
31982L0434[1]
A Bizottság második irányelve (1982. május 14.) a kozmetikai termékek összetételének ellenőrzéséhez szükséges vizsgálati módszerekre vonatkozó tagállami jogszabályok közelítéséről
A BIZOTTSÁG MÁSODIK IRÁNYELVE
(1982. május 14.)
a kozmetikai termékek összetételének ellenőrzéséhez szükséges vizsgálati módszerekre vonatkozó tagállami jogszabályok közelítéséről
(82/434/EGK)
AZ EURÓPAI KÖZÖSSÉGEK BIZOTTSÁGA,
tekintettel az Európai Gazdasági Közösséget létrehozó szerződésre,
tekintettel a 79/661/EGK irányelvvel ( 1 ) módosított, a kozmetikai termékekre vonatkozó tagállami jogszabályok közelítéséről szóló, 1976. július 27-i 76/768/EGK tanácsi irányelvre ( 2 ) és különösen annak 8. cikke (1) bekezdésére,
mivel a 76/768/EGK irányelv rendelkezik a kozmetikai termékek hivatalos vizsgálatáról azzal a céllal, hogy biztosítsa a kozmetikai termékek összetételére vonatkozó közösségi rendelkezések által előírt feltételek teljesülését;
mivel minden szükséges vizsgálati módszert a lehető leggyorsabban meg kell határozni; mivel e cél elérése érdekében az első lépés már megvalósult, bizonyos módszereknek a 80/1335/EGK ( 3 ) bizottsági irányelvben történt meghatározásával, a második lépés néhány oxidálószer azonosításának és a hidrogén-peroxid kozmetikai hajápolási termékekben történő mennyiségi meghatározásának, bizonyos oxidáló színezékek hajfestékekben történő azonosításának és félkvantitatív meghatározásának, a nitrit azonosításának és mennyiségi meghatározásának, a szabad formaldehid azonosításának és mennyiségi meghatározásának, a rezorcin samponokban és hajszeszekben történő mennyiségi meghatározásának és a metanol etanolra vagy propán-2-olra vonatkoztatott mennyiségi meghatározásának a módszer-meghatározások közé történő felvételéből áll;
mivel az ezen irányelv által megállapított rendelkezések összhangban vannak a 76/768/EGK irányelvnek a műszaki fejlődéshez való hozzáigazításával foglalkozó bizottság véleményével,
ELFOGADTA EZT AZ IRÁNYELVET:
1. cikk
A tagállamok megtesznek minden szükséges intézkedést, hogy a kozmetikai termékek hivatalos vizsgálata során:
- az oxidálószerek azonosítása és a hidrogén-peroxid hajápolási termékekben történő mennyiségi meghatározása,
- bizonyos oxidáló színezékek hajfestékekben történő azonosítása és félkvantitatív meghatározása,
- nitrit azonosítása és mennyiségi meghatározása,
- a szabad formaldehid azonosítása és mennyiségi meghatározása,
- a rezorcin samponokban és hajszeszekben történő mennyiségi meghatározása,
- a metanol mennyiségi meghatározása etanolra vagy propán-2-olra vonatkoztatva
a mellékletben leírt módszereknek megfelelően történjék.
2. cikk
A tagállamok hatályba léptetik azokat a törvényi, rendeleti és közigazgatási rendelkezéseket, amelyek szükségesek ahhoz, hogy ennek az irányelvnek legkésőbb 1983. december 31-ig megfeleljenek.
Erről haladéktalanul tájékoztatják a Bizottságot.
3. cikk
Ennek az irányelvnek a tagállamok a címzettjei.
MELLÉKLET
I. OXIDÁLÓSZEREK AZONOSÍTÁSA ÉS HIDROGÉN-PEROXID MEGHATÁROZÁSA HAJÁPOLÁSI TERMÉKEKBEN - CÉL ÉS ALKALMAZÁSI TERÜLET
A hidrogén-peroxid jodometriás meghatározása kozmetikumokban csak abban az esetben végezhető el, ha azok nem tartalmaznak a jodidokat jóddá redukáló más oxidálószereket. A hidrogén-peroxid jodometriás mennyiségi meghatározását ezért meg kell előznie a mintában előforduló egyéb oxidálószerek kimutatásának és azonosításának. Az azonosítás két szakaszból áll: az első a perszulfátokra, a bromátokra és a hidrogén-peroxidra, a második pedig a bárium-peroxidra vonatkozik.
A. PERSZULFÁTOK, BROMÁTOK ÉS HIDROGÉN-PEROXID AZONOSÍTÁSA
1. ALAPELV
A nátrium-perszulfátot, kálium-perszulfátot és ammónium-perszulfátot, valamint a kálium-bromátot, nátrium-bromátot és a hidrogén-peroxidot - függetlenül attól, hogy utóbbi bárium-peroxidból származik vagy sem - leszálló papírkromatográfiával azonosítjuk, amelynek során két előhívó oldószert használunk.
2. REAGENSEK
Minden reagensnek analitikai tisztaságúnak kell lennie.
2.1. A következő vegyületek 0,5 %-os (m/V) vizes referenciaoldata:
2.1.1. Nátrium-perszulfát
2.1.2. Kálium-perszulfát
2.1.3. Ammónium-perszulfát
2.1.4. Kálium-bromát
2.1.5. Nátrium-bromát
2.1.6. Hidrogén-peroxid
2.2. Előhívó oldószer A, 80 %-os (v/v) etanol
2.3. Előhívó oldószer B, benzol - metanol - 3-metil-bután-1-ol - víz (34:38:18:10 térfogatarányban)
2.4. Detektor A, kálium-jodid 10 %-os (m/V) vizes oldata
2.5. Detektor B, keményítő 1 %-os (m/V) vizes oldata
2.6. Detektor C, 10 %-os sósav (m/m)
2.7. 4N sósav
3. ESZKÖZÖK
3.1. Kromatográfiás papír (Whatman-papír 3. és 4., vagy ezekkel egyenértékű)
3.2. 1 μl-es mikropipetta
3.3. 100 ml-es mérőlombik
3.4. Redős szűrő
3.5. Leszálló papírkromatográfia eszközei
4. MINTAELŐKÉSZÍTÉS
4.1. Vízben oldódó termékek
Minden mintából két oldatot készítsünk 1, illetve 5 g termék 100 ml vízben történő feloldásával. Az 5. szakaszban leírt papír-kromatográfia végrehajtásához az oldatok mindegyikéből 1 μl-t használjunk.
4.2. Vízben korlátozottan oldódó termékek
4.2.1. Mérjünk be külön-külön 1 g és 5 g terméket, szuszpendáljuk 50 ml vízben, egészítsük ki 100 ml-re vízzel mindkettőt, és keverjük össze a mintákat. Szűrjük le a szuszpenziókat redős szűrőn (3.4.), és az 5. szakaszban leírt papír-kromatográfia végrehajtásához a szűrletek mindegyikéből 1 μl-t használjunk.
4.2.2. Szuszpendáljuk újra az 1 g és 5 g terméket 50 ml vízben, savanyítsuk meg híg sósavval (2.7.), egészítsük ki vízzel 100 ml-re, és keverjük össze. Szűrjük le a szuszpenziókat redős szűrőn (3.4.), és az 5. pontban leírt papír-kromatográfia végrehajtásához a szűrletek mindegyikéből 1 μl-t használjunk.
4.3. Krémek
Szuszpendáljunk termékenként 5 g-ot és 20 g-ot 100 ml vízben, és ezeket a szuszpenziókat használjuk az 5. szakaszban leírt papír-kromatográfia végrehajtásához.
5. ELJÁRÁS
5.1. A leszálló papírkromatográfia végrehajtásához tegyünk megfelelő mennyiségű A (2.2.) és B (2.3.) oldószert egy-egy kromatográfiás kádba. Legalább 24 órán át telítsük a kromatográfiás kádakat oldószergőzökkel.
5.2. Vigyünk fel 1-1 μl-t a 4. és 2.1. pontnak megfelelően előkészített minta- és referenciaoldatokból egy 40 cm hosszúságú és 20 cm szélességű vagy más megfelelő méretű kromatográfiás papírcsík (3.1.) (Whatman-3 vagy ezzel egyenértékű) kiindulási pontjaira, majd párologtassuk el az oldószert levegőn.
5.3. Helyezzük a kromatográfiás (5.2.) papírcsíkot az A előhívó oldószert (5.1.) tartalmazó kromatográfiás kádba, és addig fejlesszük, amíg az oldószerfront az alapvonaltól 35 cm-re távolodik (körülbelül 15 óra).
5.4. Ismételjük az 5.2. és 5.3. pontban leírt eljárást (Whatman-4 vagy ezzel egyenértékű) kromatográfiás papírral (3.1.) a B előhívó oldószerben. Kromatografáljuk, amíg az oldószerfront 35 cm-re távolodik az alapvonaltól (körülbelül 5 óra).
5.5. A előhívás után vegyük ki a kádból a kromatográf-papírcsíkokat, és szárítsuk meg levegőn.
5.6. A foltok előhívásához fújjuk le a kromatogramot sorrendben:
5.6.1. az A detektorral (2.4.), majd rövid idő múlva a B (2.5.) detektorral. Először a perszulfátok foltjai jelennek meg a kromatogramon, amelyeket a hidrogén-peroxid-foltok követnek. Jelöljük meg a foltok helyét ceruzával;
5.6.2. az 5.6.1. pont szerint kapott kromatogramokat a C detektorral (2.6.); a bromátok szürkéskék folttal jelennek meg a kromatogramon.
5.7. Az A (2.2.) és B (2.3.) előhívó oldószerekre vonatkozó, fent említett körülmények között az referenciaanyagok (2.1.) Rf-értékei hozzávetőlegesen a következők:
| előhívó oldószer A (2.2.) | előhívó oldószer B (2.3.) | |
| Nátrium-perszulfát | 0,40 | 0,10 |
| Kálium-perszulfát | 0,40 | 0,02 + 0,05 |
| Ammónium-perszulfát | 0,50 | 0,10 + 0,20 |
| Nátrium-bromát | 0,40 | 0,20 |
| Kálium-bromát | 0,40 | 0,10 + 0,20 |
| Hidrogén-peroxid | 0,80 | 0,80 |
B. BÁRIUM-PEROXID AZONOSÍTÁSA
1. ALAPELV
A bárium-peroxidot az (A.4.2.) minta savanyítása után keletkező hidrogén-peroxid segítségével és a báriumion jelenlétének kimutatásával azonosítjuk:
- ha (A) perszulfátok nincsenek jelen, híg kénsavnak a savas mintaoldat (B.4.1.) egy részéhez történő hozzáadása esetén, aminek eredményeként fehér bárium-szulfát-csapadék képződik. A báriumion jelenlétét a (B.4.1.) mintában, ebben az esetben is papírkromatográfiával igazoljuk az alábbi, 5. pontban leírt módon,
- ha a mintában egyidejűleg található bárium-peroxid és (B.4.2.) perszulfátok, a (B.4.2.) oldhatatlan maradék lúgos feltárásával és sósavban történő oldás után a bárium-ionok jelenlétét a (B.4.2.3.) olvadék oldatában papír kromatográfiával és/vagy a bárium-szulfát lecsapatásával igazoljuk.
2. REAGENSEK
2.1. Metanol
2.2. 36 %-os (m/m) tömény sósav
2.3. 6N sósav
2.4. 4N kénsav
2.5. Rodizonsav-dinátrium só
2.6. Bárium-klorid (BaCl2 ·2H2O)
2.7. Vízmentes nátrium-karbonát
2.8. Bárium-klorid 1 %-os (m/V) vizes oldata
2.9. Előhívó oldószer, amely metanolt, tömény sósavat (koncentráció 36 %) és vizet tartalmaz (80:10:10 térfogatarányban)
2.10. Detektor, rodizonsav dinátriumsójának 0,1 %-os (m/V) vizes oldata, amelyet felhasználás előtt frissen kell készíteni.
3. ESZKÖZÖK
3.1. 5 μl-es mikropipetta
3.2. Platinatégely
3.3. 100 ml-es mérőlombik
3.4. Schleicher és Schull 2043 b vagy ezzel egyenértékű kromatográfiás papír. Helyezzük a papírt egy éjszakán keresztül a (B.2.9.) előhívó oldószert tartalmazó (A.3.5.) leszálló kromatográfiás kádba, majd szárítsuk meg.
3.5. Redős szűrő
3.6. Felszálló papírkromatográfia szokásos eszközei
4. MINTA-ELŐKÉSZÍTÉS
4.1. Perszulfátokat nem tartalmazó termékek
4.1.1. Szuszpendáljunk 2 g terméket 50 ml vízben, és sósavval (B.2.3.) állítsuk be a pH-ját 1 körüli értékre.
4.1.2. Mossuk át a szuszpenziót vízzel egy 100 ml-es mérőlombikba, töltsük fel a jelig, és keverjük össze. Ezt a szuszpenziót használjuk az 5. pontban leírt papírkromatográfiás vizsgálat és a bárium-szulfát-csapadék kicsapatásán alapuló azonosítás során.
4.2. Perszulfátokat tartalmazó termékek
4.2.1. Szuszpendáljunk 2 g terméket 100 ml vízben, és szűrjük le.
4.2.2. Adjunk a szárított maradékhoz tömege hét-tízszeresének megfelelő mennyiségű nátrium-karbonátot (B.2.7.), keverjük össze, és olvasszuk a keveréket egy platinatégelyben (B.3.2.) fél órán keresztül.
4.2.3. Hűtsük le az olvadékot szoba-hőmérsékletűre, oldjuk fel 50 ml vízben, és szűrjük le (B.3.5.).
4.2.4. Oldjuk fel az olvadékból származó maradékot sósavban (B.2.3.), töltsük föl 100 ml-re vízzel. Ezt a szuszpenziót használjuk az 5. pontban leírt papírkromatográfiás vizsgálat és a bárium-szulfát-csapadék lecsapatásán alapuló azonosítás során.
5. ELJÁRÁS
5.1. Tegyünk megfelelő mennyiségű előhívó oldószert (B.2.9.) egy felszálló papírkromatográfiás kádba, és telítsük a kádat legalább 15 órán keresztül.
5.2. A B.3.4. pontban leírt módon előkészített kromatográfiás papírra három kiindulási pontban vigyünk fel 5-5 μl-t a B.4.1.2. és a B.4.2.4. pontnak megfelelően előkészített oldatokból és a B.2.8. pont szerinti referenciaoldatból.
5.3. Szárítsuk meg a minta- és referenciafoltokat levegőn. A kromatogram előhívását addig folytassuk, amíg az oldószerfront a függőleges irányban 30 cm magasságig emelkedik.
5.4. Vegyük ki a kromatogramokat a kádból, és szárítsuk meg levegőn.
5.5. A foltok előhívása céljából fújjuk le a papírt a B.2.10. előhívó szerrel. Bárium jelenlétében, körülbelül 0,10 Rf-értéknél a kromatogramon piros foltok jelennek meg.
C. HIDROGÉN-PEROXID MEGHATÁROZÁSA
1. ALAPELV
A hidrogén-peroxid jodometriás meghatározása a következő reakción alapszik:
Az átalakulás lassú folyamat, de ammónium-molibdát hozzáadásával gyorsítható. A képződő jód nátrium-tioszulfátos titrálással meghatározható, és lehetővé teszi a hidrogén-peroxid-tartalom meghatározását.
2. FOGALOMMEGHATÁROZÁS
Az alábbi módon mért hidrogén-peroxid-tartalmat a termék tömegére vonatkoztatva tömegszázalékban (% m/m) fejezzük ki.
3. REAGENSEK
Minden reagensnek analitikai tisztaságúnak kell lennie.
3.1. 2N kénsav
3.2. Kálium-jodid
3.3. Ammónium-molibdát
3.4. 0,1 N nátrium-tioszulfát
3.5. 10 %-os (m/V) kálium-jodid-oldat, közvetlenül felhasználás előtt kell készíteni
3.6. 20 %-os (m/V) ammónium-molibdát-oldat
3.7. 1 %-os (m/V) keményítőoldat
4. ESZKÖZÖK ÉS FELSZERELÉSEK
4.1. 100 ml-es főzőpohár
4.2. 50 ml-es büretta
4.3. 250 ml-es mérőlombik
4.4. 25 ml-es és 100 ml-es mérőhenger
4.5. 10 ml-es egyjelű pipetta
4.6. 250 ml-es Erlenmeyer-lombik
5. ELJÁRÁS
5.1. Mérjünk be 10 g (m), körülbelül 0,6 g hidrogén-peroxidot tartalmazó terméket egy 100 ml-es főzőpohárba. Egy kis vízzel mossuk a főzőpohár tartalmát egy 250 ml-es mérőlombikba, töltsük fel a jelig vízzel, és keverjük össze.
5.2. Pipettázzuk az (5.1.) mintaoldat 10 ml-ét egy 250 ml-es mérőlombikba (4.6.), és adjunk hozzá 100 ml 2 N kénsavat (3.1.), 20 ml kálium-jodid (3.5.) -oldatot és három csepp ammónium-molibdát (3.6.) -oldatot.
5.3. Titráljuk azonnal a keletkező jódot (3.4.) 0,1 N nátrium-tioszulfát-oldattal, közvetlenül a végpont előtt indikátorként adjunk hozzá néhány csepp keményítő (3.7.) -oldatot. Jegyezzük fel a 0,1 N nátrium-tioszulfát (3.4.) fogyását milliliterben (V).
5.4. Az 5.2. és 5.3. szakaszokban leírt módon végezzünk vakpróbát, a 10 ml mintaoldat helyett használjunk 10 ml vizet. Jegyezzük fel a 0,1 N nátrium-tioszulfát fogyását a vak meghatározásban (Vo ml).
6. SZÁMÍTÁS
Számítsuk ki a hidrogén-peroxid-tartalmat tömegszázalékban (% m/m) a következő képlet segítségével:
| % hidrogén-peroxid | = V − Vo × 1,7008 × 250 × 100m × 10 × 1 000 |
| = V − Vo × 4,252m, |
ahol:
m = a vizsgált termék mennyisége (5.1.),
Vo = a 0,1 N tioszulfát fogyása a vakpróbában (5.4.) milliliterben,
V = a 0,1 N tioszulfát fogyása a mintaoldat titrálása (5.3.) során, milliliterben.
7. MEGISMÉTELHETŐSÉG ( 4 )
A termék 6 % (m/m) körüli hidrogén-peroxid-tartalma esetén azonos mintán párhuzamosan végzett két mennyiségi meghatározás eredményei közötti különbség abszolút értékben nem haladhatja meg a 0,2 %-ot.
II. HAJFESTÉKEKBEN ELŐFORULÓ BIZONYOS OXIDÁLÓ SZÍNEZÉKEK AZONOSÍTÁSA ÉS FÉLKVANTITATÍV MEGHATÁROZÁSA
1. CÉL ÉS ALKALMAZÁSI TERÜLET
Ez a módszer alkalmas a következő anyagok azonosítására és félkvantitatív meghatározására krém vagy folyadék típusú hajfestékekben:
| Anyag megnevezése | Rövidítés |
| Fenilén-diaminok | |
| o-fenilén-diamin | (OPD) |
| m-fenilén-diamin | (MPD) |
| p-fenilén-diamin (V. melléklet) | (PPD) |
| Metil-fenilén-diaminok | |
| 4-metil-1,2-fenilén-diamin (toluol-3,4-diamin) | (OTD) |
| 4-metil-1,3-fenilén-diamin (toluol-2,4-diamin) | (MTD) |
| 2-metil-1,4-fenilén-diamin (toluol-2,5-diamin) | (PTD) |
| Diamino-fenolok | |
| 2,4-diamino-fenol | (DAP) |
| Hidrokinon | |
| 1,4-benzéndiol | (H) |
| α-naftol | (α-N) |
| Pirogallol | |
| 1,2,3-trihidroxi-benzol | (P) |
| Rezorcin | |
| 1,3-dihidroxi-benzol | (R) |
2. ALAPELV
Az oxidáló színezékeket a krém vagy folyadék típusú hajfestékekből pH 10-en 96 %-os etanollal kivonjuk, és egy- vagy kétdimenziós vékonyréteg-kromatográfiával azonosítjuk.
Az anyagok félkvantitatív meghatározása úgy történik, hogy a minták négy különböző előhívó rendszerben kapott kromatogramját összehasonlítjuk a hasonló körülmények között, velük egyidejűleg készített referenciaanyagok kromatogramjával.
3. REAGENSEK
Minden reagensnek analitikai tisztaságúnak kell lennie.
3.1. Vízmentes etanol
3.2. Aceton
3.3. 96 %-os etanol, v/v
3.4. 25 %-os ammóniaoldat ( [Kép #1] )
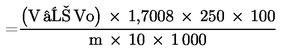
Kép #1
3.5. L(+)-aszkorbinsav
3.6. Kloroform
3.7. Ciklohexán
3.8. Technikai minőségű nitrogén
3.9. Toluol
3.10. Benzol
3.11. n-butanol
3.12. Bután-2-ol
3.13. 50 %-os (v/v) hipofoszforos savoldat
3.14. Diazo reagens. Vagy:
- 3-nitro-1-benzodiazónium-klórbenzol-szulfonát, (stabilizált só forma), mint a Red 2 JN - Francolor, vagy azzal egyenértékű,
- 2-klór-4-nitro-1-benzodiazónium-naftalin-benzoát, (stabilizált só forma), mint az NNCD reagensben - hivatkozási szám 74 150 FLUKA,
vagy azzal egyenértékű.
3.15. Ezüst-nitrát
3.16. p-dimetil-amino-benzaldehid
3.17. 2,5-dimetil-fenol
3.18. Vas-klorid-hexahidrát
3.19. 10 %-os (m/v) sósavoldat
3.20. Referenciaanyagok A referenciaanyagok felsorolását az I. cím alatt a "Cél és alkalmazási terület" tartalmazza. Amin-vegyületek esetében a referenciaanyag kizárólag hidroklorid forma (mono- vagy di-) vagy a szabad bázis.
3.21. 0,5 %-os (m/V) referenciaoldatok
Készítsük el a 3.20. pontban hivatkozott referenciaanyagok 0,5 %-os (m/v) oldatát.
Mérjünk be 50 mg ± 1 mg referenciaanyagot egy 10 ml-es mérőlombikba.
Adjunk hozzá 5 ml 96 %-os etanolt (3.3.) és 250 mg aszkorbinsavat (3.5.).
Lúgosítsuk az oldatot ammóniaoldat (3.4.) hozzáadásával, hogy a pH 10 körüli értéken legyen (ellenőrizzük indikátorpapírral).
Töltsük 10 ml-ig a lombikot 96 %-os (3.3.) etanollal, és keverjük össze.
Az oldatok fénytől védve hűvös helyen egy hétig eltarthatók.
Előfordulhat, hogy az aszkorbinsav és az ammónia hozzáadása után csapadék képződik. Ilyenkor hagyjuk kiülepedni a csapadékot, és csak ezután folytassuk az eljárást.
3.22. Előhívó oldószerek
3.22.1. Aceton - kloroform - toluol (35:25:40 térfogatarányban)
3.22.2. Kloroform - ciklohexán-abszolút etanol - 25 %-os ammónia (80:10:10:1 térfogatarányban)
3.22.3. Benzol - bután-2-ol - víz (50:25:25 térfogatarányban). Rázzuk össze erőteljesen a keveréket, és szobahőmérsékleten (20-25 °C) történő elválasztás után a felső fázist használjuk.
3.22.4. n-butanol - kloroform - M reagens (7:70:23 térfogatarányban). Óvatosan válasszuk el szobahőmérsékleten (20-25 °C), és használjuk az alsó fázist.
| Az M reagens készítése | |
| 25 %-os (v/v) ammóniaoldat, | 24 térfogat |
| 50 %-os hipofoszforos savoldat (3.13.) | 1 térfogat |
| Víz | 75 térfogat |
Megjegyzés
Az ammóniát tartalmazó előhívó oldószereket közvetlenül használat előtt alaposan fel kell rázni.
3.23. Indikátor spray-k
3.23.1. Diazo reagens
Készítsük el a kiválasztott reagens (3.14.) 5 %-os (m/v) vizes oldatát. Ezt az oldatot közvetlenül használat előtt kell készíteni.
3.23.2. Ehrlich-reagens
Oldjunk fel 2 g (3.16.) p-dimetilamino-benzaldehidet 100 ml (3.19.) sósav 10 %-os (m/v) vizes oldatában.
3.23.3. 2,5-dimetil-fenol - vas-klorid-hexahidrát
1. oldat: Oldjunk fel 1 g dimetil-fenolt (3.17.) 100 ml 96 %-os etanolban (3.3.).
2. oldat: Oldjunk fel 4 g vas-klorid-hexahidrátot (3.18.) 100 ml 96 %-os etanolban (3.3.).
Az előhíváskor ezeket az oldatokat külön kell alkalmazni, előbb az 1. oldatot, majd a 2.-at.
3.23.4. Ammóniás ezüst-nitrát
Adjunk annyi 25 %-os ammóniát (3.4.) ezüst-nitrát (3.15.) 5 %-os (m/V) vizes oldatához, hogy a csapadék éppen föloldódjon. A reagenst közvetlenül felhasználás előtt kell készíteni.
Nem tárolható.
4. ESZKÖZÖK
4.1. A vékonyréteg-kromatográfia szokásos laboratóriumi eszközei.
4.1.1. Műanyag vagy üvegfedél, amelynek kialakítása olyan, hogy a foltok felvitele és szárítása alatt a kromatográfiás lap környezetében nitrogénatmoszférát lehet létrehozni. Az óvintézkedésre bizonyos színezékek erős oxidációs hajlama miatt van szükség.
4.1.2. 10 μl-es, 0,2 μl-es beosztású mikrofecskendő négyzetes tűvel, vagy még jobb egy befogóállványon rögzített, 50 μl-es ismétlő adagoló, amely úgy van felszerelve, hogy a lapot nitrogén alatt lehessen tartani.
4.1.3. 0,25 mm vastag, 20x20 cm-es, azonnal használható, szilikagél vékonyréteg-lapok (műanyag hordozós Macherey and Nagel, Silica G-HR, vagy ezekkel egyenértékű).
4.2. 4 000 ford/perc fordulatszámú centrifuga.
4.3. 10 ml-es, PTFE bevonatú menetes kupakkal rendelkező centrifugacsövek vagy ezekkel egyenértékű csövek.
5. ELJÁRÁS
5.1. A vizsgálati minták kezelése
Ne használjuk a tubusból kinyomott krém első két-három cm-ét.
Mérjük be a következőket egy előzőleg nitrogénnel átöblített centrifugacsőbe (4.3.): 300 mg aszkorbinsavat és 3 g krémet vagy 3 g homogenizált folyadékot.
Csepegtessünk 25 %-os ammóniát (3.4.) az anyaghoz, amíg a pH a 10-et el nem éri. Töltsük fel 10 ml-re 96 %-os etanollal (3.3.).
Homogenizáljuk nitrogén (3.8.) alatt, zárjuk le, majd centrifugáljuk 4 000/perc fordulatszámon 10 percig.
Használjuk a felülúszó folyadékot.
5.2. Kromatográfia
5.2.1. A minta felvitele a lapokra
Vigyünk fel a kromatográfiás lapra a fent említett referenciaoldatokból 1-1 μl-t nitrogénatmoszféra alatt (3.8.) egy egyenes mentén, 9 pontban.
A referenciaoldatok foltjainak sorrendje a következő:
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
| R | P | H | PPD | DAP | PTD | OPD | OTD | MPD |
| MTD | α-N |
A 10-es és 11-es pontban pedig 2 μl-t cseppentsünk az 5.1. pontban kapott mintaoldatokból.
A lapot mindaddig tartsuk nitrogénatmoszféra (3.8.) alatt, amíg a kromatográfiát el nem indítjuk.
5.2.2. Előhívás
Helyezzük a lapot előzetesen nitrogénnel (3.8.) átöblített, a négy (3.22.) oldószer egyikének gőzeivel telített kádba, hívjuk elő szobahőmérsékleten (20-25 °C), fénytől védve, amíg az oldószerfront az alapvonaltól 15 cm-re távolodik.
Vegyük ki a lapot a kádból, és szárítsuk nitrogén (3.8.) alatt szobahőmérsékleten.
5.2.3. Előhívás
Azonnal fújjuk le a lapot a 3.23. pontban leírt négy előhívó egyikével.
5.2.4. Azonosítás
Hasonlítsuk össze a minta Rf-értékét és színét a párhuzamosan kromatografált referenciaanyagok hasonló jellemzőivel.
Az 1. táblázat példaként megadja valamennyi anyag Rf-értékét és színét az összes oldószerre és indikátorra.
Ha az azonosítás eredménye nem egyértelmű, sikerre vezethet a ráméréses módszer, amikor a vizsgálati mintához hozzáadjuk a megfelelő referenciaanyagot.
5.2.5. Félkvantitatív mérési módszer
Szemrevételezéssel hasonlítsuk össze az 5.2.4. pontban azonosított minden egyes anyag foltjának intenzitását és a referenciaoldatokkal a megfelelő koncentráció-tartományban felvett kromatogramok foltjainak intenzitását.
Ha a mintában előforduló egy vagy több anyag koncentrációja túlságosan nagynak mutatkozik, hígítsuk a mintakivonatot, és ismételjük meg a mérést.
I. TÁBLÁZAT
A foltok Rf értéke és színe közvetlenül a permetezés után
| (3.20) Referenciaanyag (3.20.) | Előhívó oldószerek | Indikátor permetezők | ||||||
| Rf-értékek | Foltok színe | |||||||
| (3.22.1.) | (3.22.2.) | (3.22.3.) | (3.22.4.) | Diazo (3.23.1.) | Ehrlich (3.23.2.) | Dimetil-fenol (3.23.3.) | AgNO3 (3.23.4.) | |
| OPD | 0,62 | 0,60 | 0,30 | 0,57 | világosbarna | – | – | világosbarna |
| MPD | 0,40 | 0,60 | 0,47 | 0,48 | ibolyabarna (*) | sárga | világosbarna | világosbarna |
| PPD | 0,20 | 0,50 | 0,30 | 0,48 | barna | élénkpiros (*) | ibolya | szürke |
| OTD | 0,60 | 0,60 | 0,53 | 0,60 | barna (*) | halvány narancs | világosbarna | szürkésbarna |
| MTD | 0,40 | 0,67 | 0,45 | 0,60 | vörösesbarna (*) | sárga | barna | fekete |
| PTD | 0,33 | 0,65 | 0,37 | 0,70 | barna | narancs | ibolya (*) | szürke |
| DAP | 0,07 | – | 0 | 0,05 | barna (*) | narancs | ibolya | barna |
| H | 0,50 | 0,35 | 0,80 | 0,20 | – | narancs | ibolya | fekete (*) |
| α-N | 0,90 | 0,80 | 0,90 | 0,75 | narancsbarna | – | ibolya (*) | fekete |
| P | 0,37 | – | 0,67 | 0,05 | barna | nagyon halvány ibolya | nagyon halvány barna | barna (*) |
| R | 0,50 | 0,37 | 0,80 | 0,17 | narancs (*) | halványibolya | nagyon halvány barna | világosbarna |
| Megjegyzések: 1. Az OPD csak gyengén látszik; a (3.22.3.) oldószereleggyel kell egyértelműen elválasztani az OTD-től. 2. (*) A legjobban előhívható színt jelzi. | ||||||||
6. VIZSGÁLAT KÉTDIMENZIÓS VÉKONYRÉTEG-KROMATOGRÁFIÁVAL
A kétdimenziós kromatográfiás eljárás végrehajtásához további standardok és reagensek használata szükséges.
6.1. Kiegészítő referenciaoldatok és -anyagok
6.1.1. β-naftol (β-N)
6.1.2. 2-amino-fenol (OAP)
6.1.3. 3-amino-fenol (MAP)
6.1.4. 4-amino-fenol (PAP)
6.1.5. 2-nitro-1,4-fenilén-diamin (2-NPPD)
6.1.6. 4-nitro-1,2-fenilén-diamin (4-NOPD)
Készítsük el a kiegészítő referenciaanyagok 0,5 %-os (m/V) oldatát a 3.21. pontban leírt módon.
6.2. Előhívó oldószer
6.2.1. Etil-acetát - ciklohexán - 25 %-os ammónia-oldat, (65:30:0,5 térfogatarányban)
6.3. Jelzőrendszer
Helyezzünk egy üvegedényt egy vékonyréteg-kromatográfiás előhívó kádba, mérjünk be körülbelül 2 g kristályos jódot, és zárjuk le a kádat egy fedéllel.
6.4. Kromatográfia
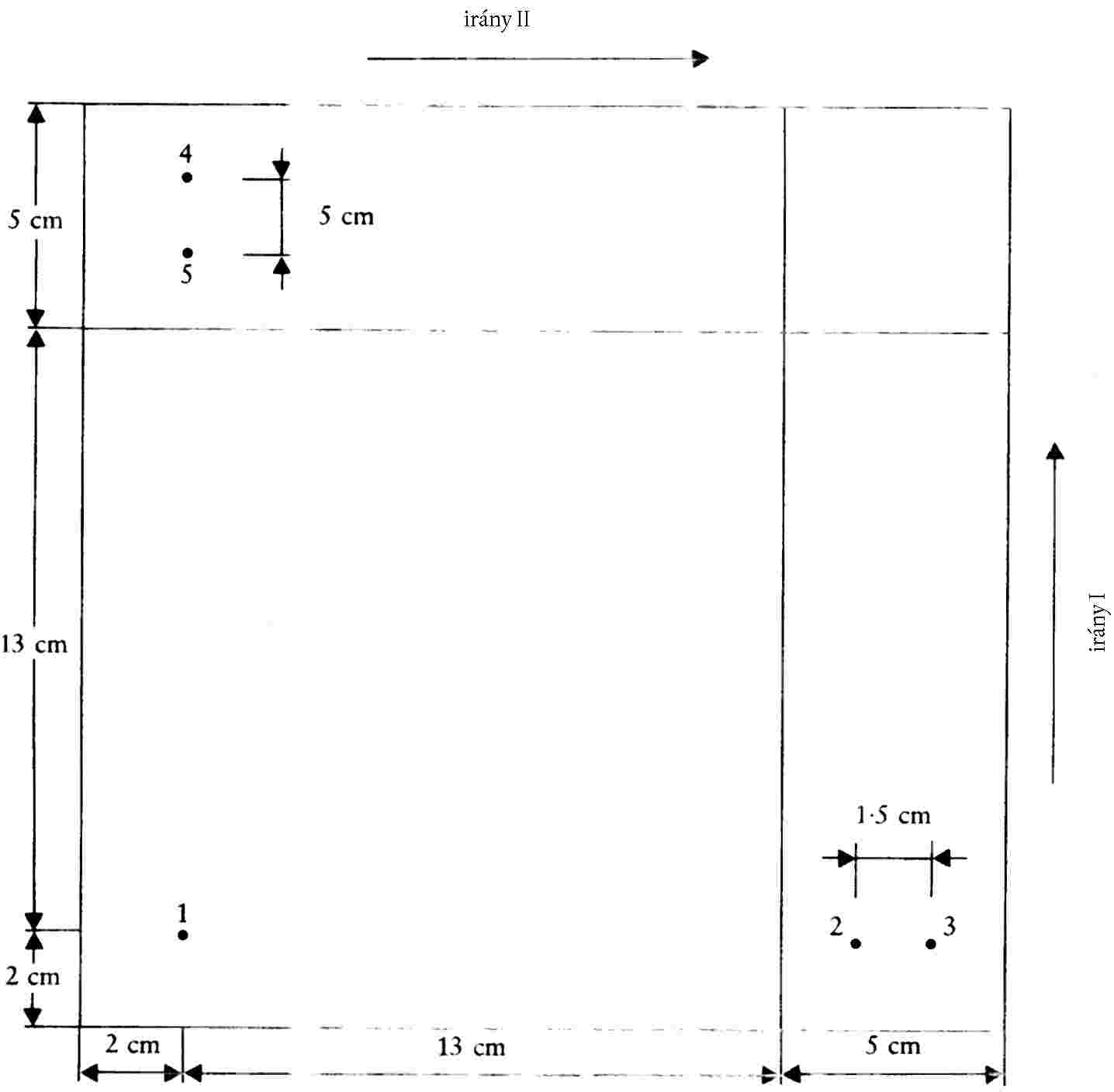
6.4.1. Az 1. ábrán látható módon húzzunk két merőleges vonalat a vékonyréteg-lapon (4.1.3.), az abszorbens felületén.
6.4.2. A lapot nitrogén-atmoszféra (4.1.1.) alatt tartva vigyünk fel az 1. ábra szerinti, az 1 jelű kiindulási pontba 1-4 μl kivonatot (5.1.). A kivonat mennyisége az 5.2. szakaszban kapott kromatogramokon megjelenő foltok intenzitásától függ.
6.4.3. Osszuk el a 2 és 3 jelű pont között (1. ábra) az 5.2. szakaszban azonosított vagy az 5.2. szakasz alapján feltételezett oxidáló színezékeket (a pontok közötti távolság 1,5 cm). Vigyünk fel valamennyi referenciaoldatból 2-2 μl-t a DAP kivételével, amelyből 6 μl-t vigyünk fel a lapra. A műveletet nitrogén (6.4.2.) alatt végezzük.
6.4.4. Ismételjük meg a 6.4.3. szakaszban leírt műveletet a 4 és 5 jelű kiindulási pontokban (1. ábra) (a pontok közötti távolság 1,5 cm), és tartsuk a lapot nitrogén alatt, amíg a kromatográfia meg nem kezdődik.
6.4.5. Öblítsünk át egy kromatográfiás kádat nitrogénnel (3.8.), majd öntsünk bele megfelelő mennyiségű 3.22.2. előhívó oldószert. Tegyük a (6.4.4.) lapot a kádba, és hívjuk elő fénytől védve az első elúciós irányba (1. ábra).
A kromatografálást addig végezzük, amíg az oldószerfront eltávolodott körülbelül 13 cm-re.
6.4.6. Vegyük ki a lapot a kádból, helyezzük egy előzetesen nitrogénnel átöblített kádba, és szárítsuk legalább 60 percen át, hogy az eluáló oldószer elpárologjon.
6.4.7. Egy térfogatmérésre alkalmas próbacsővel mérjünk be megfelelő mennyiségű eluáló oldószert (6.2.1.) egy előzetesen nitrogénnel (3.8.) átöblített kádba, tegyük be a (6.4.6.) kádba a lapot az előző helyzetéhez képest 90°-kal elforgatva, és addig végezzük a kromatografálást a másik irányban ugyancsak fénytől védve, amíg az oldószerfront el nem éri az abszorbens felületén húzott vonalat. Vegyük ki a lapot a kádból, és szárítsuk meg levegőn.
6.4.8. Tegyük be a lapot 10 percre a jódgőzökkel (6.3.) telített kromatográfiás kádba, és értékeljük a kétdimenziós kromatogramot az egyidejűleg kromatografált referenciaanyagok Rf értéke és színe alapján. (A II. táblázat tájékoztató jelleggel ismerteti az Rf-értékeket és a színeket).
Megjegyzés:
A foltok színének maximális intenzitása úgy biztosítható, ha a kromatogramot az előhívás után fél órán keresztül hagyjuk levegőn állni.
6.4.9. A 6.4.8. pontban azonosított oxidáló színezékek jelenléte egyértelműen igazolható, ha a 6.4.1.-6.4.8. pontban írt műveletsort úgy ismételjük meg, hogy a 6.4.2. pontban az 1 jelű kiindulási pontban felvitt mennyiségen felül 1-1 μl-t felviszünk a 6.4.8. pontban azonosított színezékek referenciaoldatából is. Ha ebben a műveletsorban a 6.4.8. ponthoz képest nem jelenik meg új folt, a kromatogramnak a 6.4.8. szerinti értékelése helyes.
II. TÁBLÁZAT
Referenciaoldatok színe a kromatográfia és a jódgőzökkel történő előhívás után
| Referenciaanyag | Szín, jódgőzökkel történő futtatás után |
| R | bézs |
| P | barna |
| α-N | ibolya |
| β-N | világosbarna |
| H | ibolyabarna |
| MPD | sárgásbarna |
| PPD | ibolyabarna |
| MTD | sötétbarna |
| PTD | sárgásbarna |
| DAP | sötétbarna |
| OAP | narancs |
| MAP | sárgásbarna |
| PAP | ibolyabarna |
| 2-NPPD | barna |
| 4-NOPD | narancs |
1. ábra
III. NITRIT AZONOSÍTÁSA ÉS MENNYISÉGI MEGHATÁROZÁSA
A. AZONOSÍTÁS
1. CÉL ÉS ALKALMAZÁSI TERÜLET
Ez a módszer a nitrit kozmetikai termékekben, különösen krémekben és pasztákban történő azonosítására alkalmas.
2. ALAPELV
A nitrit jelenlétét a 2-amino-benzaldehid-fenilhidrazonnal (Nitrine R) képzett színes származékának keletkezése jelzi.
3. REAGENSEK
Minden reagensnek analitikai tisztaságúnak kell lennie.
3.1. Hígított kénsav: hígítsunk 2 ml tömény kénsavat ( [Kép #2] ) 11 ml desztillált vízzel.
Kép #2
3.2. Hígított sósav: hígítsunk 1 ml tömény sósavat ( [Kép #3] ) 11 ml desztillált vízzel.
Kép #3
3.3. Metanol
3.4. 2-amino-benzaldehid-fenilhidrazon (Nitrine R reagens) metanolos oldata.
Mérjünk ki pontosan 2,0 g Nitrine R-t, vigyük át veszteség nélkül egy 100 ml-es mérőlombikba. Csepegtessünk hozzá 4 ml hígított sósavat (3.2.), és rázzuk össze. Töltsük fel a jelig metanollal, és addig keverjük, amíg az oldat teljesen ki nem tisztul. Az oldatot barna üvegpalackban (4.3.) tároljuk.
4. ESZKÖZÖK
4.1. 50 ml-es főzőpohár
4.2. 100 ml-es mérőlombik
4.3. 125 ml-es barna üvegpalack
4.4. 10 x 10 cm-es üveglap
4.5. Műanyag spatula
4.6. 10 x 10 cm-es szűrőpapír,
5. ELJÁRÁS
5.1. Egyenletesen terítsük szét a vizsgálandó minta egy részét egy üveglapon (4.4.) ügyelve arra, hogy a lap felületét legfeljebb 1 cm vastagságban borítsa be.
5.2. Itassunk át egy szűrőpapír (4.6.) -lapot desztillált vízzel. Helyezzük a szűrőpapírt a mintára, és nyomkodjuk le a műanyag spatulával (4.5.).
5.3. Várjunk körülbelül egy percet, majd a szűrőpapír közepére vigyünk fel:
- két csepp hígított kénsavat (3.1.), majd
- két csepp Nitrine R (3.4.) oldatot.
5.4. Öt-tíz másodperc múlva vegyük le a szűrőpapírt, és vizsgáljuk meg a fénnyel szemben tartva. A nitrit jelenlétét vörösesbíbor színeződés jelzi.
Ha a minta nitrittartalma alacsony, a vörösesbíbor szín öt-tizenöt másodperc után sárgára változik. Nagyobb mennyiségű nitrit jelenlétében ez a színátmenet csak egy-két perc múltán megy végbe.
6. MEGJEGYZÉS
A vörösesbíbor szín intenzitásából és a sárgába történő színátmenet időtartamából a minta nitrittartalmára lehet következtetni.
B. MENNYISÉGI MEGHATÁROZÁS
1. CÉL
A módszer a nitrit kozmetikai termékekben történő mennyiségi meghatározását írja le.
2. MEGHATÁROZÁS
A minta nitrittartalmát e módszerrel határozzuk meg, és a nátrium-nitrit tömegszázalékában fejezzük ki.
3. ALAPELV
A minta vízzel történő hígítása és derítése után a jelenlevő nitritet szulfanil-amiddal és N-1-naftil-etilén-diaminnal reagáltatjuk, és mérjük a keletkező szín optikai sűrűségét 538 nm-en.
4. REAGENSEK
Minden reagensnek analitikai tisztaságúnak kell lennie.
4.1. Derítő reagensek: ezek a reagensek készítésük után legföljebb egy hétig használhatók.
4.1.1. Carrez I reagens:
Oldjunk fel 106 g kálium-[hexaciano-ferrátot(II)], K4Fe(CN)6·3H2O-t desztillált vízben, és hígítsuk vízzel 1 000 ml-re.
4.1.2. Carrez II reagens:
Oldjunk fel 219,5 g cink-acetátot, Zn(CH3COO)2 ·2H2O-t és 30 ml jégecetet desztillált vízben, és hígítsuk vízzel 1 000 ml-re.
4.2. Nátrium-nitrit-oldat:
Oldjunk fel 0,500 g nátrium-nitritet desztillált vízben egy 1 000 ml-es mérőlombikban, és vízzel töltsük fel a jelig. Az így elkészített standard törzsoldat 10,0 ml-ét hígítsuk 500 ml-re; utóbbi oldat egy ml-e 10 mikrogramm NaNO2-ot tartalmaz.
4.3. 1N nátrium-hidroxid-oldat
4.4. 0,2 % szulfanil-amid-hidroklorid-oldat:
Oldjunk fel 2,0 g szulfanil-amidot 800 ml vízben melegítés közben. Hűtsük le, és keverés közben adjunk hozzá 100 ml tömény sósavat. Hígítsuk vízzel 1 000 ml-re.
4.5. 5N sósav
4.6. N-1-naftil reagens
Ezt az oldatot a felhasználás napján kell készíteni. Oldjunk fel 0,1 g N-1-naftil-etilén-diamin-dihidrokloridot vízben, és hígítsuk vízzel 100 ml-re.
5. ESZKÖZÖK
5.1. Analitikai mérleg
5.2. 100, 250, 500 és 1 000 ml-es mérőlombik
5.3. Hasas vagy mérőpipetta
5.4. 100 ml-es mérőhenger
5.5. 15 cm átmérőjű, nitritmentes, redős szűrőpapír
5.6. Vízfürdő
5.7. Spektrofotométer 1 cm-es úthosszúságú optikai cellával
5.8. pH-mérő
5.9. 10 ml-es mikrobüretta
5.10. 250 ml-es főzőpohár
6. ELJÁRÁS
6.1. Mérjünk ki körülbelül 0,5 g-ot (m) 0,1 mg pontossággal a homogenizált mintából, forró desztillált vízzel veszteség nélkül mossuk át egy 250 ml-es főzőpohárba (5.10.), majd forró desztillált vízzel egészítsük ki körülbelül 150 ml-re. Tegyük a főzőpoharat (5.10.) fél órára 80 °C-os (5.6.) vízfürdőbe. Közben időnként rázzuk össze a pohár tartalmát.
6.2. Hűtsük le szobahőmérsékletre, és ezután keverés közben adjunk hozzá 2 ml Carrez I (4.1.1.) reagenst és 2 ml Carrez II reagenst (4.1.2.).
6.3. 1N nátrium-hidroxiddal (4.3.) állítsuk be az anyag pH-ját 8,3-ra. Használjuk a pH-mérőt (5.8.). Vigyük át veszteség nélkül egy 250 ml-es mérőlombikba (5.2.), és töltsük fel a jelig desztillált vízzel.
6.4. Keverjük össze a lombik tartalmát, és redős szűrőn (5.5.) szűrjük a mintát.
6.5. A tiszta szűrletből pipettázzunk (5.3.) megfelelő mennyiséget, de legföljebb 25 ml-t egy 100 ml-es mérőlombikba (5.2.), és desztillált vízzel egészítsük ki a térfogatát 60 ml-re.
6.6. Az összekeverést követően adjunk hozzá 10,0 ml szulfanil-amid-hidroklorid-oldatot (4.4.), majd 6,0 ml 5N sósavat (4.5.). Keverjük össze, és hagyjuk állni az oldatot öt percig. Adjunk hozzá 2,0 ml N-1-naftil reagenst (4.6.), keverjük össze, és hagyjuk állni három percig. Hígítsuk vízzel jelig, és keverjük össze.
6.7. A vakpróba készítéséhez ismételjük a 6.5. és 6.6. műveletet az N-1-naftil reagensnek (4.6.) az oldathoz történő hozzáadása nélkül.
6.8. Mérjük (5.7.) a 6.6. műveletben kapott oldat optikai sűrűségét 538 nm-en, referenciaként a vakoldatot (6.7.) használjuk.
6.9. A kalibrációs görbéről (6.10.) olvassuk le a minta 6.8. pontban mért optikai sűrűségnek megfelelő nátrium-nitrit-tartalmat mikrogramm/100 ml koncentráció egységben (m1 mikrogramm).
6.10. A 10 μg/ml koncentrációjú nátrium-nitrit (4.2.) -oldat felhasználásával készítsünk 0, 20, 40, 60, 80, 100 μg nátrium-nitrát/100 ml koncentrációjú oldatokat, és vegyük fel a nátrium-nitrit kalibrációs egyenesét.
7. SZÁMÍTÁS
Számítsuk ki a minta nátrium-nitrit-tartalmát tömegszázalékban a következő képlet segítségével:
ahol:
m = a vizsgálatra kivett minta tömege grammban (6.1.),
m1 = a 6.9. pontban meghatározott nátrium-nitrit-tartalom mikrogrammban,
V = a méréshez (6.5.) felhasznált szűrlet térfogata ml-ben.
8. MEGISMÉTELHETŐSÉG ( 5 )
0,2 % (m/m) körüli nátrium-nitrit-tartalom esetén azonos mintán, párhuzamosan végzett két mennyiségi meghatározás eredményei közötti különbség abszolút értéke nem haladhatja meg a 0,005 %-ot.
IV. A SZABAD FORMALDEHID AZONOSÍTÁSA ÉS MENNYISÉGI MEGHATÁROZÁSA
1. CÉL ÉS ALKALMAZÁSI TERÜLET
Ez a módszer az azonosítást és a formaldehid-donorok jelenlétének, illetve távollétének megfelelően két mennyiségi meghatározást ír le. Minden kozmetikai termék esetében alkalmazható.
1.1. Azonosítás
1.2. Általános kolorimetriás meghatározás 2,4-pentándionnal
Ez a módszer akkor alkalmazható, ha a formaldehid egymagában van jelen vagy olyan más tartósítószerekkel együtt, amelyek egyike sem formaldehid-donor.
Ellenkező esetben, illetve ha az eredmény meghaladja az engedélyezett legnagyobb töménységet, a következő ellenőrzési módszert kell alkalmazni.
1.3. Mennyiségi meghatározás formaldehid-donorok jelenlétében
A fent említett módszerben (1.2.) a származékképzés során a formaldehid-donorok elhasadnak és túl magas eredményhez vezetnek (kötött és polimerizált formaldehid).
A szabad formaldehidet el kell választani folyadék-kromatográfiával.
2. FOGALOMMEGHATÁROZÁS
A mintának ezzel a módszerrel meghatározott szabad formaldehidtartalmát tömegszázalékban fejezzük ki.
3. AZONOSÍTÁS
3.1. Alapelv
A szabad és kötött formaldehid kénsavas közegben a Schiff reagenst rózsaszínűre vagy mályvaszínűre változtatja.
3.2. Reagensek
Minden reagensnek analitikai tisztaságúnak, a víznek pedig ioncseréltnek kell lennie.
3.2.1. Fukszin
3.2.2. 7 H2O-val hidratált nátrium-szulfit
3.2.3. Tömény sósav (d = 1,19)
3.2.4. Kénsav, kb. 1 M
3.2.5. Schiff reagens:
Mérjünk be 100 mg fukszint (3.2.1.) egy főzőpohárba és oldjuk fel 75 ml vízben, 80°C-on. Lehűlés után adjunk hozzá 2,5 g nátrium-szulfitot (3.2.2.). Töltsük fel 100 ml-re.
Két hétig eltartható.
3.3. Eljárás
3.3.1. Mérjünk be 2 g mintát egy 10 ml-es főzőpohárba.
3.3.2. Adjunk hozzá két csepp kénsavat (3.2.4.) és 2 ml Schiff reagenst (3.2.5.). Ennek a reagensnek alkalmazáskor teljesen színtelennek kell lennie.
Rázzuk össze és hagyjuk állni öt percig.
3.3.3. Ha öt perc alatt rózsaszínű vagy mályvaszínű elszíneződés észlelhető, akkor a formaldehid 0,01 %-ot meghaladó mennyiségben van jelen. Ebben az esetben a szabad és az összetett formaldehidet a (4.) módszer, valamint szükség esetén az (5.) módszer szerint kell meghatározni.
4. ÁLTALÁNOS KOLORIMETRIÁS MEGHATÁROZÁS 2,4-PENTÁNDIONNAL
4.1. Alapelv
A formaldehid 2,4-pentán dionnal, ammónium-acetát jelenlétében, 3,5-diacetil-1,4-dihidro-lutidint képez. Ezt butanán-1-ollal kivonjuk és a kivonat abszorbenciáját 410 nm-en megmérjük.
4.2. Reagensek
Minden reagensnek analitikai tisztaságúnak, a víznek pedig ioncseréltnek kell lennie.
4.2.1. Vízmentes ammónium-acetát
4.2.2. Tömény ecetsav, d20 4 = 1,05
4.2.3. Alacsony nyomáson, 25 mm Hg, 25o-on frissen desztillált 2,4-pentándion - nem mutathat semennyi abszorpciót 410nm-en.
4.2.4. Butan-1-ol
4.2.5. Sósav, 1 M
4.2.6. Sósav, kb. 0,1 M
4.2.7. Nátrium-hidroxid, 1 M
4.2.8. Frissen készített keményítő oldat, amely megfelel az Európai gyógyszerkönyv 1980-as 2. kiadása I-VII-1-1 részének (1g/50ml víz)
4.2.9. 37-40 %-os formaldehid
4.2.10. Jód mérőoldat, 0,05 M
4.2.11. Nátrium-tioszulfát mérőoldat, 0,1 M
4.2.12. 2,4-pentán-dion reagens
Oldjunk fel egy 1000ml-es mérőlombikban:
- 150g ammónium-acetátot (4.2.1. pont),
- 2ml 2,4-pentán-diont (4.2.3. pont),
- 3ml ecetsavat (4.2.2. pont).
Töltsük fel 1000ml-re vízzel (az oldat pH-ja 6,4 körül van).
Ezt a reagenst frissen kell készíteni
4.2.13. 2,4-pentándion nélküli reagens (4.2.12. pont)
4.2.14. Standard formaldehid: törzsoldat
Öntsünk 5 g formaldehidet (4.2.9. pont) egy 1000 ml-es mérőlombikba és töltsük fel vízzel 1000 ml-re.
Állapítsuk meg az oldat erősségét a következőképpen:
Vegyünk ki 10,00 ml-t; adjunk hozzá 25,00 ml jód mérőoldatot (4.2.10. pont) és 10,00 ml nátrium-hidroxid oldatot (4.2.7. pont).
Hagyjuk állni öt percig.
Savanyítsuk meg 11,00 ml HCl oldattal (4.2.5. pont) és határozzuk meg a jódfelesleget nátrium-tioszulfát oldattal (4.2.11. pont) keményítő oldat (4.2.8. pont), mint indikátor jelenlétében.
1 ml 0,05 M jód (4.2.10. pont) fogyása megfelel 1,5 mg formaldehidnek.
4.2.15. Standard formaldehid: hígított oldat
Hígítsuk fel a formaldehid törzsoldatot először 1/20 majd 1/100 arányban vízzel.
Ez az oldatnak milliliterenként körülbelül 1 μg formaldehidet tartalmaz.
Számítsuk ki a pontos tartalmat.
4.3. Eszközök
4.3.1. Szokásos laboratóriumi felszerelés
4.3.2. Fáziselválasztó szűrő, Whatman 1 PS (vagy ezzel egyenértékű)
4.3.3. Centrifuga
4.3.4. Vízfürdő készlet 60°C-ra állítva
4.3.5. Spektrofotométer
4.3.6. Üveg küvetta, 1 cm-es optikai úthosszal.
4.4. Eljárás
4.4.1. Mintaoldat
Egy 100ml-es mérőlombikba a vizsgálati mintából 0,001 gramm pontossággal mérjünk be kb. 150 μg formaldehid-mennyiségnek megfelelő (grammban) mennyiséget.
Töltsük fel 100 ml-re vízzel és keverjük össze (S oldat).
(Ellenőrizzük, hogy a pH 6-hoz közeli; ha nem, akkor hígítsuk a sósavoldattal (4.2.6. pont).)
Tegyük egy 50 ml-es Erlenmeyer lombikba a következőket:
- 10,00 ml S oldat,
- 5,00 ml 2,4-pentándion reagens (4.2.12.),
- annyi ioncserélt vizet, hogy a végső térfogat 30 ml legyen.
4.4.2. Referencia oldat
A referencia oldat használatával a vizsgálati minta háttérszíne által okozott esetleges zavaró hatás kiküszöbölhető:
Tegyük egy 50 ml-es Erlenmeyer lombikba a következőket:
- 10,00 ml S oldat,
- 5,00 ml reagens (4.2.13.),
- annyi ioncserélt víz, hogy a végső térfogat 30 ml legyen.
4.4.3. Vakpróba
Tegyük egy 50 ml-es Erlenmeyer lombikba a következőket:
- 5,00 ml 2,4-pentán-dion reagens (4.2.12.),
- annyi ioncserélt víz, hogy a végső térfogat 30 ml legyen.
4.4.4. Mennyiségi meghatározás
4.4.4.1. A 4.4.1., 4.4.2. és a 4.4.3. pont szerinti keverékeket rázzuk össze. Állítsuk az Erlenmeyer lombikokat 60oC-os vízfürdőbe pontosan tíz percre. Hagyjuk hűlni két percig jeges vízzel teli fürdőben.
4.4.4.2. Vigyük át egy 50 ml-es választótölcsérbe, amely 10 ml 1-butanolt (4.2.4. pont) tartalmaz. Öblítsük át mindegyik lombikot 3-5 ml vízzel. Rázzuk erősen a keveréket pontosan 30 másodpercig. Hagyjuk a fázisokat szétválni.
4.4.4.3. Szűrjük le a bután-1-olos fázist a mérőlombikba (4.3.2. pont) fáziselválasztó szűrőn keresztül. Centrifugálás (3000 g, 5 perc) is alkalmazható.
4.4.4.4. Mérjük meg a 4.4.1. pont szerinti mintaoldat kivonatának A1 abszorbanciáját 410 nm-nél a 4.4.2. pont szerinti referencia oldat kivonatához képest.
4.4.4.5. Hasonlóképpen mérjük meg a 4.4.3. szerinti vakoldat kivonatának A2 abszorbanciáját bután -1-olhoz képest.
MEGJEGYZÉS: Az összes fenti műveletet attól a pillanattól számított 25 percen belül kell elvégezni, amikor az Erlenmeyer lombikot a 60oC-os vízfürdőbe helyeztük.
4.4.5. Kalibrációs görbe
4.4.5.1. Tegyük egy 50 ml-es Erlenmeyer lombikba a következőket:
- 5,00 ml hígított standard oldat a 4.2.15. pont szerint,
- 5,00 ml 2,4-pentán-dion reagens (4.2.12.),
- annyi ioncserélt vizet, hogy a végső térfogat 30 ml legyen.
4.4.5.2. Folytassuk a 4.4.4. pontban leírtaknak megfelelően és mérjük meg az abszorbanciát bután -1-olhoz (4.2.4.) képest.
4.4.5.3. Ismételjük meg az eljárást 10, 15, 20 és 25 ml hígított standard oldat (4.2.15.) felhasználásával.
4.4.5.4. A nullapont meghatározásához (amely a reagensek színezettségének felel meg) járjunk el a 4.4.4.5. pontban megadottak szerint.
4.4.5.5. Szerkesszük meg a kalibrációs görbét, miután kivontuk a nullapont értékét a 4.4.5.1 és 4.4.5.3. pont szerint kapott egyes abszorbanciákból. A Beer törvény 30 μg formaldehid mennyiségig érvényes.
4.5. Számítás
4.5.1. Vonjuk ki A2-t A1-ből és olvassuk le a kalibrációs görbéről (4.4.5.5. pont) azt a μg-ban kifejezett C mennyiséget, amely a mintaoldatban jelen lévő formaldehidnek felel meg (4.4.1.).
4.5.2. Számítsuk ki a mintaformaldehid tartalmát (% m/m) a következő képlet segítségével:
ahol:
m = a vizsgálati részlet tömege a g-ban kifejezve.
4.6. Megismételhetőség ( 6 )
0,2 %-os formaldehidtartalom esetén azonos mintán, párhuzamosan végzett két mennyiségi meghatározás eredményei közötti eltérés nem haladhatja meg a 0,005 %-ot a 2,4-pentán-dionos kolorimetriás meghatározás esetén.
Ha a szabad formaldehidtartalom mennyiségi meghatározása a 76/768/EGK irányelvben meghatározott legnagyobb koncentrációt meghaladó eredményt ad, azaz:
a) 0,05 %- és 0,2 %- között van egy címkézetlen termékben;
b) 0,2 %- felett van, akár címkézett akár címkézetlen termékben,
akkor az alábbi, 5. pontban meghatározott eljárást kell alkalmazni.
5. MENNYISÉGI MEGHATÁROZÁS FORMALDEHID-DONOROK JELENLÉTÉBEN
5.1. Alapelv
A különálló formaldehidet sárga lutidin származékká alakítjuk 2,4-pentán-dionos reakció segítségével egy oszlopkimenethez illeszthető reaktorban és a keletkező származék abszorbanciáját 420 nm-en megmérjük.
5.2. Reagensek
Minden reagensnek analitikai tisztaságúnak, a víznek pedig ioncseréltnek kell lennie.
5.2.1. HPLC tisztaságú vagy azzal megegyező minőségű víz
5.2.2. Vízmentes ammónium-acetát
5.2.3. Tömény ecetsav
5.2.4. 2,4-pentándion (4oC-on tartva)
5.2.5. Vízmentes dinátrium-foszfát
5.2.6. 85 %-os ortofoszforsav (d = 1,7)
5.2.7. HPLC minőségű metanol
5.2.8. Diklór-metán
5.2.9. 37-40 %-os (w/v) formaldehid
5.2.10. Nátrium-hidroxid, 1 M
5.2.11. Sósav, 1 M
5.2.12. Sósav, 0,002 M
5.2.13. Frissen készített keményítő oldat, amely megfelel az Európai gyógyszerkönyvnek (lásd 4.2.8.pont)
5.2.14. Jód mérőoldat, 0,05 M
5.2.15. Nátrium-tioszulfát mérőoldat, 0,1 M
5.2.16. Mozgó fázis: Dinátrium-foszfát (5.2.5.) vizes oldata, 0,006 M, ortofoszforsavval (5.2.6.) pH 2,1 értékre beállított;
5.2.17. Oszlop utáni reagens:
Oldjunk fel egy 1000 ml-es mérőlombikban:
- 62,5 g ammónium-acetátot (5.2.2.),
- 7,5 ml ecetsavat (5.2.3.),
- 5 ml 2,4-pentán-diont (5.2.4.).
Töltsük fel 1000 ml-re vízzel (5.2.1.).
Ezt a reagenst tartsuk távol a fénytől.
Eltarthatóság 25oC-on legfeljebb 3 nap.
Az oldat színe nem változhat meg;
5.2.18. Standard formaldehid: törzsoldat
Öntsünk 10 g formaldehidet (5.2.9.) egy 1000 ml-es mérőlombikba és töltsük fel 1000 ml-re vízzel.
Állapítsuk meg ennek az oldatnak az erősségét a következőképpen:
Vegyünk ki 5,00 ml-t; adjunk hozzá 25,00 ml jód mérőoldatot (5.2.14.) és 10 ml nátrium-hidroxid oldatot (5.2.10.).
Hagyjuk állni öt percig.
Savanyítsuk meg 11,00 ml HCl oldattal (5.2.11.) és határozzuk meg a jód mérőoldat feleslegét nátrium-tioszulfát oldattal (5.2.15.) titrálva, használjunk keményítő oldat (5.2.13.) indikátorként.
1 ml jód oldat (5.2.14.) megfelel 1,5 mg formaldehidnek.
5.2.19. Standard formaldehid: hígított oldat
Hígítsuk fel a törzsoldatot kezdeti erősségéhez képest 1/100 arányban a mozgó fázissal.
Ennek az oldatnak 1 ml-e kb. 37 mg formaldehidet tartalmaz.
Számítsuk ki a pontos tartalmat.
5.3. Eszközök
5.3.1. Szokásos laboratóriumi felszerelés
5.3.2. Ingadozásmentes HPLC pumpa
5.3.3. Alacsony nyomású ingadozásmentes pumpa a reagens számára (vagy egy második HPLC pumpa)
5.3.4. Befecskendező szelep 10 μl-es hurokkal
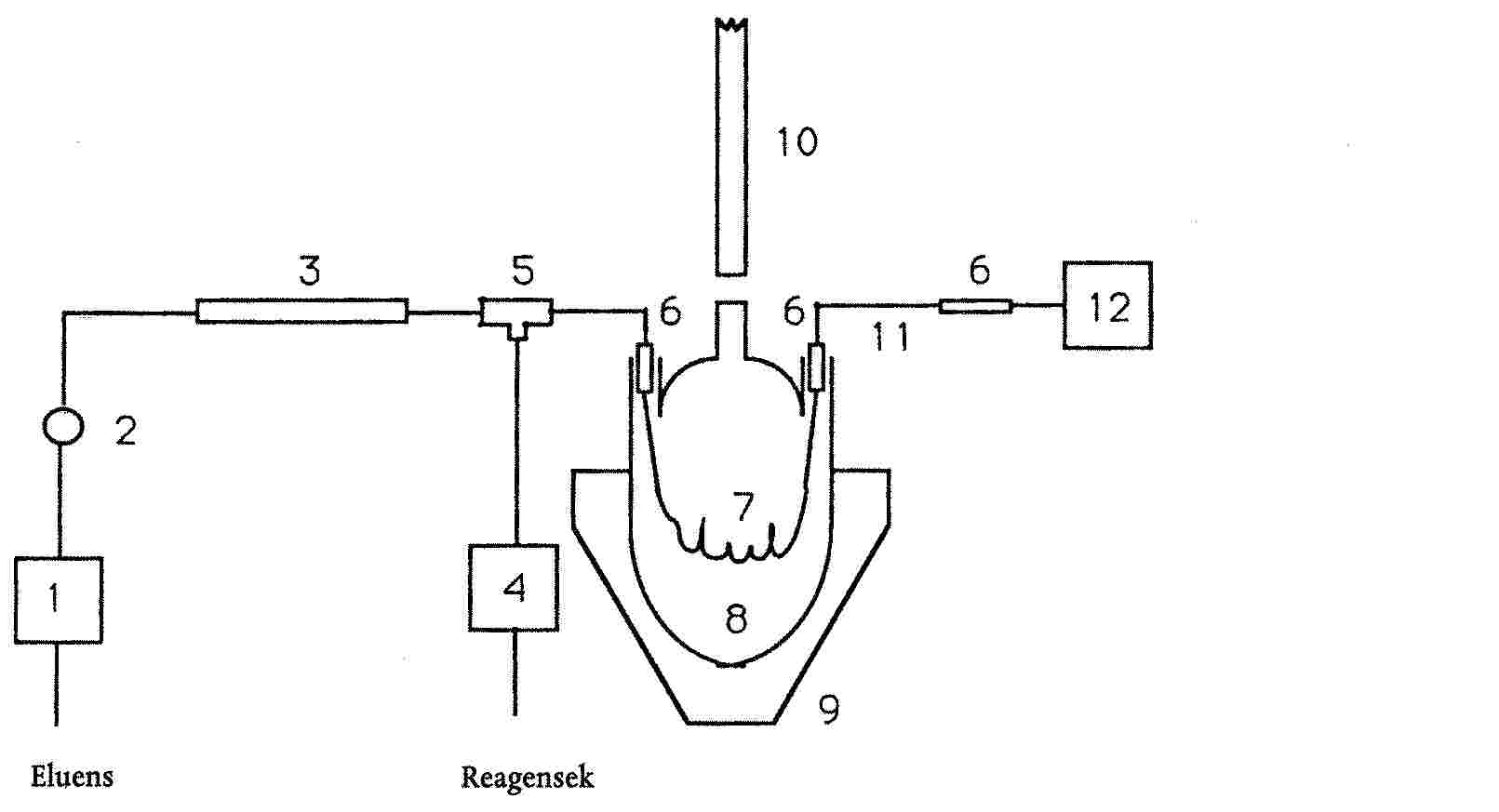
5.3.5. Oszlopkimenethez illeszthető reaktor a következő összetevőkkel:
+ egy 1 literes háromnyakú lombik;
+ egy melegítőkráter 1 literes lombikhoz;
+ két Vigreux oszlop, legalább 10 lappal, kettő léghűtéses;
+ 1,6 mm-es rozsdamentes acélcső (a hőcseréléshez) - belső átmérő 0,23 mm, hossz = 400 mm;
+ 1,6 mm-es tefloncső - belső átmérő, 0,30 mm, hossz = 5 m (csomózás) lásd 1. függelék);
+ egy T-darab, holttérfogat nélkül (Valco vagy azzal egyenértékű);
+ három csatlakozó holttérfogat nélkül
Vagy: egy Applied Biosystems PCRS 520 oszlop utáni modul, vagy azzal egyenértékű, 1 ml-es reaktorral felszerelve;
5.3.6. Membránszűrő, 0,45 μm-es pórusmérettel
5.3.7. SEP-PAKR C18 vagy azzal egyenértékű töltet
5.3.8. Használatra kész oszlopok:
- Bischoff hypersil RP 18 (NC típus, C 25,461805 hivatkozás)
- (5 μm, hossz = 250 mm, belső átmérő = 4,6 mm),
- vagy Doupont, Zorbax ODS
- (5 μm, hossz = 250 mm, belső átmérő = 4,6 mm),
- vagy Phase SEP, spherisorb ODS 2
- (5 μm, hossz = 250 mm, belső átmérő = 4,6 mm).
5.3.9. Előoszlop
Bischoff K1 hypersil RP 18 (K1 G 6301 1805 hivatkozás)
(5 μm, hossz = 10 mm, vagy ezzel egyenértékű).
5.3.10. Az oszlop és az előoszlop Ecotube-rendszer segítségével van összekötve (A 15020508 Bischoff hivatkozás) vagy ezzel egyenértékű.
5.3.11. Szereljük össze a készüléket (5.3.5.) a 2. függelék diagramján bemutatottak szerint.
A befecskendező érték utáni csatlakozásokat a lehető legrövidebbre kell méretezni. Ebben az esetben a reaktor kivezetése és a detektor bevezetése közötti rozsdamentes acélcső a keverék mérés előtti hűtése céljából van elhelyezve, és a hőmérséklet a detektorban ismeretlen, de állandó.
5.3.12. UV látható detektor
5.3.13. Adatrögzítő
5.3.14. Centrifuga
5.3.15. Ultrahangos fürdő
5.3.16. Rotációs keverő (vortex vagy azzal egyenértékű)
5.4. Eljárás
5.4.1. Kalibrációs görbe
Ezt a görbét a hígított standard formaldehid oldat koncentrációjának függvényében felrajzolt csúcsmagasságok adják meg.
Készítsük el a mérőoldatokat, a standard formaldehid oldatnak (5.2.19.) a mozgó fázissal (5.2.16.) történő hígításával:
- 1,00 ml oldat (5.2.19.) hígítva 20,00ml-re (kb. 185 μg/100ml),
- 2,00 ml oldat (5.2.19.) hígítva 20,00ml-re (kb. 370 μg/100ml),
- 5,00 ml oldat (5.2.19.) hígítva 25,00ml-re (kb. 740 μg/100ml),
- 5,00 ml oldat (5.2.19.) hígítva 20,00ml-re (kb. 925 μg/100ml),
A mérőoldatokat tartsuk laboratóriumi hőmérsékleten egy órán keresztül, és frissen készítsük el.
A kalibrációs görbe linearitása az 1,00 és 15,00 μg/ml közötti koncentráció-tartományban megfelelő.
5.4.2. Mintaelőkészítés
5.4.2.1. Emulziók (krémek, alapozók, szemkihúzók)
Mérjünk be egy dugóval ellátott 100 ml-es lombikba 0,001 g-os pontossággal 100 μg formaldehidnek megfelelő mennyiséget (grammban) a vizsgálati mintából. Adjunk hozzá pontosan bemért 20,00 ml diklór-metánt (5.2.8.) és 20,00 ml sósavat (5.2.12.). Keverjük össze a rezgő keverő (5.3.16.) és az ultrahangos fürdő (5.2.15.) segítségével. Válasszuk el a két fázist centrifugálással (3000 gn két percig). Eközben mossuk át a patront (5.3.7.) 2 ml metanollal (5.2.7.), majd kondicionáljuk 5 ml vízzel (5.2.1.).
Hajtsunk át 4 ml-t a kivonat vizes fázisából a kondicionált patronon, öntsük el az első 2 ml-t és tegyük el az azt követő részletet.
5.4.2.2. Arc- és testápolók, samponok
Mérjünk be egy dugóval ellátott 100 ml-es lombikba 0,001 g-os pontossággal egy 500 μg formaldehidnek megfelelő mennyiséget (grammban) a vizsgálati mintából.
Töltsük fel 100 ml-re a mozgó fázissal (5.2.16.).
Szűrjük le az oldatot egy szűrőn (5.3.6.) keresztül, és fecskendezzük be, vagy vezessük át egy olyan tölteten (5.3.7.), amelyet előzetesen a fenti módon (5.4.2.1.) kondicionáltunk. Minden oldatot közvetlenül az elkészítése után be kell fecskendezni.
5.4.3. Kromatográfiás körülmények
- A mozgó fázis átfolyási sebessége: 1ml/perc,
- A reagens átfolyási sebessége: 0,5 ml/perc,
- A detektor kimenetén a teljes átfolyási sebesség: 1,5 ml/perc,
- Befecskendezett térfogat: 10 μl,
- Oldási hőmérséklet: nehéz elválasztások esetén merítsük az oszlopot olvadó jeges fürdőbe: várjuk meg amíg a hőmérséklet állandósul (15 - 20 perc).
- Az oszlop utáni reakció hőmérséklete: 100°C,
- Detektálás: 420 nm-en.
MEGJEGYZÉS: A teljes kromatográfiás rendszert és az utóoszlopot is használat után át kell mosni vízzel (5.2.1.). Ha a rendszert két napnál hosszabb ideig nem használják, akkor az átmosás után metanolos (5.2.7.) mosást is alkalmazni kell. Újrakondicionálás előtt a rendszeren vizet kell átnyomni az átkristályosodás elkerülése érdekében.
5.5. Számítás
Emulziók esetében: (5.4.2.1.):
Formaldehidtartalom %-ban (m/m,)
Arc- és testápolók, samponok esetében (5.4.2.2.):
Formaldehidtartalom:
ahol:
m = a vizsgálati minta tömege g-ban (5.4.2.1.),
C = formaldehid koncentráció μg/100ml-ben a kalibrációs görbéről leolvasva (5.4.1.).
5.6. Megismételhetőség ( 7 )
0,05 %-os formaldehidtartalom esetén azonos mintán, párhuzamosan végzett két mennyiségi meghatározás közötti különbség nem haladhatja meg a 0,001 %-ot.
0,2 %-os formaldehidtartalom esetén azonos mintán, párhuzamosan végzett két mennyiségi meghatározás közötti eltérés nem haladhatja meg a 0,005 %-ot.
V. REZORCIN MEGHATÁROZÁSA SAMPONOKBAN ÉS HAJSZESZEKBEN
1. CÉL ÉS ALKALMAZÁSI TERÜLET
Ez a módszer rezorcin samponokban és hajszeszekben történő gázkromatográfiás meghatározását írja le. A módszer a minta tömegére vonatkoztatva 0,1-2,0 % rezorcin meghatározására alkalmas.
2. MEGHATÁTOZÁS
Az ezzel a módszerrel meghatározott rezorcintartalmat a minta tömegére vonatkoztatva tömegszázalékban adjuk meg.
3. ALAPELV
A rezorcint és a belső standardként adott 3,5-dihidroxi-toluolt a mintától vékonyréteg-kromatográfiával választjuk el. A két vegyületet a vékonyréteg-lapról lekaparva és metanollal kivonva izoláljuk. Végül a kivont vegyületeket megszárítjuk, szililezzük, és gázkromatografáljuk.
4. REAGENSEK
Minden reagensnek analitikai tisztaságúnak kell lennie.
4.1. 25 %-os (m/m) sósav
4.2. Metanol
4.3. 96 %-os (v/v) etanol
4.4. Készre gyártott szilikagél VRK lapok (műanyag vagy alumínium) fluoreszcens indikátorral. Hatástalanítsuk a következők szerint: fújjuk le a közönséges, előre bevont szilikagél lapokat vízzel, amíg fényessé nem válnak. Hagyjuk szobahőmérsékleten egy-három órán keresztül száradni a lefújt lapokat.
Megjegyzés:
A lap hatástalanítása nélkül a rezorcin szilikagélen történő irreverzibilis abszorpciója miatt veszteség léphet föl.
4.5. Előhívó oldószer: aceton - kloroform - ecetsav (20:75:5 térfogatarányban).
4.6. Rezorcin-standard-oldat: oldjunk fel 400 mg rezorcint 100 ml 96 %-os etanolban (4.3.) (1 ml 4 000 μg rezorcint tartalmaz).
4.7. Belső standard oldat: oldjunk fel 400 mg 3,5-dihidroxi-toluolt (DHT) 100 ml 96 %-os etanolban (4.3.) (1 ml 4 000 μg DHT-t tartalmaz).
4.8. Standard elegy: elegyítsünk 10 ml 4.6. és 10 ml 4.7. szerinti oldatot egy 100 ml-es mérőlombikban, töltsük a jelig 96 %-os etanollal (4.3.), és keverjük össze (1 ml 400 μg rezorcint és 400 μg DHT-t tartalmaz).
4.9. Szililezőszerek:
4.9.1. N,O-bis-(trimetil-szilil-)trifluoro-acetamid (BSTFA)
4.9.2. Hexametil-diszilazán (HMDS)
4.9.3. Trimetil-klórszilán (TMCS)
5. ESZKÖZÖK
5.1. A vékonyréteg-kromatográfia és a gázkromatográfia szokásos felszerelései
5.2. Üvegeszközök
6. ELJÁRÁS
6.1. Minta-előkészítés
6.1.1. Egy 150 ml-es főzőpohárba a termékből mérjünk be annyi vizsgálati mintát (m gramm), amely körülbelül 20-50 mg rezorcint tartalmaz.
6.1.2. Adjunk hozzá sósavat (4.1.), amíg a keverék savassá nem válik (mintegy 2-4 ml szükséges), és adjunk hozzá 10 ml (40 mg DHT) belső standard oldatot (4.7.), és keverjük össze. Etanollal (4.3.) mossuk át egy 100 ml-es mérőlombikba, töltsük fel a jelig etanollal, és keverjük össze.
6.1.3. Vigyünk fel 250 μl (6.1.2) oldatot egy deaktivált szilikagél lapra (4.4.) egy 8 cm hosszú, folyamatos egyenes mentén. Ügyeljünk arra, hogy az egyenes minél vékonyabb legyen.
6.1.4. Ugyanígy vigyünk fel a standard elegyből (4.8.) 250 μl-t ugyanarra a lapra (6.1.3.).
6.1.5. Az előhívást követő azonosítás egyszerűsítése céljából ugyanazon a lapon vigyünk fel párhuzamosan az alapvonalra két pontban 5-5 μl-t a 4.6. és a 4.7. oldatból.
6.1.6. Fejlesszük ki a lapot az előhívó szerrel (4.5.) megtöltött telítetlen kádban, amíg az oldószerfront el nem távolodik az alapvonaltól 12 cm-re, ez általában körülbelül 45 percet vesz igénybe. Szárítsuk meg levegőn a lapot, és állapítsuk meg a rezorcin/DHT-zóna helyét rövidhullámú UV fényben (254 nm). A két vegyület Rf-értéke nagyjából megegyezik. A sávok sötét külső határvonalától két mm-re egy ceruzával rajzoljuk körbe a sávokat. Kaparjuk le ezeket a zónákat, és az egyes sávokat tartalmazó abszorbenst külön-külön 10 ml-es palackokban gyűjtsük.
6.1.7. Vonjuk ki a mintát és a standard elegyet tartalmazó abszorbenst a következőképpen:
adjunk hozzá 2 ml metanolt (4.2.), és állandó keverés mellett egy órán keresztül vonjuk ki. Szűrjük a keveréket, majd ismételjük meg a műveletet 2 ml metanollal 15 percig extrahálva.
6.1.8. Egyesítsük a metanolos kivonatokat, és megfelelő szárítószerrel töltött vákuum-deszikkátorban egy éjszakán át szárítva párologtassuk el az oldószert. Semmiképpen ne melegítsük a mintákat.
6.1.9. A maradékkal végezzük el (6.1.8.) a szililezését a 6.1.9.1. vagy pedig a 6.1.9.2. alatt leírt módon.
6.1.9.1. Egy mikrofecskendővel adjunk hozzá 200 μl BSTFA-t (4.9.1.), és egy zárt edényben hagyjuk állni a keveréket 12 órán keresztül, szobahőmérsékleten.
6.1.9.2. Adjunk hozzá egymás után 200 μl HMDS-t (4.9.2.) és 100 μl TMCS-t (4.9.3.) egy mikrofecskendővel, és egy zárt edényben melegítsük a keveréket 30 percen át 60 °C-on. Hűtsük le a keveréket.
6.2. Gázkromatográfia
6.2.1. Gázkromatográfiás körülmények
Az oszlop a következő megoldást eredményezi: R nem lehet kisebb, mint 1,5,
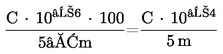
ahol:
r1 és r2 = két csúcs retenciós ideje percben,
w1 és w2 = ugyanezen csúcsoknak a magasság felénél mért szélessége mm-ben,
d' = a diagrampapír sebessége mm/percben.
Ez a felbontás a következő beállítások mellett érhető el:
| Oszlop | anyaga: | rozsdamentes acél |
| hossza: | 200 cm | |
| belső átmérője: | ˜ 3 mm | |
| töltete: | 10 % OV-17 100–120 mesh CHROMOSORB WAW-on | |
| Lángionizációs detektor | ||
| Hőmérsékletek: | ||
| oszlop: | 185 °C (izoterm) | |
| detektor: | 250 °C | |
| injektor: | 250 °C | |
| Vivőgáz: | nitrogén | |
| térfogatáram: | 45 ml/perc | |
A hidrogén és levegő térfogatáramának beállításával kapcsolatban kövessük a gyártó utasításait.
6.2.2. Fecskendezzünk a 6.1.9. pont szerint elkészített oldatokból 1-3 μl-t a gázkromatográfra. Minden (6.1.9.) oldatból ötször fecskendezzünk, mérjük az egy anyaghoz tartozó csúcsterületeket, átlagoljuk ezeket, és számítsuk ki a csúcsterületarányt, S-t. S = rezorcin-csúcsterület/DHT-csúcsterület.
7. SZÁMÍTÁS
A mintában a rezorcin koncentrációját tömegszázalékban (% m/m) a következő képlet fejezi ki:
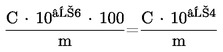
ahol:
M = vizsgálati minta tömege grammban (6.1.1.),
Sminta = a 6.2.2. alapján számított, a mintaoldatra vonatkozó átlag csúcsterületarány,
Sstandard elegy = a 6.2.2. alapján számított, a standard elegyre vonatkozó átlag csúcsterületarány.
8. MEGISMÉTELHETŐSÉG ( 8 )
0,5 % körüli rezorcintartalom esetén azonos mintán, párhuzamosan végzett két mennyiségi meghatározás eredményei közötti különbség abszolút értéke nem haladhatja meg a 0,025 %-ot.
VI. METANOL MENNYISÉGI MEGHATÁROZÁSA ETANOLRA VAGY PROPÁN-2-OLRA VONATKOZTATVA
1. CÉL ÉS ALKALMAZÁSI TERÜLET
Ez a módszer metanol gázkromatográfiás meghatározását írja le valamennyi kozmetikai termékfajtában (ideértve az aeroszolokat is).
A módszerrel 0-10 % koncentrációk határozhatók meg.
2. MEGHATÁROZÁS
A módszerrel meghatározott metanoltartalmat etanolra vagy propán-2-olra vonatkoztatva tömegszázalékban adjuk meg.
3. ALAPELV
A meghatározás gázkromatográfiával történik.
4. REAGENSEK
Használjunk analitikai minőségű reagenseket.
4.1. Metanol
4.2. Abszolút etanol
4.3. Propán-2-ol
4.4. Vízzel alkoholmentesített kloroform
5. ESZKÖZÖK
5.1. Gázkromatográf:
hővezetési detektor aeroszol mintákra,
lángionizációs detektor nem aeroszol mintákra.
5.2. 100 ml-es mérőlombikok
5.3. 2 ml-es, 20 ml-es, és 0-1 ml-es tartományú pipetták
5.4. 0-100 μl-es és 0-5 μl-es mikrofecskendők
és (csak aeroszol minták adagolására) különleges gázzáró fecskendő tolattyúval (lásd a mintavételi eljárásról szóló 5. ábrát ( 9 )).
6. ELJÁRÁS
6.1. Minta-előkészítés
6.1.1. Az aeroszol készítményeket az 1980. december 22-i 80/1335/EGK ( 10 ) bizottsági irányelv mellékletének II. fejezete alapján mintázzuk, majd gázkromatográfiásan vizsgáljuk a 6.2.1. pontban leírt körülményeknek megfelelően.
6.1.2. Az említett II. fejezetnek megfelelően mintavételezett nem aeroszol termékeket vízzel 1-2 %-os etanol- vagy propán-2-ol-tartalomra hígítjuk, majd gázkromatográfiásan vizsgáljuk a 6.2.2. pontban leírt körülményeknek megfelelően.
6.2. Gázkromatográfia
6.2.1. Aeroszol minták esetén hővezetési detektort használunk.
6.2.1.1. Az oszlop töltete 10 % Hallcomid M18, 100-200 mesh CHROMOSORB WAW hordozón.
6.2.1.2. Az oszlop a következő megoldást eredményezi: R nem lehet kisebb, mint 1,5,
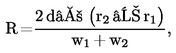
ahol:
r1 és r2 = két csúcs retenciós ideje percben,
w1 és w2 = ugyanezen csúcsoknak a magasság felénél mért szélessége mm-ben,
d' = a papírdiagram sebessége mm/percben.
6.2.1.3. Ez a felbontás az alábbi beállítások mellett érhető el:
| Oszlop | anyaga: | rozsdamentes acél |
| hossza: | 3,5 m | |
| belső átmérője: | 3 mm | |
| Hővezetési detektor híd árama: | 150 mA | |
| vivőgáz: | hélium | |
| nyomás: | 2,5 bar | |
| térfogatáram: | 45 ml/perc | |
| Hőmérsékletek: | ||
| injektor: | 150 °C | |
| detektor: | 150 °C | |
| oszlop: | 65 °C | |
A csúcsterületmérés pontossága elektronikus integrálással javítható.
6.2.2. Nem aeroszol minták
6.2.2.1. Az oszlop Chromosorb 105 vagy Porapak QS töltetű, és a lángionizációs detektort használjuk.
6.2.2.2. Az oszlop a következő megoldást eredményezi: R nem lehet kisebb, mint 1,5,
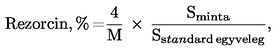
ahol:
r1 és r2 = két csúcs retenciós ideje percben,
w1 és w2 = ugyanezen csúcsoknak a magasság felénél mért szélessége mm-ben,
d' = a papírdiagram sebessége mm/percben.
6.2.2.3. Ez a felbontás az alábbi beállítások mellett érhető el:
| Oszlop | anyaga: | rozsdamentes acél |
| hossza | 2 m | |
| belső átmérője: | 3 mm | |
| Elektrométer érzékenysége: | 8 x 10-10 A | |
| Gázok: | ||
| vivőgáz: | nitrogén | |
| nyomás | 2,1 bar | |
| térfogatáram: | 40 ml/perc | |
| Detektor gázok: | hidrogén | |
| nyomás: | 1,5 bar | |
| térfogatáram: | 20 ml/perc | |
| Hőmérsékletek: | ||
| injektor | 150 °C | |
| detektor: | 230 °C | |
| oszlop | 120–130 °C | |
7. KALIBRÁCIÓS GÖRBE
7.1. A 6.2.1. szakasz szerinti gázkromatográfiás eljárás végrehajtása (Hallcomid M18 oszlop) esetén készítsük el az alábbi táblázatban felsorolt standard elegyeket. A készítés során a komponenseket pipettával adjuk az elegyhez, de a pontos bemérést úgy határozzuk meg, hogy minden egyes hozzáadást követően lemérjük a pipettát vagy a lombikot.
| Relatív tartalom (m/m %) | Metanol (ml) | Etanol vagy propán-2-ol (ml) | Kloroformmal töltve |
| körülbelül 2,5 % | 0,5 | 20 | 100 ml-re |
| körülbelül 5,0 % | 1,0 | 20 | 100 ml-re |
| körülbelül 7,5 % | 1,5 | 20 | 100 ml-re |
| körülbelül 10,0 % | 2,0 | 20 | 100 ml-re |
Injektáljunk 2-3 μl-t a gázkromatográfra a 6.2.1. szerinti körülmények beállítása mellett.
Számítsuk ki valamennyi elegyre a (metanol/etanol-) vagy a (metanol/propán-2-ol-) csúcsterületarányt. Ábrázoljuk a standard görbét a következőket feltüntetve:
| X-tengely: | % metanol/etanolra vagy propán-2-olra vonatkoztatva, |
| Y-tengely: | (metanol/etanol-) vagy (metanol/propán-2-ol-) csúcsterületarány. |
7.2. A 6.2.2. pontban leírt gázkromatográfiás eljárás végrehajtása (Porapak QS vagy Chromosorb 105) esetén készítsük el az alábbi táblázatban felsorolt standard elegyeket. A készítés során a komponenseket pipettával vagy mikrofecskendővel adjuk az elegyhez, de a pontos mennyiséget minden esetben úgy határozzuk meg, hogy a hozzáadást követően lemérjük a pipettát vagy a lombikot.
| Relatív tartalom (m/m %) | Metanol (μl) | Etanol vagy propán-2-ol (ml) | Hozzáadott víz térfogata |
| körülbelül 2,5 % | 50 | 2 | 100 ml |
| körülbelül 5,0 % | 100 | 2 | 100 ml |
| körülbelül 7,5 % | 150 | 2 | 100 ml |
| körülbelül 10,0 % | 200 | 2 | 100 ml |
Injektáljunk 2-3 μl-t a gázkromatográfra a 6.2.2. pontban írt körülmények beállítása mellett.
Számítsuk ki valamennyi elegyre a (metanol/etanol-) vagy a (metanol/propán-2-ol-) csúcsterület arányt. Ábrázoljuk a standard görbén a következőket feltüntetve:
| X-tengely: | % metanol/etanolra vagy propán-2-olra vonatkoztatva, |
| Y-tengely: | (metanol/etanol-) vagy (metanol/propán-2-ol-) csúcsterületarány. |
7.3. A standard kalibrációs görbének lineárisnak kell lennie.
8. MEGISMÉTELHETŐSÉG ( 11 )
A termék etanolra vagy propán-2-olra vonatkoztatott 5 % körüli metanoltartalma esetén azonos mintán, párhuzamosan végzett két mennyiségi meghatározás eredményei közötti különbség nem haladhatja meg a 0,25 %-ot.
1. függelék
ELŐÍRÁSOK A "CSOMÓZÁSHOZ"
ELŐÍRT TARTOZÉKOK
- Egy faorsó:
- külső átmérő 5 cm egy 1,5 cm átmérőjű lyukkal, amely a közepén megy keresztül. Helyezzünk bele négy acélszöget (amint azt az 1. és 2. ábra mutatja). Minden két szög között a távolság 1,8 cm legyen és a szögek 0,5 cm-re legyenek a lyuktól,
- egy merev tű (horgolótű típusú) a tefloncső felcsavarására,
- 5 m hosszú 1,6 mm-es tefloncső, belső átmérő 0,3 mm.
ELJÁRÁS
A "csomózás" megindításához a tefloncsövet be kell fűzni az orsó teteje felől és ki kell vezetni alul, a középső lyukon keresztül (kb. 10 cm túlnyúlást kell hagyni a csőnek az orsó aljához képest, annak érdekében, hogy a láncot keresztül lehessen húzni csomózás közben); ezután tekerjük a csövet a négy szög köré, ahogyan a 3. ábra mutatja.
A csomózás alját és tetejét fémgyűrűkkel és szorítócsavarokkal kell megvédeni; vigyázva arra, hogy a tefloncső ne törjön el a szorosra húzás során. Tekerjük körbe a csövet még egyszer minden szög körül és készítsük el az "öltést" a következőképpen:
- emeljük az alsó csövet a felső fölé (lásd 4. ábra). Ismételjük meg ezt az eljárást minden egyes szög esetén (1, 2, 3, 4 az óramutató járásával ellentétes irányban), amíg az 5 m-t vagy a megkívánt hosszúságot el nem érjük.
Hagyjunk ki kb. 10 cm csövet a lánc lezárásához. Fűzzük át a csövet a négy hurok mindegyikén és húzzuk meg óvatosan, hogy a lánc vége összeszoruljon.
MEGJEGYZÉS: Oszlopkimenethez illeszthető reaktorok számára készített csomózás kereskedelmi forgalomban kapható (Supelco).
Az orsó sematikus rajza
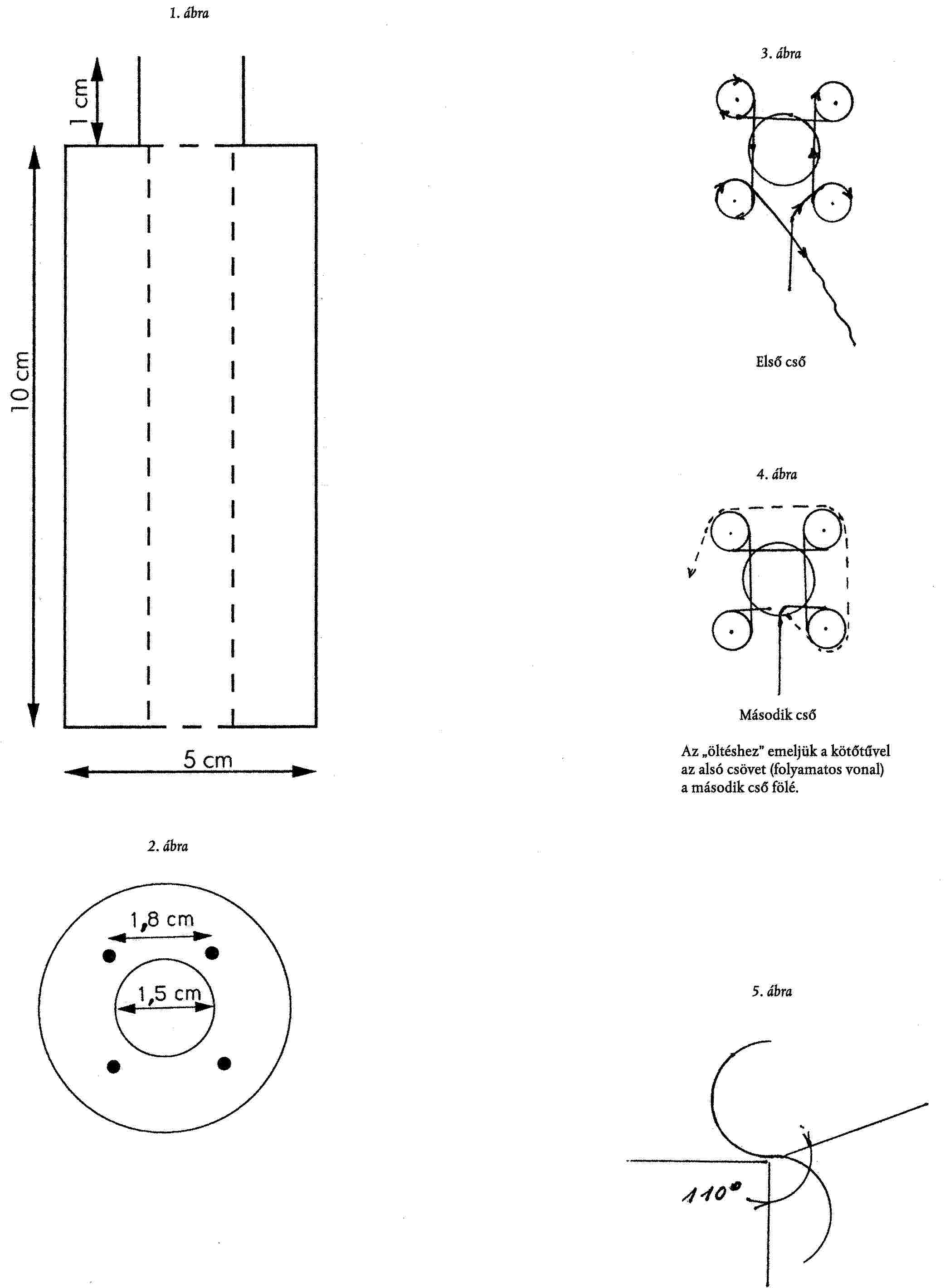
2. függelék
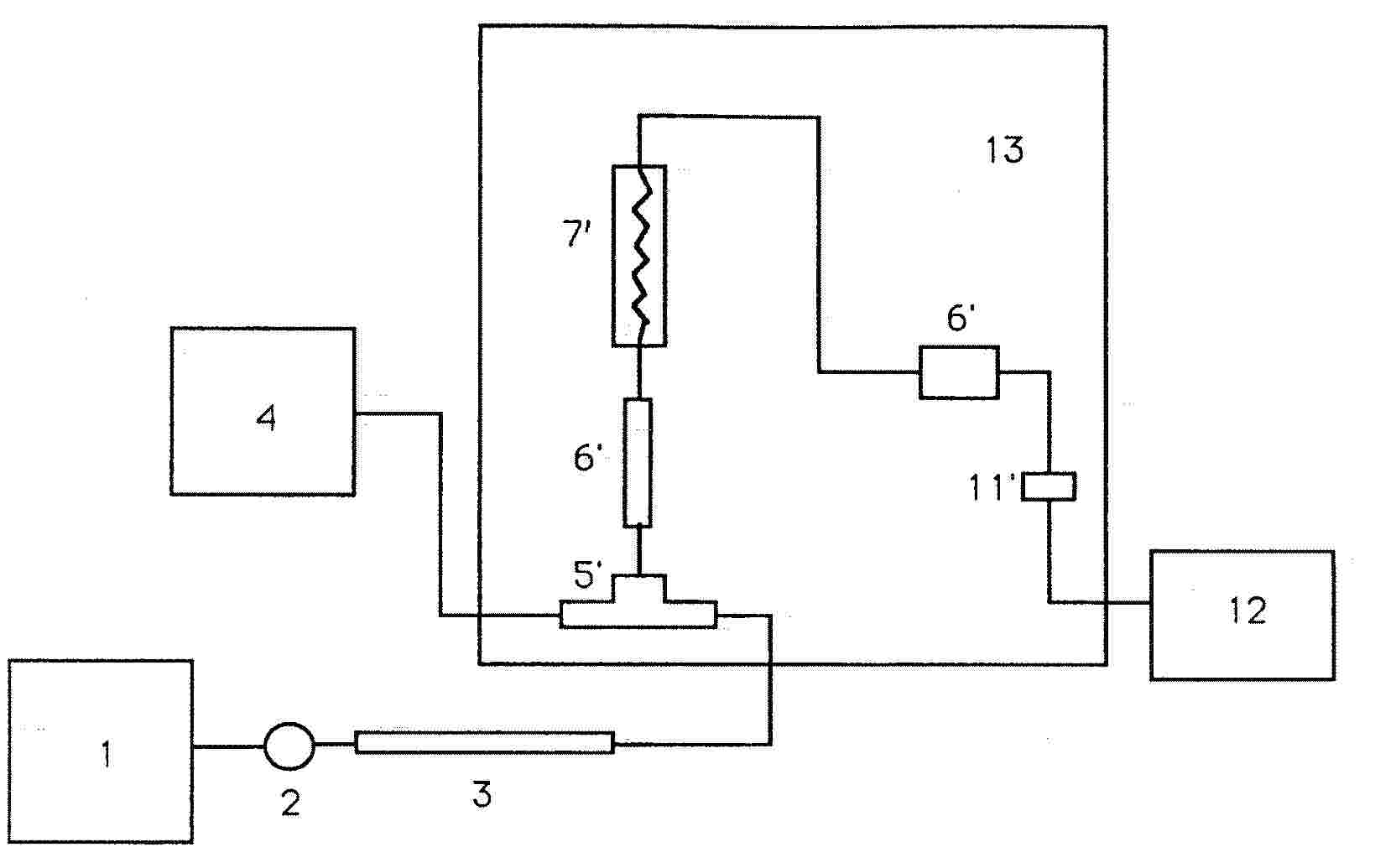
1 = HPLC pumpa
2 = Befecskendező szelep
3 = Oszlop előtét oszloppal
4 = Reagens pumpa
5 = T-darab holttérfogat nélkül
5' = T-darab (Vortex)
6-6' = Csatlakozó holttérfogat nélkül
7 = "Csomózás"
7' = Reaktor
8 = Háromnyakú lombik forró vízzel
9 = Melegítőkráter
10 = Hűtő
11 = Hőcserélő cső rozsdamentes acélból
11' = Hőcserélő
12 = Látható-UV detektor
13 = PCRS 520 oszlop utáni modul
13 = PCRS 520 utóoszlop modul
( 1 ) HL L 192., 1979.7.31., 35. o.
( 2 ) HL L 262., 1976.9.27., 169. o.
( 3 ) HL L 383., 1980.12.31., 27. o.
( 4 ) ISO 5725 szabvány szerint.
( 5 ) ISO 5725 szabvány szerint.
( 6 ) ISO 5725 szabvány szerint
( 7 ) ISO 5725 szabvány szerint
( 8 ) ISO 5725 szabvány szerint.
( 9 ) HL L 383., 1980.12.31., 27. o.
( 11 ) ISO 5725 szabvány szerint.
Lábjegyzetek:
[1] A dokumentum eredetije megtekinthető CELEX: 31982L0434 - https://eur-lex.europa.eu/legal-content/HU/ALL/?uri=CELEX:31982L0434&locale=hu Utolsó elérhető, magyar nyelvű konszolidált változat CELEX: 01982L0434-19900410 - https://eur-lex.europa.eu/legal-content/HU/ALL/?uri=CELEX:01982L0434-19900410&locale=hu