
31971L0250[1]
A Bizottság első irányelve (1971. június 15.) a takarmányok hatósági ellenőrzésére szolgáló közösségi analitikai módszerek meghatározásáról
A BIZOTTSÁG ELSŐ IRÁNYELVE
(1971. június 15.)
a takarmányok hatósági ellenőrzésére szolgáló közösségi analitikai módszerek meghatározásáról
(71/250/EGK)
AZ EURÓPAI KÖZÖSSÉGEK BIZOTTSÁGA,
tekintettel az Európai Gazdasági Közösséget létrehozó szerződésre,
tekintettel a takarmányok hatósági ellenőrzésénél alkalmazandó közösségi mintavételi és analitikai módszerek bevezetéséről szóló, 1970. július 20-i 70/373/EGK tanácsi irányelvre ( 1 ) és különösen annak 2. cikkére;
mivel, az említett irányelv előírja, hogy a takarmányok hatósági ellenőrzését, amelynek célja a takarmányok minőségére és összetételére vonatkozó törvényi, rendeleti és közigazgatási rendelkezésekből eredő követelmények betartásának ellenőrzése, közösségi mintavételi és analitikai módszerek alkalmazásával kell végrehajtani;
mivel minden szükséges analitikai módszert a lehető leghamarabb ki kell dolgozni; mivel a hidrocián-sav, a kalcium, a karbonátok, a nyershamu, a sósavban oldhatatlan hamu, a kloridokból származó klór, a mustárolaj, a laktóz, a kálium, a nátrium, a cukrok, a teobromin és a karbamid meghatározása, a lupinin-alkaloidok meghatározása, valamint a szójából készült termékek karbamid-aktivitásának meghatározása az első fázis ezen folyamatban;
mivel az ezen irányelvben meghatározott intézkedések összhangban vannak a Takarmányok Állandó Bizottsága véleményével,
ELFOGADTA EZT AZ IRÁNYELVET:
1. cikk
A tagállamok előírják, hogy a takarmányok hatósági ellenőrzését szolgáló azon analitikai vizsgálatokat, amelyek a takarmányokban lévő hidrocián-sav, kalcium, karbonátok, nyershamu, sósavban oldhatatlan hamu, kloridokból származó klór, -----laktóz, kálium, nátrium, cukrok, -----és karbamid szintjének meghatározására, -----valamint a szójából származó termékek karbamid-aktivitásának meghatározására vonatkoznak, ezen irányelv mellékletében rögzített módszerek alkalmazásával kell végrehajtani.
A 70/373/EGK tanácsi irányelv alapján elfogadott analitikai módszerekre a melléklet 1. részében leírt általános szabályokat kell alkalmazni.
A 2002/32/EK európai parlamenti és tanácsiirányelv ( 2 ) értelmében vett nemkívánatos anyagokra vonatkozóan - a dioxinokat és dioxinjellegű PCB-ket is ideértve - az ezen irányelv melléklete 1. része C. pontjának 3. alpontja alkalmazandó.
2. cikk
A tagállamok legkésőbb 1972. július 1-ig hatályba léptetik azokat a törvényi, rendeleti és közigazgatási rendelkezéseket, amelyek szükségesek ahhoz, hogy ennek az irányelvnek megfeleljenek. Erről haladéktalanul tájékoztatják a Bizottságot.
3. cikk
Ennek az irányelvnek a tagállamok a címzettjei.
MELLÉKLET
A TAKARMÁNYOK ALKOTÓELEMEIRE VONATKOZÓ ANALITIKAI MÓDSZEREK
1. A TAKARMÁNYANALITIKAI MÓDSZEREKRE VONATKOZÓ ÁLTALÁNOS RENDELKEZÉSEK
A. MINTÁK ANALITIKAI VIZSGÁLATRA TÖRTÉNŐ ELŐKÉSZÍTÉSE
1. Cél
Az alábbiakban leírt vizsgálati módszerek, a takarmányok hatósági ellenőrzésére szolgáló közösségi mintavételezési módszerek meghatározásáról szóló, 1976. március 1-jei 76/371/EGK első bizottsági irányelvben ( 3 ) meghatározott rendelkezéseknek megfelelő mintavételezés után, az ellenőrző laboratóriumoknak megküldött végső minták analitikai vizsgálatának előkészítésére vonatkoznak.
A mintákat oly módon kell előkészíteni, hogy az analitikai módszerekben előírt kimért mennyiségek homogének és a végső mintákra jellemzőek legyenek.
2. Szükséges óvintézkedések
Minden szükséges műveletet oly módon kell elvégezni, hogy minimális legyen a minta beszennyeződésének és összetétele megváltozásának lehetősége. Az őrlést, keverést és rostálást a lehető leggyorsabban kell végrehajtani, hogy a minta a lehető legrövidebb ideig érintkezzen a levegővel és a fénnyel. Kerülni kell az olyan daráló- és őrlőgépek használatát, amelyek a mintát érzékelhetően felmelegítik. A különösen hőérzékeny takarmányok esetében a kézi őrlés alkalmazása javasolt. Figyelmet kell fordítani annak biztosítására is, hogy az eszköz maga ne lehessen nyomelemekkel történő szennyezés forrása.
Ha az előkészítést nem lehet a minta nedvességtartalmának jelentős megváltozása nélkül elvégezni, akkor a nedvességtartalmat az előkészítés előtt és után is meg kell határozni, az 1972. december 5-i 73/47/EGK ( 4 ) bizottsági irányelvvel módosított, a takarmányok hatósági ellenőrzésére szolgáló közösségi analitikai módszerek meghatározásáról szóló, 1971. november 18-i 71/393/EGK ( 5 ) második bizottsági irányelv mellékletének 1. részében meghatározott módszer szerint.
3. A vizsgálat módja
A végső mintát géppel vagy kézzel alaposan keverjük össze. Osszuk a mintát két egyenlő részre (adott esetben a negyedeléses módszert alkalmazzuk). Az egyik részt tegyük egy megfelelően tiszta, száraz, légmentesen záródó edénybe, majd készítsük elő a másik részt vagy annak egy reprezentatív részét, legalább 100 grammot, az alábbiak szerint.
3.1. Előkészítés nélkül őrölhető takarmányok
Amennyiben az analitikai módszerekben másképp nem írják elő, szükség esetén őrlés után, rostáljuk át a minta teljes mennyiségét egy 1 mm szemátmérőjű, négyzetes szemű rostán (az ISO R565 ajánlásának megfelelően). Kerüljük a túlőrlést.
Keverjük össze az átrostált mintát, és vegyük fel egy megfelelően tiszta, száraz, légmentesen záródó edénybe. Közvetlenül az analízisre szánt mennyiség kimérése előtt keverjük össze újra.
3.2. Szárítás után őrölhető takarmányok
Amennyiben az analitikai módszerekben másképp nem írják elő, a 2. szakaszban említett nedvességmeghatározási módszer 4.3. pontja szerint leírt előzetes szárítási eljárásnak megfelelően szárítsuk ki a mintát 8-12 %-os nedvességtartalmúra. Ezt követően a 3.1. szakaszban leírtak szerint járjunk el.
3.3. Folyékony vagy félfolyékony takarmányok
Vegyük fel a mintát egy megfelelően tiszta, száraz, légmentesen záródó edénybe. Közvetlenül az analízisre szánt mennyiség kimérése előtt alaposan keverjük össze.
3.4. Egyéb takarmányok
Azok a minták, amelyek a fent említett eljárások egyikével sem készíthetők elő, bármely egyéb, olyan eljárással kezelhetők, amelyek biztosítják, hogy az analízisre kimért mennyiségek homogének és a végső mintákra jellemzőek legyenek.
4. A minták tárolása
A mintákat olyan hőmérsékleten kell tárolni, amely nem változtatja meg az összetételüket. A vitaminok vagy fényre különösen érzékeny anyagok analízisére szánt mintákat barna üvegedényben kell tárolni.
B. AZ ANALITIKAI MÓDSZEREK SORÁN HASZNÁLT REAGENSEKRE ÉS ESZKÖZÖKRE VONATKOZÓ RENDELKEZÉSEK
1. Amennyiben az analitikai módszerekben másképp nem írják elő, minden analitikai reagensnek analitikai tisztaságúnak (a. t.) kell lennie. Nyomelemek meghatározásakor a reagensek tisztaságát vakpróbával kell ellenőrizni. Az így kapott eredmények a reagensek esetleges további tisztítását teszik szükségessé.
2. Az analitikai módszerek leírásakor említett bármely oldatkészítési, hígítási, öblítési vagy mosási művelet esetében, ahol az alkalmazott oldószer vagy hígítószer fajtáját nem tüntetik fel, vizet kell használni. Általános szabályként ioncserélt vagy desztillált vizet kell használni. Az analitikai módszerekben jelzett különleges esetekben a vizet speciális tisztítási eljárásokkal kell kezelni.
3. Az ellenőrző laboratóriumokban megtalálható alapfelszerelésre való tekintettel, az analitikai módszerek leírásakor csak a speciális vagy speciális használatot megkövetelő eszközök és készülékek kerülnek említésre. Ezeknek tisztának kell lenniük, különösen nagyon kis anyagmennyiségek meghatározása esetén.
C. AZ ANALITIKAI MÓDSZEREK ALKALMAZÁSA ÉS AZ EREDMÉNYEK KIFEJEZÉSE
1. Általában a takarmányokban található minden anyag meghatározására egyedi analitikai módszer kerül bevezetésre. Amennyiben több módszer adott, akkor az ellenőrző laboratórium által alkalmazott konkrét módszert fel kell tüntetni az analitikai bizonylaton.
2. Az analitikai bizonylatban közölt eredménynek, a minta elkülönített részein, kielégítő ismételhetőséggel elvégzett, legalább két meghatározásból nyert átlagértéknek kell lennie.
Ezt az eredményt az analitikai módszerben meghatározott módon és megfelelő helyiérték-számmal kell kifejezni, és ha szükséges, akkor a végső minta előkészítés előtti nedvességtartalmára kell korrigálni.
3. A 2002/32/EK irányelv értelmében vett nemkívánatos anyagokra vonatkozóan - a dioxinokat és dioxinjellegű PCB-ket is ideértve - valamely takarmánynak szánt termék akkor számít nem megfelelőnek a megállapított maximális tartalom tekintetében, ha úgy ítélik meg, hogy az analitikai eredmény meghaladja a maximális tartalmat, figyelembe véve a kiterjesztett mérési bizonytalanságot és a visszanyerési korrekciót. Az elemzett koncentrációt, amelyet visszanyerésre korrigáltak és amelyből kivonták a kiterjesztett mérési bizonytalanságot, alkalmazzák a megfelelősség megállapítására. Ez utóbbit csak azokban az esetekben kell alkalmazni, amikor az analitikai módszer lehetővé teszi a mérési bizonytalanság és a visszanyerési korrekció becslését (pl. mikroszkópos elemzésnél nem lehetséges).
Az analitikai eredményt a következőképpen kell jelenteni (amennyiben az alkalmazott analitikai módszer lehetővé teszi a mérési bizonytalanság és a visszanyerési korrekció becslését):
a) visszanyerésre korrigálva vagy nem korrigálva, feltüntetve a jelentés módját és a visszanyerési szintet,
b) "x +/- U" formában, ahol x az analitikai eredmény és U a kiterjesztett mérési bizonytalanság, 2 kiterjesztési tényező alkalmazásával, amelynek megbízhatósága megközelítőleg 95 %.
2. A HIDROCIÁN-SAV MEGHATÁROZÁSA
1. Cél és alkalmazási terület
Ezen módszer lehetővé teszi a takarmányokban, különösen a lenmagból, maniókalisztből és bizonyos babfajokból származó termékekben szabadon és glikozidok alkotórészeként előforduló hidrocián-sav szintjének meghatározását.
2. Vizsgálati alapelv
A mintát vízben szuszpendáljuk. Enzimtevékenység révén felszabadítjuk a hidrocián-savat, vízgőz-desztillációval visszük be, és savasított ezüst-nitrát oldat egy meghatározott mennyiségében gyűjtjük össze. Az ezüst-cianidot szűréssel választjuk el, a többlet ezüst-nitrátot pedig ammónium-tiocianát oldattal titráljuk.
3. Reagensek
3.1. Édesmandula szuszpenziója: törjünk össze húsz hámozott édesmandulát 100 ml, 37-40 °C-os vízben. Nátriumpikrát-papír használatával vagy az 5. pont utolsó bekezdésében leírt vakpróba elvégzésével ellenőrizzük 10 ml szuszpenzión, hogy nem tartalmaz-e hidrocián-savat.
3.2. 10 %-os, fenolftalein-semleges nátrium-acetát oldat.
3.3. Habzásgátló emulzió (pl. szilikon).
3.4. Salétromsav, d: 1,40.
3.5. 0,02 N ezüstnitrát oldat.
3.6. 0,02 N ammónium-tiocianát oldat.
3.7. Telített ammónium-vasszulfát oldat.
3.8. Szalmiákszesz: d: 0,958.
4. Eszközök
4.1. 38 °C-ra beállított hőfokszabályozós kemence.
4.2. Vízgőz-desztilláló készülék, hajlított kiegészítő elemmel ellátott hűtővel.
4.3. Csiszolt üvegdugóval ellátott, 1 000 ml-es lapos fenekű lombik.
4.4. Olajfürdő.
4.5. 1/20 ml-re kalibrált büretta.
5. A vizsgálat módja
Mérjünk le 20 g-ot a mintából 5 mg-nyi pontossággal, helyezzük egyliteres lapos fenekű lombikba, és adjunk hozzá 50 ml vizet, valamint 10 ml édesmandula szuszpenziót (3.1.). Zárjuk le a lombikot, és helyezzük tizenhat órára a 38 °C-os kemencébe. Ezt követően hűtsük le szobahőmérsékletre, és adjunk hozzá 80 ml vizet, 10 ml nátrium-acetát oldatot (3.2.) és egy csepp habzásgátló emulziót (3.3.).
Kössük a lombikot a vízgőz-desztilláló készülékhez, és helyezzük egy olajfürdőbe, amelyet előzetesen kevéssel 100 °C fölé melegítettünk. Desztilláljunk 200-300 ml folyadékot a lombikon átvezetett erős gőzáramlattal és az olajfürdőt enyhén melegítsük. Gyűjtsük a párlatot egy fénytől védett és pontosan 50 ml 0,02 N ezüst-nitrát oldatot (3.5.) és 1 ml salétromsavat (3.4.) tartalmazó Erlenmeyer-lombikba. Bizonyosodjunk meg arról, hogy a hűtő kiegészítő része elmerül az ezüst-nitrát oldatban.
Helyezzük át az Erlenmeyer-lombik tartalmát egy 500 ml űrtartalmú lombikba, töltsük fel térfogatig vízzel, keverjük meg és szűrjük le. Vegyünk el 250 ml-t a szűrletből, adjunk hozzá kb. 1 ml ammónium-vasszulfát oldatot (3.7.), és az 1/20 ml-re kalibrált bürettából vett 0,02 N ammónium-tiocianát oldattal (3.6.) visszatitráljuk az ezüst-nitrát többletet.
Amennyiben szükséges, vakpróbát végezhetünk ugyanennek az eljárásnak 10 ml-nyi édesmandula szuszpenzióra (3.1.) történő alkalmazásával, az elemzendő minta kihagyásával.
6. Az eredmények kiszámítása
Ha a vakpróba azt jelzi, hogy 0,02 N ezüst-nitrát oldat került felhasználásra, akkor annak az értékét vonjuk ki a minta párlata által felvett mennyiségből. 1 ml 0,02 N AgNO3 0,54 mg HCN-nak felel meg. Az eredményt a mintára vonatkoztatva, százalékosan fejezzük ki.
7. Észrevétel
Ha a minta nagy mennyiségű szulfidot tartalmaz (pl. babfélék), fekete ezüst-szulfid üledék csapadék, amely az ezüst-cianid üledékkel együtt szűrődik. Ezen csapadék képződése 0,02 N ezüst-nitrát oldat veszteséget okoz, amelynek mennyiségét ki kell vonni a HCN-tartalom kiszámításához használt mennyiségből. Ehhez az alábbiak szerint járjunk el:
Az ezüst-cianid feloldása érdekében kezeljük a szűrőn maradt üledéket 50 ml szalmiákszesszel (3.8.). Mossuk a maradékot hígított szalmiákszeszben, majd határozzuk meg ezüsttartalmát. A kapott eredményt alakítsuk át 0,02 N ezüst-nitrát oldat ml-ére.
A minta HCN-tartalma is meghatározható a savasított ammóniás szűrlet salétromsavval történő titrálásával.
3. KALCIUM MEGHATÁROZÁSA
1. Cél és alkalmazási terület
A módszer lehetővé teszi a takarmányok teljes kalciumtartalmának meghatározását.
2. Vizsgálati alapelv
A kalciumot hamvasztjuk, a hamut sósavval kezeljük, a kalcium pedig kalcium-oxalátként csapódik ki. A csapadékot kénsavban oldjuk, és a kialakuló oxálsavat kálium-permanganát oldattal titráljuk.
3. Reagensek
3.1. Sósav a.r., d: 1,14.
3.2. Salétromsav a.r., d: 1,40.
3.3. Kénsav a.r., d: 1,13.
3.4. Szalmiákszesz a.r., d: 0,98.
3.5. Ammónium-oxalát hideg, telített oldata a.r.
3.6. 30 %-os (w/v) citromsav oldat a.r.
3.7. 5 %-os (w/v) ammónium-klorid oldat a.r.
3.8. 0,04 %-os (w/v) zöld bróm-krezol oldat.
3.9. 0,1 N kálium-permanganát oldat.
4. Eszközök
4.1. Levegőkeringéses és hőfokszabályozós elektromos tokos kemence.
4.2. Platina-, kvarc- vagy porcelán hamvasztótégely.
4.3. G4-es porozitású üveg szűrőtégely.
5. A vizsgálat módja
Mérjünk le a mintából kb. 5 g-ot (vagy szükség esetén többet) mg-nyi pontossággal, kalcináljuk 550 °C-on, és helyezzük a hamut egy 250 ml-es főzőpohárba.
Adjunk hozzá 40 ml sósavat (3.1.), 60 ml vizet és néhány csepp salétromsavat (3.2.). Forraljuk fel, és tartsuk a forrásponton 30 percig. Hűtsük le, és helyezzük az oldatot egy 250 ml űrtartalmú lombikba. Öblögessük, töltsük fel jelig vízzel, homogenizáljuk, és szűrjük le.
Pipettázzunk át a feltételezett kalciumtartalom szerint 10-40 mg kalciumot tartalmazó aliquot részt egy 250 ml-es főzőedénybe. Adjunk hozzá 1 ml citromsav oldatot (3.6.) és 5 ml ammónium-klorid oldatot (3.7.).
Töltsük fel a térfogatot vízzel körülbelül 100 ml-re. Forraljuk fel, adjunk hozzá nyolc-tíz csepp bróm-krezol zöld oldatot (3.8.) és 30 ml meleg ammónium-oxalát oldatot (3.5). Ha csapadék képződik, oldjuk fel néhány csepp sósav (3.1.) hozzáadásával.
Folyamatos kevergetés közben nagyon lassan semlegesítsünk szalmiákszesszel (3.4.), amíg 4,4-4,6 közötti pH-értéket kapunk (azaz, amíg az indikátor színe meg nem változik). Helyezzük a főzőpoharat egy forrásban lévő vízfürdőbe, és tartsuk ott harminc percig, hogy a képződött csapadék leülepedhessen. Vegyük ki a főzőpoharat a vízfürdőből. Hagyjuk állni egy óráig, majd szűrjük át egy G4-es szűrő tégelyen.
Mossuk vízzel a főzőpoharat és a tégelyt mindaddig, amíg az ammónium-oxalát felesleg teljesen el nem tűnik (a klorid hiánya a mosóvízben jelzi, hogy a mosás megfelelően lett elvégezve).
Oldjuk fel a szűrőn lévő csapadékot 50 ml meleg kénsavban (3.3.). Öblítsük a tégelyt meleg vízzel, és töltsük fel a szűrletet kb. 100 ml-re. Emeljük a hőmérsékletet 70-80 °C-ra, és cseppenként titráljuk kálium-permanganát oldattal (3.9.) egészen addig, amíg egy percen tartó rózsaszín elszíneződést nem kapunk.
6. Az eredmények kiszámítása
1 ml 0,1 N kálium-permanganát 2,004 mg kalciumnak felel meg. Az eredményt a mintára vonatkoztatva, százalékosan fejezzük ki.
7. Észrevételek
7.1. Nagyon alacsony kalciumtartalom esetén a következők szerint járjunk el: a kalcium-oxalát csapadékot szűrjük keresztül egy hamumentes szűrőpapíron. Mosás után szárítsuk meg a szűrőt, és kalcináljuk 550 °C-on egy platinatégelyben. Oldjuk fel újra a maradékot néhány csepp kénsavban (3.3.), párologtassuk, amíg száraz nem lesz, kalcináljuk újra 550 °C-on, majd mérjük meg. Ha W jelöli az így kapott kalcium-szulfát tömegét, akkor a mintaként vett aliquot mennyiség kalciumtartalma = W × 0,2944.
7.2. Ha a minta kizárólag ásványi anyagokból áll, előzetes hamvasztás nélkül oldjuk fel sósavban. Az olyan termékek esetében, mint a kalcium-alumínium foszfát, amelyeket nehéz savban feloldani, oldás előtt a következő lúgképző eljárással olvasszuk: platinatégelyben alaposan keverjük el az elemzésre szánt mintát egy ötször akkora tömegű keverékkel, amely egyenlő mennyiségben tartalmaz kálium-karbonátot és nátrium-karbonátot. Óvatosan hevítsük, amíg az elegy teljesen meg nem olvad. Hűtsük le, és oldjuk fel sósavban.
7.3. Ha a minta magnéziumtartalma magas, csapassuk ki a kalcium-oxalátot másodszor is.
4. KARBONÁTOK MEGHATÁROZÁSA
1. Cél és alkalmazási terület
Ez a módszer lehetővé teszi a karbonátok, hagyományosan kalcium-karbonátban kifejezett mennyiségének meghatározását a legtöbb takarmányban. Bizonyos esetekben (például a vas-karbonát esetében) speciális módszert kell alkalmazni.
2. Vizsgálati alapelv
A karbonátokat sósavban bontjuk szét; a kibocsátott szén-dioxidot egy kalibrált csőben gyűjtjük össze, és térfogatát összehasonlítjuk az azonos körülmények között a kalcium-karbonát a.r. ismert mennyisége által kibocsátott mennyiséggel.
3. Reagensek
3.1. Sósav a.r., d: 1,10.
3.2. Kalcium-karbonát, a.r.
3.3. Kb. 0,1 N kénsav, metilvörös indikátorral megszínezve.
4. Eszközök
Scheibler-Dietrich készülék (lásd az ábrát) vagy annak megfelelő készülék.
5. A vizsgálat módja
A minta karbonát-tartalmától függően mérjünk ki egy részmintát az alábbiak szerint:
0,5 g az 50-100 % karbonát-tartalmú termékek esetén, kalcium-karbonátban kifejezve;
1 g a 40-50 % karbonát-tartalmú termékek esetén, kalcium-karbonátban kifejezve;
2-3 g egyéb termékek esetében.
Helyezzük a részmintát a készülék speciális lombikjába (4), amelyet 10 ml sósavat (3.1.) tartalmazó, törhetetlen anyagból készült, kisméretű csővel szereltek fel, és kapcsoljuk a lombikot a készülékhez. Fordítsuk el a háromfázisú csapot (5), hogy a cső (1) a külső résszel kapcsolódjon. A színezett kénsavval (3.3.) töltött és kalibrált csőhöz (1) kapcsolt mozgatható cső (2) segítségével igazítsuk a folyadék szintjét a nulla jelhez. Az (1) és a (3) csövek összekapcsolásához fordítsuk el a csapot (5), és ellenőrizzük, hogy a szint nullán áll-e.
A lombik (4) megdöntésével folyassunk lassan sósavat (3.1.) a részmintára. A (2) cső leengedésével egyenlítsük ki a nyomást. Rázzuk a (4) lombikot egészen addig, amíg a szén-dioxid kibocsátás teljesen meg nem szűnik.
Az (1) és a (2) csőben lévő folyadék egyenlő szintre igazításával állítsuk vissza a nyomást. Néhány perc múlva, amikor a gáz mennyisége állandósul, olvassuk le az eredményt.
Végezzünk ellenőrző vizsgálatot ugyanilyen körülmények között 0,5 g kalcium-karbonáton (3.2.).
6. Az eredmények kiszámítása
A kalcium karbonát formájában, grammban kifejezett karbonát-tartalom a mintára vonatkoztatva, százalékosan következő képlet segítségével számítható ki:
ahol:
V = a részminta által kibocsátott szén-dioxid, ml-ben.
T = 0,5 g CaCO3 a.r. által kibocsátott szén-dioxid, ml-ben.
W = a részminta grammban kifejezett tömege.
7. Észrevételek
7.1. Ha a részminta 2 g-nál nehezebb, először töltsünk 15 ml desztillált vizet a (4) lombikba, és keverjük össze a vizsgálat megkezdése előtt. Az ellenőrző vizsgálat esetében ugyanezt a vízmennyiséget használjuk.
7.2. Ha a használt készülék űrtartalma eltér a Scheibler-Dietrich készülékétől, a mintából és az kontrollanyagból vett részt és az eredmények kiszámítását annak megfelelően kell korrigálni.
A SZÉN-DIOXID MEGHATÁROZÁSÁRA SZOLGÁLÓ SCHEIBER-DIETRICH KÉSZÜLÉK
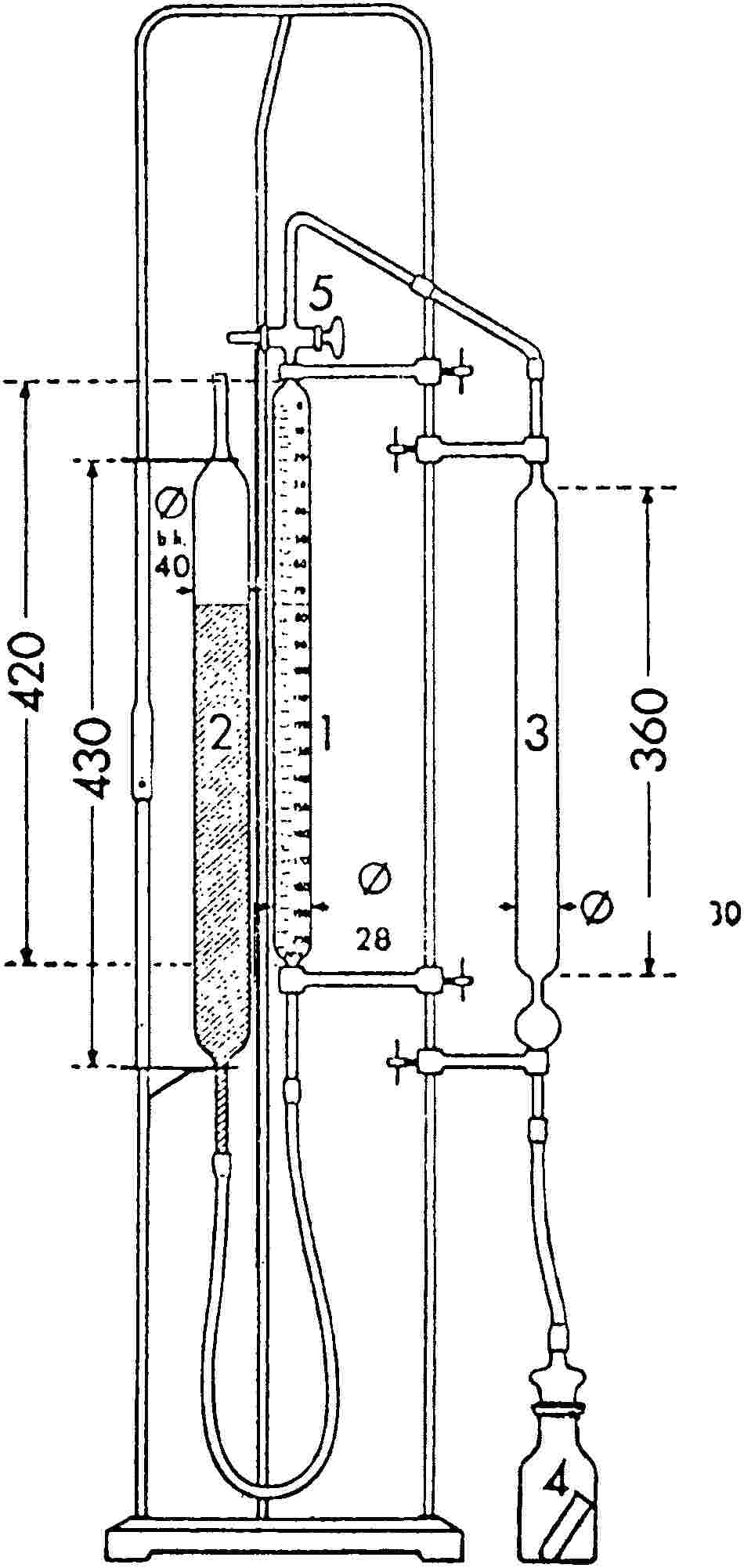
Arány:
(mm)
5. NYERSHAMU MEGHATÁROZÁSA
1. Cél és alkalmazási terület
A módszer lehetővé teszi a takarmányok nyershamutartalmának meghatározását.
2. Vizsgálati alapelv
A mintát 550 °C-on hamvasztjuk; a maradékanyagot megmérjük.
3. Reagensek
20 %-os (w/v) ammónium-nitrát oldat.
4. Eszközök
4.1. Főzőlap.
4.2. Hőfokszabályzós, elektromos tokos kemence.
4.3. Platinából vagy platina és arany (10 % Pt, 90 % Au) ötvözetéből készült négyszögletes- (60 × 40 × 25 mm) vagy köralakú (átmérő: 60-75 mm, magasság: 20-25 mm) hamvasztótégelyek.
5. A vizsgálat módja
Mérjünk ki a mintából 5 g-ot mg-os pontossággal (2,5 g-ot a duzzadóképes anyagból származó termékek esetében), és helyezzük egy hamvasztótégelybe, amelyet előzetesen kalcináltunk és táráztunk. Helyezzük a tégelyt a főzőlapra, és fokozatosan addig hevítsük, amíg az anyag el nem szenesedik. Helyezzük a tégelyt az 550 °C ± 5 °C-ra beállított tokos kemencébe. Tartsuk ezt a hőmérsékletet egészen addig, amíg széntartalmú részecskéktől mentesnek tűnő fehér, halványszürke vagy vöröses hamut nem nyerünk. Helyezzük a tégelyt egy szárítóba, hagyjuk kihűlni, majd azonnal mérjük meg.
6. Az eredmények kiszámítása
A maradék tömegét a táratömeg kivonásával számoljuk ki.
Az eredményt a mintára vonatkoztatva, százalékosan fejezzük ki.
7. Észrevételek
7.1. A nehezen hamvasztható anyagok hamuját legalább három órán keresztül előzetesen hamvasztani kell, le kell hűteni, majd néhány csepp 20 %-os ammónium-nitrát oldatot kell hozzáadni (ügyelve arra, hogy elkerüljük a hamu szétszóródását vagy a csomóképződést). Kemence kiszárítása után folytassuk a kalcinálást. Szükség szerint ismételjük a műveletet a teljes elhamvadásig.
7.2. A 7.1. pontban ismertetett eljárással szemben ellenálló anyagok esetében a következőképpen járjunk el: háromórás hamvasztás után helyezzük a hamut meleg vízbe, és egy kisméretű, hamumentes szűrőn szűrjük át. Hamvasszuk el a szűrőt és annak tartalmát az eredeti tégelyben. Helyezzük a szűrletet a lehűtött tégelybe, párologtassuk kiszáradásig, hamvasszuk, és mérjük meg.
7.3. Olajok és zsírok esetében mérjünk ki pontosan egy kb. 25 g-os mintát egy megfelelő méretű tégelybe. Hamumentes szűrőpapír-csíkkal meggyújtva, szenesítsük az anyagot. A gyulladás után a lehető legkevesebb vízzel nedvesítsük meg. Szárítsuk, és hamvasszuk az 5. pontban ismertetett módszerrel.
6. SÓSAVBAN OLDHATATLAN HAMU MEGHATÁROZÁSA
1. Cél és alkalmazási terület
A módszer lehetővé teszi a takarmányokban lévő sósavban oldhatatlan ásványi anyagok szintjének meghatározását. A minta jellegétől függően két módszer alkalmazható.
1.1. A-módszer: az egynemű, szerves takarmányok és a legtöbb összetett takarmány esetében alkalmazható.
1.2. B-módszer: olyan ásványi vegyületek és keverékek, valamint összetett takarmányok esetében alkalmazható, amelyeknek az A-módszer szerint meghatározott, sósavban nem oldható anyagtartalma 1 %-nál magasabb.
2. Vizsgálati alapelv
2.1. A-módszer: a mintát hamvasztjuk, a hamut sósavban forraljuk, az oldhatatlan maradékanyagot pedig szűrjük és megmérjük.
2.2. B-módszer: a mintát sósavval kezeljük. Az oldatot szűrjük, a maradékanyagot hamvasztjuk, az így kapott hamut pedig az A-módszer szerint kezeljük.
3. Reagensek
3.1. 3 N sósav.
3.2. 20 %-os (w/v) triklór-ecetsav oldat.
3.3. 1 %-os (w/v) triklór-ecetsav oldat.
4. Eszközök
4.1. Főzőlap.
4.2. Hőfokszabályzós, elektromos tokos kemence.
4.3. Platinából vagy platina és arany (10 % Pt, 90 % Au) ötvözetéből készült négyszögletes- (60 × 40 × 25 mm) vagy köralakú (átmérő: 60-75 mm, magasság: 20-25 mm) hamvasztótégelyek.
5. A vizsgálat módja
5.1. A-módszer:
Hamvasszuk a mintát a nyershamu meghatározásánál ismertetett módszer szerint. Használhatjuk az analízis során kapott hamut is.
75 ml 3 N sósav (3.1.) felhasználásával helyezzük a hamut egy 250-400 ml-es főzőpohárba. Lassan forraljuk, és tartsuk forrásban óvatosan tizenöt percig. Szűrjük át a meleg oldatot egy hamumentes szűrőpapíron, és mossuk a maradékot meleg vízzel, mindaddig, amíg a savreakció már nem észlelhető. Szárítsuk ki a maradékot tartalmazó szűrőt és hamvasszuk egy tárázott tégelyben legalább 550 °C és legfeljebb 700 °C hőmérsékleten. Hűtsük le egy szárítóban, és mérjük meg.
5.2. B-módszer:
Mérjünk le a mintából 5 g-ot mg-nyi pontossággal, és helyezzük egy 250-400 ml-es főzőpohárba. Adjunk hozzá egymást követően 25 ml vizet, és 25 ml 3 N sósavat (3.1.), keverjük össze, és várjunk a pezsgés elállásáig. Adjunk hozzá további 50 ml 3 N sósavat (3.1.). Várjunk az esetleges gázkibocsátás megszűnéséig, majd helyezzük a főzőpoharat forró vízfürdőbe, és tartsuk ott harminc percig vagy szükség esetén tovább, az esetlegesen jelentkező bármely keményítő teljes hidrolízise érdekében.
Még meleg állapotban szűrjük át egy hamumentes szűrőn, és mossuk a szűrőt 50 ml meleg vízbe (lásd 7. megjegyzés). Helyezzük a maradékanyagot tartalmazó szűrőt hamvasztótégelybe, szárítsuk ki, és hamvasszuk legalább 550 °C és legfeljebb 700 °C hőmérsékleten. 75 ml 3 N sósav (3.1.) felhasználásával helyezzük a hamut egy 250-400 ml-es főzőpohárba; kövessük az 5.1. második albekezdésében leírtakat.
6. Az eredmények kiszámítása
A maradék tömegét a táratömeg kivonásával számítjuk ki. Az eredményt a mintára vonatkoztatva, százalékosan fejezzük ki.
7. Észrevétel
Ha a szűrés nehéznek bizonyul, kezdjük újra az analízist úgy, hogy az 50 ml 3 N sósavat (3.1.) 50 ml 20 %-os triklór-ecetsavval (3.2.) helyettesítjük és a szűrőt 1 %-os meleg triklór-ecetsavban oldatban (3.3.) mossuk.
7. KLORIDOKBÓL SZÁRMAZÓ KLÓR MEGHATÁROZÁSA
1. Cél és alkalmazási terület
Ez a módszer lehetővé teszi a vízben oldható kloridokban található, hagyományosan nátrium-kloridban kifejezett klór mennyiségének meghatározását. A módszer minden takarmány esetében alkalmazható.
2. Vizsgálati alapelv
A kloridokat vízben oldjuk. Ha a termék szerves anyagot tartalmaz, tisztítjuk. Az oldatot salétromsavval enyhén savasítjuk, és a kloridokat ezüst-klorid formájában csapjuk ki ezüst-nitrát oldat felhasználásával. Az ezüst-nitrát felesleget ammónium-tiocianát oldattal, Volhard módszere szerint titráljuk.
3. Reagensek
3.1. 0,1 N ammónium-tiocianát oldat.
3.2. 0,1 N ezüst-nitrát oldat.
3.3. Telített ammónium-vasszulfát oldat.
3.4. Salétromsav, d: 1,38.
3.5. Dietiléter a.r.
3.6. Aceton a.r.
3.7. Carrez I. oldat: vízben oldjunk fel 21,9 g cink-acetátot, Zn(CH3COO)2 !!! error character !!! Β· 2H2O és 3 g jégecetet. Töltsük fel vízzel 100 ml-re.
3.8. Carrez II. oldat: vízben oldjunk fel 10,6 g kálium-vascianidot K4Fe(CN)6 !!! error character !!! Β· 3H2O. Töltsük fel vízzel 100 ml-re.
3.9. Aktív szén a.r., klorid-mentes, azokat nem abszorbáló.
4. Eszközök
Keverőgép (forgó dobos): kb. 35-40 rpm fordulatszámmal.
5. A vizsgálat módja
5.1. Az oldat elkészítése:
A minta jellegétől függően készítsünk oldatot az 5.1.1., az 5.1.2. vagy az 5.1.3. pontban foglaltak szerint.
Ezzel egy időben végezzünk vakpróbát az analizálandó minta kihagyásával.
5.1.1. Szerves anyagtól mentes minták
Mg-os pontossággal mérjünk le a mintából 10 g-nál nem többet, amely 3 g-nál kevesebb klórt tartalmaz kloridok formájában. 400 ml vízzel helyezzük egy 500 ml-es mérőlombikba kb. 20 °C-on. Keverjük harminc percig a keverőgépen, töltsük fel térfogatra, homogenizáljuk, és szűrjük.
5.1.2. Szerves anyagot tartalmazó minták, az 5.1.3. pontban felsorolt termékek kivételével
Mérjünk le a mintából 5 g-ot mg-os pontossággal, és 1 g aktív szénnel együtt helyezzük egy 500 ml-es mérőhengerbe. Adjunk hozzá 400 ml, körülbelül 20 °C-os vizet és 5 ml Carrez I. oldatot (3.7.), keverjük össze, majd adjunk hozzá 5 ml Carrez II. oldatot (3.8.). Keverjük harminc percig a keverőgépen, töltsük fel térfogatra, homogenizáljuk, és szűrjük.
5.1.3. Főzött takarmányok, lenpogácsák és lenliszt, lenlisztben gazdag termékek, valamint más, nyákban és kolloid anyagokban (például dextrinált keményítő) gazdag termékek
Készítsük el az oldatot az 5.1.2. pontban foglaltak szerint, de ne szűrjük. Dekantáljuk, (ha szükséges centrifugáljuk), vegyünk el 100 ml-t a felülúszó folyadékból, és öntsük át egy 200 ml-es mérőlombikba. Keverjük össze acetonnal (3.6.), és töltsük fel térfogatra ezzel az oldószerrel, homogenizáljuk, és szűrjük.
5.2. Titrálás
Pipettázzunk egy Erlenmeyer-lombikba 25-100 ml-t (a feltételezett klórtartalom szerint) a szűrletből, amelyet az 5.1.1., az 5.1.2. vagy az 5.1.3. pontban foglaltak szerint nyertünk. Az aliquot rész nem tartalmazhat 150 mg-nál több klórt (Cl). Szükség szerint hígítsuk vízzel, legalább 50 ml-ig, adjunk hozzá 5 ml salétromsavat (3.4.), 20 ml telített ammónium-vasszulfát oldatot (3.3.) és két csepp ammónium-tiocianát oldatot (3.1.) egy nulla jelig töltött büretta segítségével. Büretta segítségével helyezzük át az ezüst-nitrát oldatot (3.2.) úgy, hogy 5 ml-nyi többletet kapjunk. Adjunk hozzá 5 ml dietilétert (3.5.), és rázzuk fel erősen, a csapadék koagulálásáig.
Titráljuk az ezüst-nitrát többletet az ammónium-tiocianát oldattal (3.1.), amíg egy percen át tartó vörösesbarna színárnyalatot nem kapunk.
6. Az eredmények kiszámítása
A titrálásra került szűrlet mennyiségében jelen lévő, nátrium-kloridban kifejezett klór mennyiségét (w) a következő képlet segítségével számíthatjuk ki:
ahol:
V1 = a hozzáadott C,1 N ezüst-nitrát oldat mennyisége, ml-ben
V2 = a titráláshoz használt 0,1 N ammónium-tiocianát oldat mennyisége, ml-ben.
Ha a vakpróba azt jelzi, hogy 0,1 N ezüst-nitrát oldat került felhasználásra, vonjuk ki ezt az értéket a (V1 - V2) mennyiségből.
7. Észrevételek
7.1. A titrálást potenciometrikus módszerrel is el lehet végezni;
7.2. Olajban és zsírban rendkívül gazdag termékek esetében először dietiléterrel vagy könnyűolajjal végezzünk zsírtalanítást;
7.3. Halliszt esetében a titrálás Mohr módszerével is végezhető.
9. LAKTÓZ MEGHATÁROZÁSA
1. Cél és alkalmazási terület
Ez a módszer lehetővé teszi a 0,5 %-nál magasabb laktóztartalmú takarmányok laktózszintjének meghatározását.
2. Vizsgálati alapelv
A cukrokat vízben oldjuk. Az oldatot Saccharomyces cerevisiae élesztővel erjesztjük, amely a laktózt érintetlenül hagyja. Derítés és szűrés után a szűrlet laktóztartalmát Luff-Schoorl módszerrel határozzuk meg.
3. Reagensek
3.1. Saccharomyces cerevisiae szuszpenzió: szuszpendáljunk 25 g friss élesztőt 100 ml vízben. A szuszpenzió hűtőben legfeljebb egy hétig áll el.
3.2. Carrez I. oldat: vízben oldjunk fel 21,9 g cink-acetátot, Zn(CH3COO)2 ·2H2O és 3 g jégecetet. Töltsük fel vízzel 100 ml-re.
3.3. Carrez II. oldat: vízben oldjunk fel 10,6 g kálium-vascianidot, K4Fe(CN)6 ·3H2O. Töltsük fel vízzel 100 ml-re.
3.4. Luff-Schoorl reagens:
Óvatosan kevergetve öntsük a citromsavoldatot (3.4.2.) a nátrium-karbonát-oldatba (3.4.3.). Adjuk hozzá a réz-szulfát-oldatot (3.4.1.), és töltsük fel egy 1 l-re vízzel. Hagyjuk másnapig ülepedni, majd szűrjük. Ellenőrizzük az így kapott vegyszer szabályosságát (0,1 N Cu; 2 N Na2CO3). Az oldat pH-értékének körülbelül 9,4-nek kell lenni.
3.4.1. Réz-szulfát-oldat: 100 ml vízben oldjunk fel 25 g vasmentes rézszulfátot a.r. CuSO4 · 5H2O.
3.4.2. Citromsavoldat: 50 ml vízben oldjunk fel 50 g citromsavat C6H8O7 · H2O.
3.4.3. Nátrium-karbonát-oldat: kb. 300 ml meleg vízben oldjunk fel 143,8 g dehidratált nátrium-karbonátot a.r. Hagyjuk hűlni.
3.5. Sósavban forralt, vízben mosott és szárított, granulált habkő.
3.6. 30 %-os (w/v) kálium-jodid.
3.7. 6 N kénsav.
3.8. 0,1 N nátrium-tioszulfát oldat.
3.9. Keményítőoldat: adjunk 30 ml vízben elkevert 5 g oldható keményítőt 1 liter forrásban lévő vízhez. Forraljuk 3 percig, hagyjuk hűlni, és amennyiben szükséges, adjunk hozzá 10 mg higany-jodidot tartósítószerként.
4. Eszközök
38-40 °C-ra beállított, hőfokszabályzóval ellátott vízfürdő.
5. A vizsgálat módja
Mérjünk ki a mintából 1 g-ot mg-os pontossággal, és ezt a részmintát helyezzük egy 100 ml-es mérőlombikba. Adjunk hozzá 25-30 ml vizet. Helyezzük a lombikot harminc percre forró vízfürdőbe, majd hűtsük le kb. 35 °C-ra. Adjunk hozzá 5 ml élesztő szuszpenziót (3.1.), és homogenizáljuk. Hagyjuk a lombikot két órán keresztül, 38-40 °C-on állni a vízfürdőben. Hűtsük le kb. 20 °C-ra.
Adjunk hozzá 2,5 ml Carrez I. oldatot (3.2.), és kevergessük harminc másodpercig, majd adjunk hozzá 2,5 ml Carrez II. oldatot (3.3.), és újra kevergessük harminc másodpercig. Töltsük fel vízzel 100 ml-re, keverjük össze, és szűrjük. Pipetta segítségével vegyünk fel a szűrletből egy részt, amely nem haladja meg a 25 ml-t, és amely lehetőleg 40-80 mg laktózt tartalmaz, és helyezzük át egy 300 ml-es Erlenmeyer-lombikba. Amennyiben szükséges, töltsük fel 25 ml-re vízzel.
Végezzünk vakpróbát ugyanilyen módon 5 ml élesztő oldattal (3.1.).
A laktóz-tartalmat a Luff-Schoorl módszer szerint, a következőképpen határozzuk meg: adjunk az oldathoz pontosan 25 ml Luff-Schoorl reagenst (3.4.) és két szemcsényi habkövet (3.5.). Közepes méretű, szabad lángon történő melegítés közben kézzel kevergessük, és kb. két perc alatt forraljuk fel a folyadékot. Az Erlenmeyer-lombikot azonnal helyezzük egy 6 cm átmérőjű lyukkal ellátott azbesztes dróthálóra, amely alatt előzőleg lángot gyújtunk. A lángot úgy kell szabályoznunk, hogy az Erlenmeyer-lombiknak csak az alját melegítse. Illesszünk egy visszafolyós hűtőt az Erlenmeyer-lombikhoz. Forraljuk pontosan tíz percig. Hűtsük le azonnal hideg vízben, és kb. öt perc elteltével a következők szerint titráljuk:
Adjunk hozzá 10 ml kálium-jodid oldatot (3.6.), és rögtön utána (óvatosan, a túlzott habképződés veszélye miatt) adjunk hozzá 25 ml 6 N kénsavat (3.7.). Titráljuk 0,1 N nátrium-tioszulfát oldattal (3.8.) amíg fakósárga színt nem kapunk, adjuk hozzá a keményítőindikátort (3.9.), és fejezzük be a titrálást.
Forralás nélkül, 10 ml kálium-jodid oldat (3.6.) és 25 ml 6 N kénsav (3.7) hozzáadása után végezzük el ugyanezt a titrálást egy pontosan kimért, 25 ml Luff-Schoorl reagenst (3.4.) és 25 ml vizet tartalmazó keveréken.
6. Az eredmények kiszámítása
A csatolt táblázat segítségével határozzuk meg a laktóz mennyiségét mg-ban, ami a két titrálás eredménye közti különbségnek felel meg, 0,1 N nátrium-tioszulfát ml-ében kifejezve.
Az eredményt, mint a dehidratált laktóz részét, a mintára vonatkoztatva százalékosan fejezzük ki.
7. Észrevételek
A 40 %-nál több erjeszthető cukrot tartalmazó termékek esetében, 5 ml-nél nagyobb mennyiségű élesztő szuszpenziót (3.1.) használjunk.
25 ml Luff-Schoorl reagenshez tartozó értékek táblázata
0,1 N Na2S2O3, ml-e, kétpercnyi melegítés, tízpercnyi forralás
| Na2S2O30,1 N | Glükóz, fruktóz,invertcukor C6H12O6 | Laktóz C12H22O11 | Maltóz C12H22O11 | Na2S2O30,1 N | |||
| ml | mg | különbség | mg | különbség | mg | különbség | ml |
| 1 | 2,4 | 2,4 | 3,6 | 3,7 | 3,9 | 3,9 | 1 |
| 2 | 4,8 | 2,4 | 7,3 | 3,7 | 7,8 | 3,9 | 2 |
| 3 | 7,2 | 2,5 | 11,0 | 3,7 | 11,7 | 3,9 | 3 |
| 4 | 9,7 | 2,5 | 14,7 | 3,7 | 15,6 | 4,0 | 4 |
| 5 | 12,2 | 2,5 | 18,4 | 3,7 | 19,6 | 3,9 | 5 |
| 6 | 14,7 | 2,5 | 22,1 | 3,7 | 23,5 | 4,0 | 6 |
| 7 | 17,2 | 2,6 | 25,8 | 3,7 | 27,5 | 4,0 | 7 |
| 8 | 19,8 | 2,6 | 29,5 | 3,7 | 31,5 | 4,0 | 8 |
| 9 | 22,4 | 2,6 | 33,2 | 3,8 | 35,5 | 4,0 | 9 |
| 10 | 25,0 | 2,6 | 37,0 | 3,8 | 39,5 | 4,0 | 10 |
| 11 | 27,6 | 2,7 | 40,8 | 3,8 | 43,5 | 4,0 | 11 |
| 12 | 30,3 | 2,7 | 44,6 | 3,8 | 47,5 | 4,1 | 12 |
| 13 | 33,0 | 2,7 | 48,4 | 3,8 | 51,6 | 4,1 | 13 |
| 14 | 35,7 | 2,8 | 52,2 | 3,8 | 55,7 | 4,1 | 14 |
| 15 | 38,5 | 2,8 | 56,0 | 3,9 | 59,8 | 4,1 | 15 |
| 16 | 41,3 | 2,9 | 59,9 | 3,9 | 63,9 | 4,1 | 16 |
| 17 | 44,2 | 2,9 | 63,8 | 3,9 | 68,0 | 4,2 | 17 |
| 18 | 47,1 | 2,9 | 67,7 | 4,0 | 72,2 | 4,3 | 18 |
| 19 | 50,0 | 3,0 | 71,7 | 4,0 | 76,5 | 4,4 | 19 |
| 20 | 53,0 | 3,0 | 75,7 | 4,1 | 80,9 | 4,5 | 20 |
| 21 | 56,0 | 3,1 | 79,8 | 4,1 | 85,4 | 4,6 | 21 |
| 22 | 59,1 | 3,1 | 83,9 | 4,1 | 90,0 | 4,6 | 22 |
| 23 | 62,2 | 3,1 | 88,0 | 94,6 | 23 | ||
10. KÁLIUM MEGHATÁROZÁSA
1. Cél és alkalmazási terület
Ez a módszer lehetővé teszi a takarmányokban lévő kálium szintjének meghatározását.
2. Vizsgálati alapelv
A mintát hamvasztjuk, a hamut pedig sósavban oldjuk. Az oldat nátriumtartalmát lángfotometriával határozzuk meg cézium-klorid és alumínium-nitrát jelenlétében. Ezeknek az anyagoknak a hozzáadása nagymértékben kiküszöböli a zavaró elemek okozta interferenciát.
3. Reagensek
3.1. Sósav a.r., d: 1,12.
3.2. Cézium-klorid a.r.
3.3. Alumínium-nitrát Al(NO3)3 · 9H2O, vegytiszta.
3.4. Kálium-klorid a.r., dehidratált.
3.5. Pufferoldat: oldjunk fel vízben 50 g cézium-kloridot (3.2.) és 250 g alumínium-nitrátot (3.3.), töltsük fel vízzel 1 literre, és homogenizáljuk. Tároljuk műanyag palackban.
3.6. Kálium-standard-oldat: oldjunk fel vízben 1,907 g kálium-kloridot (3.4.), adjunk hozzá 5 ml sósavat (3.1.), töltsük fel vízzel 1 literre, és homogenizáljuk. Tároljuk műanyag palackban. Ezen oldat 1 ml-je 1,00 mg káliumot tartalmaz.
4. Eszközök
4.1. Platina-, kvarc- vagy porcelán hamvasztótégelyek, szükség esetén fedéllel ellátva.
4.2. Hőfokszabályzós, elektromos tokos kemence.
4.3. Lángfotométer.
5. A vizsgálat módja
5.1. A minta elemzése
Az általános szabály szerint, mérjünk ki a mintából 10 g-ot 10 mg-nyi pontossággal, helyezzük egy tégelybe, és 450 °C-on hamvasszuk három órán keresztül. Lehűtés után helyezzük át a hamut mennyiségileg egy 500 ml-es mérőlombikba 250-300 ml víz, majd 50 ml sósav (3.1.) felhasználásával. Amikor minden szén-dioxid kibocsátás megszűnik, hevítsük az oldatot, és alkalmanként megkeverve, tartsuk kb. 90 °C-os hőmérsékleten két órán keresztül. Szobahőmérsékletre való lehűtés után töltsük fel jelig vízzel, rázzuk össze, és szűrjük. A szűrlet egy, legfeljebb 1,0 mg káliumot tartalmazó aliquot részét helyezzük át egy 100 ml-es mérőlombikba, adjunk hozzá 10,0 ml pufferoldatot (3.5.), töltsük fel jelig vízzel, és homogenizáljuk. Magasabb káliumszint esetén, a pufferoldat hozzáadása előtt, megfelelő arányban hígítsuk fel az elemzendő oldatot.
| A minta feltételezett káliumtartalma (% K) | Hígítási arány | Aliquot rész az oldat ml-ében |
| 0,1-ig | –– | 50 |
| 0,1-től 0,5-ig | –– | 10 |
| 0,5-től 1,0-ig | –– | 5 |
| 1,0-től 5,0-ig | 1: 10 | 10 |
| 5,0-től 10,0-ig | 1: 10 | 5 |
| 10,0-től 20,0-ig | 1: 20 | 5 |
A mérést lángfotometriával, 768 nm-es hullámhosszon végezzük. Az eredményt kalibrációs görbével számítsuk ki.
5.2. Kalibrációs görbe
Helyezzünk pontosan 10 ml szabványoldatot (3.6.) egy 250 ml-es mérőlombikba, töltsük fel jelig vízzel, és homogenizáljuk. Töltsünk ezen oldatból pontosan 5, 10, 15, 20 és 25 ml-nyi mennyiséget 100 ml-es mérőlombikokba, melyek egyenként 0,2; 0,4; 0,6; 0,8 és 1,0 mg káliumnak felelnek meg. Egészítsük ki a sorozatot egy szabványoldatot nem tartalmazó, összehasonlító lombikkal. Öntsünk 10 ml pufferoldatot (3.5.) minden egyes lombikba, töltsük fel őket jelig vízzel, és homogenizáljuk. Végezzük el az 5.1. bekezdésben feltüntetett méréseket. A kalibrációs görbe 1 mg-os kálium koncentrációig 100 ml oldatban általában lineáris.
6. Az eredmények kiszámítása
Az eredményt a mintára vonatkoztatva, százalékosan fejezzük ki.
7. Észrevételek
A zavaró elemek okozta interferencia kiküszöbölése érdekében nem minden esetben szükséges pufferoldat (3.5.) hozzáadása.
11. NÁTRIUM MEGHATÁROZÁSA
1. Cél és alkalmazási terület
Ez a módszer lehetővé teszi a takarmányokban lévő nátrium szintjének meghatározását.
2. Vizsgálati alapelv
A mintát hamvasztjuk, a hamut pedig sósavban oldjuk. Az oldat nátriumtartalmát lángfotometriával határozzuk meg, cézium-klorid és alumínium-nitrát jelenlétében. Ezen anyagok hozzáadása nagymértékben kiküszöböli a zavaró elemek okozta interferenciát.
3. Reagensek
3.1. Sósav a.r., d: 1,12.
3.2. Cézium-klorid a.r.
3.3. Alumínium-nitrát Al(NO3)3 · 9H2O, vegytiszta.
3.4. Nátrium-klorid a.r., dehidratált.
3.5. Pufferoldat: oldjunk fel vízben 50 g cézium-kloridot (3.2.) és 250 g alumínium-nitrátot (3.3.), töltsük fel vízzel 1 literre, és homogenizáljuk. Tároljuk műanyag palackban.
3.6. Nátrium-szabványoldat: oldjunk fel vízben 2,542 g nátrium-kloridot (3.4.), adjunk hozzá 5 ml sósavat (3.1.), töltsük fel vízzel 1 literre, és homogenizáljuk. Tároljuk műanyag palackban, ezen oldat 1 ml-je 1,00 mg nátriumot tartalmaz.
4. Eszközök
4.1. Platina, kvarc- vagy porcelán hamvasztótégelyek, szükség esetén fedéllel ellátva.
4.2. Hőfokszabályzós, elektromos tokos kemence.
4.3. Lángfotométer.
5. A vizsgálat módja
5.1. A minta elemzése
Általános szabály szerint, mérjünk le a mintából 10 g-ot 10 mg-nyi pontossággal, helyezzük egy tégelybe (4.2.), és 450 °C-on hamvasszuk három órán keresztül. Kerüljük a túlhevítést (gyulladást). Lehűtés után öntsük át a hamut mennyiségileg egy 500 ml-es mérőlombikba, 250-300 ml víz, majd 50 ml sósav (3.1.) felhasználásával. Amikor minden szén-dioxid kibocsátás megszűnik, hevítsük az oldatot, és alkalmanként megkeverve, tartsuk kb. 90 °C-os hőmérsékleten két órán keresztül. Szobahőmérsékletre való lehűtés után töltsük fel jelig vízzel, rázzuk fel, és szűrjük. A szűrlet, egy legfeljebb 1,0 mg nátriumot tartalmazó aliquot részét öntsük át egy 100 ml-es mérőlombikba, adjunk hozzá 10,0 ml pufferoldatot (3.5.), töltsük fel jelig vízzel, és homogenizáljuk. Magasabb nátriumszint esetén, a pufferoldat hozzáadása előtt megfelelő arányban hígítsuk fel az elemzendő oldatot.
| A minta feltételezett nátriumtartalma (% Na) | Hígítási arány | Aliquot rész az oldat ml-ében |
| 0,1-ig | –– | 50 |
| 0,1-től 0,5-ig | –– | 10 |
| 0,5-től 1,0-ig | –– | 5 |
| 1,0-től 5,0-ig | 1: 10 | 10 |
| 5,0-től 10,0-ig | 1: 10 | 5 |
| 10,0-től 20,0-ig | 1: 20 | 5 |
A mérést lángfotometriával, 589 nm-es hullámhosszon végezzük. Az eredményt egy kalibrációs görbével számítsuk ki.
5.2. Kalibrációs görbe
Helyezzünk pontosan 10 ml szabványoldatot (3.6.) egy 250 ml-es mérőlombikba, töltsük fel jelig vízzel, és homogenizáljuk. Töltsünk ezen oldatból pontosan 5, 10, 15, 20 és 25 ml-nyi mennyiséget 100 ml-es mérőlombikokba, amelyek egyenként 0,2; 0,4; 0,6; 0,8 és 1,0 mg nátriumnak felelnek meg. Egészítsük ki a sorozatot egy szabványoldatot nem tartalmazó összehasonlító lombikkal. Öntsünk 10 ml pufferoldatot (3.5.) minden egyes lombikba, töltsük fel őket jelig vízzel, és homogenizáljuk. Végezzük el az 5.1. bekezdésben feltüntetett méréseket. A kalibrációs görbe 1 mg-os nátrium koncentrációig 100 ml oldatban általában lineáris.
6. Az eredmények kiszámítása
Az eredményt a mintára vonatkoztatva, százalékosan fejezzük ki.
7. Észrevételek
7.1. A 4 %-nál több nátriumot tartalmazó termékek esetén, előnyösebb az anyagot két órán keresztül zárható tégelyben hamvasztani. Lehűtés után adjunk hozzá vizet, platinadrót segítségével szuszpenzáljuk a hamut, szárítsuk ki, és hamvasszuk újból két órán keresztül zárható tégelyben.
7.2. Ha a minta kizárólag ásványi anyagokat tartalmaz, előzetes hamvasztás nélkül oldjuk fel.
12. CUKOR MEGHATÁROZÁSA
1. Cél és alkalmazási terület
A módszer lehetővé teszi a redukálócukor és az invertálás utáni összes cukor glükózban vagy, adott esetben, szacharózban kifejezett szintjének meghatározását, 0,95-os tényezővel konvertálva. A módszer összetett takarmányok esetében alkalmazható. Egyéb takarmányokra különleges módszerek vonatkoznak. Adott esetben, a laktózt külön kell megmérni, és figyelembe venni az eredmények kiszámításánál.
2. Vizsgálati alapelv
A cukrot hígított etanolban extraháljuk; az oldatot Carrez I. és II. oldattal derítjük. Az etanol kizárása után az invertálás előtti és utáni mennyiség Luff-Schoorl módszerrel kerül meghatározásra.
3. Reagensek
3.1. 40 %-os (v/v) etanol d: 0,948, 20°C-on, fenolftaleinhez semlegesítve.
3.2. Carrez I. oldat: vízben oldjunk fel 21,9 g cink-acetátot, Zn(CH3COO)2 · 2H2O és 3 g jégecetet. Töltsük fel vízzel 100 ml-re.
3.3. Carrez II. oldat: vízben oldjunk fel 10,6 g kálium-vascianidot, K4Fe(CN)6 · 3H2O. Töltsük fel vízzel 100 ml-re.
3.4. 0,1 %-os (w/v) metilnarancs oldat.
3.5. 4 N sósav.
3.6. 0,1 N sósav.
3.7. 0,1 N nátrium-hidroxid oldat.
3.8. Luff-Schoorl reagens:
Óvatosan kevergetve öntsük a citromsavoldatot (3.8.2.) a nátrium-karbonát-oldatba (3.8.3.). Adjuk hozzá a réz-szulfát oldatot (3.8.1.), és öntsük fel 1 literig vízzel. Hagyjuk az éjszaka folyamán ülepedni, majd szűrjük. Ellenőrizzük az így kapott reagens szabályosságát (0,1 N Cu; 2 N Na2CO3). Az oldat pH-értékének kb. 9,4-nek kell lennie.
3.8.1. Réz-szulfát-oldat: 100 ml vízben oldjunk fel 25 g vasmentes rézszulfátot a.r. CuSO4 · 5H2O.
3.8.2. Citromsavoldat: 50 ml vízben oldjunk fel 50 g citromsavat a.r. C6H8O7 · H2O.
3.8.3. Nátrium-karbonát-oldat: kb. 300 ml meleg vízben oldjunk fel 143,8 g dehidratált nátrium-karbonátot a.r. Hagyjuk kihűlni.
3.9. 0,1 N nátrium-tioszulfát-oldat.
3.10. Keményítőoldat: adjunk 30 ml vízben elkevert 5 g oldható keményítőt 1 liter forrásban lévő vízhez. Forraljuk három percen keresztül, hagyjuk kihűlni, szükség esetén adjunk hozzá 10 mg higany-jodidot tartósítószerként.
3.11. 6 N kénsav.
3.12. 30 %-os (w/v) kálium-jodid.
3.13. Sósavban forralt, vízben mosott és szárított, szemcsés horzsakő.
3.14. 3-metilbután-1-ol.
4. Eszközök
Keverőgép (forgó dobos): kb. 35-40 rpm fordulatszámmal.
5. A vizsgálat módja
5.1. A minta extrahálása
Mérjünk ki mg-os pontossággal 2,5 g-ot a mintából, és helyezzük egy 250 ml-es mérőlombikba. Adjunk hozzá 200 ml etanolt (3.1.), és keverjük a keverőgépen egy órán keresztül. Adjunk hozzá 5 ml Carrez I. oldatot (3.2), és kevergessük egy percig. Adjunk hozzá 5 ml Carrez II. oldatot (3.3), és kevergessük újra egy percig. Töltsük fel a térfogatra etanollal (3.1.), homogenizáljuk, és szűrjük. Vegyünk el 200 ml-t a szűrletből, és az etanol nagy részének eltávolítása céljából pároljuk térfogatának kb. felére. Helyezzük a bepárlás utáni maradékot mennyiségileg egy 200 ml-es mérőlombikba meleg vízzel, hűtsük, töltsük fel térfogatra vízzel, homogenizáljuk, és szükség esetén szűrjük. Ezen oldatot használjuk a redukálócukor és invertálás utáni összes cukor mennyiségének meghatározására.
5.2. A redukálócukor meghatározása
Pipetta segítségével vegyünk fel legfeljebb 25 ml oldatot, amely 60 mg-nál kevesebb, glükózban kifejezett redukálócukrot tartalmaz. Szükség esetén töltsük fel az oldatot desztillált vízzel 25 ml-re, és határozzuk meg redukálócukor tartalmát a Luff-Schoorl módszerrel. Az eredményt a minta glükóztartalmára vonatkoztatva, százalékosan fejezzük ki.
5.3. Az invertálás utáni összes cukor meghatározása
Pipetta segítségével vegyünk fel az oldatból 50 ml-t, és öntsük át egy 100 ml-es mérőlombikba. Adjunk hozzá néhány csepp metilnarancs oldatot (3.4.), majd óvatosan és folyamatos kevergetés közben, adjunk hozzá 4 N sósavat (3.5.), amíg a folyadék határozottan vörössé nem válik. Adjunk hozzá 15 ml 0,1 N sósavat (3.6.), merítsük a lombikot egy gyorsan forró vízfürdőbe, és tartsuk ott harminc percen keresztül. Hűtsük le gyorsan kb. 20 °C-ra, és adjunk hozzá 15 ml 0,1 N nátrium-hidroxid-oldatot (3.7.). Töltsük fel vízzel 100 ml-re, és homogenizáljuk. Vegyünk el legfeljebb 25 ml oldatot, amely 60 mg-nál kevesebb, glükózban kifejezett redukálócukrot tartalmaz. Szükség esetén töltsük fel az oldatot desztillált vízzel 25 ml-re, és határozzuk meg redukálócukor tartalmát a Luff-Schoorl módszerrel. Az eredményt a glükózra vagy, adott esetben, a szacharózra vonatkoztatva, százalékosan fejezzük ki 0,95-os tényezővel szorozva.
5.4. Titrálás a Luff-Schoorl módszerrel
Pipetta segítségével vegyünk fel 25 ml Luff-Schoorl reagenst (3.8.), és helyezzük át egy 300 ml-es Erlenmeyer-lombikba; adjunk hozzá pontosan 25 ml-t a derített cukoroldatból. Adjunk hozzá két szemcsényi habkövet (3.13.), hevítsük közepes méretű, szabad lángon, közben kézzel kevergessük, és kb. két perc alatt forraljuk fel a folyadékot. Helyezzük rögtön az Erlenmeyer-lombikot egy 6 cm átmérőjű lyukkal ellátott azbesztes dróthálóra, amely alatt lángot gyújtottunk. A lángot úgy kell szabályoznunk, hogy az Erlenmeyer-lombiknak csak az alját melegítse. Illesszünk visszafolyós hűtőt az Erlenmeyer-lombikhoz. Forraljuk a folyadékot pontosan tíz percig. Hűtsük le azonnal hideg vízben, és kb. öt perc elteltével a következők szerint titráljuk:
Adjunk hozzá 10 ml kálium-jodid-oldatot (3.12.), és közvetlenül ezután (óvatosan, a túlzott habképződés veszélye miatt) adjunk hozzá 25 ml 6 N kénsavat (3.11.). Titráljuk 0,1 N nátrium-tioszulfát-oldattal (3.9.) a fakósárga szín megjelenéséig, adjuk hozzá a keményítő indikátort (3.10.), és fejezzük be a titrálást.
Forralás nélkül, 10 ml kálium-jodid-oldat (3.12.) és 25 ml 6 N kénsav (3.11.) hozzáadása után végezzük el ugyanezt a titrálást egy pontosan kimért, 25 ml Luff-Schoorl reagenst (3.8.) és 25 ml vizet tartalmazó elegyen.
6. Az eredmények kiszámítása
A táblázat segítségével határozzuk meg a glükóz mennyiségét mg-ban, ami a két titrálás eredménye közti különbségnek felel meg 0,1 N nátrium-tioszulfát mg-jában kifejezve.
Az eredményt a mintára vonatkoztatva, százalékosan fejezzük ki.
7. Különleges vizsgálati módok
7.1. A melaszban gazdag, valamint egyéb, kevésbé homogén összetételű takarmányok esetében mérjünk ki 20 g-ot, és helyezzük egy 1 literes mérőlombikba 500 ml vízzel felöntve. Keverjük forgó dobon egy órán keresztül. Az 5.1. pontban leírtak szerint derítsük Carrez I. (3.2.) és Carrez II. (3.3.) reagensek használatával, ebben az esetben azonban négyszeres mennyiséget használjunk az egyes reagensekből. Töltsük fel térfogatra 80 %-os (v/v) etanollal.
Homogenizáljuk és szűrjük. Vonjuk ki az etanolt az 5.1. pontban leírtak szerint. Ha dextrinált keményítő nincs jelen, töltsük fel térfogatra desztillált vízzel.
7.2. Melaszok, valamint cukorban gazdag és gyakorlatilag keményítőmentes egynemű takarmányok (szentjánoskenyér, szárított cukorrépa szeletek stb.) esetében mérjünk ki 5 g-ot, és helyezzük egy 250 ml-es mérőlombikba, adjunk hozzá 200 ml desztillált vizet, és keverjük forgó dobon egy órán keresztül vagy szükség esetén tovább. Az 5.1. pontban leírtak szerint derítsük Carrez I. (3.2.) és Carrez II. (3.3.) reagensekkel. Töltsük fel térfogatra hideg vízzel, homogenizáljuk, és szűrjük. Az összes cukortartalom meghatározásához az 5.3. pontban leírtak szerint folytassuk a vizsgálatot.
8. Észrevételek
8.1. A habképződés elkerülése érdekében a Luff-Schoorl reagenssel történő forralás előtt tanácsos (a mennyiségtől függetlenül) kb. 1 ml 3-metilbután-1-ol (3.14.) hozzáadása.
8.2. A glükózban kifejezett, invertálás utáni összes cukortartalom és a glükózban kifejezett, redukálócukor-tartalom különbsége, 0,95-tel szorozva, megadja a szacharóztartalomra vonatkoztatott százalékos arányt.
8.3. A redukálócukor-tartalom meghatározására, laktóz kivételével, két módszer alkalmazható:
8.3.1. közelítő számításhoz, szorozzuk meg 0,675-tel a laktóztartalmat, amely egy másik elemzési módszerrel került megállapításra, és vonjuk ki a kapott eredményt a redukálócukor-tartalomból.
8.3.2. A redukálócukor pontos kiszámításához, a laktóz kivételével, ugyanazt a mintát kell használni a két végső meghatározáshoz. Az egyik vizsgálatot az 5.1. pontban foglaltak szerint nyert oldaton végezzük, a másikat pedig a laktóz meghatározása során nyert oldaton az abból a célból meghatározott módszer szerint (más típusú cukrok erjesztése és derítés után).
A jelen lévő cukormennyiség meghatározása mindkét esetben a Luff-Schoorl módszerrel történik, és a glükóz mg-ban kerül kiszámításra. Az egyik értéket kivonjuk a másikból, és az eredményt a mintára vonatkoztatva, százalékosan fejezzük ki.
A kiválasztott két mennyiség az egyes meghatározások esetében 250 mg-os mintának felel meg.
Az első esetben 17 ml 0,1 N nátrium-tioszulfát oldat 44,2 mg fogyott glükóznak felel meg; a második esetben 11 ml felel meg 27,6 mg glükóznak.
A különbség 16,6 mg glükóz.
A redukálócukor-tartalom (laktóz kivételével), glükózban számítva így:
25 ml Luff-Schoorl reagenshez tartozó értékek táblázata
0,1 N Na2S2O3 ml-e, kétpercnyi melegítés, tízpercnyi forralás
| 0,1 N Na2S2O3 | Glükóz, fruktóz,invertcukor C6H12O6 | Laktóz C12H22O11 | Maltóz C12H22O11 | 0,1 N Na2S2O3 | |||
| ml | mg | különbség | mg | különbség | mg | különbség | ml |
| 1 | 2,4 | 2,4 | 3,6 | 3,7 | 3,9 | 3,9 | 1 |
| 2 | 4,8 | 2,4 | 7,3 | 3,7 | 7,8 | 3,9 | 2 |
| 3 | 7,2 | 2,5 | 11,0 | 3,7 | 11,7 | 3,9 | 3 |
| 4 | 9,7 | 2,5 | 14,7 | 3,7 | 15,6 | 4,0 | 4 |
| 5 | 12,2 | 2,5 | 18,4 | 2,5 | 19,6 | 3,9 | 5 |
| 6 | 14,7 | 2,5 | 22,1 | 3,7 | 23,5 | 4,0 | 6 |
| 7 | 17,2 | 2,6 | 25,8 | 3,7 | 27,5 | 4,0 | 7 |
| 8 | 19,8 | 2,6 | 29,5 | 3,7 | 31,5 | 4,0 | 8 |
| 9 | 22,4 | 2,6 | 33,2 | 3,8 | 35,5 | 4,0 | 9 |
| 10 | 25,0 | 2,6 | 37,0 | 3,8 | 39,5 | 4,0 | 10 |
| 11 | 27,6 | 2,7 | 40,8 | 3,8 | 43,5 | 4,0 | 11 |
| 12 | 30,3 | 2,7 | 44,6 | 2,7 | 47,5 | 4,1 | 12 |
| 13 | 33,0 | 2,7 | 48,4 | 3,8 | 51,6 | 4,1 | 13 |
| 14 | 35,7 | 2,8 | 52,2 | 3,8 | 55,7 | 4,1 | 14 |
| 15 | 38,5 | 2,8 | 56,0 | 3,9 | 59,8 | 4,1 | 15 |
| 16 | 41,3 | 2,9 | 59,9 | 3,9 | 63,9 | 4,1 | 16 |
| 17 | 44,2 | 2,9 | 63,8 | 3,9 | 68,0 | 4,2 | 17 |
| 18 | 47,1 | 2,9 | 67,7 | 4,0 | 72,2 | 4,3 | 18 |
| 19 | 50,0 | 3,0 | 71,7 | 4,0 | 76,5 | 4,4 | 19 |
| 20 | 53,0 | 3,0 | 75,7 | 4,1 | 80,9 | 4,5 | 20 |
| 21 | 56,0 | 3,1 | 79,8 | 4,1 | 85,4 | 4,6 | 21 |
| 22 | 59,1 | 3,1 | 83,9 | 4,1 | 90,0 | 4,6 | 22 |
| 23 | 62,2 | 88,0 | 94,6 | 23 | |||
14. KARBAMID MEGHATÁROZÁSA
1. Cél és alkalmazási terület
Ez a módszer lehetővé teszi a takarmányokban lévő karbamid szintjének meghatározását.
2. Vizsgálati alapelv
A mintát vízben szuszpendáljuk egy derítőszerrel. A szuszpenziót leszűrjük. A szűrlet karbamid-tartalmát, 4-dimetil-amino-benzaldehid (4-DMAB) hozzáadása után, az optikai sűrűség 420 nm-es hullámhosszon való megmérésével határozzuk meg.
3. Reagensek
3.1. 4-dimetil-amino-benzaldehid-oldat: oldjunk fel 1,6 g 4-DMAB a.r.-t 100 ml 96 %-os etanolban, és adjunk hozzá 10 ml sósavat a.r. (d: 1,19). Ezen reagens legfeljebb kéthetes időtartamon keresztül áll el.
3.2. Carrez I. oldat: vízben oldjunk fel 21,9 g cink-acetátot, Zn(CH3COO) · 2H2O és 3 g jégecetet. Töltsük fel vízzel 100 ml-re.
3.3. Carrez II. oldat: vízben oldjunk fel 10,6 g kálium-hexaciáno-ferrát-ot, K4Fe(CN)6 · 3H2O. Töltsük fel vízzel 100 ml-re.
3.4. Aktív szén a.r., amely nem abszorbálja a karbamidot (ezt ellenőrizni kell).
3.5. 0,1 %-os (w/v) karbamid a.r.
4. Eszközök
4.1. Keverőgép (forgó dobos): kb. 35-40 rpm fordulatszámmal
4.2. Kémcsövek: 160 × 16 mm, csiszolt üvegdugóval.
4.3. Spektrofotométer.
5. A vizsgálat módja
5.1. A minta analízise
Mérjünk ki a mintából 2 g-ot mg-nyi pontossággal, és 1 g aktív szénnel együtt helyezzük egy 500 ml-es mérőlombikba. Adjunk hozzá 400 ml vizet és 5 ml Carrez I. (3.2.) és II. (3.3.) oldatot. Keverjük a forgó dobon harminc percig. Töltsük térfogatra vízzel, rázzuk fel, és szűrjük.
Vegyünk el 5 ml-t az áttetsző, színtelen szűrletekből, öntsük csiszolt üvegdugóval ellátott kémcsövekbe, adjunk hozzájuk 5 ml 4-DMAB oldatot (3.1.), és keverjük fel. Helyezzük a kémcsöveket forróvíz-fürdőbe 20 °C-on. Tizenöt perc elteltével a spektrofotométer segítségével, 420 nm-en mérjük meg a mintaoldat optikai sűrűségét, és ezt hasonlítsuk össze a vakpróba során a reagensekből készített oldattal.
5.2. Kalibrációs görbe
Vegyünk el a karbamid oldatból (3.5.) 1, 2, 4, 5 és 10 ml-t, öntsük 100 ml-es mérőlombikokba, és töltsük fel őket térfogatra vízzel. Vegyünk el 5 ml-t minden egyes oldatból, és mindegyikhez adjunk 5 ml 4-DMAB oldatot (3.1.), homogenizáljuk, és a fentiek szerint mérjük meg az optikai sűrűséget összehasonlítva az 5 ml 4-DMAB oldatot és 5 ml vizet tartalmazó karbamidmentes kontrolloldattal. Szerkesszük meg a kalibrációs görbét.
6. Az eredmények kiszámítása
A kalibrációs görbe segítségével határozzuk meg a mintában lévő karbamidmennyiséget.
Az eredményt a mintára vonatkoztatva, százalékosan fejezzük ki.
7. Észrevételek
7.1. 3 %-ot meghaladó karbamidtartalom esetén, csökkentsük a minta nagyságát 1 g-ra vagy hígítsuk az eredeti oldatot úgy, hogy 500 ml-ben ne legyen több 50 mg karbamidnál.
7.2. Alacsony karbamidtartalom esetén növeljük a minta nagyságát mindaddig, amíg a szűrlet áttetsző és színtelen marad.
7.3. Ha a minta olyan egyszerű nitrogéntartalmú elegyeket tartalmaz, mint az aminosavak, akkor az optikai sűrűséget 435 nm-en kell mérni.
16. SZÓJÁBÓL KÉSZÜLT TERMÉKEK KARBAMIDAKTIVITÁSÁNAK MEGHATÁROZÁSA
1. Cél és alkalmazási terület
Ez a módszer lehetővé teszi a szójából készült termékek karbamid-aktivitásának meghatározását és ezen termékek nem megfelelő elkészítési módjának kimutatását.
2. Vizsgálati alapelv
A karbamid-aktivitást egy karbamidoldatból 30 °C-on 1 g termékenként felszabaduló ammóniás nitrogén mennyisége alapján becsüljük.
3. Reagensek
3.1. 0,1 N sósav.
3.2. 0,1 N nátrium-hidroxid oldat.
3.3. 0,05 M foszfát-pufferoldat, amely 1000 ml-enként 4,45 g dinátrium-hidrogén-foszfátot (NA2HPO4 · 2H2O) és 3,40 g kálium-dihidrogén-foszfátot (KH2PO4) tartalmaz.
3.4. Frissen készített karbamid-pufferoldat, amelyben 1 000 ml pufferoldat (3.3.) 30,0 g karbamidot tartalmaz; pH 6,9-7,0.
4. Eszközök
4.1. Potenciometrikus titráló berendezés vagy nagy érzékenységű pH-mérő (0,02 pH) mágneses keverővel.
4.2. Pontosan 30 °C-ra beállított, hőfokszabályzós vízfürdő.
4.3. Kémcsövek csiszolt üvegdugóval, 150 × 18 mm.
5. A vizsgálat módja
Morzsoljunk szét (például egy kávédarálóban) 10 g mintát úgy, hogy keresztülmenjen egy 0,2 mm-es szemátmérőjű szűrőn. Mérjünk ki 0,2 g-ot mg-nyi pontossággal a szétmorzsolt mintából, helyezzük egy csiszolt üvegdugóval ellátott kémcsőbe, és adjunk hozzá 10 ml karbamid-pufferoldatot (3.4.). Zárjuk le azonnal, és erőteljesen rázzuk fel. Helyezzük a kémcsövet egy pontosan 30 °C-ra beállított vízfürdőbe, és rázzuk fel erőteljesen. Helyezzük a kémcsövet egy pontosan 30 °C-ra beállított vízfürdőbe és tartsuk ott pontosan harminc percig. Adjunk hozzá azonnal 10 ml 0,1 N sósavat (3.1.), gyorsan hűtsük le 20 °C-ra, és öntsük át a kémcső teljes tartalmát egyforma adagokban egy titrálóedénybe, 5 ml vízzel kétszer öblítve. Egy üvegelektródával (4.1.) titráljuk 0,1 N nátrium-hidroxid oldattal (3.2.). azonnal és gyorsan 4,7-es pH-értékre, elektrometria alkalmazásával.
Végezzünk vakpróbát a következőképpen:
Mg-nyi pontossággal kimért 0,2 g mintát gyorsan helyezzünk egy csiszolt üvegdugóval ellátott kémcsőbe, adjunk hozzá 10 ml 0,1 N sósavat (3.1.), majd 10 ml karbamid pufferoldatot (3.4.). Azonnal hűtsük le a kémcsövet jeges vízben, és hagyjuk ott harminc percre. A fent leírtaknak megfelelően, öntsük át a kémcső tartalmát a titrálóedénybe, és a 0,1 N nátrium-hidroxid oldattal (3.2.) titráljuk 4,7-es pH-értékre.
6. Számítás
A karbamid-aktivitást a következő képlettel számítjuk ki:
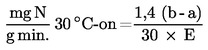
ahol:
a = a minta által felvett 0,1 N nátrium-hidroxid-oldat, ml-ben,
b = a vakpróba során felvett 0,1 N nátrium-hidroxid-oldat, ml-ben,
E = a minta tömege, g-ban.
7. Észrevételek
7.1. Ezen módszer 30 °C-on 1 mg N/g/min-es karbamid tevékenységnél alkalmazható. Aktívabb termékek esetében a minta mérete 50 mg-ra csökkenthető.
7.2. A 10 %-nál több nyerszsírt tartalmazó termékeket először hidegen zsírtalanítani kell.
( 1 ) HL L 170., 1970.8.3., 2. o.
( 2 ) HL L 140., 2002.5.30., 10. o.
( 3 ) HL L 102., 1976.4.15., 1. o.
( 4 ) HL L 83., 1973.3.30., 35. o.
( 5 ) HL L 279., 1971.12.20., 7. o.
Lábjegyzetek:
[1] A dokumentum eredetije megtekinthető CELEX: 31971L0250 - https://eur-lex.europa.eu/legal-content/HU/ALL/?uri=CELEX:31971L0250&locale=hu Utolsó elérhető, magyar nyelvű konszolidált változat CELEX: 01971L0250-20050216 - https://eur-lex.europa.eu/legal-content/HU/ALL/?uri=CELEX:01971L0250-20050216&locale=hu