosszipolt nyerjünk, amely lehetővé teszi a precíz fotometriás mérést. A gyapotmag, a gyapotmagliszt és a gyapotmag-pogácsa szabad gosszipoltartalmának mérésére a minta mennyisége ne haladja meg az 1 g-ot, összetett takarmányok vizsgálata során a minta mennyisége akár 5 g is lehet. A szűrlet 10 ml-nyi aliquot része a legtöbb esetben megfelelő: ez kb. 50-100 μg gosszipolt tartalmaz. Az összes gosszipoltartalom meghatározásához a minta mennyisége 0,5 és 5 g között legyen, így a szűrlet 2 ml-nyi aliquot része kb. 40-200 μg gosszipolt fog tartalmazni.
A vizsgálatokat kb. 20 °C-os szobahőmérsékleten kell végezni.
5.2. A szabad gosszipoltartalom meghatározása
Tegyük a vizsgálandó mintát egy 250 ml-es, csiszolatos lombikba, amelynek az alján üvegzúzalékot helyeztünk el. Pipettázzunk 50 ml A-oldószert (3.2) a lombikba, zárjuk le a lombikot, és keverjük egy órán át a keverőben. Szűrjük át egy száraz szűrőn, és gyűjtsük össze a szűrletet egy kis, csiszolatos lombikban. Szűrés közben fedjük be a tölcsért egy óraüveggel. Pipettázzunk azonos aliquot mennyiségű, 50-100 μg gosszipolt tartalmazó szűrletet két, 25 ml-es mérőlombikba (A- és B-jelű). Ha szükséges, töltsük fel a mintát 10 ml-re az A-oldószerrel (3.2). Ezután töltsük fel térfogatra az A-jelű lombikot propán-2-ol-hexán keverékkel (3.1). Ezt az oldatot referenciaoldatként használjuk, mellyel szemben a mintaoldatot mérjük.
Pipettázzunk 10-10 ml A-oldószert (3.2) két másik, 25 ml-es mérőlombikba (C- és D-jelű). Töltsük fel a C-jelű lombik tartalmát térfogatra propán-2-ol-hexán keverékkel (3.1). Ezt az oldatot referenciaoldatként használjuk, mellyel szemben a vakpróbaoldatot mérjük.
Adjunk 2-2 ml anilint (3.4) a D- és a B-jelű lombikhoz. Hevítsük ezeket 30 percen át forró vízfürdő fölött, hogy tartalmuk elszíneződjön. Hűtsük le szobahőmérsékletűre és töltsük fel tartalmukat térfogatra propán-2-ol-hexán keverékkel (3.1), homogenizáljuk, majd hagyjuk állni egy órán át.
Határozzuk meg a vakpróbaoldat (D) optikai sűrűségét a referenciaoldattal (C) összehasonlítva, és a mintaoldat (B) optikai sűrűségét a referenciaoldattal (A) összehasonlítva egy spektrofotométerrel, 440 nm-nél, 1 cm-es üvegküvettákat használva.
Vonjuk ki a vakpróbaoldat optikai sűrűségét a mintaoldat optikai sűrűségéből (= korrigált optikai sűrűség). Ebből az értékből a 6. pontban leírtak szerint számítsuk ki a szabad gosszipoltartalmat.
5.3. Az összes gosszipoltartalom meghatározása
Tegyünk egy 1-5 mg gosszipolt tartalmazó mintát egy 50 ml-es mérőlombikba, és adjunk hozzá 10 ml B-oldószert (3.3). Készítsünk ezzel párhuzamosan egy vakpróbaoldatot is úgy, hogy 10 ml B-oldószert (3.3) teszünk egy másik, 50 ml-es mérőlombikba. Hevítsük a két lombikot forró vízfürdő fölött 30 percen át. Hűtsük le a lombikokat szobahőmérsékletűre, és töltsük fel tartalmukat térfogatra propán-2-ol-hexán keverékkel (3.1). Homogenizáljuk, és hagyjuk ülepedni 10-15 percig, utána szűrjük át, majd gyűjtsük össze a szűrletet csiszolatos lombikokba.
A mintaszűrletből pipettázzunk 2-2 ml-t két, 25 ml-es mérőlombikba, és a vakpróbaszűrletből is pipettázzunk 2-2 ml-t két másik, 25 ml-es lombikba. Mindkét sorozatban az egyik lombikot töltsük fel 25 ml-re a propán-2-ol-hexán keverékkel (3.1). Ezeket az oldatokat használjuk referenciaoldatként.
A másik két lombik tartalmához adjunk 2-2 ml anilint (3.4). Hevítsük ezeket 30 percen át forró vízfürdő fölött, hogy tartalmuk elszíneződjön. Utána hűtsük le őket szobahőmérsékletűre, töltsük fel tartalmukat propán-2-ol-hexán keverékkel 25 ml-re (3.1), homogenizáljuk, és hagyjuk állni egy órán át.
Határozzuk meg az optikai sűrűséget az 5.2 pontban, a szabad gosszipoltartalomra vonatkozóan ismertetett módon. Ebből az értékből számítsuk ki az összes gosszipoltartalmat a 6. pont útmutatása alapján.
6. Az eredmények kiszámítása
Az eredmény kiszámítható akár a fajlagos optikai sűrűségből (6.1), akár egy kalibrációs görbe segítségével (6.2).
6.1. A fajlagos optikai sűrűségből
A fajlagos optikai sűrűség a leírt feltételek esetén az alábbiak szerint alakul:
| Szabad gosszipoltartalom: | [Kép #2] |
| Összes gosszipoltartalom: | [Kép #3] |
Kép #2
Kép #3
A minta szabad vagy összes gosszipoltartalmát a következő képlet alapján számítjuk ki:
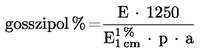
ahol:
E = az 5.2 pont szerint meghatározott korrigált optikai sűrűség,
p = a minta mennyisége, g-ban;
a = a szűrlet mennyisége, ml-ben.
6.2. Kalibrációs görbéből
6.2.1. Szabad gosszipoltartalom
Készítsünk elő két, egyenként 5 db 25 ml-es mérőlombikból álló sorozatot. Pipettázzunk mindkét sorozat lombikjaiba 2,0, 4,0, 6,0, 8,0 és 10,0 ml gosszipol-A standard oldatot (3.5). Töltsük fel valamennyi lombikot 10 ml-re az A-oldószerrel (3.2). Egészítsük ki mindkét sorozatot egy-egy, 25 ml-es, de csak 10 ml A-oldószert (3.2) tartalmazó lombikkal (vakpróba).
Töltsük fel 25 ml-re az egyik sorozat lombikjait (beleértve az egyik vakpróba lombikját is) propán-2-ol-hexán keverékkel (3.1) (referenciasorozat).
Adjunk a másik sorozat mindegyik lombikjának tartalmához 2 ml anilint (3.4), beleértve a másik vakpróba lombikját is. Hevítsük ezeket a lombikokat 30 percen át forró vízfürdő fölött, hogy tartalmuk elszíneződjön. Ezután hűtsük le őket szobahőmérsékletűre, töltsük fel térfogatra propán-2-ol-hexán keverékkel (3.1), homogenizáljuk, és hagyjuk állni egy órán át (standard sorozat).
Határozzuk meg a standard oldatok optikai sűrűségét az 5.2 pont szerint, összehasonlítva a referencia-sorozat oldataival. Szerkesszük meg a kalibrációs görbét úgy, hogy az optikai sűrűséget a gosszipol μg-ban kifejezett mennyiségének függvényében ábrázoljuk.
6.2.2. Összes gosszipoltartalom
Készítsünk elő 6 db 50 ml-es mérőlombikot. Tegyünk az első lombikba 10 ml-t a B-oldószerből (3.3), a többibe pedig sorban 2,0, 4,0, 6,0, 8,0 és 10,0 ml gosszipol-B standard oldatot (3.6). Töltsük fel valamennyi lombik tartalmát 10 ml-re a B-oldószerrel (3.3). Hevítsük a lombikokat forró vízfürdő fölött 30 percen át. Hűtsük le őket szobahőmérsékletűre, töltsük fel térfogatra a propán-2-ol-hexán keverékkel (3.1), és homogenizáljuk a tartalmukat.
Az elkészített oldatokból tegyünk 2-2 ml-t a két sorozatban előkészített, 6-6 db 25 ml-es mérőlombikba. Töltsük fel az első sorozat lombikjainak tartalmát 25 ml-re propán-2-ol-hexán keverékkel (3.1) (referenciasorozat).
A másik sorozat mindegyik lombikjába tegyünk 2 ml anilint (3.4). Hevítsük a lombikokat forró vízfürdő fölött 30 percen át. Ezután hűtsük le őket szobahőmérsékletűre, töltsük fel térfogatra propán-2-ol-hexán keverékkel (3.1), homogenizáljuk, és hagyjuk állni egy órán át (standard-sorozat).
Határozzuk meg a standard oldatok optikai sűrűségét az 5.2 pont szerint, összehasonlítva a referenciasorozat oldataival. Szerkesszük meg a kalibrációs görbét úgy, hogy az optikai sűrűséget a gosszipol μg-ban kifejezett mennyiségének függvényében ábrázoljuk.
6.3. Ismételhetőség
Az ugyanazon a mintán végzett két párhuzamos meghatározás eredménye közötti különbség nem haladhatja meg:
- az 500 ppm-nél kevesebb gosszipolt tartalmazó minták esetében a 15 %-ot, relatív értékben,
- az 500 ppm-nél nem kevesebb és 750 ppm-nél nem több gosszipolt tartalmazó minták esetében a 75 ppm-et, abszolút értékben,
- a 750 ppm-nél több gosszipolt tartalmazó minták esetében a 10 %-ot, relatív értékben.
II. MELLÉKLET
4. A TILOZIN MEGHATÁROZÁSA
- agardiffúzióval -
1. Cél és alkalmazási terület
Ez a módszer a takarmányok, a koncentrátumok és az előkeverékek tilozintartalmának meghatározását teszi lehetővé, amennyiben a tilozin 2 ppm-nél nagyobb mennyiségben van jelen.
2. Vizsgálati alapelv
A mintát 80 °C-ra előmelegített, 8 pH-jú foszfátpuffer-oldattal kezeljük, majd metil-alkohollal extraháljuk. Centrifugálás után a kivonatot hígítjuk, és antibiotikum-aktivitását a tilozin, Sarcina lutea-val beoltott agartáptalajban való diffúziójának mérésével határozzuk meg. A diffúziót a mikroorganizmus jelenlétében gátlási zónák kialakulása jelzi. E zónák átmérője egyenesen arányos az antibiotikum-koncentráció logaritmusával.
3. Mikroorganizmus: Sarcina lutea ATCC No 9341
3.1. Az eredeti törzs fenntartása
Oltsunk be Sarcina lutea-val egy ferde agarcsövet, amelyet a (4.1) táptalajból vettünk, a pH-t állítsuk 7,0-re. Inkubáljuk egy éjszakán át kb. 35 °C-on. Tegyük a tenyészetet hűtőbe, és havonta oltsuk át vele a ferde agart.
3.2. A baktériumszuszpenzió elkészítése
Kb. 2-3 ml fiziológiás konyhasóoldattal (4.4) mossuk le a baktériumokat egy frissen készített ferde agarcsőről (3.1). Oltsunk be ezzel a szuszpenzióval egy Roux-palackot, amely 250 ml, pH 7,0-re beállított táptalajt (4.1) tartalmaz. Inkubáljuk 35 °C-on 24 órán át, majd vegyük fel a baktériumokat 25 ml fiziológiás konyhasóoldatban (4.4). Homogenizáljuk, és hígítsuk fel ezt a szuszpenziót úgy, hogy 650 nm-nél, kb. 75 %-os fényáteresztést kapjunk.
Hűtőben tárolva ez a szuszpenzió egy hétig használható.
A meghatározáshoz használt alaptáptalajjal (4.1) öntött lemezeken végzett előzetes vizsgálatokkal állapítsuk meg az inokulumnak azt a mennyiségét, amely az alkalmazott különböző tilozinkoncentrációk esetén a lehető legnagyobb, még tiszta, gátlási zónákat adja. A táptalajt 48-50 °C-on kell beoltani.
4. Táptalajok és reagensek
4.1. Alaptáptalaj a meghatározáshoz ( 5 )
| Glükóz | 1 g |
| Triptines pepton | 10 g |
| Húskivonat | 1,5 g |
| Élesztőkivonat | 3 g |
| Agar, minőségtől függően | 10–20 g |
| Desztillált vízzel feltöltve | 1 000 ml-re |
Közvetlenül használat előtt állítsuk a pH-t, az eredeti törzs fenntartásához és baktériumszuszpenzió elkészítéséhez 7,0-re, a meghatározáshoz 8,0-ra.
4.2. Foszfátpuffer-oldat, pH 8
| Kálium-dihidrogén-foszfát KH2PO4 a. r. | 0,523 g |
| Dikálium-hidrogén-foszfát K2HPO4 a. r. | 16,730 g |
| Desztillált vízzel feltöltve | 1 000 ml-re |
4.3. Foszfátpuffer-oldat, pH 7
| Kálium-dihidrogén-foszfát KH2PO4 A. R. | 5,5 g |
| Dikálium-hidrogén-foszfát K2HPO4 A. R. | 13,6 g |
| Desztillált víz | ad 1 000 ml |
4.4. Steril fiziológiás konyhasóoldat.
4.5. Tiszta metil-alkohol.
4.6. 40 %-os metil-alkohol.
4.7. Foszfátpuffer-oldat (4.2)/tiszta metanol 60/40 térfogategység arányú keveréke.
4.8. Standard anyag: ismert aktivitású tilozin.
5. Standard oldatok
Szárítsuk a standard anyagot (4.8) 3 órán át 60 °C-on egy vákuumkemencében (5 mm higany). Mérjünk ki belőle 10-50 mg-ot egy mérőlombikba, oldjuk fel 5 ml metil-alkoholban (4.5), és hígítsuk az oldatot pH 7-es foszfátpuffer-oldattal (4.3), hogy 1000 μg/ml tilozin-bázis koncentrációt kapjunk.
A (4.7) keverékkel történő hígítással készítsünk ebből a törzsoldatból egy 2 μg/ml tilozin-bázist tartalmazó S8 standard munkaoldatot.
Ezután a (4.7) keverék felhasználásával végzett sorozatos hígítással (1 + 1) készítsük el a következő koncentrációjú oldatokat:
| S4 | 1 | μg/ml |
| S2 | 0,5 | μg/ml |
| S1 | 0,25 | μg/ml |
6. Extrahálás
Mérjünk ki koncentrátumok esetén 10 g, előkeverékek és takarmányok esetén 20 g vizsgálati mintát. Adjunk a mintához 60 ml, előzetesen 80 °C-ra melegített, pH 8-as foszfátpuffer-oldatot (4.2), és homogenizáljuk 2 percen át (háztartási keverőgéppel vagy Ultra-turrax-szal, stb.).
Hagyjuk állni 10 percen át, adjunk hozzá 40 ml metil-alkoholt (4.5), és homogenizáljuk 5 percig. Centrifugáljuk, majd vegyünk egy aliquot részt a kivonatból, és hígítsuk fel a (4.7) oldattal úgy, hogy 2 μg/ml-es feltételezett tilozin-koncentrációt kapjunk (= U8). Ezután a (4.7) keverékkel végzett sorozatos hígítással (1 + 1) készítsük el az U4, U2 és U1 koncentrációjú oldatokat.
10 ppm-nél alacsonyabb tartalom esetén pároljuk a mintát szárazra egy rotációs bepárlókészüléken, 35 °C-on, majd oldjuk fel a maradékot 40 %-os metil-alkoholban (4.6).
7. Meghatározási módszer
7.1. A táptalaj beoltása
A meghatározáshoz használt, pH 8,0-re beállított alaptáptalajt (4.1) oltsuk be 48-50 °C-on a baktériumszuszpenzióval (3.2).
7.2. A tálcák előkészítése
Az agardiffúziót tálcákon hajtjuk végre, a standard oldat 4 koncentrációját (S8, S4, S2, S1) és a kivonat 4 koncentrációját (U8, U4, U2, U1) felhasználva. A standard oldat és a kivonat 4-4 koncentrációját minden tálcára fel kell vinni.
Ezért olyan nagyságú tálcát használjunk, amely lehetővé teszi, hogy a rajta elhelyezett agaron legalább 8, egyenként 10-13 mm átmérőjű lyukat fúrhassunk. Számítsuk ki a beoltott táptalaj (7.1) szükséges mennyiségét úgy, hogy kb. 2 mm vastagságú, egyenletes réteget kapjunk. Leginkább olyan tálcák felelnek meg a célnak, amelyek tökéletesen vízszintes, 200 mm átmérőjű és 20 mm magas alumínium vagy műanyag karimagyűrűvel ellátott sík üveglemezből állnak.
A lyukakba pipettázzunk 0,10-0,15 ml pontosan kimért antibiotikum-oldatot a lyukak átmérőjétől függően.
Minden egyes minta esetében legalább négyszer ismételjük meg a diffúziót, hogy minden egyes meghatározás összesen 32 gátlási zóna értékelésén alapuljon.
7.3. Inkubálás
Inkubáljuk a tálcákat 35-37 °C-on egy éjszakán át.
8. Értékelés
Mérjük meg a gátlási zónák átmérőjét, lehetőleg kivetítő segítségével. Jegyezzük fel a mérési eredményeket féllogaritmikus papíron, a koncentrációk logaritmusát a gátlási zónák átmérőjének függvényében ábrázolva. Rajzoljuk meg a standard oldat és a kivonat vonalát. Ha nem áll fenn interferencia, a két vonal párhuzamos lesz.
A relatív aktivitás logaritmusát a következő képlet segítségével számíthatjuk ki:
valódi aktivitás = feltételezett aktivitás × relatív aktivitás
9. Ismételhetőség
Az ugyanazon a mintán végzett két párhuzamos meghatározás eredménye közötti különbség relatív értékben kifejezve nem haladhatja meg a 10 %-ot.
5. VIRGINIAMICIN MEGHATÁROZÁSA
- agar táptalajon történő diffúzióval -
1. Cél és alkalmazási terület
A módszer a virginiamicin takarmányokban és előkeverékekben történő meghatározására szolgál. A meghatározás alsó határa 2 mg/kg (2 ppm) ( 6 ).
2. Vizsgálati alapelv
A mintát Tween 80 metilalkoholos oldatával extraháljuk. A kivonatot dekantáljuk vagy centrifugáljuk, majd felhígítjuk. Antibiotikus aktivitását a virginiamicin Micrococcus luteussal beoltott agar táptalajban történő diffúziójának mérésével határozzuk meg. A diffúziót a mikroorganizmus gátlási zónáinak képződése jelzi. Ezeknek a zónáknak az átmérője egyenes arányosnak tekinthető az antibiotikum-koncentrációnak az alkalmazott antibiotikum-koncentrációk tartományával szembeni logaritmusával.
3. Mikroorganizmus: Micrococcus luteus ATCC 9341 (NCTC 8340, NCIB 8553)
3.1. A törzstenyészet fenntartása
Oltsunk be ferde táptalajokat (4.1) tartalmazó csöveket Micrococcus luteussal, majd inkubáljuk 30 °C-on 24 órán át. A tenyészetet hűtőgépben tároljuk, 4 oC körüli hőmérsékleten. Kéthetenként oltsuk át.
3.2. A baktériumszuszpenzió elkészítése ( 7 )
2-3 ml nátrium-klorid oldat (4.3) segítségével gyűjtsük be egy nemrégiben elkészített ferde agarról (3.1) a növekményt. Ezzel a szuszpenzióval oltsunk be 250 ml, Roux lombikban lévő táptalajt (4.1) és inkubáljuk 30 °C-on 18-20 órán át. Gyűjtsük be a növekményt 25 ml nátrium-klorid (4.3) oldatban, majd keverjük össze. Hígítsuk fel a szuszpenziót nátrium-klorid oldattal (4.3) 1/10 arányban. A szuszpenziónak a 650 nm-en, 1 cm-es cellában mért fényáteresztő képessége a nátrium-klorid (4.3) ellenében 75 % körüli kell, hogy legyen. Ez a szuszpenzió 4 °C körüli hőmérsékleten egy hétig tárolható.
4. Táptalajok és reagensek
4.1. Alap- és vizsgálati táptalaj ( 8 )
| Hús pepton | 6,0 g |
| Tripton | 4,0 g |
| Élesztőkivonat | 3,0 g |
| Húskivonat | 1,5 g |
| Glükóz | 1,0 g |
| Agar | 10,0–20,0 g |
| Víz | 1 000 ml |
| pH 6,5 (sterilizálás után) |
4.2. Foszfátpuffer, pH 6
| Kálium-hidrogén-foszfát K2HPO4 | 2,0 g |
| Kálium-dihidrogén-foszfát KH2PO4 | 8,0 g |
| Vízzel kiegészítve | 1 000 ml-re |
4.3. 0,8 %-os (w/v) nátrium-klorid oldat: oldjunk fel vízben 8 g nátrium-kloridot, majd hígítsuk 1 000 ml-re; sterilizáljuk.
4.4. Metil-alkohol.
4.5. Foszfátpuffer (4.2)/metil-alkohol (4.4) keveréke: 80/20 (v/v)
4.6. Tween 80 0,5 %-os (w/v) metil-alkoholos oldata: oldjunk fel metil-alkoholban 5 g Tween 80-at, majd metil-alkohollal hígítsuk 1 000 ml-re.
4.7. Standard anyag: ismert aktivitású virginiamicin.
5. Standard oldatok
Oldjuk fel a standard anyag (4.7) pontosan kimért mennyiségét metil-alkoholban (4.4) majd hígítsuk fel metil-alkohollal, hogy 1 000 μg/ml virginiamicint tartalmazó törzsoldatot kapjunk.
Lezárt lombikban 4 °C-on tárolva az oldat 5 napig eltartható.
Ebből a törzsoldatból, a keverékkel (4.5) történő egymást követő hígításokkal készítsük el a következő oldatokat:
| s8 | 1 | μg/ml |
| s4 | 0,5 | μg/ml |
| s2 | 0,25 | μg/ml |
| s1 | 0,125 | μg/ml |
6. A kivonat és a vizsgálati oldatok elkészítése
6.1. Extrakció
6.1.1. 100 mg/kg vagy az alatti virginiamicintartalmú termékek.
Mérjünk ki 50 g mintát, adjunk hozzá 200 ml oldatot (4.6), majd rázzuk 30 percig. Hagyjuk leülepedni vagy centrifugáljuk, majd vegyünk ki a felülúszó oldatból 20 ml-t és azt egy rotációs bepárlóban, 40 °C-ot meg nem haladó hőmérsékleten pároljuk be körülbelül 5 ml-re. A maradékot hígítsuk fel a keverékkel (4.5), hogy megközelítőleg 1 μg/ml virginiamicin-tartalmat (= u8) kapjunk.
6.1.2. 100 mg/kg-nál nagyobb virginiamicintartalmú termékek.
Mérjünk ki 10,0 g-ot meg nem haladó mintamennyiséget a mintából, amely 1 és 50 mg közötti virginiamicint tartalmaz, adjunk hozzá 100 ml oldatot (4.6), majd rázzuk 30 percig. Hagyjuk leülepedni vagy centrifugáljuk, majd a felülúszó oldatot hígítsuk fel a keverékkel (4.5), hogy megközelítőleg 1 μg/ml virginiamicintartalmat (= u8) kapjunk.
6.2. Vizsgálati oldatok
Az u8 oldatból a keverékkel (4.5) történő egymást követő hígítással (1 + 1) készítsünk u4 (várható tartalom: 0,5 μg/ml), u2 (várható tartalom: 0,25 μg/ml) és u1 (várható tartalom: 0,125 μg/ml) oldatokat.
7. A vizsgálat módja
7.1. A vizsgálati táptalaj beoltása
Oltsuk be a vizsgálati táptalajt (4.1) a baktériumszuszpenzióval (3.2), körülbelül 50 °C-on. A vizsgálati táptalajt (4.1) tartalmazó lemezeken végzett előzetes vizsgálatokkal határozzuk meg azt a szükséges baktériumszuszpenzió-mennyiséget, amely a különböző virginiamicinkoncentrációkkal a legnagyobb és legtisztább gátlási zónákat adja.
7.2. A lemezek elkészítése
Az agardiffúziót a négy standard oldat koncentrációjával (s8, s4, s2, és s1) és a négy vizsgálati oldat koncentrációjával (u8, u4, u2, u1) öntött lemezeken végezzük. A standard és a kivonat ezen négy-négy koncentrációját minden egyes lemezre fel kell vinni. Ezért megfelelő nagyságú lemezeket kell választanunk ahhoz, hogy az agar táptalajban legalább nyolc darab, 10-13 mm átmérőjű, egymástól legalább 30 mm távolságra lévő lyukat alakíthassunk ki. A vizsgálat olyan lemezeken végezhető el, amelyek egy üveglapból és egy annak tetejére helyezett, 200 mm átmérőjű és 20 mm magas csiszolt alumínium vagy műanyag karimagyűrűből állnak.
A lemezekre öntsünk annyi, a 7.1 pont szerint beoltott táptalajt (4.1), hogy az egy körülbelül 2 mm vastagságú réteget alkosson (200 mm átmérőjű lemez esetében ez 60 ml). Hagyjuk egyenletesen eloszlani, fúrjuk ki a lyukakat, és helyezzük el bennük a vizsgálati és standard oldatok pontosan kimért mennyiségeit (az átmérő függvényében lyukanként 0,10 és 0,15 ml közötti mennyiség). Minden koncentrációt legalább négyszer alkalmazzunk, hogy minden meghatározás 32 gátlási zóna kiértékelése alapján történjen.
7.3. Inkubáció
Inkubáljuk a lemezeket 30 ± 2 °C-on, 16-18 órán át.
8. Kiértékelés
Mérjük meg a gátlási zónák átmérőjét 0,1 mm-es pontossággal. Fél-logaritmikus milliméterpapírra jegyezzük fel az egyes koncentrációknál mért eredmények középértékeit, jelezve a koncentrációk logaritmusát a gátlási zónák átmérőivel összefüggésben. Szerkesszük meg mind a standard oldat, mind a kivonat regreszsziós vonalát, például az alábbiak szerint:
Határozzuk meg a standard legalacsonyabb szint (SA) regressziós pontját a következő képlet segítségével:
(a)
Határozzuk meg a standard legmagasabb szint (SM) regressziós pontját a következő képlet segítségével:
(b)
Hasonló módon számítsuk ki a regressziós pontokat a kivonat legalacsonyabb szintjére (UA) és a kivonat legmagasabb szintjére (UM), a fenti képletekben az s1, s2, s4 és s8 értékek helyett az u1, u2, u4 és u8 értékeket használva.
Jegyezzük fel a kiszámított SA és SM értékeket ugyanarra a milliméterpapírra és kössük össze őket, hogy megkapjuk a standard oldat regressziós vonalát. Hasonló módon jegyezzük fel az UA és UM értékeket és kössük össze őket, hogy megkapjuk a kivonat regressziós vonalát.
Ha interferencia nem lép fel, a vonalaknak párhuzamosnak kell lenniük. Gyakorlati megfontolásból a vonalakat párhuzamosnak tekinthetjük, ha az (SM - SA) és (UM - UA) érték középértékeiktől való eltérése nem haladja meg a 10 %-ot.
Ha a vonalak nem tekinthetőek párhuzamosnak, akkor akár az u1 és s1, akár az u8 és s8 elhagyható, az SA, SM, UA és UM értékek az alternatív képletekkel számolhatók ki, hogy megkapjuk az alternatív regressziós vonalakat:
| (a′) SA = | [Kép #4] | vagy | [Kép #5] |
| (b′) SM = | [Kép #6] | vagy | [Kép #7] |
Kép #4
Kép #5
Kép #6
Kép #7
és hasonló módon az UA-ra és UM-ra vonatkozóan. Ugyanazt a párhuzamossági feltételt kell teljesíteni. Azt a tényt, hogy az eredmény három tényező alapján került kiszámításra, fel kell tüntetni a zárójelentésben.
Ha a vonalak párhuzamosnak tekinthetőek, akkor számoljuk ki a relatív aktivitás logaritmusát (log A) a következő képletek egyikének felhasználásával, attól függően, hogy három vagy négy tényezőből történt-e a párhuzamosság megállapítása.
Négy tényező esetén
(c)
Három tényező esetén
(d)
vagy
(d ′ )
A mintakivonat aktivitása = a vonatkozó standard aktivitása × A
(u8 = s8 × A)
Ha a relatív aktivitás a 0,5 és 2,0 közötti tartományon kívül esik, akkor ismételjük meg a vizsgálatot a kivonat koncentrációin, vagy ahol ez nem lehetséges, ott a standard oldatokon végzett megfelelő módosításokkal. Ha a relatív aktivitást nem tudjuk az említett tartományon belülre korrigálni, akkor bármely kapott eredményt közelítő eredménynek kell tekinteni, és ezt fel kell tüntetni a zárójelentésben.
Ha a vonalak nem tekinthetőek párhuzamosnak, ismételjük meg a meghatározást. Ha a párhuzamosságot még ezután nem sikerült elérni, akkor a meghatározást nem kielégítőnek kell tekinteni.
Az eredményt milligrammban kifejezett virginiamicinre és kilogrammban kifejezett takarmányra vonatkoztatva fejezzük ki.
9. Ismételhetőség
Az ugyanazon mintán, ugyanazon analitikus által párhuzamosan végrehajtott két meghatározás eredménye közötti különbség nem haladhatja meg:
- a 10 mg/kg alatti virginiamicin-tartalom esetén a 2 mg/kg-ot, abszolút értékben,
- a 10 és 25 mg/kg közötti tartalom esetén a legmagasabb érték 20 %-át,
- a 25 és 50 mg/kg közötti tartalom esetén az 5 mg/kg-ot, abszolút értékben,
- az 50 mg/kg feletti tartalom esetén a legmagasabb érték 10 %-át.
( 1 ) HL L 170., 1970.8.3., 2. o.
( 2 ) HL L 155., 1971.7.12., 13. o.
( 3 ) HL L 279., 1971.12.20., 7. o.
( 4 ) A nitrogéntartalmat a szemi-mikro Kjeldahl-módszer elméleti 17,7 %-os nitrogéntartalma alapján határozzuk meg.
( 5 ) Bármely hasonló összetételű és ugyanazt az eredményt adó, a kereskedelmi forgalomban kapható táptalaj használható.
( 6 ) 1 mg virginiamycin 1 000 UK egységnek felel meg.
( 7 ) Más módszerek is alkalmazhatók feltéve, hogy megállapítást nyert, hogy azok hasonló baktériumszuszpenziókat eredményeznek.
( 8 ) Minden hasonló összetételű és ugyanezeket az eredményeket adó kereskedelmi táptalaj felhasználható.
Lábjegyzetek:
[1] A dokumentum eredetije megtekinthető CELEX: 31972L0199 - https://eur-lex.europa.eu/legal-content/HU/ALL/?uri=CELEX:31972L0199&locale=hu Utolsó elérhető, magyar nyelvű konszolidált változat CELEX: 01972L0199-19990827 - https://eur-lex.europa.eu/legal-content/HU/ALL/?uri=CELEX:01972L0199-19990827&locale=hu