
32007R0702[1]
A Bizottság 702/2007/EK rendelete ( 2007. június 21. ) az olívaolaj és az olívamaradék-olaj jellemzőiről és az ezekre vonatkozó elemzési módszerekről szóló 2568/91/EGK rendelet módosításáról
A BIZOTTSÁG 702/2007/EK RENDELETE
(2007. június 21.)
az olívaolaj és az olívamaradék-olaj jellemzőiről és az ezekre vonatkozó elemzési módszerekről szóló 2568/91/EGK rendelet módosításáról
AZ EURÓPAI KÖZÖSSÉGEK BIZOTTSÁGA,
tekintettel az Európai Közösséget létrehozó szerződésre,
tekintettel az olívaolaj és az étkezési olajbogyó piacának közös szervezéséről és a 827/68/EGK rendelet módosításáról szóló, 2004. április 29-i 865/2004/EK tanácsi rendeletre (1) és különösen annak 5. cikke (3) bekezdésére,
mivel:
(1) A 2568/91/EGK bizottsági rendelet (2) meghatározza az olíva- és olívamaradék-olajok fizikai és kémiai jellemzőit, és e jellemzők értékelésének módszereit. Ezeket a módszereket, illetve a jellemzőkre vonatkozó határértékeket - a vegyészeti szakértők véleményét figyelembe véve, és a Nemzetközi Olívaolaj Tanács keretében végzett munka eredményeivel összhangban - ki kell igazítani.
(2) A szakértők nevezetesen úgy vélték, hogy a 2-gliceril monopalmitát százalék meghatározása alkalmas az észterezett olajok pontosabb kimutatására. A szűz olívaolajokban található sztigmasztadién határértékének leszállítása szintén elősegíti a szűz, illetve finomított olívaolajok megfelelőbb szétválasztását.
(3) Az új előírásokhoz való alkalmazkodáshoz és az alkalmazásukat lehetővé tévő eszközök kialakításához szükséges idő biztosítása, illetve a kereskedelmi ügyletekben előálló zavarok elkerülése érdekében e rendeletet csak 2008. január 1-jétől kell alkalmazni. Ugyanezen okokból rendelkezni kell arról is, hogy a fenti időpontot megelőzően a Közösség területén jogszerűen előállított és címkézett, illetve a Közösségbe jogszerűen importált és szabad forgalomba bocsátott olívaolajok és olívamaradék-olajok a készletek kimerüléséig kereskedelmi forgalomban maradhassanak.
(4) Az e rendeletben előírt intézkedések összhangban állnak az Olívaolaj- és Étkezésiolajbogyó-piaci Irányítóbizottság véleményével,
ELFOGADTA EZT A RENDELETET:
1. cikk
A 2568/91/EGK rendelet a következőképpen módosul:
1. A 2. cikk (1) bekezdésének hatodik francia bekezdése helyébe a következő szöveg lép:
"- a 2-gliceril monopalmitát százalékának meghatározása a VII. melléklet szerinti módszerrel,".
2. A mellékletek az e rendelet mellékletében foglaltak szerint módosulnak.
2. cikk
Ez a rendelet az Európai Unió Hivatalos Lapjában való kihirdetését követő harmadik napon lép hatályba.
Ezt a rendeletet 2008. január 1-jétől kell alkalmazni.
Azonban azok a termékek, amelyeket 2008. január 1-je előtt jogszerűen gyártottak és címkéztek az Európai Közösségben, illetve jogszerűen hoztak be az Európai Közösségbe és bocsátottak szabad forgalomba, a készletek kimerüléséig forgalomba hozhatók.
Ez a rendelet teljes egészében kötelező és közvetlenül alkalmazandó valamennyi tagállamban.
Kelt Brüsszelben, 2007. június 21-én.
a Bizottság részéről
Mariann FISCHER BOEL
a Bizottság tagja
(1) HL L 161., 2004.4.30., 97. o.
(2) HL L 248., 1991.9.5., 1. o. A legutóbb az 1989/2003/EK rendelettel (HL L 295., 2003.11.13., 57. o.) módosított rendelet.
MELLÉKLET
A 2568/91/EGK rendelet mellékletei a következőképpen módosulnak:
1. Az összefoglalás a következőképpen módosul:
a) a II. melléklet címe helyébe a következő cím lép:
"A szabad zsírsavak meghatározása hideg injektálásos módszerrel"
b) a VII. melléklet címe helyébe a következő cím lép:
"A 2-gliceril monopalmitát százalékának meghatározása";
2. Az I. melléklet helyébe a következő melléklet lép:
"I. MELLÉKLET
AZ OLÍVALAJ JELLEMZŐI
Megjegyzések:
(a) Az eredményeket ugyanannyi tizedesjegy pontossággal kell megadni, mint amennyi az egyes jellemzők esetében meg van adva.
Az utolsó értékes számjegyet fel kell kerekíteni a következő számjegyre, ha az ezt követő, nem értékes számjegy 4-nél nagyobb.
(b) Ha a jellemzői közül bármelyik kívül esik a megállapított határértékeken, az olajat egy másik kategóriába kell sorolni, vagy a tisztaság tekintetében nem megfelelőnek kell minősíteni.
(c) Az olaj minőségével kapcsolatos, csillaggal (*) jelzett jellemzők azt jelentik, hogy:
- a lampante olívaolaj esetében ezeket a határértékeket nem kell egyidejűleg betartani;
- egyéb szűz olívaolajok esetében ezek közül a határértékek közül legalább egynek a be nem tartása a szűz olívaolajok kategóriáján belüli átsorolással jár, bár az olívaolaj továbbra is a szűz olívaolaj kategóriák valamelyikéhez tartozik.
(d) A két csillaggal (**) jelölt jellemzők az olaj minőségére vonatkoznak, és azt jelentik, hogy egyik érintett olívamaradék-olaj esetében sem kell egyidejűleg betartani ezeket a határokat."
| SZÖVEG HIÁNYZIK |
| SZÖVEG HIÁNYZIK |
3. Az 1. függelék a következőképpen módosul:
a) az első francia bekezdés helyébe a következő szöveg lép:
| SZÖVEG HIÁNYZIK |
b) a harmadik francia bekezdés helyébe a következő szöveg lép:
| SZÖVEG HIÁNYZIK |
4. A II. melléklet címe helyébe a következő szöveg lép:
5. A IV. melléklet helyébe a következő szöveg lép:
"
IV. MELLÉKLET
A VIASZTARTALOM MEGHATÁROZÁSA KAPILLÁRIS GÁZKROMATOGRÁFIÁVAL
1. CÉL
Ez a módszer az olívaolajok viasztartalmának meghatározására szolgáló eljárást írja le. A viaszok leválasztása a szénatomok száma alapján történik. A módszer alkalmas a sajtolással és az extrakcióval kinyert olívaolajok (olívamaradék-olajok) megkülönböztetésére.
2. ALAPELV
A zsírszerű anyag oszlopkromatográfiás elválasztása, a megfelelő belső standard hozzáadásával, hidratáltszilikagél-oszlopon; a tesztkörülmények között elsőként eluálódott (a trigliceridekénél kisebb polaritású) frakció leválasztása, majd kapilláris gázkromatográfia segítségével történő analízise.
3. BERENDEZÉS
3.1. 25 ml-es térfogatú Erlenmeyer-lombik.
3.2. 15,0 mm belső átmérőjű, 30-40 cm hosszú kromatográfiás üvegoszlop csappal.
3.3. Gázkromatográf kapilláris oszloppal történő használatra, a következőkből álló, közvetlenül az oszlopra történő injektálásra alkalmas rendszerrel felszerelve:
3.3.1. Termosztatikus kamra az oszlopok számára (oszlopkemence), hőmérséklet-programozott fűtéssel.
3.3.2. Közvetlenül az oszlopba vezetett, hideg befecskendező rendszer.
3.3.3. Láng-ionizációs detektor és konverter erősítő.
3.3.4. Regisztráló-integrátor berendezés a konverter erősítővel (3.3.3.) történő használatra, amelynek a válaszideje nem haladja meg az egy másodpercet, és változtatható papírsebességű. (Lehetséges olyan informatizált rendszerek használata is, amelyek alkalmasak a gázkromatográfiai adatok számítógépes feldolgozására.)
3.3.5. Üvegből vagy ömlesztett szilícium-dioxidból készült 8-12 mm hosszúságú, 0,25-0,32 mm belső átmérőjű kapilláris oszlop, belülről egyenletesen 0,10-0,30 μm rétegvastagságú folyadékfázissal borítva. (a folyadékfázis a célnak megfelelő, kereskedelmi forgalomban SE-52 vagy SE-54 jelzésű lehet.)
3.4. 10 μl térfogatú mikrofecskendő közvetlenül az oszlopra történő injektáláshoz, keményített tűvel.
3.5. Vibráló elektromos keverő.
3.6. Forgó párologtató.
3.7. Görgős kemence.
3.8. Analitikai mérleg, + 0,1 mg-os mérési pontossággal.
3.9. Szokványos laboratóriumi üvegeszközök.
4. REAGENSEK
4.1. 60 és 200 μm közötti szemcsenagyságú szilikagél.
A szilikagélt 500 °C-on legalább négy órán keresztül melegíteni kell a kemencében. A kihűlést követően a felhasznált szilikagél-mennyiséghez képest 2 % vizet kell hozzáadni. Keverje át a keveréket megfelelő módon, amíg egyenletes masszát kap. Felhasználás előtt legalább 12 órán keresztül tartsa sötét helységben.
4.2. n-hexán, kromatográfiai minőségű.
4.3. Etil-éter, kromatográfiai minőségű.
4.4. n-heptán, kromatográfiai minőségű.
4.5. Hexános lauril-arachidát standardoldat, 0,1 % (m/V) oldat (belső standard). (Palmitil palmitát vagy mirisztil sztearát is megfelel.)
4.5.1. Szudán 1 (1-fenilazo-2-naftol).
4.6. Vivőgáz: hidrogén és hélium, gázkromatográfiai felhasználáshoz megfelelő tisztaságú.
4.7. Segédgázok:
- hidrogén, gázkromatográfiai felhasználáshoz megfelelő tisztaságú,
- levegő, gázkromatográfiai felhasználáshoz megfelelő tisztaságú.
5. ELJÁRÁS
5.1. A kromatográfiás oszlop előkészítése
Szuszpendáljon 15 g szilikagélt (4.1.) az n-hexánban (4.2.), és töltse az oszlopba (3.2.). A teljes ülepedés után elektromos rázóberendezéssel (3.5.) tömörítse, hogy homogénebb kromatográfiás réteget kapjon. 30 ml n-hexánnal mossa át az oszlopot, az esetleges szennyeződések eltávolítása érdekében. Mérjen le pontosan a mérleggel (3.8.) 500 mg-ot a mintából a 25 ml térfogatú Erlenmeyer-lombikba (3.1.), majd adja hozzá a megfelelő mennyiségű belső standardot (4.5.), a feltételezett viasztartalomnak megfelelően. Például olívaolaj esetében 0,1 mg lauril-arachidát oldatot, olívamaradék-olaj esetében 0,25-0,5 mg lauril-arachidát oldatot adjon hozzá. Az ily módon előkészített mintát 2 x 2 ml n-hexán segítségével vigye fel a kromatográfiás oszlopra (4.2.).
Hagyja lefutni az oldatot 1 mm-rel az abszorbens felszíne fölé, majd további 70 ml n-hexánnal mossa át az oszlopot, a természetesen jelen lévő n-alkánok eltávolítása érdekében. Kezdje el a kromatográfiás eluálást, és gyűjtsön össze 180 ml 99:1 arányú n-hexán-etil-éter elegyet úgy, hogy 10 másodpercenként megközelítőleg 15 csepp folyjon át. A minta eluálását 22 + 4 °C -os szobahőmérsékleten kell elvégezni.
Megjegyzések:
- A 99:1 arányú n-hexán/etil-éter keveréket minden nap frissen kell elkészíteni.
- A viaszok megfelelő eluálódásának vizuális ellenőrzése érdekében a mintaoldathoz hozzáadhat 100μl szudánt, az eluáló elegy 1 %-a arányában. Mivel a színezőanyag retenciós ideje a viaszok és a trigliceridek között helyezkedik el, amikor a színezék eléri a kromatográfiás oszlop alját, függessze fel az eluálást, ekkorra ugyanis valamennyi viasz eluálódott.
Az így kapott frakciót a rotációs bepárlóban (3.6.) párolja szárazra, amíg az oldószer gyakorlatilag teljesen eltűnik belőle. Az oldószer utolsó 2 ml-ét gyenge nitrogénárammal távolítsa el; majd adjon hozzá 2-4 ml n-heptánt.
5.2. Gázkromatográfiás elemzés
5.2.1. Előzetes műveletek
A kapilláris oszlopot illessze be a gázkromatográfba (3.3) úgy, hogy az oszlop bemenetét az oszlopra szerelt (»on-column«) rendszerhez, az oszlop kimenetét pedig a detektorhoz csatlakoztassa. Végezze el a gázkromatográfiai rendszer összeszerelésének általános ellenőrzését (gázszerelvények szorossága, a detektor hatékonysága, a regisztráló rendszer hatékonysága stb.).
Az első alkalommal használt kapilláris oszlopokat kondicionálni kell. Fúvasson át egy kevés vivőgázt a kapilláris oszlopon, majd kapcsolja be a gázkromatográfiás berendezést. Melegítse fokozatosan addig, amíg körülbelül 4 óra elteltével eléri a 350 °C hőmérsékletet. Ezt a hőmérsékletet tartsa legalább két órán keresztül, majd hozza a berendezést üzemi körülmények közé (gázáram szabályozása, bontóláng begyújtása, csatlakoztatás az elektronikus íróhoz (3.3.4.), a kapilláris oszlop kemencéje, a detektor hőmérsékletének beállítása stb.) és állítsa be a jelet az analízis során tervezett legmagasabb szintnél legalább kétszer nagyobb érzékenységre. Az alapvonalnak lineárisnak kell lennie, illetve mindennemű csúcstól és ingadozástól mentesnek.
A negatív egyenes vonalú drift az oszlop illesztékeinek tökéletlen tömítettségét, míg a pozitív drift az oszlop nem megfelelő kondicionálását jelzi.
5.2.2. Az üzemi körülmények megválasztása
Általában véve az irányadó üzemi körülmények a következők:
- oszlophőmérséklet:
| 20 °C/perc | 5 °C/perc | 20 °C/perc | ||||
| kiindulási hőmérséklet: 80 °C (1′) | → | 240 °C | → | 325 °C (6′) | → | 340 °C (10′) |
- a detektor hőmérséklete: 350 °C,
- a befecskendezett anyag mennyisége: 1 μl az n-heptánoldatból (2-4 ml),
- vivőgáz: hélium vagy hidrogén, a kiválasztott gáz számára optimális lineáris sebességgel (lásd a függeléket),
- a berendezés érzékenysége: az alábbi körülményeknek megfelelően:
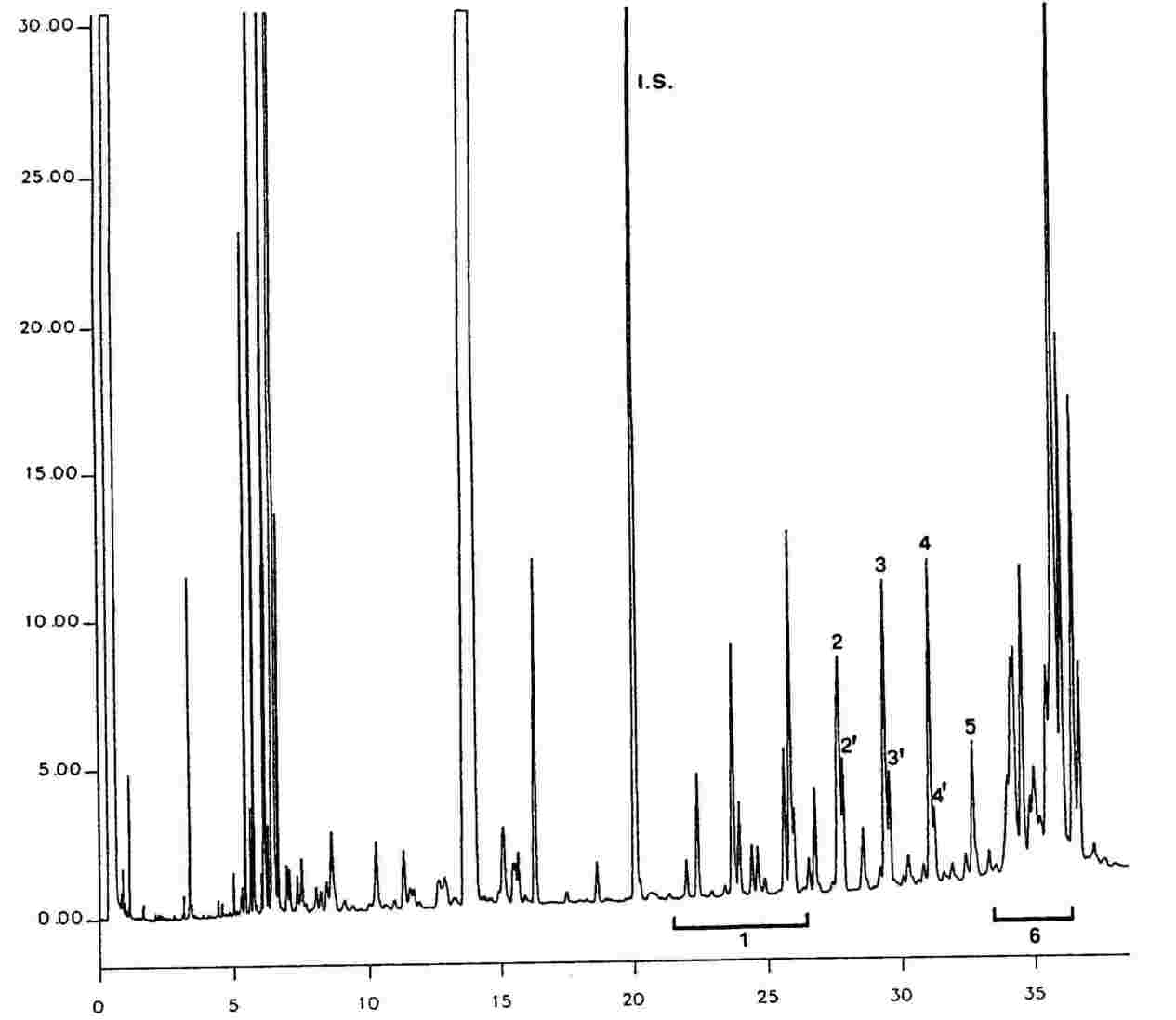
A fenti feltételek az oszlop és a gázkromatográf jellemzőinek megfelelően módosíthatók, hogy a kapott kromatogramok lehetővé tegyék valamennyi viasz leválasztását, a csúcsok kielégítő felbontásban láthatók legyenek (lásd az ábrát), miközben a C32 belső standard retenciós ideje 18 ± 3 perc. A viaszok legreprezentatívabb csúcsa a teljes skálaérték minimum 60 %-a legyen.
A csúcsok integrálásához a paramétereket úgy kell beállítani, hogy az lehetővé tegye az érintett csúcsok területeinek pontos becslését.
Megjegyzés: Tekintettel a magas véghőmérsékletre, pozitív drift előfordulása elfogadható, amely azonban nem haladhatja meg a teljes skálaérték 10 %-át.
5.3. Az elemzés végrehajtása
A 10 μl térfogatú mikrofecskendő segítségével szívjon fel 1 μl oldatot; a mikrofecskendő dugattyúját felfelé mozgatva ürítse ki a tűt. A tűt vezesse be a befecskendező berendezés válaszfalán, majd egy-két másodperc elmúltával fecskendezze be gyorsan az oldatot, és öt másodperc múlva lassan húzza ki a tűt.
Addig folytassa a rögzítést, ameddig a viaszok teljesen eluálódtak.
Az alapvonalnak minden esetben meg kell felelnie az előírt feltételeknek.
5.4. A csúcsok azonosítása
Az egyes csúcsok azonosítását a retenciós idő alapján és az azonos körülmények között analizált, ismert retenciós idejű viaszkeverékekkel összehasonlítva végezze.
A szűz olívaolajban található viaszok kromatogramja az ábrán látható.
5.5. Mennyiségi értékelés
A belső standard és a nyílt szénláncú C40-C46 észterek csúcsainak területét elektronikus integrálással számítsa ki.
Az egyes észterek mg/kg zsírszerű anyagban kifejezett viasztartalmát a következő képlet segítségével lehet kiszámítani:
ahol:
Ax = az egyes észterek csúcsának területe négyzetmilliméterben;
As = a belső standard csúcsának területe négyzetmilliméterben;
ms = a hozzáadott belső standard tömege milligrammban;
m = a meghatározni kívánt minta tömege grammban.
6. AZ EREDMÉNYEK KIFEJEZÉSE
Jegyezze fel a különböző C40-C46 viasztartalmak összegét mg/kg zsírszerű anyagban megadva (ppm).
Megjegyzés: A meghatározandó összetevők a C40-es és a C46-os észterek közötti tartományban lévő páros szénatomok csúcsainak felelnek meg, az alábbi ábrán látható olívaolajviasz-kromatogram példájának megfelelően. Ha a C46-os észter duplán jelenik meg, az azonosítás érdekében tanácsos elvégezni egy olyan olívamaradék-olaj viaszfrakcióinak az analízisét, amelyen a C46-os csúcs könnyen felismerhető, mivel tisztán kiemelkedik.
Az eredményeket egy tizedesjegy pontosságig kell megadni.
Ábra
Egy olívaolaj viasztartalmainak kromatogramja (9)
Függelék
A gáz lineáris sebességének meghatározása
Fecskendezzen be 1-3 μ metánt (vagy propánt) a normál üzemi körülményekre beállított gázkromatográfba. Stopperóra segítségével mérje meg a metán vagy propán oszlopon történő átáramlásának idejét, a befecskendezés pillanatától a csúcs eluálásának pillanatáig (tM).
A lineáris sebességet - cm/s-ben - az L/tM összefüggés adja meg, ahol L az oszlop hosszúsága cm-ben, tM pedig a stopperórával mért idő másodpercben.
";
6. A VII. melléklet helyébe a következő szöveg lép:
"VII. MELLÉKLET
A 2-GLICERIL MONOPALMITÁT SZÁZALÉKÁNAK MEGHATÁROZÁSA
1. CÉL ÉS ALKALMAZÁSI TERÜLET
Ez a módszer a palmitinsav-tartalom meghatározásának analitikus eljárását írja le a trigliceridek 2-pozícióján, a 2-gliceril monopalmitát százalékának értékelésének segítségével.
A módszer a szobahőmérsékletű (20 °C), folyékony növényi olajokra alkalmazható.
2. ALAPELV
Az előkészítés után az olajminta reakcióba lép a pankreatin-lipázzal: a triglicerid molekula 1- és 3-pozícióján végbemenő részleges és specifikus hidrolízis nyomán elválasztódnak a 2-pozíción lévő monogliceridek. A monoglicerid-frakción belül a 2-gliceril monopalmitát százalékát - a szililesedés után - kapilláris gázkromatográfia segítségével határozzuk meg.
3. BERENDEZÉS ÉS LABORATÓRIUMI ESZKÖZÖK
3.1. 25 ml térfogatú Erlenmeyer-lombik
3.2. 100, 250 és 300 ml térfogatú főzőpoharak
3.3. 21-23 mm belső átmérőjű, 400 mm hosszú kromatográfiás üvegoszlop összesütött üveglemezzel a csapnál
3.4. 10, 50, 100 és 200 ml térfogatú kalibrált kémcsövek
3.5. 100 és 250 ml térfogatú gömbölyű fenekű lombik
3.6. Forgó párologtató
3.7. 10 ml térfogatú, kúpos aljú centrifugakémcső csiszolt üvegdugóval
3.8. Laboratóriumi centrifuga 10 és 100 ml térfogatú kémcsövekkel
3.9. A hőmérsékletet 40 °C + 0,5 °C-on tartó termosztát
3.10. 1 és 2 ml térfogatú mérőpipetták
3.11. 1 ml-es fecskendő
3.12. 100 μl-es mikrofecskendő
3.13. 1 000 ml-es tölcsér
3.14. Gázkromatográf kapilláris oszloppal, az alábbiakból álló, közvetlenül az oszlopra történő injektálásra alkalmas»on column« rendszerrel, valamint a kiválasztott hőmérsékletet 1 °C pontossággal tartó kemencével felszerelve
3.15. Közvetlenül az oszlopba vezetett, »on column« hideg befecskendező rendszer
3.16. Lángionizációs detektor és elektrométer
3.17. Regisztráló-integrátor berendezés az elektrométerrel történő használatra, amelynek a válaszideje nem haladja meg az egy másodpercet, és változtatható papírsebességű
3.18. Üvegből vagy ömlesztett szilícium-dioxidból készült, 8-12 méter hosszúságú, 0,25-0,32 mm belső átmérőjű, 5 % 0,10-0,30 μm rétegvastagságú metil-polisziloxánnal vagy fenil-metil-polisziloxánnal bélelt kapilláris oszlop, amely 370 °C-on használható
3.19. 10 μl-es, legalább 7,5 cm hosszú mikrofecskendő keményített tűvel, közvetlenül az oszlopra történő injektáláshoz.
4. REAGENSEK
4.1. 0,063 és 0,200 mm közötti szemcsenagyságú szilikagél (70/280 mesh), a következőképpen elkészítve: helyezze a szilikagélt egy porcelán csészébe, 160 °C-os kemencében szárítsa ki 4 órán keresztül, majd hagyja kihűlni szobahőmérsékleten egy szárítóban. Adjon hozzá a szilikagél súlya 5 %-ának megfelelő vízmennyiséget a következőképpen: egy 500 ml térfogatú Erlenmeyer-lombikban mérjen le 152 g szilikagélt, adjon hozzá 8 g desztillált vizet, dugaszolja be, majd óvatosan rázza fel, hogy a víz egyenletesen oszoljon el. Felhasználás előtt hagyja állni legalább 12 óráig.
4.2. n-hexán (kromatográfiai minőségű)
4.3. Izopropanol
4.4. Izopropanol, vízoldat 1/1 (V/V)
4.5. Pankreatin lipáz. A felhasznált lipáznak 2,0 és 10 lipáz egység/mg lipáztevékenységet kell mutatnia (Kereskedelmi forgalomban is kaphatók 2 és 10 lipáz egység/enzim mg lipáztevékenységet mutató pankreatin lipázok.)
4.6. Tris-hidroximetilaminometán-pufferoldat: 1 M vizes oldatát állítsa be pH 8 értékűre koncentrált HCl (sósav) (1/1 V/V) hozzáadásával (ellenőrizze potenciométerrel)
4.7. Nátrim-kolát (enzim minőségű), 0,1 %-os vizes oldat (ezt az oldatot az elkészítését követő 2 héten belül fel kell használni)
4.8. Kalcium-klorid, 22 %-os vizes oldat
4.9. Dietil-éter kromatográfiához
4.10. Kifejlesztőoldat: n-hexán/dietil-éter keverék (87/13) (V/V)
4.11. Nátrium-hidroxid, (súlyra) 12 %-os oldat
4.12. Fenolftalein, 1 %-os etanol oldat
4.13. Vivőgáz: hidrogén vagy hélium, gázkromatográfiai felhasználáshoz megfelelő tisztaságú
4.14. Segédgázok: hidrogén, minimum 99 %-os, nedvességtől és szerves szennyeződésektől mentes, illetve levegő, szintén gázkromatográfiai felhasználáshoz megfelelő tisztaságú
4.15. Szilanizációs reagensek: 9/3/1 (V/V/V) arányú piridin-hexametildiszilizin-trimetilkloroszilán keverék. A használatra kész oldatok kereskedelmi forgalomban kaphatók; egyéb szilanizációs reagensek is felhasználhatók, mint például N, 0-bis (trimetilszilil) trifluoroacetamid + 1 % trimetilkloroszilán azonos térfogatú víztelen piridinnel való keveréshez.
4.16. Referenciaminták: tiszta monogliceridek, vagy olyan monoglicerid-keverékek, amelyeknek ismert, a mintához hasonló százalékos összetételük van.
5. ELJÁRÁS
5.1. A minta előkészítése
5.1.1. A 3 %-nál kisebb szabad savasságú olajokat nem szükséges semlegesíteni a szilikagéllel elvégzett oszlopkromatográfia előtt. A 3 %-nál nagyobb szabad savasságú olajokat az 5.1.1.1. pontnak megfelelően semlegesíteni kell.
5.1.1.1. Töltsön az 1 000 ml-es tölcsérbe (3.13.) 50 g olajat és 200 ml n-hexánt. Adjon hozzá 100 ml izopropanolt, és az olaj 5 %-kal megemelt szabad savasságának megfelelő mennyiségű 12 %-os nátrium-hidroxid oldatot (4.11.). Egy percen keresztül rázza erőteljesen. Adjon hozzá 100 ml desztillált vizet, majd rázza fel ismét, ezután hagyja leülepedni.
A leválasztást követően távolítsa el az alsó szappanréteget. Távolítsa el az esetleges többi réteget (nyálka, oldhatatlan anyag) is. Mossa át a semlegesített olaj hexánoldatát többször 50-60 ml 1/1 (V/V) izopropanol/víz oldatban (4.4.), amíg a fenolftalein rózsaszín árnyalata el nem tűnik.
A hexán nagyobbik részét távolítsa el vákuum alatt végzett bepárlással (használjon például forgó párologtatót), és töltse át az olajat egy 100 ml-es gömbölyű lombikba (3.5.). Szárítsa ki az olajat vákuumban, amíg az oldószer teljesen el nem távozik.
Az eljárás végén az olaj savasságának 0,5 %-nál alacsonyabbnak kell lennie.
5.1.2. A fent leírtak szerint előkészített olajból 1,0 g-ot töltsön egy 25 ml térfogatú Erlenmeyer-lombikba (3.1.), és oldja fel 10 ml kifejlesztőoldatban (4.10.). Hagyja leülepedni az oldatot legalább 15 percen keresztül, a szilikagéllel végzett oszlop-kromatográfia előtt.
Ha az oldat zavaros, centrifugázza ki, a kromatográfiához szükséges optimális feltételeket teremtve. (Használatra kész, 500 mg-os SPE szilikagél hüvelyek is felhasználhatók a célra).
5.1.3. A kromatográfiás oszlop előkészítése
Öntsön az oszlopba (3.3.) megközelítőleg 30 ml kifejlesztőoldatot (4.10.), helyezzen az oszlop alsó felébe üvegrúd segítségével egy vattadarabot; nyomkodja meg, hogy a levegő távozzon belőle.
Egy főzőpohárban készítsen szuszpenziót, 25 g szilikagélt (4.1.) megközelítőleg 80 ml kifejlesztőoldatban feloldva, és egy tölcsér segítségével öntse az oszlopba.
Ellenőrizze, hogy az egész szilikagél be lett töltve az oszlopba; mossa át a kifejlesztőoldattal (4.10.), nyissa ki a csapot, és hagyja, hogy a folyadék szintje érjen fel megközelítőleg 2 mm-rel a szilikagél felső szintje fölé.
5.1.4. Oszlopkromatográfia
Egy 25 ml térfogatú Erlenmeyer-lombikban (3.1.) mérjen le pontosan 1,0 g, az 5.1. pontnak megfelelően előkészített mintát.
Oldja fel a mintát 10 ml kifejlesztőoldatban (4.10.). Az így kapott oldatot öntse az 5.1.3. pontnak megfelelően előkészített kromatográfiás oszlopba. Kerülje az oszlop felszínének mozgatását.
Nyissa meg a csapot, és hagyja átfolyni a mintaoldatot, amíg az el nem éri a szilikagél szintjét. Fejlessze ki 150 ml kifejlesztőoldattal. Állítsa a térfogatáramot 2 ml/percre (úgy, hogy körülbelül 60-70 perc alatt 150 ml oldat folyjon át az oszlopba).
Az eluátumot fogja fel egy előzetesen lemért, 250 ml térfogatú gömbölyű lombikban. Vákuum alatt párologtassa el az oldószert, az utolsó nyomokat nitrogénáram segítségével távolítva el.
Mérje le a gömbölyű lombikot, és számítsa ki a kinyert kivonatot.
(Használatra kész SPE szilikagél hüvelyek használata esetén a következőképpen járjon el: Öntsön 1 ml oldatot (5.1.2.) az előzetesen 3 ml n-hexánnal előkészített hüvelyekbe.)
Az átmosás után fejlessze ki az oldatot 4 ml 9/1 (V/V) arányú n-hexán/dietil-éter keverékkel.
Fogja fel az eluátumot egy 10 ml térfogatú kémcsőben, és párologtassa el nitrogénáram segítségével a teljes kiszáradásig.
A kiszárított maradékot tegye ki a pankreatin-lipáz hatásának (5.2.). Rendkívül fontos, hogy SPE hüvelyen való áthaladás előtt, illetve után egyaránt ellenőrizze a zsírsavösszetételt).
5.2. Hidrolízis pankreatin-lipázzal
5.2.1. Mérjen a centrifugakémcsőbe 0,1 g, az 5.1. pontnak megfelelően előkészített olajat. Adjon hozzá 2 ml pufferoldatot (4.6.), 0,5 ml nátrium-kolát-oldatot (4.7.) és 0,2 ml kalcium-klorid-oldatot; minden hozzáadás után alaposan rázza fel a keveréket. Zárja le a kémcsövet a csiszolt dugóval, majd tegye a 40 ± 0,5 °C hőmérsékleten tartott termosztátba (4.17.).
5.2.2. Adjon hozzá 20 mg lipázt, rázza fel alaposan (kerülje a dugó benedvesedését), majd tegye a kémcsövet a termosztátba pontosan 2 percre. Ezután vegye ki, rázza fel erőteljesen pontosan 1 percig, és hagyja kihűlni.
5.2.3. Adjon hozzá 1 ml dietil-étert, zárja le a dugóval és erőteljesen rázza fel, majd centrifugálás után az éteroldatot mikrofecskendő segítségével öntse át egy tiszta és száraz kémcsőbe.
5.3. A szilanizált származékok és a gázkromatográfia előkészítése
5.3.1. Egy mikrofecskendő segítségével töltsön 100 μl oldatot (5.2.3.) egy 10 ml térfogatú kúpos fenekű kémcsőbe.
5.3.2. Enyhe nitrogénárammal párologtassa el az oldószert, adjon hozzá 200 μl szilanizációs reagenst (4.15.), dugaszolja be a kémcsövet, és hagyja ülepedni 20 percig.
5.3.3. A 20 perc leteltével adjon hozzá 1-5 ml n-hexánt (a kromatográfiai körülmények függvényében): az így kapott oldat készen áll a gázkromatográfiás eljárásra.
5.4. Gázkromatográfia
Az üzemi körülmények a következők:
- az injektor hőmérséklete (oszlopra szerelt »on column« injektor): az oldószer forráspontjánál (68 °C) alacsonyabb,
- a detektor hőmérséklete: 350 °C,
- az oszlop hőmérséklete: a kemence hőmérsékletének programozása: 1 percig 60 °C, percenként 15 °C-kal emelve 180 °C-ig, majd percenként 5 °C-kal 340 °C-ig, majd 13 percig 340 °C,
- vivőgáz: hidrogén vagy hélium, az 1. ábrán tükröződő felbontáshoz szükséges lineáris sebességre beállítva. A C54 triglicerid retenciós idejének 40 ± 5 percnek kell lennie (lásd a 2. ábrát). (A fent leírt üzemi körülmények tájékoztató jellegűek. Minden esetben optimalizálni kell őket a kívánt felbontás elérése érdekében. A 2-gliceril monopalmitátnak megfelelő csúcs minimális magasságának a regisztráló berendezés skálája 10 %-ának kell lennie),
- a befecskendezett anyag mennyisége: 0,5-1 μl n-hexán oldat (5 ml) (5.3.3.).
5.4.1. A csúcsok azonosítása
Az egyes monogliceridek azonosítását a retenciós idők alapján, és az azonos körülmények között analizált standard monoglicerid-keverékek retenciós idejével összehasonlítva kell elvégezni.
5.4.2. Mennyiségi értékelés
Az egyes csúcsok területét elektronikus integrálással számítsa ki.
6. AZ EREDMÉNYEK KIFEJEZÉSE
A gliceril monopalmitát százalékát az ehhez tartozó csúcs területe, illetve az összes monoglicerid csúcsok területének összege közötti arány alapján számítsa ki (lásd a 2. ábrát), a következő képlet alapján:
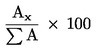
ahol:
Ax = a gliceril monopalmitáthoz tartozó csúcs területe
ΣA = az összes monogliceridcsúcs területének összege
Az eredményt egy tizedesjegy pontosságig kell megadni.
7. ANALÍZIS JEGYZŐKÖNYV
Az analízis jegyzőkönyvnek a következőket kell tartalmaznia:
- a fenti módszerre való hivatkozás,
- a minta maradéktalan azonosításához szükséges mindenfajta információ,
- az analízis eredménye,
- a fenti módszertől, akár az érintett felek döntéséből, akár egyéb okból kifolyólag történő mindennemű eltérés,
- a laboratórium részletes azonosítása, az analízis időpontja és az analízisért felelős személyek aláírása.
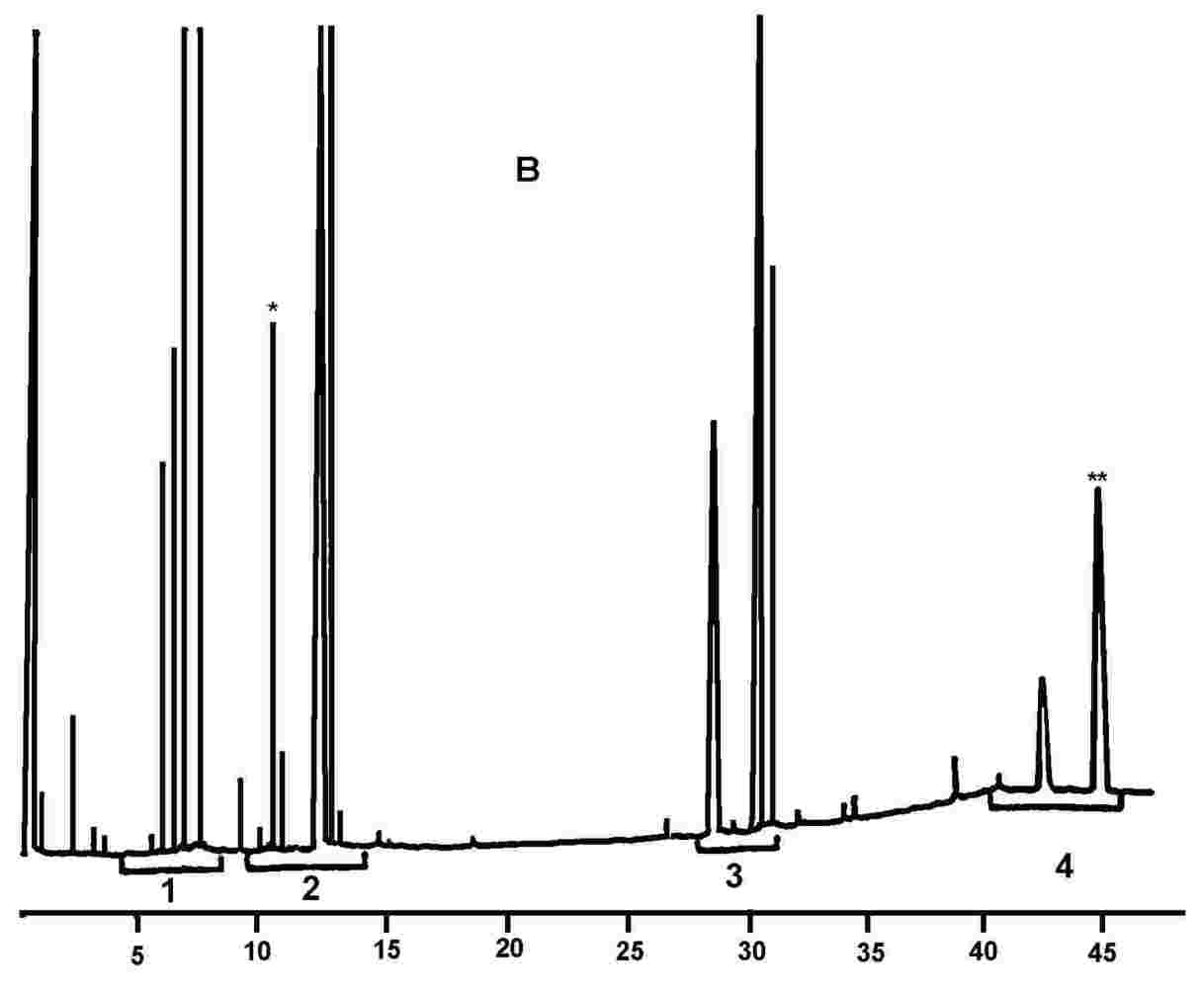
1. ábra
A 20 % észterezett olajjal (100 %) kevert finomított olívaolajra kifejtett lipázhatás nyomán keletkezett szilanizációs reakciótermékek kromatogramja.
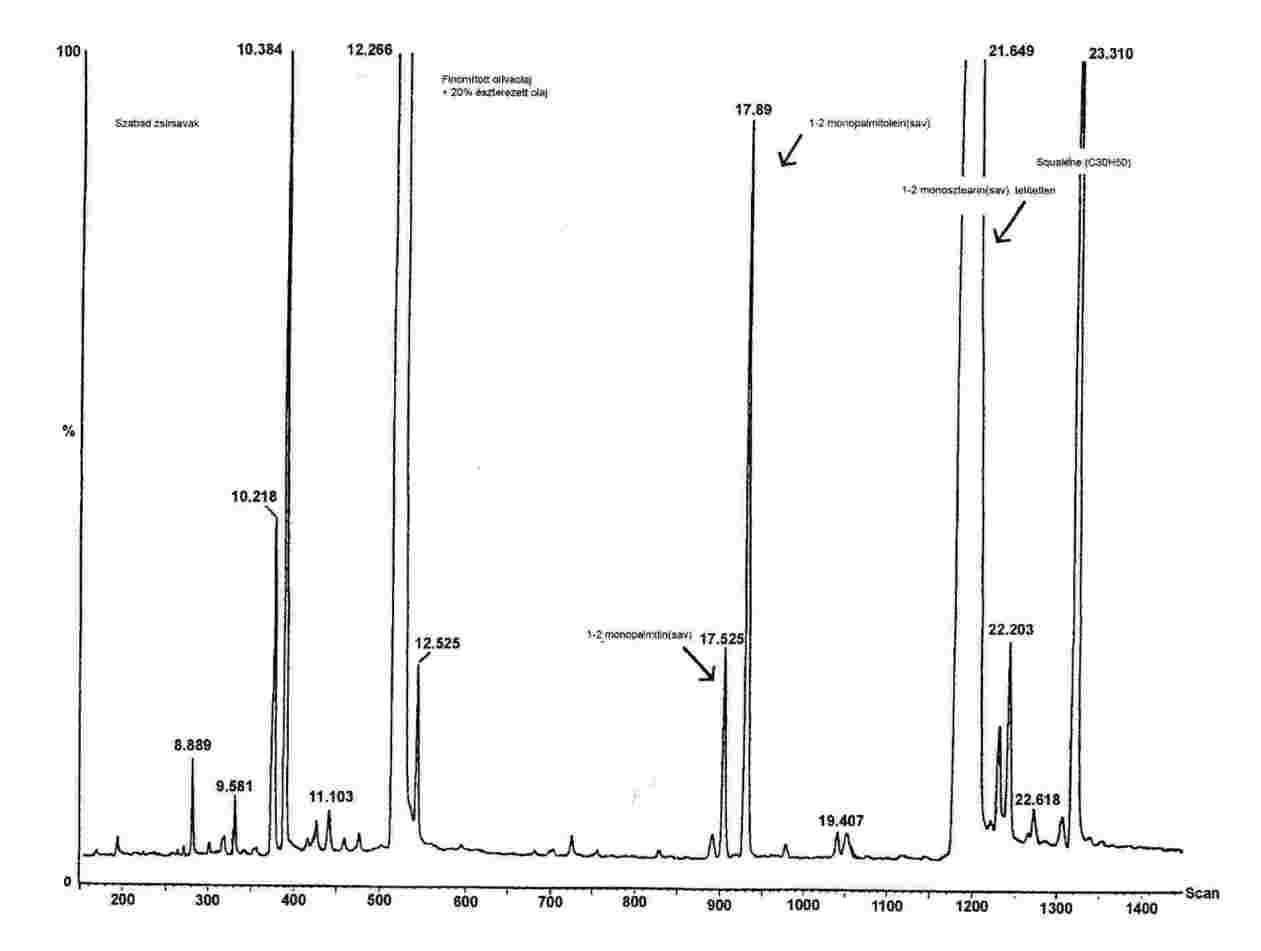
2. ábra
Kromatogram:
(A) nem észterezett olívaolaj, a lipázreakció után; a szilanizáció után; ilyen körülmények között (8-12 m-es kapilláris oszlop) a viaszfrakció a digliceridfrakcióval egy időben, vagy kevéssel utána eluálódik.
A lipázreakció után a triglicerid-tartalom nem haladhatja meg a 15 %-ot.
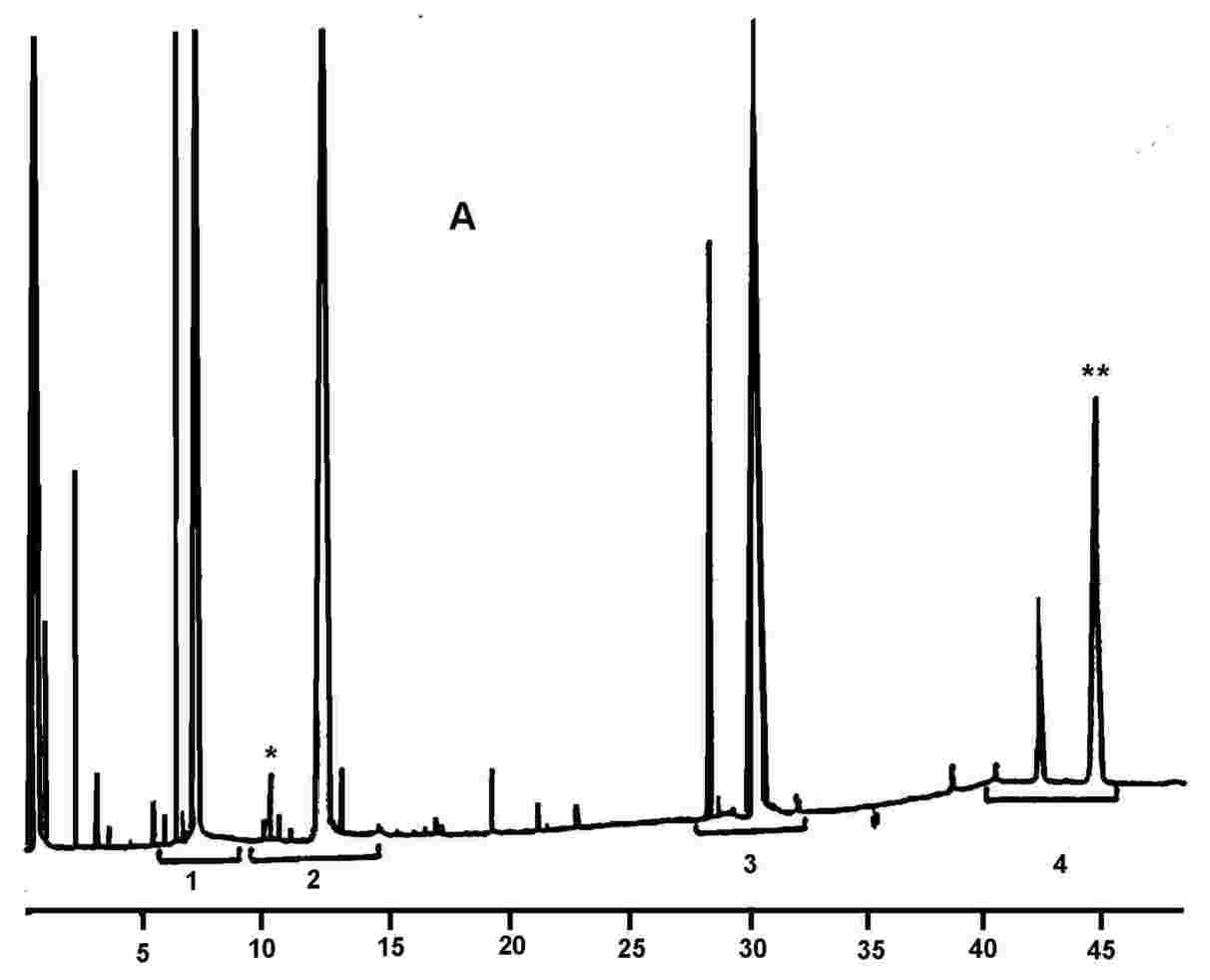
Kromatogram:
(B) észterezett olaj a lipázreakció után; a szilanizáció után; ilyen körülmények között (8-12 m-es kapilláris oszlop) a viaszfrakció a digliceridfrakcióval egy időben, vagy kevéssel utána eluálódik.
A lipázreakció után a triglicerid-tartalom nem haladhatja meg a 15 %-ot.
8. MEGJEGYZÉSEK
1. megjegyzés: A LIPÁZ ELKÉSZÍTÉSE
Kielégítő lipázaktivitású lipázok kaphatók kereskedelmi forgalomban, de lehetséges laboratóriumi elkészítésük is a következők szerint:
Hűtsön le 5 kg friss sertés-hasnyálmirigyet 0 °C hőmérsékletre. Távolítsa el a környező szilárd zsiradékot és a kötőszövetet, majd dörzsölje szét egy keverőgépben, hogy pépes folyadékot kapjon. A keverővel keverje össze a pépet 2,5 l vízmentes acetonnal 4-6 órán keresztül, majd centrifugálja. Extrahálja a maradékot még háromszor, azonos mennyiségű acetonnal, majd két alkalommal aceton és dietil-éter 1/1 (V/V) keverékével, majd kétszer dietil-éterrel.
Szárítsa a maradékot in vacuo 48 órán keresztül, hogy stabil port kapjon, amely hűtőszekrényben és nedvességtől védve hosszan tárolható.
2. megjegyzés: A LIPÁZ AKTIVITÁSÁNAK BEÁLLÍTÁSA
Készítsen olívaolaj-emulziót a következőképpen:
Megfelelő keverőben 10 percen keresztül rázza össze a 165 ml 100 g/l-es gumiarábikum-oldatból, 15 g jégzúzalékból és 20 ml előzetesen semlegesített olívaolajból álló keveréket.
Egy főzőpohárba tegyen 10 ml-t ebből az emulzióból, majd ezután 0,3 ml 0,2 g/ml-es nátrium-kolát oldatot és 20 ml desztillált vizet.
Tegye a főzőpoharat egy 37 °C hőmérsékleten tartott termosztátba; helyezze bele egy pH-mérő elektródáit és egy keverőspirált.
Egy büretta segítségével cseppenként adagoljon nátrium-hidroxid-oldatot (0,1 N), amíg a pH-érték 8,3 nem lesz.
Adjon hozzá megfelelő mennyiséget a lipáz vizes szuszpenziójából (0,1 g/ml lipáz). Amint a pH-mérő 8,3-at mutat, indítsa el a stopperórát, és olyan sebességgel csepegtesse a nátrium-hidroxid-oldatot, hogy a 8,3 pH-érték megmaradjon. Minden percben olvassa le a felhasznált lúgoldat térfogatát.
A megfigyeléseket rögzítse grafikonon, az időértékeket használva abszcisszaként, az állandó pH-érték fenntartásához szükséges 0,1 N-es lúgoldat ml-ben kifejezett térfogatát pedig ordinátaként. Lineáris grafikont kell kapnia.
A felhasznált por lipáz egység/mg-ban kifejezett aktivitása a következő képlettel határozható meg:
ahol:
A a lipáz egység/mg-ban kifejezett aktivitása,
V a percenként felhasznált nátrium-hidroxid-oldat ml-ek száma (a grafikonból kiszámítva),
N a nátrium-hidroxid-oldat moláris koncentrációja,
m a lipáz por vizsgált mennyiségének tömege mg-ban.
A lipázegység definíciója a következő: az az enzimmennyiség, amely percenként 10 μ-savekvivalenst szabadít fel.";
7. A X. A. melléklet 6.2. pontja helyébe a következő szöveg lép:
"6.2. A metil-észterek előkészítése a X B. mellékletben leírt B eljárás szerint történik. A 3 %-nál magasabb szabad savtartalmú zsírszerű anyagokat előzetesen semlegesíteni kell a VII. melléklet 5.1.1. pontjában meghatározott módszer szerint."
(1) Kapilláris oszloppal szétválasztható (vagy nem szétválasztható) izomerek összege.
(2) Ahol a hibamedián nem lehet 2,5-nél nagyobb, a gyümölcsösségi medián pedig 0-val egyenlő.
(3) A 300 mg/kg és 350 mg/kg közötti viasztartalmú olajok lampante olívaolajnak minősülnek, ha a nyílt szénláncú alkohol tartalma nem haladja meg a 350 mg/kg-ot, és az eritrodiol- és uvaoltartalom legfeljebb 3,5 %.
(4) A 300 mg/kg és 350 mg/kg közötti viasztartalmú olajok nyers olívamaradék-olajnak minősülnek, ha a nyílt szénláncú alkoholok összértéke meghaladja a 350 mg/kg-ot, és az eritrodiol- és uvaoltartalom nagyobb mint 3,5 %.
(5) Egyéb zsírsavtartalom (%): palmitin: 7,5-20,0; palmitolein: 0,3-3,5; heptadekán: ≤ 0,3; heptadecen: ≤ 0,3; sztearin: 0,5-5,0; olaj: 55,0-83,0; linolén: 3,5-21,0.
(6) Összesen: Delta-5-23-Sztigmasztadienol+Kleroszterin+Béta-Szitoszterin+Szitosztanol+Delta-5-Avenaszterin+Delta-5-24-Sztigmasztadienol.
(7) A 300 mg/kg és 350 mg/kg közötti viasztartalmú olajok lampante olívaolajnak minősülnek, ha a nyílt szénláncú alkohol tartalom nem haladja meg a 350 mg/kg-ot, vagy az eritrodiol- és uvaoltartalom legfeljebb 3,5 %.
(8) A 300 mg/kg és 350 mg/kg közötti viasztartalmú olajok nyers olívamaradék-olajnak minősülnek, ha a nyílt szénláncú alkoholok összértéke meghaladja a 350 mg/kg-ot, és az eritrodiol- és uvaoltartalom nagyobb, mint 3,5 %.
(9) A szterinészterek eluálódása után a kromatográfiás vonalon nem mutatkozhatnak jelentős csúcsok (trigliceridek).
Lábjegyzetek:
[1] A dokumentum eredetije megtekinthető CELEX: 32007R0702 - https://eur-lex.europa.eu/legal-content/HU/ALL/?uri=CELEX:32007R0702&locale=hu