
31971L0393[1]
A Bizottság második irányelve (1971. november 18.) a takarmányok hatósági ellenőrzésére szolgáló közösségi analitikai módszerek meghatározásáról
A BIZOTTSÁG MÁSODIK IRÁNYELVE
(1971. november 18.)
a takarmányok hatósági ellenőrzésére szolgáló közösségi analitikai módszerek meghatározásáról
(71/393/EGK)
AZ EURÓPAI KÖZÖSSÉGEK BIZOTTSÁGA,
tekintettel az Európai Gazdasági Közösséget létrehozó szerződésre,
tekintettel a takarmányok hatósági ellenőrzésénél alkalmazandó közösségi mintavételi és analitikai módszerek bevezetéséről szóló, 1970. július 20-i tanácsi irányelvre ( 1 ) és különösen annak 2. cikkére,
mivel az említett irányelv előírja, hogy a takarmányok hatósági ellenőrzését, amelynek célja a takarmányok minőségére és összetételére vonatkozó törvényi, rendeleti és közigazgatási rendelkezésekből eredő követelmények betartásának ellen- őrzése, közösségi mintavételi és analitikai módszerek alkalmazásával kell végrehajtani;
mivel az 1971. június 15-i 71/250/EGK bizottsági irányelv ( 2 ) már megállapított néhány közösségi analitikai módszert; mivel az azóta eltelt időszakban, az adott területen történt előrelépésekre való tekintettel, tanácsos egy második módszersorozat elfogadása;
mivel az ezen irányelvben előírt intézkedések összhangban vannak a Takarmányok Állandó Bizottsága véleményével,
ELFOGADTA EZT AZ IRÁNYELVET:
1. cikk
A tagállamok előírják, hogy a takarmányok hatósági ellenőrzésére szolgáló, azok nedvességtartalmának, illó nitrogénbázis-tartalmának, összes foszfor-, nyersolaj- és zsírtartalmának meghatározására irányuló analitikai vizsgálatokat az ezen irányelv mellékletében leírt módszerek szerint kell végrehajtani.
2. cikk
A tagállamok legkésőbb 1973. január 1-jéig hatályba léptetik azokat a törvényi, rendeleti és közigazgatási rendelkezéseket, amelyek szükségesek ahhoz, hogy ennek az irányelvnek megfeleljenek. Erről haladéktalanul tájékoztatják a Bizottságot.
3. cikk
Ennek az irányelvnek a tagállamok a címzettjei.
MELLÉKLET
I. A NEDVESSÉGTARTALOM MEGHATÁROZÁSA
1. Cél és alkalmazási terület
Ez a módszer lehetővé teszi a takarmányok nedvességtartalmának meghatározását. Nem vonatkozik a tejtermékek, mint közvetlen takarmányok elemzésére, az ásványi anyagok és a túlnyomórészt ásványi anyagokból álló keverékek analízisére, az állati és növényi zsírok, illetve olajok analízisre, sem pedig az olaj- és zsírpiac közös szervezésének létrehozásáról szóló, 1966. szeptember 22-i 136/66/EGK tanácsi rendeletben meghatározott olajos magvak és olajtartalmú gyümölcsök analízisére.
Az olajos magvak és olajtartalmú gyümölcsök nedvességtartalmának meghatározását az olajos magvakból történő mintavételről, a minták aprításáról, valamint az olaj-, a szennyezőanyag- és a nedvességtartalom meghatározásáról szóló, 1968. szeptember 23-i 1470/68/EGK bizottsági rendelet ( 3 ) III. melléklete írja le.
2. Vizsgálati alapelv
A mintát, a takarmány jellege szerint, meghatározott körülmények között szárítjuk. A tömegveszteséget méréssel határozzuk meg. Amennyiben magas nedvességtartalmú, szilárd takarmányokkal dolgozunk, előzetes szárítást kell végezni.
3. Eszközök
3.1. Nem nedvszívó anyagból készült, könnyen tisztítható aprítógép, amelynek használata gyors és egynemű őrlést tesz lehetővé anélkül, hogy észrevehető mennyiségű hőtermeléssel járna, a lehető legnagyobb mértékben megakadályozza a külső levegővel történő érintkezést, és megfelel a 4.1.1. és 4.1.2. pontban foglalt követelményeknek (pl. kalapácsos törő vagy vízhűtéses mikro-aprítók, összecsukható kúpos malmok, finommozgású-lassú fordulatú vagy fogaskerekes aprítók).
3.2. 0,5 mg pontosságú analitikai mérleg.
3.3. Nem rozsdásodó fémből vagy üvegből készült, légmentes záródást biztosító fedéllel ellátott száraz tartályok; megfelelő munkafelület, amely lehetővé teszi a minta 0,3 g/cm2 körüli mennyiségben történő terítését.
3.4. Elektromos fűtésű izotermikus, megfelelően szellőztethető kemence (± 1 °C), amely lehetővé teszi a gyors hőfokszabályozást ( 4 ).
3.5. Szabályozható, elektromos fűtésű vákuumkemence, olajszivattyúval és forró, száraz levegő vagy szárítóanyag (pl. kalcium-oxid) bejuttatására alkalmas szerkezettel ellátva.
3.6. Megfelelő szárítóanyagot tartalmazó, vastag, perforált fém- vagy porcelánlemezes szárítóberendezés.
4. A vizsgálat módja
Megjegyzés:
Az ebben a szakaszban leírt műveleteket a mintacsomagok kibontása után azonnal el kell végezni. Az elemzést legalább egyszer meg kell ismételni.
4.1. Előkészítés
4.1.1. A 4.1.2. és a 4.1.3. pontban nem szereplő takarmányok
Vegyünk legalább 50 g-ot a mintából. Szükség esetén aprítsuk fel vagy oszlassuk el úgy, hogy a nedvességtartalma egyenletes legyen (lásd 6. pont).
4.1.2. Gabona- és darafélék
Vegyünk legalább 50 g-ot a mintából. Őröljük le úgy, hogy a kapott részecskék legalább 50 %-a átjusson egy 0,5 mm-es szembőségű szitán, és ne maradjon vissza 10 %-nál több egy kerek szemű, 1 mm-es szembőségű szitán.
4.1.3. Folyékony vagy kenőcsös állagú takarmányok, túlnyomórészt olajokból és zsírokból álló takarmányok
Mérjünk ki 25 g mintát 10 mg pontossággal, adjunk hozzá megfelelő mennyiségű vízmentes homokot, 10 mg pontossággal kimérve, és keverjük addig, amíg homogén masszát nem kapunk.
4.2. Szárítás
4.2.1. A 4.2.2. és a 4.2.3. pont alatt nem szereplő takarmányok
Fedelével együtt mérjünk le egy tartályt (3.3.) 0,5 mg pontossággal. Mérjünk a lemért tartályba 5 g mintát 1 mg pontossággal, és terítsük el egyenletesen. Helyezzük a tartályt fedél nélkül a 103 °C-ra előmelegített kemencébe. Annak megakadályozása érdekében, hogy a kemence hőfoka a kívánatos érték alá süllyedjen, a tartályt a lehető leggyorsabban helyezzük a kemencébe. Hagyjuk száradni négy órán át attól az időponttól számítva, amikor a kemence hőfoka újból elérte a 103 °C-ot. Helyezzük vissza a tartályra a fedelet, a tartályt vegyük ki a kemencéből, hagyjuk hűlni 30-45 percig a szárítóberendezésben (3.6.), és mérjük le 1 mg pontossággal.
Túlnyomórészt olajokból és zsírokból álló takarmányok esetén szárítsuk további 30 percen át 130 °C-on a kemencében. A két mérés különbsége nem haladhatja meg a nedvességtartalom 0,1 %-át.
4.2.2. Gabonafélék, liszt, darafélék és derce
Fedelével együtt mérjünk le egy tartályt (3.3.) 0,5 mg pontossággal. Mérjünk a lemért tartályba 5 g aprított mintát 1 mg pontossággal, és terítsük el egyenletesen. Helyezzük a tartályt fedél nélkül a 130 °C-ra előmelegített kemencébe. Annak megakadályozása érdekében, hogy a kemence hőfoka a kívánatos érték alá süllyedjen, a tartályt a lehető leggyorsabban helyezzük a kemencébe. Hagyjuk száradni két órán át, attól az időponttól számítva, amikor a kemence hőfoka újból elérte a 130 °C-ot. Helyezzük vissza a tartályra a fedelét, a tartályt vegyük ki a kemencéből, hagyjuk hűlni 30-45 percig a szárítóberendezésben (3.6.), és mérjük le 1 mg pontossággal.
4.2.3. Több mint 4 % szacharózt vagy laktózt tartalmazó összetett takarmányok: egynemű takarmányok, mint a szentjánoskenyér, a hidrolizált gabonatermékek, a malátamagvak, a szárított répaszelet, a hal- és cukorlevek; a 25 %-nál több ásványi sót (beleértve a kristályvíztartalmat is) tartalmazó összetett takarmányok.
Fedéllel együtt mérjünk le egy tartályt (3.3.) 0,5 mg pontossággal. Mérjünk a lemért tartályba kb. 5 g mintát 1 mg pontossággal, és terítsük el egyenletesen. Helyezzük a tartályt fedél nélkül a vákuumkemencébe (3.5), amelyet 80-85 °C-ra előmelegítettünk. Annak megakadályozása érdekében, hogy a kemence hőfoka a kívánatos érték alá süllyedjen, a tartályt a lehető leggyorsabban helyezzük a kemencébe.
Emeljük a nyomást 100 torr-ra, és hagyjuk a mintát száradni ezen a nyomáson négy órán át száraz, forró levegő áramoltatásával vagy szárítóanyag (20 mintához kb. 300 g) felhasználásával. Utóbbi esetben a kívánt nyomás elérésekor kapcsoljuk le a vákuumszivattyút. A szárítás időtartamát attól az időponttól számítsuk, amikor a kemence hőfoka újra elérte a 80-85 °C-ot. Óvatosan szüntessük meg a kemence túlnyomását. Nyissuk ki a kemencét, azonnal helyezzük vissza a tartály fedelét, vegyük ki a tartályt a kemencéből, hagyjuk hűlni 30-45 percen át a szárítóberendezésben (3.6.), és mérjük le 1 mg-os pontossággal. Szárítsuk további 30 percen át a vákuumkemencében 80-85 °C-on, és mérjük le újra. A két mérés különbsége nem haladhatja meg a nedvességtartalom 0,1 %-át.
4.3. Előszárítás
4.3.1. A 4.3.2. pontban nem szereplő takarmányok
A magas nedvességtartalmú szilárd takarmányokat, amelyek nehezen apríthatók fel, előszárításnak kell alávetni az alábbiak szerint:
Mérjünk le 50 g-ot az aprítatlan mintából 10 mg pontossággal (a préselt vagy agglomerált mintákat kissé fel is lazíthatjuk, ha szükséges) egy megfelelő tartályba (pl. 20×12 cm-es, 0,5 cm-es peremmel ellátott alumíniumtálca). Hagyjuk száradni 60-70 °C-os kemencében, amíg a nedvességtartalom 8-12 % körüli értékre csökken. Vegyük ki a kemencéből, hagyjuk fedél nélkül hűlni a laboratóriumban egy órán át, és mérjük le 10 mg pontossággal. Ezután aprítsuk fel azonnal a 4.1.1. pontban felrtűntetett módon, és szárítsuk a 4.2.1. vagy a 4.2.3. pontban feltűntetett módon, a takarmány jellege szerint.
4.3.2. Gabonafélék
A 17 %-nál magasabb nedvességtartalmú gabonaféléket előszárításnak kell alávetni az alábbiak szerint:
Mérjünk le 50 g-ot az aprítatlan gabonából 10 mg pontossággal egy megfelelő tartályba (pl. 20×12 cm-es, 0,5 cm-es peremmel ellátott alumíniumtálca). Hagyjuk száradni 5-7 percen át 130 °C-os kemencében. Vegyük ki a kemencéből, hagyjuk hűlni fedél nélkül a laboratóriumban két órán át, és mérjük le 10 mg pontossággal. Őröljük meg azonnal a 4.1.2. pontban feltűntetett módon, és szárítsuk a 4.2.2. pontban feltűntetett módon.
5. Az eredmények kiszámítása
A minta százalékosan kifejezett nedvességtartalmát az alábbi képletek segítségével számítjuk ki:
5.1. Szárítás előszárítás nélkül
ahol:
E = a vizsgálandó minta eredeti tömege, grammban
m = a száraz vizsgálandó minta tömege, grammban.
5.2. Szárítás előszárítással
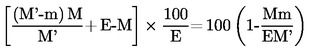
ahol:
E = a vizsgálati minta eredeti tömege, grammban
M = a vizsgálati minta tömege, grammban, előszárítás után
M′ = a vizsgálati minta tömege, grammban, aprítás vagy őrlés után
m = a száraz vizsgálati minta tömege, grammban.
5.3. Ismételhetőség
Az ugyanazon mintán elvégzett két párhuzamos meghatározás eredménye közötti különbség nem haladhatja meg a 0,2 % nedvességtartalmat.
6. Észrevétel
Ha az aprítás szükségesnek bizonyul, és úgy tűnik, hogy ez befolyásolja a termék nedvességtartalmát, a takarmány-alkotóelemek analízisének eredményét az eredeti minta nedvességtartalmának megfelelően korrigálni kell.
II. AZ ILLÓ NITROGÉNBÁZISOK MEGHATÁROZÁSA
A. MIKRODIFFÚZIÓVAL
1. Cél és alkalmazási terület
Ez a módszer lehetővé teszi a takarmányok ammóniában kifejezett illó nitrogénbázis-tartalmának meghatározását.
2. Vizsgálati alapelv
A mintát vízzel extraháljuk, majd az oldatot derítjük és szűrjük. Az illó nitrogénbázisokat mikrodiffúzióval, kálium-karbonát oldat használatával kiszorítjuk, bórsavas oldatban fogjuk fel és kénsavval titráljuk.
3. Reagensek
3.1. 20 %-os (w/v) triklór-ecetsav oldat.
3.2. Indikátor: oldjunk fel 33 mg brómkrezol-zöldet és 65 mg metilvöröst 100 ml 95-96 %-os (v/v) etanolban.
3.3. Bórsavas oldat: 1 literes mérőlombikban oldjunk fel 10 g a.r. bórsavat 200 ml, 95-96 %-os (v/v) etanolban és 700 ml vízben. Adjunk hozzá 10 ml indikátort (3.2.). Keverjük össze, és ha szükséges, állítsuk be az oldat színét világos vörösre nátrium-hidroxid oldat hozzáadásával. Ezen oldat 1 ml-je maximum 300 μg NH3-t fog megkötni.
3.4. Telített kálium-karbonát oldat: oldjunk fel 100 g a.r. kálium-karbonátot 100 ml forrásban lévő vízben. Hagyjuk lehűlni, majd szűrjük le.
3.5. 0,02 N kénsav.
4. Eszközök
4.1. Billenődobos keverőgép: kb. 35-40 fordulatszám/perc teljesítménnyel.
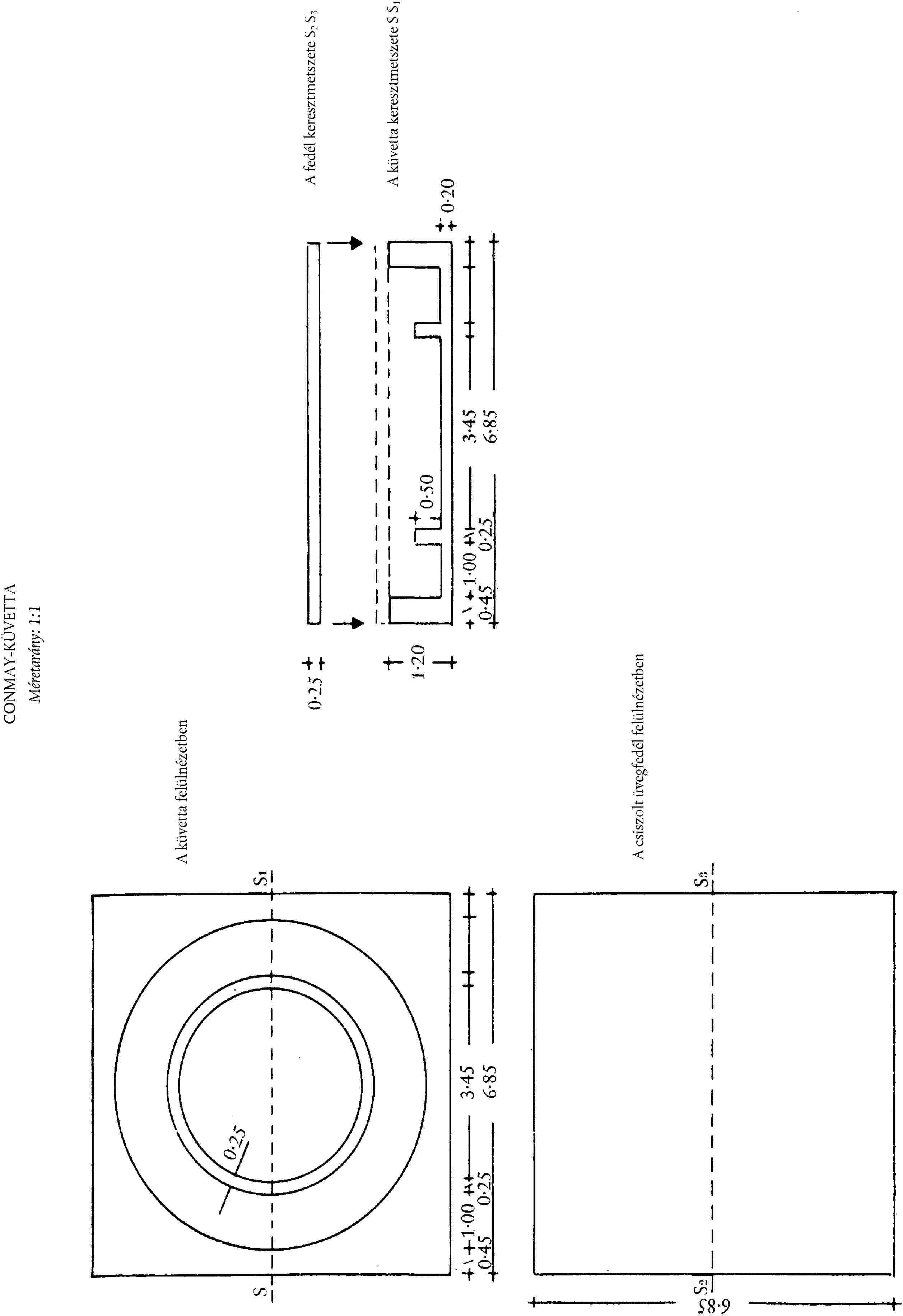
4.2. Üveg vagy műanyag Conway-küvetták (lásd az ábrát).
4.3. 1/100 ml-es fokbeosztásos mikrobüretták.
5. A vizsgálat módja
Mérjünk le 10 g mintát 1 mg pontossággal és tegyük egy 100 ml vizet tartalmazó 200 ml-es mérőlombikba. Keverjük a keverőgépben 30 percen át. Adjunk hozzá 50 ml triklór-ecetsavas oldatot (3.1.), töltsük fel térfogatra vízzel, rázzuk össze alaposan, majd szűrjük át hajtogatott szűrőn.
Pipettázzunk 1 ml bórsavas oldatot (3.3.) a Conway-küvetta középső részébe és 1 ml mintaszűrletet a küvetta koronarészébe. Fedjük le részlegesen a bezsírozott fedéllel. Tegyünk 1 ml telített kálium-karbonát oldatot (3.4.) gyorsan a küvetta koronarészébe, majd zárjuk le légmentesen a küvettát a fedéllel. Forgassuk el a küvettát óvatosan a vízszintes síkban úgy, hogy a két reagens összekeveredjen. Hagyjuk inkubálódni legalább négy órán át szobahőmérsékleten, vagy egy órán át 40 °C-on.
Mikrobüretta (4.3.) segítségével titráljuk az illó bázisokat a bórsavoldatban 0,02 N kénsavval (3.5.).
Végezzünk vakpróbát ugyanezzel az eljárással, de az analizálandó minta hozzáadása nélkül.
6. Az eredmények kiszámítása
1 ml 0,02 N H2SO4 0,34 mg ammóniának felel meg.
Az eredményt a mintára vonatkoztatva, százalékosan fejezzük ki.
Ismételhetőség
Az ugyanazon mintán végzett két párhuzamos meghatározás eredményének különbsége nem haladhatja meg:
relatív értékben a 10 %-ot az 1,0 %-nál alacsonyabb ammóniatartalom esetében;
abszolút értékben a 0,1 %-ot az 1,0 %-nál magasabb ammóniatartalom esetében.
7. Észrevétel
Ha a minta ammóniatartalma meghaladja a 0,6 %-ot, az eredeti szűrletet hígítani kell.
B. DESZTILLÁLÁSSAL
1. Cél és alkalmazási terület
Ez a módszer lehetővé teszi a karbamidot gyakorlatilag nem tartalmazó halliszt, ammóniában kifejezett illó nitrogénbázis-tartalmának meghatározását. Csak 0,25 %-nál alacsonyabb ammóniatartalom esetén alkalmazható.
2. Vizsgálati alapelv
A mintát vízzel extraháljuk, majd az oldatot derítjük és szűrjük. Az illó nitrogénbázist forrásponton szorítjuk ki magnézium-oxid hozzáadásával, és meghatározott mennyiségű kénsavban fogjuk fel, amelynek feleslegét nátrium-hidroxid oldattal titráljuk vissza.
3. Reagensek
3.1. 20 %-os (w/v) triklór-ecetsav oldat.
3.2. Magnézium-oxid, a.r.
3.3. Habzásgátló emulzió (pl. szilikon).
3.4. 0,1 N kénsav.
3.5. 0,1 N nátrium-hidroxid oldat.
3.6. 0,3 %-os (w/v) metilvörös oldat, 95-96 %-os (v/v) etanolban.
4. Eszközök
4.1. Billenődobos keverőgép: kb. 35-40 fordulatszám/perc teljesítménnyel.
4.2. Kjeldahl-féle lepárlókészülék.
5. A vizsgálat módja
Mérjünk le 10 g mintát 1 mg pontossággal és tegyük egy 100 ml vizet tartalmazó 200 ml-es mérőlombikba. Keverjük a keverőgépben 30 percen át. Adjunk hozzá 50 ml triklór-ecetsav oldatot (3.1.), töltsük fel térfogatra vízzel, rázzuk össze alaposan, majd szűrjük át hajtogatott szűrőn.
Vegyünk a tiszta szűrletből elegendő mennyiséget, hogy a feltételezett illó nitrogénbázis-tartalmat meg tudjuk határozni (100 ml mennyiség rendszerint megfelel a célnak). Hígítsuk fel 200 ml-re és adjunk hozzá 2 g magnézium-oxidot (3.2.), valamint néhány csepp habzásgátló emulziót (3.3.). Az oldatnak lakmuszpapírral mérve lúgos kémhatásúnak kell lennie; ha nem az, adjunk hozzá egy kevés magnézium-oxidot (3.2.). Pároljunk be kb. 150 ml-t az oldatból a Kjeldahl-készülékben, és fogjuk fel a párlatot egy pontosan kimért (25-50 ml) térfogatú, 0,1 N kénsavat (3.4.) tartalmazó Erlenmeyer-lombikban. Bepárlás közben kerüljük el az oldalak túlhevítését. Forraljuk a kénsavoldatot két percen át, hűtsük le és titráljuk vissza a fölös mennyiségű kénsavat 0,1 N nátrium-hidroxid oldattal (3.5.), metilvörös indikátor (3.6.) jelenlétében.
Végezzünk vakpróbát ugyanezzel az eljárással, de az analizálandó minta nélkül.
6. Az eredmények kiszámítása
1 ml 0,1 N H2SO4 1,7 mg ammóniánakfelel meg.
Az eredményt a mintára vonatkoztatva, százalékosan fejezzük ki.
Ismételhetőség
Az ugyanazon mintán végzett két párhuzamos meghatározás eredménye közötti különbség nem haladhatja meg relatív értékben az ammóniatartalom 10 %-át.
III. AZ ÖSSZES FOSZFOR MEGHATÁROZÁSA
Fotometriás eljárással
1. Cél és alkalmazási terület
Ez a módszer lehetővé teszi a takarmányok összes foszfortartalmának meghatározását. Különösen alkalmas az alacsony foszfortartalmú termékek elemzésére. Bizonyos esetekben (foszforban gazdag termékeknél), gravimetriás módszert használhatunk.
2. Vizsgálati alapelv
A mintát száraz égetéssel (szerves takarmányok esetén) vagy savas emésztéssel (ásványi vegyületek és folyékony takarmányok esetén) mineralizáljuk és savas oldatba helyezzük. Az oldatot molibdén-vanadát reagenssel kezeljük. Az így keletkező sárga oldat optikai sűrűségét 430 nm-nél mérjük egy spektrofotométeren.
3. Reagensek
3.1. Kalcium-karbonát, a.r.
3.2. Sósav, a.r., d: 1,1 (kb. 6 N).
3.3. Salétromsav, a.r., d: 1,045.
3.4. Salétromsav, a.r., d: 1,38-1,42.
3.5. Kénsav, a.r., d: 1,84.
3.6. Molibdén-vanadát reagens: 1 literes mérőlombikban keverjünk össze 200 ml ammónium-heptamolibdát oldatot (3.6.1.), 200 ml ammónium-monovanadát oldatot (3.6.2.) és 134 ml salétromsavat (3.4.). Töltsük fel vízzel térfogatra.
3.6.1. Ammónium-heptamolibdát oldat: oldjunk fel forró vízben 100 g ammónium-heptamolibdátot a.r. (NH4)6MO7O24.4H2O. Adjunk hozzá 10 ml ammóniát (d: 0,91) és töltsük fel vízzel 1 literre.
3.6.2. Ammónium-monovanadát oldat: 2,35 g ammónium-monovanadátot a.r. (NH4VO3) oldjunk fel 400 ml forró vízben. Állandó keverés közben, lassan adjunk hozzá 20 ml hígított salétromsavat (7 ml HNO3 (3.4.) + 13 ml H2O) és töltsük fel vízzel 1 literre.
3.7. Standard, 1 mg/ml foszfortartalmú oldat: oldjunk fel 4,387 g kálium-dihidrogén-foszfátot (KG2PO4, A.R.) vízben. Töltsük fel vízzel 1 literre.
4. Eszközök
4.1. Kvarc vagy porcelán hamvasztótégelyek.
4.2. Elektromos tokos kemence, 550 °C-ra beállított termosztáttal.
4.3. 250 ml-es Kjeldahl-lombik.
4.4. Mérőlombikok és precíziós pipetták.
4.5. Spektrofotométer.
4.6. Kb. 16 mm átmérőjű kémcsövek, 14,5 mm átmérőjűre keskenyedő dugóval; űrtartalom: 25-30 ml.
5. A vizsgálat módja
5.1. Oldatkészítés
A minta jellege szerint készítsünk oldatot az 5.1.1. vagy az 5.1.2. pontban leírtak szerint.
5.1.1. Általános módszer
Mérjünk le kb. 1 g-ot vagy valamivel többet a mintából, 1 mg pontossággal. Tegyük a mintát egy Kjeldahl-lombikba és adjunk hozzá 20 ml kénsavat (3.5.), rázzuk össze, hogy az anyag teljesen telítődjön a savval és ne tapadjon semmi a lombik oldalára, hevítsük és tartsuk forrásponton 10 percen át. Hagyjuk kissé lehűlni, adjunk hozzá 2 ml salétromsavat (3.4.), lassan hevítsük, majd hagyjuk újra kissé lehűlni, ezt követően adjunk hozzá kevés salétromsavat (3.4.) és forraljuk fel újra. Addíg ismételjük ezen műveletet, amíg színtelen oldatot nem kapunk. Hagyjuk hűlni, adjunk hozzá egy kevés vizet, majd dekantáljuk a folyadékot 500 ml-es mérőlombikba, és öblítsük ki a Kjeldahl-lombikot forró vízzel. Hagyjuk hűlni, töltsük fel vízzel térfogatra, homogenizáljuk és szűrjük.
5.1.2. Szerves anyagokat tartalmazó, kalcium- és magnézium-dihidrogén-foszfátoktól mentes minták
Mérjünk kb. 2,5 g-ot a mintából 1 mg pontossággal egy hamvasztótégelybe. Keverjük össze a vizsgálati mintát 1 g kalcium-karbonáttal (3.1.), amíg teljesen egyneművé nem válik. Hamvasszuk a kemencében 550 °C-on (± 5 °C), amíg fehér vagy szürke hamut nem kapunk (egy kis szénmaradvány nem számít). Tegyük a hamut 250 ml-es főzőpohárba. Adjunk hozzá 20 ml vizet és sósavat (3.2.), amíg a pezsgés meg nem szűnik. Adjunk hozzá még 10 ml sósavat (3.2.). Tegyük a főzőpoharat homokfürdőbe és hagyjuk teljesen elpárologni, hogy a kvarc szárazzá és oldhatatlanná váljon. Oldjuk fel a maradékot 10 ml salétromsavban (3.3.) és forraljuk a homokfürdőben 5 percen át anélkül, hogy teljesen beszárítanánk. Dekantáljuk a folyadékot egy 500 ml-es mérőlombikba, majd öblítsük ki a főzőpoharat néhányszor forró vízzel. Hagyjuk hűlni, töltsük fel vízzel térfogatra, homogenizáljuk és szűrjük.
5.2. Elszíneződés kialakulása és az optikai sűrűség mérése
Hígítsuk az 5.1.1. vagy az 5.1.2. pont szerint nyert szűrlet aliquot részét úgy, hogy 40 μg/ml-nél nem nagyobb foszforkoncentrációt kapjunk. Tegyünk 10 ml-t ebből az oldatból kémcsőbe (4.6.) és adjunk hozzá 10 ml molibdén-vanadát reagenst (3.6.). Homogenizáljuk és hagyjuk állni legalább 10 percen át 20 °C-on. Mérjük meg az optikai sűrűséget 430 nm-nél spektrofotométeren egy olyan oldattal szemben, amelyet 10 ml molibdén-vanadát reagens (3.6.) és 10 ml víz hozzáadásával nyertünk.
5.3. Kalibrációs görbe
A standard oldatból (3.7.) készítsünk egyenként 5, 10, 20, 30 és 40 μg/ml foszfort tartalmazó oldatokat. Vegyünk 10 ml-t ezekből az oldatokból, és adjunk hozzá mindegyikhez 10 ml molibdén-vanadát reagenst (3.6.). Homogenizáljuk és hagyjuk állni legalább 10 percen át 20 °C-on. Mérjük meg az optikai sűrűséget az 5.2. pontban leírt módon.
Szerkesszük meg a kalibrációs görbét úgy, hogy az optikai sűrűséget a megfelelő foszformennyiség függvényében ábrázoljuk. A 0 és 40 μg/ml közötti koncentrációk esetében a görbe lineáris lesz.
6. Az eredmények kiszámítása
A vizsgálati minta foszfortartalmát a kalibrációs görbe segítségével határozzuk meg.
Az eredményt a mintára vonatkoztatva, százalékosan fejezzük ki.
Ismételhetőség
Az ugyanazon mintán végzett két párhuzamos meghatározás eredménye közötti különbség nem haladhatja meg:
relatív értékben a 3 %-ot, 5 %-osnál alacsonyabb foszfortartalom esetén;
abszolút értékben a 0,15 %-ot, az 5 %-os vagy annál magasabb foszfortartalom esetén.
IV. A NYERSOLAJOK ÉS ZSÍROK MEGHATÁROZÁSA
1. Cél és alkalmazási terület
Ez a módszer az állati takarmányokban lévő nyersolajok és zsírok meghatározására szolgál. Nem vonatkozik az 1966. szeptember 22-i, 136/66/EGK tanácsi rendeletben meghatározott olajos magvakra és olajtartalmú gyümölcsökre.
A továbbiakban leírt két módszer használata a takarmány jellegétől és összetételétől, valamint az analitikai vizsgálat céljától függ.
1.1. A-eljárás - Közvetlenül extrahálható nyersolajok és zsírok
Ez a módszer a növényi eredetű takarmányok esetében alkalmazható, kivéve azokat, amelyek a B-módszer tárgykörébe esnek.
1.2. B-eljárás - Összes nyersolaj és zsír
Ez a módszer az állati eredetű takarmányok és minden összetett takarmány esetében alkalmazható. Mindazon anyagokra alkalmazandó, amelyekből az olajok és a zsírok teljesen nem extrahálhatók előzetes hidrolízis nélkül (pl. sikér, élesztő, burgonyafehérjék és extrudálás, a pelyhesítés és a melegítés révén készített termékek).
1.3. Az eredmények értelmezése
Mindazon esetekben, amikor magasabb eredményt kapunk a B-eljárással, mint az A-eljárással, a B-eljárással kapott eredményt tekintsük a helyes értéknek.
2. Vizsgálati alapelv
2.1. A-módszer
A mintát petroléterrel extraháljuk. Az oldószert bepároljuk, a maradékanyagot megszárítjuk, és megmérjük.
2.2. B-módszer
A mintát melegítés közben sósavval kezeljük. A keveréket lehűtjük, és leszűrjük. A maradékanyagot kimossuk, megszárítjuk és a meghatározást az A-módszer szerint végezzük el.
3. Reagensek
3.1. Petroléter, forráspont tartománya: 40-60 °C. Brómszáma kisebb legyen 1-nél, és a bepárlási maradékanyaga kisebb legyen, mint 2 mg/100 ml.
3.2. Nátrium-szulfát, vízmentes.
3.3. Sósav, k = 3 mol HCl/l.
3.4. Szűrési segédanyag, pl. Kieselguhr, Hyflo-supercel.
4. Eszközök
4.1. Extraháló készülék. Amennyiben szifonnal ellátott készülékről van szó (Soxhlet-készülék) a visszafolyás mértéke körülbelül óránként 10 ciklus legyen; ha nem szifonos típust használunk, a visszafolyás mértéke körülbelül percenként 10 ml legyen.
4.2. Extraháló tégely, petroléterben oldható anyagoktól mentes, a 4.1 pont követelményeinek megfelelő porozitással.
4.3. Szárítókemence, vagy 75 °C ± 3 °C-ra beállított vákuumkemence, vagy 100 °C ± 3 °C-ra beállított légkeveréses kemence.
5. A vizsgálat módja
5.1. A-módszer (lásd 8.1. pont)
1 mg pontossággal mérjünk ki 5 g mintát, töltsük át egy extraháló tégelybe (4.2.), és fedjük be egy zsírmentes üvegvatta-tamponnal.
Tegyük az extraháló tégelyt az extraháló készülékbe (4.1.), és extraháljuk hat órán keresztül petroléterrel (3.1.). A petroléter-kivonatot egy horzsakődarabokat ( 5 ) tartalmazó, száraz, lemért lombikba gyűjtsük.
Pároljuk be az oldószert. A lombikot másfél órán át tartsuk a szárítókemencében (4.3.), hogy a maradékanyag megszáradjon. Hagyjuk kihűlni egy szárítóberendezésben, és mérjük meg a súlyát. Ismét szárítsuk 30 percig annak biztosítása érdekében, hogy az olajok és zsírok tömege stabilizálódjon (a tömegveszteségnek a két egymást követő mérés között 1 mg-nál kisebbnek kell lennie).
5.2. B-módszer
1 mg pontossággal mérjünk ki 2,5 g mintát (lásd 8.2. pont), tegyük egy 400 ml-es főzőpohárba vagy egy 300 ml-es Erlenmeyer-lombikba, adjunk hozzá 100 ml sósavat (3.3.) és horzsakődarabokat. Fedjük le a főzőpoharat egy óraüveggel, vagy csatlakoztassunk az Erlenmeyer-lombikhoz egy visszafolyós hűtőt. Lassú lángon vagy főzőlapon enyhén forraljuk fel, és tartsuk így egy órán keresztül. Ne hagyjuk, hogy az anyag odatapadjon az edény falához.
Hűtsük le, és adjunk hozzá annyi szűrési segédanyagot (3.4.), amely elég ahhoz, hogy megakadályozza a szűrés folyamán keletkező olaj- vagy zsírveszteséget. Szűrjük át egy nedvesített, zsírmentes, kettős szűrőpapíron. Addig mossuk hideg vízzel a maradékanyagot, amíg semleges szűrletet nem kapunk. Ellenőrizzük, hogy a szűrlet nem tartalmaz olajat vagy zsírokat. Ezek jelenléte azt jelzi, hogy a mintát, a hidrolízis előtt petroléterrel extrahálni kell az A-módszer szerint.
Tegyük a maradékanyagot tartalmazó kettős szűrőpapírt egy óraüvegre, és szárítsuk a légkeveréses kemencében (4.3.) másfél órán keresztül, 100 °C ± 3 °C-on.
Tegyük a száraz maradékanyagot tartalmazó kettős szűrőpapírt egy extraháló tégelybe (4.2.), és fedjük le egy zsírmentes üvegvatta-tamponnal. Helyezzük az extraháló tégelyt egy extraháló készülékbe (4.1.), és folytassuk a műveletet az 5.1. pont második és harmadik bekezdése szerint.
6. Az eredmény kifejezése
A maradékanyag tömegét a mintára vonatkoztatva, százalékosan fejezzük ki.
7. Ismételhetőség
Ugyanazon a mintán, ugyanazon analitikus által végzett két párhuzamos meghatározás eredménye közötti különbség nem haladhatja meg a:
- 0,2 %-ot abszolút értékben, 5 %-nál alacsonyabb nyersolaj- és zsírtartalom esetében,
- 4,0 %-ot a legmagasabb eredményhez viszonyítva, 5-10 % közötti olaj- és zsírtartalom esetében,
- 0,4 %-ot abszolút értékben, 10 % feletti olaj- és zsírtartalom esetében.
8. Észrevételek
8.1. Magas olaj- és zsírtartalmú termékeknél, amelyeket nehéz felaprítani, vagy amelyek nem alkalmasak arra, hogy homogén vizsgálati mintát képezzünk belőlük, a következőképpen járjunk el:
1 mg pontossággal mérjünk ki 20 g mintát, és keverjük össze 10 g vagy annál több vízmentes nátriumszulfáttal (3.2.). Extraháljuk petroléterrel (3.1.) az 5.1. pont szerint. A kapott kivonatot töltsük fel petroléterrel (3.1.) 500 ml-re, és keverjük össze. Vegyünk ki az oldatból 50 ml-t, és tegyük egy horzsakődarabokat ( 6 ) tartalmazó kicsi, száraz, lemért lombikba. Pároljuk be az oldószert, szárítsuk ki, és az 5.1. pont utolsó bekezdésében leírt módon folytassuk a műveletet.
Távolítsuk el az oldószert az extraháló tégelyben hagyott maradékanyagból, aprítsuk fel a maradékanyagot 1 mm-es finomságúra, tegyük vissza az extraháló tégelybe (ne adjunk hozzá nátriumszulfátot), és az 5.1. pont második és harmadik bekezdésében leírtak szerint folytassuk a műveletet.
Az olaj- és zsírtartalmat a mintára vonatkoztatva, százalékosan számítsuk ki a következő képlet segítségével:
(10 a + b) x 5
ahol:
a = a maradékanyag tömege grammban az első extrahálás után (a kivonat aliquot része),
b = a maradékanyag tömege grammban a második extrahálás után.
8.2. Az olajokban és zsírokban szegény termékek esetében a vizsgálati mintát 5 g-ra lehet növelni.
8.3. A magas víztartalmú társállateledeleket vízmentes nátriumszulfáttal kell keverni a B-módszerben szerinti hidrolízis és extrahálás előtt.
8.4. Az 5.2. pontban hatásosabb lehet forró víz használata hideg víz helyett a maradékanyag szűrés utáni mosásához.
8.5. Az 1,5 órás szárítási időt ki kell terjeszteni néhány takarmány esetében. El kell kerülni a túlzott szárítást, mivel az alacsony eredményekhez vezethet. Mikrohullámú sütő is használható.
8.6. Az A-módszer szerinti előzetes extrahálás a hidrolízis előtt és a B-módszer szerinti ismételt extrahálás akkor javasolt, ha a nyersolaj- és zsírtartalom nagyobb, mint 15 %. Bizonyos mértékig ez a takarmány természetétől és a takarmányban lévő olaj és zsír jellegétől függ.
( 1 ) HL L 170., 1970.8.3., 2. o.
( 2 ) HL L 155., 1971.7.12., 13. o.
( 3 ) HL L 239., 1968.9.28., 2. o.
( 4 ) A gabonafélék, lisztek, darafélék és derce szárítására szolgáló kemence olyan hőkapacitású legyen, hogy 131 °C-ra beállítva kevesebb mint 45 perc alatt visszaálljon a beállított hőfokra azt követően, hogy a maximális számú vizsgálati mintát elhelyezték benne egyidejű szárítás céljából. A szellőztetése olyan legyen, hogy amikor a maximális befogadóképességnek megfelelő számú közönséges búzamintát elhelyezzük benne és két órán át szárítjuk, akkor az így kapott eredmény kevesebb mint 0,15 %-kal térjen csak el attól az eredménytől, amelyet négy órán át folytatott szárítással kapunk.
( 5 ) Amennyiben az olajon vagy a zsíron ezt követően minőségellenőrzést végeznek, a horzsakődarabokat üveggyöngyökkel helyettesítsük.
( 6 ) Hal az olaj vagy a zsír ezt követően minőségvizsgálaton megy keresztül, a horzsakődarabokat üveggyöngyökkel helyettesítsük.
Lábjegyzetek:
[1] A dokumentum eredetije megtekinthető CELEX: 31971L0393 - https://eur-lex.europa.eu/legal-content/HU/ALL/?uri=CELEX:31971L0393&locale=hu Utolsó elérhető, magyar nyelvű konszolidált változat CELEX: 01971L0393-19981009 - https://eur-lex.europa.eu/legal-content/HU/ALL/?uri=CELEX:01971L0393-19981009&locale=hu