
31991R2568[1]
A Bizottság 2568/91/EGK rendelete (1991. július 11.) az olívaolaj és az olívamaradék-olaj jellemzőiről és az ezekre vonatkozó elemzési módszerekről
A BIZOTTSÁG 2568/91/EGK RENDELETE
(1991. július 11.)
az olívaolaj és az olívamaradék-olaj jellemzőiről és az ezekre vonatkozó elemzési módszerekről
1. cikk
(1) Azok az olajok, amelyeknek jellemzői megegyeznek az e rendelet I. mellékletének 1. és 2. pontja szerintiekkel, szűz olívaolajnak minősülnek a 136/66/EGK rendelet mellékletének 1. a) és b) pontja értelmében.
(2) Az az olaj, amelynek jellemzői megegyeznek az e rendelet I. mellékletének 3. pontja szerintiekkel, lampante olívaolajnak minősül a 136/66/EGK rendelet melléklete 1. c) pontja értelmében.
(3) Az az olaj, amelynek jellemzői megegyeznek az e rendelet I. mellékletének 4. pontja szerintiekkel, finomított olívaolajnak minősül a 136/66/EGK rendelet mellékletének 2. pontja értelmében.
(4) Az az olaj, amelynek jellemzői megegyeznek az e rendelet I. mellékletének 5. pontja szerintiekkel, finomított olívaolajokból és szűz olívaolajokból álló olívaolajnak minősül a 136/66/EGK rendelet mellékletének 3. pontja értelmében.
(5) Az az olaj, amelynek jellemzői megegyeznek az e rendelet I. mellékletének 6. pontja szerintiekkel, nyers olívamaradék-olajnak minősül a 136/66/EGK rendelet mellékletének 4. pontja értelmében.
(6) Az az olaj, amelynek jellemzői megegyeznek az e rendelet I. mellékletének 7. pontja szerintiekkel, finomított olívamaradék-olajnak minősül a 136/66/EGK rendelet mellékletének 5. pontja értelmében.
(7) Az az olaj, amelynek jellemzői megegyeznek az e rendelet I. mellékletének 8. pontja szerintiekkel, olívamaradék-olajnak minősül a 136/66/EGK rendelet mellékletének 6. pontja értelmében.
2. cikk
(1) Az olajok I. melléklet szerinti jellemzőinek megállapítása a következő elemzési módszerekkel történik:
a) a szabad zsírsavak mennyiségének az olajsav százalékban történő meghatározása a II. melléklet szerinti módszerrel;
b) a peroxid-index meghatározása a III. melléklet szerinti módszerrel;
c) a viasztartalom meghatározása a IV. melléklet szerinti módszerrel;
d) a szterinek és a triterpén dialkoholok összetételének és mennyiségének kapilláris oszlopos gázkromatográfiával történő meghatározása az V. melléklet szerinti módszerrel;
e) a 2-gliceril monopalmitát százalékának meghatározása a VII. melléklet szerinti módszerrel;
f) a spektrofotometriás elemzés a IX. melléklet szerinti módszerrel;
g) a zsírsavösszetétel meghatározása a X. melléklet szerinti módszerrel;
h) az illékony halogénezett oldószerek meghatározása a XI. melléklet szerinti módszerrel;
i) a szűz olívaolaj érzékszervi jellemzőinek értékelése a XII. melléklet szerinti módszerrel;
j) a sztigmasztadiének mennyiségének meghatározása a XVII. melléklet szerinti módszerrel;
k) az ECN42 triglicerid-tartalom meghatározása a III. melléklet szerinti módszerrel;
l) a szterinösszetétel és -tartalom meghatározása, valamint az alkoholos vegyületek meghatározása kapilláris oszlopos gázkromatográfiával, a XIX. melléklet szerinti módszerrel;
m) a viasz-, valamint a zsírsav-metilészter- és zsírsav-etilészter-tartalom meghatározása a XX. melléklet szerinti módszerrel.
(2) A szűz olívaolajok érzékszervi jellemzőinek nemzeti hatóságok vagy képviselőik által történő ellenőrzését a tagállamok által jóváhagyott kóstolói csoportok végzik.
Valamely olajnak az első albekezdésben említett érzékszervi jellemzői akkor tekintendők a bejelentett kategóriának megfelelőnek, ha a tagállamok által jóváhagyott kóstolói csoport megerősíti az osztályozást.
Amennyiben a csoport az érzékszervi jellemzők tekintetében nem erősíti meg a bejelentett kategóriát, az érdekelt fél kérésére a nemzeti hatóságok vagy képviselőik haladéktalanul elvégeztetnek két ellenőrző értékelést más jóváhagyott csoportokkal. A csoportok közül legalább az egyiknek az érintett termelő tagállam által jóváhagyott csoportnak kell lennie. Az érintett jellemzők akkor tekintendők a bejelentett jellemzőkkel megegyezőknek, ha mindkét ellenőrző értékelés megerősíti a bejelentett osztályozást. Ha nem ez a helyzet, az ellenőrző értékelések során megállapított hibák típusától függetlenül az osztályozást a jellemzőkkel össze nem egyeztethetőnek kell nyilvánítani, és az ellenőrző értékelések költségei az érdekelt felet terhelik.
(3) Az olaj jellemzőinek a nemzeti hatóságok vagy képviselőik által az (1) bekezdésben előírtaknak megfelelően történő ellenőrzése során a mintavételezés a vizsgálati minták előkészítésére vonatkozó EN ISO 661, valamint a mintavételezésre vonatkozó EN ISO 5555 nemzetközi szabvány szerint történik. Azonban az EN ISO 5555 szabvány 6.8 pontja ellenére, a közvetlen csomagolású olajok esetében a mintavételezés e rendelet Ia. melléklete szerint történik. Az ömlesztett olajok esetében, amelyekre nem alkalmazható az EN ISO 5555 szabvány szerinti mintavétel, a mintavételezés a tagállam illetékes hatósága által adott utasítások szerint történik.
Az EN ISO 5555 szabvány és az EN ISO 661 szabvány 6. fejezetének sérelme nélkül a levett mintákat a lehető leggyorsabban sötét helyen, hőhatástól távol kell elhelyezni, és legkésőbb a mintavételezést követő ötödik munkanapig be kell küldeni elemzésre; eltérő esetben úgy kell tárolni a mintákat, hogy azok a laboratóriumba küldésük előtt a szállítás vagy a tárolás során ne sérüljenek vagy károsodjanak.
(4) A (3) bekezdésben említett ellenőrzések céljára a II., III., IX., XII. és XX. mellékletben említett elemzéseket és - ha a nemzeti törvények előírják - az ellenvizsgálatokat csomagolt termékek esetében a minőségmegőrzési idő lejárta előtt kell elvégezni. Az ömlesztett olajok mintavételezése esetében az elemzéseket legkésőbb a mintavételt követő hatodik hónapban el kell végezni.
Az e rendeletben előírt egyéb elemzések vonatkozásában semmiféle határidőt nem kell alkalmazni.
Hacsak a mintavétel nem kevesebb mint két hónappal a minőségmegőrzési idő lejárta előtt történik, akkor amennyiben az elemzések eredményei nem felelnek meg a bejelentett kategóriájú olívaolaj és olívapogácsa-olaj jellemzőinek, az érintett felet legkésőbb egy hónappal az első albekezdésben meghatározott időtartam lejárta előtt értesíteni kell.
(5) Az olívaolajok jellemzőinek az (1) bekezdés első albekezdésében előírt módszerek szerinti meghatározása céljából az elemzés eredményeit közvetlenül össze kell vetni az e rendeletben meghatározott határértékekkel.
2a. cikk
(1) E cikk alkalmazásában a "forgalmazott olívaolaj" egy adott tagállamból származó olívaolaj és olívapogácsa-olaj teljes mennyisége, amely a szóban forgó tagállamban kerül fogyasztásra, vagy amelyet ebből a tagállamból exportálnak.
(2) A tagállamok gondoskodnak arról, hogy a megfelelőségi ellenőrzések szelektív módon, kockázatelemzésre alapozva és megfelelő gyakorisággal történjenek annak biztosítására, hogy a forgalmazott olívaolaj megfelel a bejelentett kategóriának.
(3) A kockázatelemzési szempontok között szerepelhet:
a) az olaj kategóriája, az előállítás időszaka, az olajok más növényi olajokkal összevetett ára, a keverési és csomagolási eljárások, a tárolási létesítmények és feltételek, a származási ország, a célország, a szállítási eszközök vagy a tétel volumene;
b) a gazdasági szereplők értékesítési láncban elfoglalt helye, az általuk forgalmazott volumen és/vagy érték, az általuk forgalmazott olajkategóriák skálája, az olyan elvégzett munkatípusok, mint a kinyerés, tárolás, finomítás, keverés, csomagolás vagy kiskereskedelmi értékesítés;
c) korábbi ellenőrzések eredményei, beleértve a feltárt hiányosságok számát és típusát, a forgalmazott olajok általános minőségét és a használt technikai eszközök teljesítményét;
d) a gazdasági szereplők által használt, a forgalmazási előírásoknak való megfeleléssel kapcsolatos minőségbiztosítási rendszereknek vagy önellenőrzési rendszereknek a megbízhatósága;
e) az ellenőrzés végrehajtásának helyszíne, különösen, ha az Unióba való első belépési pontról, az Unióból való utolsó kilépési pontról vagy az olajok előállításának, csomagolásának, berakodásának vagy a végső fogyasztó számára történő értékesítésének helyéről van szó;
f) minden egyéb olyan információ, amely a megfelelés hiányának kockázatára utalhat.
(4) A tagállamok előre meghatározzák a következőket:
a) a tételek megfelelésének hiányára vonatkozó kockázatelemzés szempontjai;
b) az egyes kockázati kategóriák kockázatelemzése alapján azon gazdasági szereplők vagy tételek és/vagy mennyiségek minimális száma, amelyeknél megfelelőségi ellenőrzést kell végezni.
A tagállamban forgalmazott olívaolaj minden ezer tonnájára évente legalább egy megfelelőségi ellenőrzést kell végezni.
(5) A tagállamoknak ellenőrizniük kell a megfelelést úgy, hogy:
a) bármilyen sorrendben végrehajtják az I. mellékletben előírt elemzéseket; vagy
b) az Ib. melléklet folyamatábráján előírt sorrendet követve hajtják végre azokat, amíg el nem jutnak a folyamatábrában szereplő valamelyik döntésig.
3. cikk
Amennyiben megállapítást nyer, hogy egy olaj nem felel meg kategórialeírásának, az érintett tagállam - egyéb szankciók sérelme nélkül - hatékony, arányos és visszatartó erejű, a feltárt szabálytalanság súlyosságának megfelelően meghatározott szankciókat alkalmaz.
Ha az ellenőrzések jelentős szabálytalanságokat tárnak fel, a tagállamok növelik a forgalmazás szakaszára, az olajkategóriára, a származásra vagy egyéb szempontokra vonatkozó ellenőrzések gyakoriságát.
4. cikk
(1) The Member States may approve assessment panels so that national authorities or their representatives can assess and verify organoleptic characteristics.
The terms of approval shall be set by Member States and ensure that:
- the requirements of Annex XII.4 are met,
- the panel head is given training recognised for this purpose by the Member State,
- continued approval depends on performance in annual checks arranged by the Member State.
Member States shall notify to the Commission a list of approved panels and the action taken under this paragraph.
(2) Amennyiben a tagállamok számára problémát jelent ezeknek a csoportoknak a felállítása a saját területükön, felkérhetnek egy másik tagállamban elfogadott kóstolói csoportot.
(3) Valamennyi tagállam listát készít a szakmai- vagy ágazati szervezetek által az (1) bekezdésben meghatározott feltételekkel összhangban felállított kóstolói csoportokról, továbbá biztosítja e feltételek betartását.
6. cikk
(1) Az olívaolaj kinyeréséből származó olajpogácsa és egyéb maradékanyagok (KN-kód: 2306 90 11 és 2306 90 19 ) olajtartalmának meghatározása a XV. mellékletben szabályozott módszerrel történik.
(2) Az (1) bekezdésben említett olajtartalmat az olaj súlyának a szárazanyag súly százalékos arányában fejezik ki.
7. cikk
A szennyező anyagok jelenlétére vonatkozó közösségi rendelkezéséket alkalmazni kell.
A halogénezett oldószerek esetében a következő határértékek vonatkoznak minden olívaolaj-kategóriára:
- a maximális oldószertartalom minden egyes kimutatott halogénezett oldószer esetében: 0,1 mg/kg,
- a maximális összesített oldószertartalom az összes észlelt halogénezett oldószer tekintetében: 0,2 mg/kg.
7a. cikk
Azon természetes vagy jogi személyek és azok csoportjai, amelyek - akár szakmai, akár kereskedelmi célból - a sajtolólétesítményben történő olajkinyeréstől a palackozási stádiumig tartó (ez utóbbit is magában foglaló) folyamat bármely fázisában lévő olívaolajat és olívapogácsa-olajat tárolnak, minden olajkategória tekintetében nyilvántartást vezetnek a belépő és a kilépő tételekről.
A tagállamok biztosítják, hogy a gazdasági szereplők az első bekezdésben megállapított kötelezettségnek teljes mértékben megfelelnek.
8. cikk
(1) A tagállamok értesítik a Bizottságot az e rendelet végrehajtására hozott intézkedéseikről. Tájékoztatják a Bizottságot a későbbiekben bekövetkezett minden változásról.
(2) A tagállamok minden évben legkésőbb május 31-ig jelentést nyújtanak be a Bizottsághoz az e rendeletnek az előző naptári év során történő alkalmazásáról. A jelentés mindenképpen tartalmazza az olívaolajokon végzett megfelelőségi ellenőrzéseknek a XXI. mellékletben szereplő táblázat szerinti eredményeit.
(3) Az e rendeletben említett értesítéseket a 792/2009/EK bizottsági rendeletnek ( 1 ) megfelelően kell megküldeni.
9. cikk
A 1058/77/EGK rendelet hatályát veszti.
10. cikk
(1) Ez a rendelet az Európai Közösségek Hivatalos Lapjában való kihirdetését követő harmadik napon lép hatályba.
A XII. melléklet szerinti módszer azonban 1992. november 1.-jétől hatályos, kivéve az intervenciós rendszer működésével kapcsolatos műveleteket.
Ez a módszer nem vonatkozik az 1992. november 1. előtti forgalombahozatal céljára készített szűz olívaolajra.
(2) E rendeletet nem kell alkalmazni a hatálybalépését megelőzően csomagolt és 1992. október 31-ig forgalomba hozott olívaolajra és olívamaradék-olajra.
Ez a rendelet teljes egészében kötelező és közvetlenül alkalmazandó valamennyi tagállamban.
MELLÉKLETEK
ÖSSZEFOGLALÁS
I. MELLÉKLET
AZ OLÍVAOLAJ JELLEMZŐI
Minőségi jellemzők
| Kategória | Savasság (%) (*) | Peroxidszám (milliekvivalens O2/kg) | K232 | K268 vagy K270 | Delta-K | Érzékszervi értékelés | Zsírsav-etilészterek (mg/kg) | |
| Hibamedián (Hm) (*) | Gyümölcsösségi medián (Gym) | |||||||
| 1. Extra szűz olívaolaj | ≤ 0,80 | ≤ 20,0 | ≤ 2,50 | ≤ 0,22 | ≤ 0,01 | Md = 0,0 | Mf > 0,0 | ≤ 35 |
| 2. Szűz olívaolaj | ≤ 2,0 | ≤ 20,0 | ≤ 2,60 | ≤ 0,25 | ≤ 0,01 | Md ≤ 3,5 | Mf > 0,0 | — |
| 3. Lampante olívaolaj | > 2,0 | — | — | — | — | Md > 3,5 (1) | — | — |
| 4. Finomított olívaolaj | ≤ 0,30 | ≤ 5,0 | — | ≤ 1,25 | ≤ 0,16 | — | — | |
| 5. Finomított olívaolajból és szűz olívaolajból álló olívaolaj | ≤ 1,00 | ≤ 15,0 | — | ≤ 1,15 | ≤ 0,15 | — | — | |
| 6. Nyers olívapogácsa-olaj | — | — | — | — | — | — | — | |
| 7. Finomított olívapogácsa-olaj | ≤ 0,30 | ≤ 5,0 | — | ≤ 2,00 | ≤ 0,20 | — | — | |
| 8. Olívapogácsa-olaj | ≤ 1,00 | ≤ 15,0 | — | ≤ 1,70 | ≤ 0,18 | — | — | |
| (1) A hibamedián legfeljebb 3,5 lehet, ha a gyümölcsösségi medián 0,0. | ||||||||
Tisztasági jellemzők
| Kategória | Zsírsavösszetétel (1) | Transzolaj-izomerek összege (%) | Transzlinol- + transzlinolén-izomerek összege (%) | Sztigma-sztadiének (mg/kg) (2) | A HPLC-módszerrel kapott ECN42-érték és az elméleti számítás útján kapott ECN42-érték különbsége | 2-gliceril-monopalmitát (%) | |||||
| Mirisztinsav (%) | Linolénsav (%) | Arachinsav (%) | Eikozénsav (%) | Behénsav (%) | sav (%) | ||||||
| 1. Extra szűz olívaolaj | ≤ 0,03 | ≤ 1,00 | ≤ 0,60 | ≤ 0,50 | ≤ 0,20 | ≤ 0,20 | ≤ 0,05 | ≤ 0,05 | ≤ 0,05 | ≤ |0,20| | ≤ 0,9, ha a palmitinsav összaránya ≤ 14,00 % |
| ≤ 1,0, ha a palmitinsav összaránya > 14,00 % | |||||||||||
| 2. Szűz olívaolaj | ≤ 0,03 | ≤ 1,00 | ≤ 0,60 | ≤ 0,50 | ≤ 0,20 | ≤ 0,20 | ≤ 0,05 | ≤ 0,05 | ≤ 0,05 | ≤ |0,20| | ≤ 0,9, ha a palmitinsav összaránya ≤ 14,00 % |
| ≤ 1,0, ha a palmitinsav összaránya > 14,00 % | |||||||||||
| 3. Lampante olívaolaj | ≤ 0,03 | ≤ 1,00 | ≤ 0,60 | ≤ 0,50 | ≤ 0,20 | ≤ 0,20 | ≤ 0,10 | ≤ 0,10 | ≤ 0,50 | ≤ |0,30| | ≤ 0,9, ha a palmitinsav összaránya ≤ 14,00 % |
| ≤ 1,1, ha a palmitinsav összaránya > 14,00 % | |||||||||||
| 4. Finomított olívaolaj | ≤ 0,03 | ≤ 1,00 | ≤ 0,60 | ≤ 0,50 | ≤ 0,20 | ≤ 0,20 | ≤ 0,20 | ≤ 0,30 | — | ≤|0,30| | ≤ 0,9, ha a palmitinsav összaránya ≤ 14,00 % |
| ≤ 1,1, ha a palmitinsav összaránya > 14,00 % | |||||||||||
| 5. Finomított olívaolajból és szűz olívaolajból álló olívaolaj | ≤ 0,03 | ≤ 1,00 | ≤ 0,60 | ≤ 0,50 | ≤ 0,20 | ≤ 0,20 | ≤ 0,20 | ≤ 0,30 | — | ≤ |0,30| | ≤ 0,9, ha a palmitinsav összaránya ≤ 14,00 % |
| ≤ 1,0, ha a palmitinsav összaránya > 14,00 % | |||||||||||
| 6. Nyers olívapogácsa-olaj | ≤ 0,03 | ≤ 1,00 | ≤ 0,60 | ≤ 0,50 | ≤ 0,30 | ≤ 0,20 | ≤ 0,20 | ≤ 0,10 | — | ≤ |0,60| | ≤ 1,4 |
| 7. Finomított olívapogácsa-olaj | ≤ 0,03 | ≤ 1,00 | ≤ 0,60 | ≤ 0,50 | ≤ 0,30 | ≤ 0,20 | ≤ 0,40 | ≤ 0,35 | — | ≤ |0,50| | ≤ 1,4 |
| 8. Olívapogácsa-olaj | ≤ 0,03 | ≤ 1,00 | ≤ 0,60 | ≤ 0,50 | ≤ 0,30 | ≤ 0,20 | ≤ 0,40 | ≤ 0,35 | — | ≤ |0,50| | ≤ 1,2 |
| (1) Egyéb zsírsavtartalom (%): palmitinsav: 7,50–20,00; palmitoleinsav: 0,30–3,50; heptadekánsav: ≤ 0,40; heptadecénsav: ≤ 0,60; sztearinsav: 0,50–5,00; olajsav: 55,00–83,00; linolsav: 2,50–21,00. (2) A kapilláris kolonnával elválasztható (vagy el nem választható) izomerek összesen. | |||||||||||
| Kategória | Szterinösszetétel | Szterinek összesen (mg/kg) | Eritrodiol és uvaol (%) (**) | Viaszok (mg/kg) (**) | |||||
| Koleszterin (%) | Brasszikaszterin (%) | Kampeszterin (1) (%) | Sztigmaszterin (%) | Láth. β-szitoszterin (2) (%) | Delta-7-sztigmasztenol (1) (%) | ||||
| 1. Extra szűz olívaolaj | ≤ 0,5 | ≤ 0,1 | ≤ 4,0 | < Kamp. | ≥ 93,0 | ≤ 0,5 | ≥ 1 000 | ≤ 4,5 | C42 + C44 + C46 ≤ 150 |
| 2. Szűz olívaolaj | ≤ 0,5 | ≤ 0,1 | ≤ 4,0 | < Kamp. | ≥ 93,0 | ≤ 0,5 | ≥ 1 000 | ≤ 4,5 | C42 + C44 + C46 ≤ 150 |
| 3. Lampante olívaolaj | ≤ 0,5 | ≤ 0,1 | ≤ 4,0 | — | ≥ 93,0 | ≤ 0,5 | ≥ 1 000 | ≤ 4,5 (3) | C40 + C42 + C44 + C46 ≤ 300 (3) |
| 4. Finomított olívaolaj | ≤ 0,5 | ≤ 0,1 | ≤ 4,0 | < Kamp. | ≥ 93,0 | ≤ 0,5 | ≥ 1 000 | ≤ 4,5 | C40 + C42 + C44 + C46 ≤ 350 |
| 5. Finomított olívaolajból és szűz olívaolajból álló olívaolaj | ≤ 0,5 | ≤ 0,1 | ≤ 4,0 | < Kamp. | ≥ 93,0 | ≤ 0,5 | ≥ 1 000 | ≤ 4,5 | C40 + C42 + C44 + C46 ≤ 350 |
| 6. Nyers olívapogácsa-olaj | ≤ 0,5 | ≤ 0,2 | ≤ 4,0 | — | ≥ 93,0 | ≤ 0,5 | ≥ 2 500 | > 4,5 (4) | C40 + C42 + C44 + C46 > 350 (4) |
| 7. Finomított olívapogácsa-olaj | ≤ 0,5 | ≤ 0,2 | ≤ 4,0 | < Kamp. | ≥ 93,0 | ≤ 0,5 | ≥ 1 800 | > 4,5 | C40 + C42 + C44 + C46 > 350 |
| 8. Olívapogácsa-olaj | ≤ 0,5 | ≤ 0,2 | ≤ 4,0 | < Kamp. | ≥ 93,0 | ≤ 0,5 | ≥ 1 600 | > 4,5 | C40 + C42 + C44 + C46 > 350 |
| (1) Lásd e melléklet függelékét. (2) Láth. β-szitoszterin: Delta-5,23-sztigmasztadienol+kleroszterin+béta-szitoszterin+szitosztanol+delta-5-avenaszterin+delta-5,24-sztigmasztadienol. (3) A 300 mg/kg és 350 mg/kg közötti viasztartalmú olajok lampante olívaolajnak minősülnek, ha a teljes alifásalkohol-tartalom legfeljebb 350 mg/kg, vagy ha az eritrodiol- és uvaoltartalom legfeljebb 3,5 %. (4) A 300 mg/kg és 350 mg/kg közötti viasztartalmú olajok nyers olívapogácsa-olajnak tekintendők, ha a teljes alifásalkohol-tartalom meghaladja a 350 mg/kg-ot, és ha az eritrodiol- és uvaoltartalom 3,5 %-nál nagyobb. | |||||||||
Megjegyzések:
a) Az eredményeket ugyanannyi tizedesjeggyel kell megadni, mint amennyi az egyes jellemzőknél meg van adva. Az utolsó értékes számjegyet fel kell kerekíteni a következő számjegyre, ha az ezt követő, nem értékes számjegy 4-nél nagyobb.
b) Ha a jellemzői közül bármelyik kívül esik a megállapított határértéken, az olajat vagy egy másik kategóriába kell sorolni, vagy e rendelet alkalmazásában nem megfelelőnek kell minősíteni.
c) A lampante olívaolaj esetében a csillaggal (*) jelölt mindkét minőségi jellemző egyidejűleg eltérhet az adott kategóriára megállapított határértékektől.
d) Ha valamely jellemző két csillaggal (**) van megjelölve, az a nyers olívapogácsa-olaj esetében azt jelenti, hogy mindkét vonatkozó határérték egyidejűleg eltérhet a megadott értékektől. Az olívapogácsa-olaj és a finomított olívapogácsa-olaj esetében az egyik vonatkozó határérték térhet el a megadott értékektől.
Függelék
Döntési fák
A kampeszterinre vonatkozó döntési fa szűz és extra szűz olívaolajok esetében:
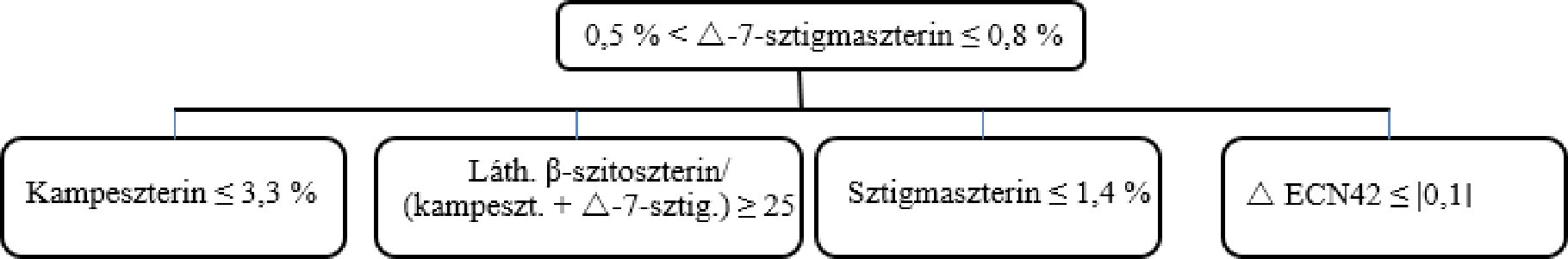
A többi paraméternek meg kell felelnie a rendeletben meghatározott határértékeknek.
A delta-7-sztigmaszterinre vonatkozó döntési fa a következők esetében:
- extra szűz és szűz olívaolajok
- [Kép #1]

Kép #1
A többi paraméternek meg kell felelnie a rendeletben meghatározott határértékeknek.
- olívapogácsa-olajok (nyers és finomított)
- [Kép #2]

Kép #2
A többi paraméternek meg kell felelnie a rendeletben meghatározott határértékeknek.
Ia MELLÉKLET
OLÍVAOLAJBÓL VAGY OLÍVAPOGÁCSA-OLAJBÓL TÖRTÉNŐ MINTAVÉTEL KÖZVETLEN CSOMAGOLÁSBAN SZÁLLÍTOTT
Ez a mintavételi módszer a közvetlen csomagolású olívaolaj- vagy olívapogácsa-olaj gyártási tételekre vonatkozik. A mintavételi módszer annak függvényében változik, hogy a közvetlen csomagolású kiszerelés meghaladja-e az 5 litert, vagy sem.
A "gyártási tétel" olyan árucikk-készlet, amit olyan körülmények között állítottak elő, dolgoztak fel és csomagoltak, hogy a minden egyes árucikk által tartalmazott olaj az összes analitikai jellemző tekintetében homogénnek mondható. Az egyes gyártási tételek azonosítását az Európai Parlament és a Tanács 2011/91/EU irányelvének megfelelően kell elvégezni ( 2 ).
Az "egyedi minta" a közvetlen csomag által tartalmazott olajmennyiség, és amit a gyártási tételből emeltek ki véletlenszerűen.
1. AZ ELSŐDLEGES MINTA TARTALMA
1.1. 5 litert meg nem haladó közvetlen csomagolás
Az "elsődleges minta" az 5 litert meg nem haladó közvetlen csomagolás esetében az egy gyártási tételből vett egyedi minták számát jelenti az 1. táblázatnak megfelelően.
1. táblázat
Az elsődleges minta minimális méretét a következők szerint kell megállapítani
| Amennyiben a közvetlen csomagolás űrtartalma | Az elsődleges minta az alábbi számú közvetlen csomagból származó olajat tartalmaz |
| (a) 1 liter vagy annál nagyobb | (a) 1 közvetlen csomag |
| (b) kisebb mint 1 liter | (b) a legalább 1 liter össztérfogatot kitevő minimális számú csomag |
Az 1. táblázatban található, az elsődleges mintát tartalmazó csomagok száma a tagállamok egyedi igényeinek megfelelően növelhető (például, ha az organoleptikus vizsgálatot nem ugyanaz a laboratórium végzi, mint a kémiai vizsgálatot, ellenőrző elemzést stb.)
1.2. 5 litert meghaladó közvetlen csomagolás
Az "elsődleges minta" az 5 litert meghaladó közvetlen csomag esetében az összes egyedi minta reprezentatív része, amit a 2. táblázatnak megfelelő redukciós eljárás szerint kell megkapni. Az elsődleges mintát különböző mintavételi elemekből kell összeállítani.
Az elsődleges minta "mintavételi eleme" azokat a csomagokat jelenti, amelyek együttesen az elsődleges mintát teszik ki.
2. táblázat
Vételezendő egyedi minták minimális száma
| Csomagok száma a tételben | A vételezendő egyedi minták minimális száma |
| Legfeljebb 10 | 1 |
| 11 és 150 között | 2 |
| 151 és 500 között | 3 |
| 501 és 1 500 között | 4 |
| 1 501 és 2 500 között | 5 |
| > 2 500 /1 000 csomag | 1 plusz egyedi minta |
Annak érdekében, hogy csökkenthető legyen a közvetlen csomagokból történő mintavétel, az egyedi minták tartalmát homogenizálni kell az elsődleges minta előállításához. A különböző egyedi minták adagjait a keverés általi homogenizáláshoz egy közös tartályba kell önteni úgy, hogy a lehető leginkább védve legyenek a levegőtől.
Az elsődleges minta tartalmát egy sorozat legkevesebb 1,0 l űrtartalmú csomagba kell önteni, ezek mindegyike az elsődleges minta egy-egy mintavételi elemét képzi.
Az elsődleges minták száma a tagállamok egyedi igényeinek megfelelően növelhető (például, ha az organoleptikus vizsgálatot nem ugyanaz a laboratórium végzi, mint a kémiai vizsgálatot, ellenőrző elemzést stb.)
Minden egyes csomagot úgy kell feltölteni, hogy a tetején a lehető legkisebb legyen a levegőréteg, majd megfelelően le kell zárni és biztosítani, hogy a termék manipulációbiztos legyen.
Ezeket a mintavételi elemeket a megfelelő azonosítás érdekében fel kell címkézni.
2. ELEMZÉSEK ÉS EREDMÉNYEK
2.1. Minden egyes elsődleges mintát az EN ISO 5555 szabvány 2.5. pontja szerint laboratóriumi mintákra kell felosztani, és az Ib. melléklet folyamatábrája szerinti vagy bármilyen más véletlenszerű sorrendben kell elemezni.
2.2. Amennyiben a vizsgálatok minden eredménye megfelel az olaj bejelentett kategóriája jellemzőinek, az egész tételt megfelelőnek kell nyilvánítani.
Amennyiben az elemzések eredményeinek valamelyike nem felel meg az olaj bejelentett kategóriája jellemzőinek, az egész tételt nem megfelelőnek kell nyilvánítani.
3. A GYÁRTÁSI TÉTEL KATEGÓRIÁJÁNAK ELLENŐRZÉSE
3.1. A gyártási tétel kategóriájának ellenőrzésére, az illetékes hatóság a következő táblázat szerint megnövelheti a gyártási tétel különböző pontjain vételezendő elsődleges minták számát:
3. táblázat
A gyártási tétel mérete által meghatározott elsődleges minták száma
| Gyártási tétel mérete (literben) | Elsődleges minták száma |
| 7 500 | 2 |
| 7 500 – 25 000 | 3 |
| 25 000 – 75 000 | 4 |
| 75 000 – 125 000 | 5 |
| ≥ 125 000 | 6 + 1 minden további 50 000 liter után |
Az elsődleges mintát adó egyedi mintákat a tételben egymás mellett elhelyezkedő közvetlen csomagokból kell vételezni; minden egyes elsődleges minta elhelyezését fel kell jegyezni, és a mintákat egyértelmű azonosítóval kell ellátni.
Minden egyes elsődleges mintát az 1.1. és az 1.2. pontokban ismertetett eljárásoknak megfelelően kell vételezni.
Ezt követően minden elsődleges mintával a 2(1) cikkben ismertetett elemzéseket kell elvégezni.
3.2. Ha a 2(1) cikkben ismertetett elemzések eredményeinek egyike legalább egy elsődleges minta esetében nem felel meg az olaj bejelentett kategóriája jellemzőinek, a teljes mintázott tételt nem megfelelőnek kell nyilvánítani.
Ib. MELLÉKLET
FOLYAMATÁBRA ANNAK ELLENŐRZÉSÉRE, HOGY EGY OLÍVAOLAJ-MINTA MEGFELEL-E A BEJELENTETT KATEGÓRIÁNAK
Áttekintő táblázat

1. táblázat - Extra szűz olívaolaj - Minőségi kritériumok

2. táblázat - Szűz olívaolaj - Minőségi kritériumok

3. táblázat - Extra szűz olívaolaj és szűz olívaolaj - Tisztasági kritériumok

4. táblázat - Lampante olívaolaj - Tisztasági kritériumok

5. táblázat - Finomított olívaolaj - Minőségi kritériumok

6. táblázat - Olívaolaj (finomított olívaolajból és szűz olívaolajból) - Minőségi kritériumok

7. táblázat - Finomított olívaolaj, valamint finomított olívaolajból és szűz olívaolajból álló olívaolaj - Tisztasági kritériumok

8. táblázat - Nyers olívapogácsa-olaj - Tisztasági kritériumok

9. táblázat - Finomított olívapogácsa-olaj - Minőségi kritériumok

10. táblázat - Olívapogácsa-olaj - Minőségi kritériumok

11. táblázat - Finomított olívapogácsa-olaj és olívapogácsa-olaj - Tisztasági kritériumok

II. MELLÉKLET
A SZABAD ZSÍRSAVAK MEGHATÁROZÁSA HIDEG INJEKTÁLÁSOS MÓDSZERREL
1. TÁRGY ÉS ALKALMAZÁSI TERÜLET
A módszer az olívaolajok és az olívapogácsa-olajok szabadzsírsav-tartalmának meghatározását ismerteti. A szabadzsírsav-tartalmat az olajsav aránya alapján kiszámított savasságként adják meg.
2. ALAPELV
A mintát feloldják oldószerek keverékében és a jelen lévő szabad zsírsavakat kálium- vagy nátrium-hidroxid oldattal titrálják.
3. REAGENSEK
Minden esetben elismert analitikai minőségű reagenseket és desztillált vagy hasonló minőségű vizet kell használni.
3.1. Dietil-éter; 95 % etanol (v/v), azonos térfogategységű keveréke.
Semlegesítse pontosan a felhasználás pillanatában kálium-hidroxid oldattal (3.2.), 100 ml keverékenként 0,3 ml fenolftalein oldat (3.3.) hozzáadásával.
1. megjegyzés: a dietil-éter kiemelten tűzveszélyes és robbanásveszélyes peroxidokat képezhet. Használata során különleges óvatosságra van szükség.
2. megjegyzés: amennyiben nincs mód dietil-éter használatára, etanolt és toluolt tartalmazó oldószerkeverék használható. Szükség esetén az etanol propanol-2-vel helyettesíthető.
3.2. Kálium- vagy nátrium-hidroxid, titrált etanolos vagy vizes oldat, c(KOH) [vagy c(NaOH)], megközelítőleg 0,1 mol/l vagy szükség esetén c(KOH) [vagy c(NaOH)], megközelítőleg 0,5 mol/l. Oldószerek kereskedelmi forgalomban kaphatók.
A kálium-hidroxid (vagy nátrium-hidroxid) oldatának pontos koncentrációját ismerni és ellenőrizni kell a felhasználás előtt. Használjon a felhasználás előtt legalább öt nappal korábban készült, barna üvegben tárolt és gumidugóval lezárt oldatot. Az oldatnak színtelennek vagy szalmaszínűnek kell lennie.
Amennyiben a kálium-hidroxid (vagy nátrium-hidroxid) vizes oldatának használata során fáziselválás észlelhető, a vizes oldatot cserélje etanolos oldatra.
3. megjegyzés: a kálium-hidroxid (vagy nátrium-hidroxid) stabil színtelen oldatát a következő módon lehet elkészíteni. Forraljon fel 1 000 ml etanolt vagy vizet 8 g kálium-hidroxiddal (vagy nátrium-hidroxiddal) és 0,5 g alumíniumreszelékkel és folytassa a forralást egy órán keresztül. Közvetlenül ezután desztillálja. A desztillátumban oldjon fel szükséges mennyiségű kálium-hidroxidot (vagy nátrium-hidroxidot). Hagyja néhány napig állni, majd öntse le az átlátszó felülúszó folyadékot a leülepedett kálium-karbonátról (vagy nátrium-karbonátról).
Az oldat desztillálás nélkül is elkészíthető a következő módon: 1 000 ml etanolhoz (vagy vízhez) adjon 4 ml alumínium-butilátot és hagyja állni a keveréket néhány napig. Öntse le a felülúszó folyadékot és oldja fel a kívánt mennyiségű kálium-hidroxidot (vagy nátrium-hidroxidot). Az oldat használatra készen áll.
3.3. Fenolftalein 10 g/l oldata 95-96 % (v/v) etanolban vagy alkálikék 6B vagy timolftalein 20 g/l oldata 95-96 % (v/v) etanolban. Az erősen színezett olajok esetén alkálikék vagy timolftalein használandó.
4. BERENDEZÉS
Szokványos laboratóriumi berendezés, beleértve a következőket:
4.1. analitikai mérleg;
4.2. 250 ml-es Erlenmeyer-lombik;
4.3. "A" osztályú, 10 ml-es büretta 0,05 ml-es beosztással, vagy egyenértékű automata büretta.
5. ELJÁRÁS
5.1. A vizsgálati minta előkészítése
Ha a minta zavaros, át kell szűrni.
5.2. Vizsgált mennyiség
A mintavételezést a várható savasságnak megfelelően végezze, a következő táblázat szerint:
| Várható savasság (olajsav g/100 g) | Minta tömege (g) | Mérési pontosság (g) |
| 0–2 | 10 | 0,02 |
| > 2–7,5 | 2,5 | 0,01 |
| > 7,5 | 0,5 | 0,001 |
Mérje meg a mintát az Erlenmeyer-lombikban (4.2.).
5.3. Meghatározás
Oldja fel a mintát (5.2.) dietil-éter és etanol (3.1.) 50-100 ml korábban semlegesített keverékében.
Keverés közben végezzen titrálást 0,1 mol/l kálium-hidroxid (vagy nátrium-hidroxid) oldattal (3.2) (lásd a 4. megjegyzést), amíg az indikátor változni kezd (az indikátor elszíneződése legalább 10 másodpercig nem múlik el).
4. megjegyzés: amennyiben a szükséges 0,1 mol/l kálium-hidroxid (vagy nátrium-hidroxid) oldat mennyisége meghaladja a 10 ml-t, használjon 0,5 mol/l oldatot, vagy változtassa meg a mintatömeget a várható szabad savasság és a táblázatban javasolt érték szerint.
5. megjegyzés: amennyiben az oldat titrálás közben zavarossá válik, adjon hozzá megfelelő mennyiségű oldószert (3.1.), hogy tiszta oldatot kapjon.
Végezzen második meghatározást, ha az első eredmény magasabb, mint az olaj kategóriája esetében megadott határérték.
6. AZ EREDMÉNYEK KIFEJEZÉSE
Az olajsav tömegszázalékában kifejezett savasság a következő:

ahol:
V = a felhasznált titrált kálium-hidroxid (vagy nátrium-hidroxid) oldat térfogata ml-ben;
c = a felhasznált titrált kálium-hidroxid (vagy nátrium-hidroxid) oldat pontos koncentrációja mol/l-ben;
M = 282 g/mol; az olajsav moláris tömege g/mol-ban;
m = a minta tömege grammban.
Az olajsavban kifejezett savasság megadása a következőképpen történik:
a) a 0-1 értékek esetén két tizedesjegy pontossággal;
b) az 1-100 értékek esetén egy tizedesjegy pontossággal.
III. MELLÉKLET
A PEROXIDSZÁM MEGHATÁROZÁSA
1. Tárgy
Ez a melléklet egy, az állati és növényi olajok és zsírok peroxidszámának meghatározására szolgáló módszert ír le.
2. Fogalommeghatározás
A peroxidszám a mintában található azon anyagok mennyisége aktív oxigén/kg milliekvivalensben kifejezve, amelyek kálium-jodidot oxidálnak a megadott üzemi körülmények között.
3. Alapelv
A vizsgált mennyiség ecetsavas és kloroformos oldatának kezelése kálium-jodid oldattal. A felszabadított jód titrálása standardizált nátrium-tioszulfát oldattal.
4. Berendezés
Minden felhasznált berendezésnek redukáló- vagy oxidálóanyagoktól mentesnek kell lennie.
1. megjegyzés: Ne zsírozza be a csiszolt felületeket.
4.1. 3 ml-es üvegkanál.
4.2. Megközelítőleg 250 ml térfogatú lombikok csiszolt nyakkal és dugókkal, előzetesen kiszárítva és tiszta, száraz inert gázzal (nitrogénnel vagy lehetőleg szén-dioxiddal) feltöltve.
4.3. 5, 10 vagy 25 ml térfogatú büretta, legalább 0,05 ml-es beosztással, lehetőleg automatikus nullpont-beállítással, vagy egyenértékű automata büretta.
4.4. Analitikai mérleg.
5. Reagensek
5.1. Analitikai reagens minőségű kloroform, amelyet mentesítettek az oxigéntől a rajta átbuborékoltatott tiszta, száraz inert gázárammal.
5.2. Analitikai reagens minőségű jégecet, amelyet mentesítettek az oxigéntől a rajta átbuborékoltatott tiszta, száraz inert gázárammal.
5.3. Kálium-jodid frissen készített, jódtól és jodátoktól mentes, telített vizes oldata. Oldjon fel megközelítőleg 14 g kálium-jodidot megközelítőleg 10 ml vízben szobahőmérsékleten.
5.4. Nátrium-tioszulfát 0,01 mol/l (0,01 N) pontosan standardizált vizes oldata, közvetlenül a felhasználás előtt standardizálva.
Készítse el naponta frissen, a felhasználás előtt a 0,01 mol/l-es nátrium-tioszulfát oldatot 0,1 mol/l nátrium-tioszulfát standard oldatból, vagy határozza meg a pontos moláris koncentrációt. A tapasztalatok azt mutatják, hogy a stabilitás korlátozott és a pH-értéktől, valamint a szabad szén-dioxid mennyiségétől függ. A hígításhoz kizárólag frissen forralt, lehetőleg nitrogénnel tisztított vizet használjon.
A következő eljárás alkalmazása javasolt a nátrium-tioszulfát oldat pontos moláris koncentrációjának meghatározására:
Mérjen ki (250 ml vagy 500 ml térfogatú) mérőlombikba 0,27-0,33 g (a legközelebbi 0,001 g-ra kerekítve) kálium-jodátot (mKIO3), és öntse fel a jelzésig frissen forralt és szobahőmérsékletre lehűtött vízzel (V2). Pipettával töltsön át 5 ml-t vagy 10 ml-t az így kapott kálium-jodát oldatból (V1) egy 250 ml térfogatú Erlenmeyer-lombikba. Adjon hozzá 60 ml frissen forralt vizet, 5 ml 4 mol/l-es sósavat, valamint 25-50 mg kálium-jodidot vagy 0,5 ml-t a telített kálium-jodid oldatból. Titrálja ezt az oldatot a nátrium-tioszulfát oldattal (V3) a nátrium-tioszulfát oldat pontos moláris koncentrációjának meghatározásához.

ahol:
mKIO3 a kálium-jodát tömege grammban
V1 a kálium-jodát oldat térfogata milliliterben (5 ml vagy 10 ml)
V2 a kálium-jodát oldat össztérfogata milliliterben (250 ml vagy 500 ml)
V3 a nátrium-tioszulfát oldat térfogata milliliterben
wKIO3 a kálium-jodát tisztasága g/100 grammban
MKIO3 a kálium-jodát molekulatömege (214 g/mol)
T a nátrium-tioszulfát oldat pontos moláris koncentrációja (mol/l).
5.5. Keményítőoldat, 10 g/l vizes diszperzió, frissen készítve természetes oldható keményítőből. Ezzel egyenértékű reagensek is használhatók.
6. Minta
Ügyeljen rá, hogy a minta a vételezése és a tárolása során fénytől védett legyen, és hűvös helyen, teljesen megtöltött üvegedényekben, csiszolt üveg- vagy parafadugóval lezárva legyen tárolva.
7. Eljárás
A tesztet szórt nappali fény vagy mesterséges megvilágítás mellett kell végezni. Üvegkanálban (4.1.) vagy ennek hiányában lombikban (4.2.) mérje ki a mintából a várt peroxidszámnak megfelelő mennyiséget a legközelebbi 0,001 g-ra kerekítve, az alábbi táblázatban foglaltak szerint:
| Várható peroxidszám (meq) | A vizsgált mennyiség súlya (g) |
| 0–12 | 5,0–2,0 |
| 12–20 | 2,0–1,2 |
| 20–30 | 1,2–0,8 |
| 30–50 | 0,8–0,5 |
| 50–90 | 0,5–0,3 |
Vegye le a lombik (4.2.) dugóját és tegye bele a vizsgált mennyiséget tartalmazó üvegkanalat. Adjon hozzá 10 ml kloroformot (5.1.). Keveréssel oldja fel gyorsan a vizsgált mennyiséget. Adjon hozzá 15 ml ecetsavat (5.2.), majd 1 ml kálium-jodid oldatot (5.3.). Tegye rá gyorsan a dugót, rázza egy percen keresztül, majd hagyja állni pontosan öt percig fénytől védve, 15-25 °C hőmérsékleten.
Adjon hozzá megközelítőleg 75 ml desztillált vizet. Titrálja a felszabadított jódot a nátrium-tioszulfát oldattal (5.4.), miközben erőteljesen rázza; indikátorként használjon keményítőoldatot (5.5.).
Végezzen a mintán két meghatározást.
Ezzel egyidejűleg végezzen vakpróbát is. Amennyiben a vakpróba eredménye meghaladja a 0,05 ml 0,01 N nátrium-tioszulfát (5.4.) értéket, cserélje ki az elszennyeződött reagenseket.
8. Az eredmények kifejezése
A peroxidszám (PV) aktív oxigén/kg milliekvivalensben kifejezve a következő képlettel határozható meg:

ahol:
V = a teszthez felhasznált standardizált nátrium-tioszulfát oldat (5.4.) ml-jeinek száma, a vakpróba figyelembevételével korrigálva.
T = a felhasznált nátrium-tioszulfát oldat (5.4.) pontos moláris koncentrációja mol/l-ben.
m = a vizsgált mennyiség súlya g-ban.
Eredményként a két elvégzett meghatározás számtani közepét kell venni.
A meghatározás eredményét egy tizedesjegy pontossággal kell megadni.
IV. MELLÉKLET
A VIASZTARTALOM MEGHATÁROZÁSA KAPILLÁRIS GÁZKROMATOGRÁFIÁVAL
1. CÉL
Ez a módszer az olívaolajok viasztartalmának meghatározására szolgáló eljárást írja le. A viaszok leválasztása a szénatomok száma alapján történik. A módszer alkalmas a sajtolással és az extrakcióval kinyert olívaolajok (olívamaradék-olajok) megkülönböztetésére.
2. ALAPELV
A zsírszerű anyag oszlopkromatográfiás elválasztása, a megfelelő belső standard hozzáadásával, hidratáltszilikagél-oszlopon; a tesztkörülmények között elsőként eluálódott (a trigliceridekénél kisebb polaritású) frakció leválasztása, majd kapilláris gázkromatográfia segítségével történő analízise.
3. BERENDEZÉS
3.1. 25 ml-es térfogatú Erlenmeyer-lombik.
3.2. 15,0 mm belső átmérőjű, 30-40 cm hosszú kromatográfiás üvegoszlop csappal.
3.3. Gázkromatográf kapilláris oszloppal történő használatra, a következőkből álló, közvetlenül az oszlopra történő injektálásra alkalmas rendszerrel felszerelve:
3.3.1. Termosztatikus kamra az oszlopok számára (oszlopkemence), hőmérséklet-programozott fűtéssel.
3.3.2. Közvetlenül az oszlopba vezetett, hideg befecskendező rendszer.
3.3.3. Láng-ionizációs detektor és konverter erősítő.
3.3.4. Regisztráló-integrátor berendezés a konverter erősítővel (3.3.3.) történő használatra, amelynek a válaszideje nem haladja meg az egy másodpercet, és változtatható papírsebességű. (Lehetséges olyan informatizált rendszerek használata is, amelyek alkalmasak a gázkromatográfiai adatok számítógépes feldolgozására.)
3.3.5. Üvegből vagy ömlesztett szilícium-dioxidból készült 8-12 mm hosszúságú, 0,25-0,32 mm belső átmérőjű kapilláris oszlop, belülről egyenletesen 0,10-0,30 μm rétegvastagságú folyadékfázissal borítva. (a folyadékfázis a célnak megfelelő, kereskedelmi forgalomban SE-52 vagy SE-54 jelzésű lehet.)
3.4. 10 μl térfogatú mikrofecskendő közvetlenül az oszlopra történő injektáláshoz, keményített tűvel.
3.5. Vibráló elektromos keverő.
3.6. Forgó párologtató.
3.7. Görgős kemence.
3.8. Analitikai mérleg, + 0,1 mg-os mérési pontossággal.
3.9. Szokványos laboratóriumi üvegeszközök.
4. REAGENSEK
4.1. 60 és 200 μm közötti szemcsenagyságú szilikagél.
A szilikagélt 500 °C-on legalább négy órán keresztül melegíteni kell a kemencében. A kihűlést követően a felhasznált szilikagél-mennyiséghez képest 2 % vizet kell hozzáadni. Keverje át a keveréket megfelelő módon, amíg egyenletes masszát kap. Felhasználás előtt legalább 12 órán keresztül tartsa sötét helységben.
4.2. n-hexán, kromatográfiai minőségű.
4.3. Etil-éter, kromatográfiai minőségű.
4.4. n-heptán, kromatográfiai minőségű.
4.5. Hexános lauril-arachidát standardoldat, 0,1 % (m/V) oldat (belső standard). (Palmitil palmitát vagy mirisztil sztearát is megfelel.)
4.5.1. Szudán 1 (1-fenilazo-2-naftol).
4.6. Vivőgáz: hidrogén és hélium, gázkromatográfiai felhasználáshoz megfelelő tisztaságú.
4.7. Segédgázok:
- hidrogén, gázkromatográfiai felhasználáshoz megfelelő tisztaságú,
- levegő, gázkromatográfiai felhasználáshoz megfelelő tisztaságú.
5. ELJÁRÁS
5.1. A kromatográfiás oszlop előkészítése
Szuszpendáljon 15 g szilikagélt (4.1.) az n-hexánban (4.2.), és töltse az oszlopba (3.2.). A teljes ülepedés után elektromos rázóberendezéssel (3.5.) tömörítse, hogy homogénebb kromatográfiás réteget kapjon. 30 ml n-hexánnal mossa át az oszlopot, az esetleges szennyeződések eltávolítása érdekében. Mérjen le pontosan a mérleggel (3.8.) 500 mg-ot a mintából a 25 ml térfogatú Erlenmeyer-lombikba (3.1.), majd adja hozzá a megfelelő mennyiségű belső standardot (4.5.), a feltételezett viasztartalomnak megfelelően. Például olívaolaj esetében 0,1 mg lauril-arachidát oldatot, olívamaradék-olaj esetében 0,25-0,5 mg lauril-arachidát oldatot adjon hozzá. Az ily módon előkészített mintát 2 x 2 ml n-hexán segítségével vigye fel a kromatográfiás oszlopra (4.2.).
Hagyja lefutni az oldatot 1 mm-rel az abszorbens felszíne fölé, majd további 70 ml n-hexánnal mossa át az oszlopot, a természetesen jelen lévő n-alkánok eltávolítása érdekében. Kezdje el a kromatográfiás eluálást, és gyűjtsön össze 180 ml 99:1 arányú n-hexán-etil-éter elegyet úgy, hogy 10 másodpercenként megközelítőleg 15 csepp folyjon át. A minta eluálását 22 + 4 °C -os szobahőmérsékleten kell elvégezni.
Megjegyzések:
- A 99:1 arányú n-hexán/etil-éter keveréket minden nap frissen kell elkészíteni.
- A viaszok megfelelő eluálódásának vizuális ellenőrzése érdekében a mintaoldathoz hozzáadhat 100μl szudánt, az eluáló elegy 1 %-a arányában. Mivel a színezőanyag retenciós ideje a viaszok és a trigliceridek között helyezkedik el, amikor a színezék eléri a kromatográfiás oszlop alját, függessze fel az eluálást, ekkorra ugyanis valamennyi viasz eluálódott.
Az így kapott frakciót a rotációs bepárlóban (3.6.) párolja szárazra, amíg az oldószer gyakorlatilag teljesen eltűnik belőle. Az oldószer utolsó 2 ml-ét gyenge nitrogénárammal távolítsa el; majd adjon hozzá 2-4 ml n-heptánt.
5.2. Gázkromatográfiás elemzés
5.2.1. Előzetes műveletek
A kapilláris oszlopot illessze be a gázkromatográfba (3.3) úgy, hogy az oszlop bemenetét az oszlopra szerelt ("on-column") rendszerhez, az oszlop kimenetét pedig a detektorhoz csatlakoztassa. Végezze el a gázkromatográfiai rendszer összeszerelésének általános ellenőrzését (gázszerelvények szorossága, a detektor hatékonysága, a regisztráló rendszer hatékonysága stb.).
Az első alkalommal használt kapilláris oszlopokat kondicionálni kell. Fúvasson át egy kevés vivőgázt a kapilláris oszlopon, majd kapcsolja be a gázkromatográfiás berendezést. Melegítse fokozatosan addig, amíg körülbelül 4 óra elteltével eléri a 350 °C hőmérsékletet. Ezt a hőmérsékletet tartsa legalább két órán keresztül, majd hozza a berendezést üzemi körülmények közé (gázáram szabályozása, bontóláng begyújtása, csatlakoztatás az elektronikus íróhoz (3.3.4.), a kapilláris oszlop kemencéje, a detektor hőmérsékletének beállítása stb.) és állítsa be a jelet az analízis során tervezett legmagasabb szintnél legalább kétszer nagyobb érzékenységre. Az alapvonalnak lineárisnak kell lennie, illetve mindennemű csúcstól és ingadozástól mentesnek.
A negatív egyenes vonalú drift az oszlop illesztékeinek tökéletlen tömítettségét, míg a pozitív drift az oszlop nem megfelelő kondicionálását jelzi.
5.2.2. Az üzemi körülmények megválasztása
Általában véve az irányadó üzemi körülmények a következők:
- oszlophőmérséklet:
-
| 20 °C/perc | 5 °C/perc | 20 °C/perc | ||||
| kiindulási hőmérséklet: 80 °C (1′) | → | 240 °C | → | 325 °C (6′) | → | 340 °C (10′) |
- a detektor hőmérséklete: 350 °C,
- a befecskendezett anyag mennyisége: 1 μl az n-heptánoldatból (2-4 ml),
- vivőgáz: hélium vagy hidrogén, a kiválasztott gáz számára optimális lineáris sebességgel (lásd a függeléket),
- a berendezés érzékenysége: az alábbi körülményeknek megfelelően:
A fenti feltételek az oszlop és a gázkromatográf jellemzőinek megfelelően módosíthatók, hogy a kapott kromatogramok lehetővé tegyék valamennyi viasz leválasztását, a csúcsok kielégítő felbontásban láthatók legyenek (lásd az ábrát), miközben a C32 belső standard retenciós ideje 18 ± 3 perc. A viaszok legreprezentatívabb csúcsa a teljes skálaérték minimum 60 %-a legyen.
A csúcsok integrálásához a paramétereket úgy kell beállítani, hogy az lehetővé tegye az érintett csúcsok területeinek pontos becslését.
Megjegyzés:Tekintettel a magas véghőmérsékletre, pozitív drift előfordulása elfogadható, amely azonban nem haladhatja meg a teljes skálaérték 10 %-át.
5.3. Az elemzés végrehajtása
A 10 μl térfogatú mikrofecskendő segítségével szívjon fel 1 μl oldatot; a mikrofecskendő dugattyúját felfelé mozgatva ürítse ki a tűt. A tűt vezesse be a befecskendező berendezés válaszfalán, majd egy-két másodperc elmúltával fecskendezze be gyorsan az oldatot, és öt másodperc múlva lassan húzza ki a tűt.
Addig folytassa a rögzítést, ameddig a viaszok teljesen eluálódtak.
Az alapvonalnak minden esetben meg kell felelnie az előírt feltételeknek.
5.4. A csúcsok azonosítása
Az egyes csúcsok azonosítását a retenciós idő alapján és az azonos körülmények között analizált, ismert retenciós idejű viaszkeverékekkel összehasonlítva végezze.
A szűz olívaolajban található viaszok kromatogramja az ábrán látható.
5.5. Mennyiségi értékelés
A belső standard és a nyílt szénláncú C40-C46 észterek csúcsainak területét elektronikus integrálással számítsa ki.
Az egyes észterek mg/kg zsírszerű anyagban kifejezett viasztartalmát a következő képlet segítségével lehet kiszámítani:

ahol:
Ax = az egyes észterek csúcsának területe négyzetmilliméterben;
As = a belső standard csúcsának területe négyzetmilliméterben;
ms = a hozzáadott belső standard tömege milligrammban;
m = a meghatározni kívánt minta tömege grammban.
6. AZ EREDMÉNYEK KIFEJEZÉSE
Jegyezze fel a különböző C40-C46 viasztartalmak összegét mg/kg zsírszerű anyagban megadva (ppm).
Megjegyzés:A meghatározandó összetevők a C40-es és a C46-os észterek közötti tartományban lévő páros szénatomok csúcsainak felelnek meg, az alábbi ábrán látható olívaolajviasz-kromatogram példájának megfelelően. Ha a C46-os észter duplán jelenik meg, az azonosítás érdekében tanácsos elvégezni egy olyan olívamaradék-olaj viaszfrakcióinak az analízisét, amelyen a C46-os csúcs könnyen felismerhető, mivel tisztán kiemelkedik.
Az eredményeket egy tizedesjegy pontosságig kell megadni.
Ábra
Egy olívaolaj viasztartalmainak kromatogramja ( 3 )

Jelmagyarázat::
I. S. = Lauril-arachidát
1. = Diterpén észterek
2 + 2′ = C40 -es észterek
3 + 3′ = C42 -es észterek
4 + 4′ = C44 -es észterek C44
5. = C46 -os észterek
6. = Szterinészterek és triterpénes alkohol.
Függelék
A gáz lineáris sebességének meghatározása
Fecskendezzen be 1-3 μ metánt (vagy propánt) a normál üzemi körülményekre beállított gázkromatográfba. Stopperóra segítségével mérje meg a metán vagy propán oszlopon történő átáramlásának idejét, a befecskendezés pillanatától a csúcs eluálásának pillanatáig (tM).
A lineáris sebességet - cm/s-ben - az L/tM összefüggés adja meg, ahol L az oszlop hosszúsága cm-ben, tM pedig a stopperórával mért idő másodpercben.
VII. MELLÉKLET
A 2-GLICERIL MONOPALMITÁT SZÁZALÉKÁNAK MEGHATÁROZÁSA
1. CÉL ÉS ALKALMAZÁSI TERÜLET
Ez a módszer a palmitinsav-tartalom meghatározásának analitikus eljárását írja le a trigliceridek 2-pozícióján, a 2-gliceril monopalmitát százalékának értékelésének segítségével.
A módszer a szobahőmérsékletű (20 °C), folyékony növényi olajokra alkalmazható.
2. ALAPELV
Az előkészítés után az olajminta reakcióba lép a pankreatin-lipázzal: a triglicerid molekula 1- és 3-pozícióján végbemenő részleges és specifikus hidrolízis nyomán elválasztódnak a 2-pozíción lévő monogliceridek. A monoglicerid-frakción belül a 2-gliceril monopalmitát százalékát - a szililesedés után - kapilláris gázkromatográfia segítségével határozzuk meg.
3. BERENDEZÉS ÉS LABORATÓRIUMI ESZKÖZÖK
3.1. 25 ml térfogatú Erlenmeyer-lombik
3.2. 100, 250 és 300 ml térfogatú főzőpoharak
3.3. 21-23 mm belső átmérőjű, 400 mm hosszú kromatográfiás üvegoszlop összesütött üveglemezzel a csapnál
3.4. 10, 50, 100 és 200 ml térfogatú kalibrált kémcsövek
3.5. 100 és 250 ml térfogatú gömbölyű fenekű lombik
3.6. Forgó párologtató
3.7. 10 ml térfogatú, kúpos aljú centrifugakémcső csiszolt üvegdugóval
3.8. Laboratóriumi centrifuga 10 és 100 ml térfogatú kémcsövekkel
3.9. A hőmérsékletet 40 °C + 0,5 °C-on tartó termosztát
3.10. 1 és 2 ml térfogatú mérőpipetták
3.11. 1 ml-es fecskendő
3.12. 100 μl-es mikrofecskendő
3.13. 1 000 ml-es tölcsér
3.14. Gázkromatográf kapilláris oszloppal, az alábbiakból álló, közvetlenül az oszlopra történő injektálásra alkalmas"on column" rendszerrel, valamint a kiválasztott hőmérsékletet 1 °C pontossággal tartó kemencével felszerelve
3.15. Közvetlenül az oszlopba vezetett, "on column" hideg befecskendező rendszer
3.16. Lángionizációs detektor és elektrométer
3.17. Regisztráló-integrátor berendezés az elektrométerrel történő használatra, amelynek a válaszideje nem haladja meg az egy másodpercet, és változtatható papírsebességű
3.18. Üvegből vagy ömlesztett szilícium-dioxidból készült, 8-12 méter hosszúságú, 0,25-0,32 mm belső átmérőjű, 5 % 0,10-0,30 μm rétegvastagságú metil-polisziloxánnal vagy fenil-metil-polisziloxánnal bélelt kapilláris oszlop, amely 370 °C-on használható
3.19. 10 μl-es, legalább 7,5 cm hosszú mikrofecskendő keményített tűvel, közvetlenül az oszlopra történő injektáláshoz.
4. REAGENSEK
4.1. 0,063 és 0,200 mm közötti szemcsenagyságú szilikagél (70/280 mesh), a következőképpen elkészítve: helyezze a szilikagélt egy porcelán csészébe, 160 °C-os kemencében szárítsa ki 4 órán keresztül, majd hagyja kihűlni szobahőmérsékleten egy szárítóban. Adjon hozzá a szilikagél súlya 5 %-ának megfelelő vízmennyiséget a következőképpen: egy 500 ml térfogatú Erlenmeyer-lombikban mérjen le 152 g szilikagélt, adjon hozzá 8 g desztillált vizet, dugaszolja be, majd óvatosan rázza fel, hogy a víz egyenletesen oszoljon el. Felhasználás előtt hagyja állni legalább 12 óráig.
4.2. n-hexán (kromatográfia céljára alkalmas minőségű). A hexán helyettesíthető izooktánnal (kromatográfia céljára alkalmas minőségű 2,2,4-trimetil-pentán), feltéve, hogy összehasonlítható pontossági értékek érhetők el.
4.3. Izopropanol
4.4. Izopropanol, vízoldat 1/1 (V/V)
4.5. Pankreatin lipáz. A felhasznált lipáznak 2,0 és 10 lipáz egység/mg lipáztevékenységet kell mutatnia (Kereskedelmi forgalomban is kaphatók 2 és 10 lipáz egység/enzim mg lipáztevékenységet mutató pankreatin lipázok.)
4.6. Tris-hidroximetilaminometán-pufferoldat: 1 M vizes oldatát állítsa be pH 8 értékűre koncentrált HCl (sósav) (1/1 V/V) hozzáadásával (ellenőrizze potenciométerrel)
4.7. Nátrim-kolát (enzim minőségű), 0,1 %-os vizes oldat (ezt az oldatot az elkészítését követő 2 héten belül fel kell használni)
4.8. Kalcium-klorid, 22 %-os vizes oldat
4.9. Dietil-éter kromatográfiához
4.10. Kifejlesztőoldat: n-hexán/dietil-éter keverék (87/13) (V/V)
4.11. Nátrium-hidroxid, (súlyra) 12 %-os oldat
4.12. Fenolftalein, 1 %-os etanol oldat
4.13. Vivőgáz: hidrogén vagy hélium, gázkromatográfiai felhasználáshoz megfelelő tisztaságú
4.14. Segédgázok: hidrogén, minimum 99 %-os, nedvességtől és szerves szennyeződésektől mentes, illetve levegő, szintén gázkromatográfiai felhasználáshoz megfelelő tisztaságú
4.15. Szilanizációs reagensek: 9/3/1 (V/V/V) arányú piridin-hexametildiszilizin-trimetilkloroszilán keverék. A használatra kész oldatok kereskedelmi forgalomban kaphatók; egyéb szilanizációs reagensek is felhasználhatók, mint például N, 0-bis (trimetilszilil) trifluoroacetamid + 1 % trimetilkloroszilán azonos térfogatú víztelen piridinnel való keveréshez.
4.16. Referenciaminták: tiszta monogliceridek, vagy olyan monoglicerid-keverékek, amelyeknek ismert, a mintához hasonló százalékos összetételük van.
5. ELJÁRÁS
5.1. A minta előkészítése
5.1.1. A 3 %-nál kisebb szabad savasságú olajokat nem szükséges semlegesíteni a szilikagéllel elvégzett oszlopkromatográfia előtt. A 3 %-nál nagyobb szabad savasságú olajokat az 5.1.1.1. pontnak megfelelően semlegesíteni kell.
5.1.1.1. Töltsön az 1 000 ml-es tölcsérbe (3.13.) 50 g olajat és 200 ml n-hexánt. Adjon hozzá 100 ml izopropanolt, és az olaj 5 %-kal megemelt szabad savasságának megfelelő mennyiségű 12 %-os nátrium-hidroxid oldatot (4.11.). Egy percen keresztül rázza erőteljesen. Adjon hozzá 100 ml desztillált vizet, majd rázza fel ismét, ezután hagyja leülepedni.
A leválasztást követően távolítsa el az alsó szappanréteget. Távolítsa el az esetleges többi réteget (nyálka, oldhatatlan anyag) is. Mossa át a semlegesített olaj hexánoldatát többször 50-60 ml 1/1 (V/V) izopropanol/víz oldatban (4.4.), amíg a fenolftalein rózsaszín árnyalata el nem tűnik.
A hexán nagyobbik részét távolítsa el vákuum alatt végzett bepárlással (használjon például forgó párologtatót), és töltse át az olajat egy 100 ml-es gömbölyű lombikba (3.5.). Szárítsa ki az olajat vákuumban, amíg az oldószer teljesen el nem távozik.
Az eljárás végén az olaj savasságának 0,5 %-nál alacsonyabbnak kell lennie.
5.1.2. A fent leírtak szerint előkészített olajból 1,0 g-ot töltsön egy 25 ml térfogatú Erlenmeyer-lombikba (3.1.), és oldja fel 10 ml kifejlesztőoldatban (4.10.). Hagyja leülepedni az oldatot legalább 15 percen keresztül, a szilikagéllel végzett oszlop-kromatográfia előtt.
Ha az oldat zavaros, centrifugázza ki, a kromatográfiához szükséges optimális feltételeket teremtve. (Használatra kész, 500 mg-os SPE szilikagél hüvelyek is felhasználhatók a célra).
5.1.3. A kromatográfiás oszlop előkészítése
Öntsön az oszlopba (3.3.) megközelítőleg 30 ml kifejlesztőoldatot (4.10.), helyezzen az oszlop alsó felébe üvegrúd segítségével egy vattadarabot; nyomkodja meg, hogy a levegő távozzon belőle.
Egy főzőpohárban készítsen szuszpenziót, 25 g szilikagélt (4.1.) megközelítőleg 80 ml kifejlesztőoldatban feloldva, és egy tölcsér segítségével öntse az oszlopba.
Ellenőrizze, hogy az egész szilikagél be lett töltve az oszlopba; mossa át a kifejlesztőoldattal (4.10.), nyissa ki a csapot, és hagyja, hogy a folyadék szintje érjen fel megközelítőleg 2 mm-rel a szilikagél felső szintje fölé.
5.1.4. Oszlopkromatográfia
Egy 25 ml térfogatú Erlenmeyer-lombikban (3.1.) mérjen le pontosan 1,0 g, az 5.1. pontnak megfelelően előkészített mintát.
Oldja fel a mintát 10 ml kifejlesztőoldatban (4.10.). Az így kapott oldatot öntse az 5.1.3. pontnak megfelelően előkészített kromatográfiás oszlopba. Kerülje az oszlop felszínének mozgatását.
Nyissa meg a csapot, és hagyja átfolyni a mintaoldatot, amíg az el nem éri a szilikagél szintjét. Fejlessze ki 150 ml kifejlesztőoldattal. Állítsa a térfogatáramot 2 ml/percre (úgy, hogy körülbelül 60-70 perc alatt 150 ml oldat folyjon át az oszlopba).
Az eluátumot fogja fel egy előzetesen lemért, 250 ml térfogatú gömbölyű lombikban. Vákuum alatt párologtassa el az oldószert, az utolsó nyomokat nitrogénáram segítségével távolítva el.
Mérje le a gömbölyű lombikot, és számítsa ki a kinyert kivonatot.
(Használatra kész SPE szilikagél hüvelyek használata esetén a következőképpen járjon el: Öntsön 1 ml oldatot (5.1.2.) az előzetesen 3 ml n-hexánnal előkészített hüvelyekbe.)
Az átmosás után fejlessze ki az oldatot 4 ml 9/1 (V/V) arányú n-hexán/dietil-éter keverékkel.
Fogja fel az eluátumot egy 10 ml térfogatú kémcsőben, és párologtassa el nitrogénáram segítségével a teljes kiszáradásig.
A kiszárított maradékot tegye ki a pankreatin-lipáz hatásának (5.2.). Rendkívül fontos, hogy SPE hüvelyen való áthaladás előtt, illetve után egyaránt ellenőrizze a zsírsavösszetételt).
5.2. Hidrolízis pankreatin-lipázzal
5.2.1. Mérjen a centrifugakémcsőbe 0,1 g, az 5.1. pontnak megfelelően előkészített olajat. Adjon hozzá 2 ml pufferoldatot (4.6.), 0,5 ml nátrium-kolát-oldatot (4.7.) és 0,2 ml kalcium-klorid-oldatot; minden hozzáadás után alaposan rázza fel a keveréket. Zárja le a kémcsövet a csiszolt dugóval, majd tegye a 40 ± 0,5 °C hőmérsékleten tartott termosztátba (4.17.).
5.2.2. Adjon hozzá 20 mg lipázt, rázza fel alaposan (kerülje a dugó benedvesedését), majd tegye a kémcsövet a termosztátba pontosan 2 percre. Ezután vegye ki, rázza fel erőteljesen pontosan 1 percig, és hagyja kihűlni.
5.2.3. Adjon hozzá 1 ml dietil-étert, zárja le a dugóval és erőteljesen rázza fel, majd centrifugálás után az éteroldatot mikrofecskendő segítségével öntse át egy tiszta és száraz kémcsőbe.
5.3. A szilanizált származékok és a gázkromatográfia előkészítése
5.3.1. Egy mikrofecskendő segítségével töltsön 100 μl oldatot (5.2.3.) egy 10 ml térfogatú kúpos fenekű kémcsőbe.
5.3.2. Enyhe nitrogénárammal párologtassa el az oldószert, adjon hozzá 200 μl szilanizációs reagenst (4.15.), dugaszolja be a kémcsövet, és hagyja ülepedni 20 percig.
5.3.3. A 20 perc leteltével adjon hozzá 1-5 ml n-hexánt (a kromatográfiai körülmények függvényében): az így kapott oldat készen áll a gázkromatográfiás eljárásra.
5.4. Gázkromatográfia
Az üzemi körülmények a következők:
- az injektor hőmérséklete (oszlopra szerelt "on column" injektor): az oldószer forráspontjánál (68 °C) alacsonyabb,
- a detektor hőmérséklete: 350 °C,
- az oszlop hőmérséklete: a kemence hőmérsékletének programozása: 1 percig 60 °C, percenként 15 °C-kal emelve 180 °C-ig, majd percenként 5 °C-kal 340 °C-ig, majd 13 percig 340 °C,
- vivőgáz: hidrogén vagy hélium, az 1. ábrán tükröződő felbontáshoz szükséges lineáris sebességre beállítva. A C54 triglicerid retenciós idejének 40 ± 5 percnek kell lennie (lásd a 2. ábrát). (A fent leírt üzemi körülmények tájékoztató jellegűek. Minden esetben optimalizálni kell őket a kívánt felbontás elérése érdekében. A 2-gliceril monopalmitátnak megfelelő csúcs minimális magasságának a regisztráló berendezés skálája 10 %-ának kell lennie),
- a befecskendezett anyag mennyisége: 0,5-1 μl n-hexán oldat (5 ml) (5.3.3.).
5.4.1. A csúcsok azonosítása
Az egyes monogliceridek azonosítását a retenciós idők alapján, és az azonos körülmények között analizált standard monoglicerid-keverékek retenciós idejével összehasonlítva kell elvégezni.
5.4.2. Mennyiségi értékelés
Az egyes csúcsok területét elektronikus integrálással számítsa ki.
6. AZ EREDMÉNYEK KIFEJEZÉSE
A gliceril monopalmitát százalékát az ehhez tartozó csúcs területe, illetve az összes monoglicerid csúcsok területének összege közötti arány alapján számítsa ki (lásd a 2. ábrát), a következő képlet alapján:

ahol:
Ax = a gliceril monopalmitáthoz tartozó csúcs területe
ΣA = az összes monogliceridcsúcs területének összege
Az eredményt egy tizedesjegy pontosságig kell megadni.
7. ANALÍZIS JEGYZŐKÖNYV
Az analízis jegyzőkönyvnek a következőket kell tartalmaznia:
- a fenti módszerre való hivatkozás,
- a minta maradéktalan azonosításához szükséges mindenfajta információ,
- az analízis eredménye,
- a fenti módszertől, akár az érintett felek döntéséből, akár egyéb okból kifolyólag történő mindennemű eltérés,
- a laboratórium részletes azonosítása, az analízis időpontja és az analízisért felelős személyek aláírása.
1. ábra
A 20 % észterezett olajjal (100 %) kevert finomított olívaolajra kifejtett lipázhatás nyomán keletkezett szilanizációs reakciótermékek kromatogramja.

2. ábra
Kromatogram:
(A) nem észterezett olívaolaj, a lipázreakció után; a szilanizáció után; ilyen körülmények között (8-12 m-es kapilláris oszlop) a viaszfrakció a digliceridfrakcióval egy időben, vagy kevéssel utána eluálódik.
A lipázreakció után a triglicerid-tartalom nem haladhatja meg a 15 %-ot.

Jelmagyarázat::
1 = Szabad zsírsavak
2 = Monogliceridek
3 = Digliceridek
4 = Trigliceridek
* = 2-monopalmitin
** = C54 triglicerid
Kromatogram:
(B) észterezett olaj a lipázreakció után; a szilanizáció után; ilyen körülmények között (8-12 m-es kapilláris oszlop) a viaszfrakció a digliceridfrakcióval egy időben, vagy kevéssel utána eluálódik.
A lipázreakció után a triglicerid-tartalom nem haladhatja meg a 15 %-ot.

Jelmagyarázat:
1 = Szabad zsírsavak
2 = Monogliceridek
3 = Digliceridek
4 = Trigliceridek
* = 2-monopalmitin
** = C54 triglicerid
8. MEGJEGYZÉSEK - A LIPÁZ ELKÉSZÍTÉSE
Kielégítő lipázaktivitású lipázok kaphatók kereskedelmi forgalomban, de lehetséges laboratóriumi elkészítésük is a következők szerint:
Hűtsön le 5 kg friss sertés-hasnyálmirigyet 0 °C hőmérsékletre. Távolítsa el a környező szilárd zsiradékot és a kötőszövetet, majd dörzsölje szét egy keverőgépben, hogy pépes folyadékot kapjon. A keverővel keverje össze a pépet 2,5 l vízmentes acetonnal 4-6 órán keresztül, majd centrifugálja. Extrahálja a maradékot még háromszor, azonos mennyiségű acetonnal, majd két alkalommal aceton és dietil-éter 1/1 (V/V) keverékével, majd kétszer dietil-éterrel.
Szárítsa a maradékot in vacuo 48 órán keresztül, hogy stabil port kapjon, amely hűtőszekrényben és nedvességtől védve hosszan tárolható.
A LIPÁZ AKTIVITÁSÁNAK BEÁLLÍTÁSA
Készítsen olívaolaj-emulziót a következőképpen:
Megfelelő keverőben 10 percen keresztül rázza össze a 165 ml 100 g/l-es gumiarábikum-oldatból, 15 g jégzúzalékból és 20 ml előzetesen semlegesített olívaolajból álló keveréket.
Egy főzőpohárba tegyen 10 ml-t ebből az emulzióból, majd ezután 0,3 ml 0,2 g/ml-es nátrium-kolát oldatot és 20 ml desztillált vizet.
Tegye a főzőpoharat egy 37 °C hőmérsékleten tartott termosztátba; helyezze bele egy pH-mérő elektródáit és egy keverőspirált.
Egy büretta segítségével cseppenként adagoljon nátrium-hidroxid-oldatot (0,1 N), amíg a pH-érték 8,3 nem lesz.
Adjon hozzá megfelelő mennyiséget a lipáz vizes szuszpenziójából (0,1 g/ml lipáz). Amint a pH-mérő 8,3-at mutat, indítsa el a stopperórát, és olyan sebességgel csepegtesse a nátrium-hidroxid-oldatot, hogy a 8,3 pH-érték megmaradjon. Minden percben olvassa le a felhasznált lúgoldat térfogatát.
A megfigyeléseket rögzítse grafikonon, az időértékeket használva abszcisszaként, az állandó pH-érték fenntartásához szükséges 0,1 N-es lúgoldat ml-ben kifejezett térfogatát pedig ordinátaként. Lineáris grafikont kell kapnia.
A felhasznált por lipáz egység/mg-ban kifejezett aktivitása a következő képlettel határozható meg:

ahol:
A a lipáz egység/mg-ban kifejezett aktivitása,
V a percenként felhasznált nátrium-hidroxid-oldat ml-ek száma (a grafikonból kiszámítva),
N a nátrium-hidroxid-oldat moláris koncentrációja,
m a lipáz por vizsgált mennyiségének tömege mg-ban.
A lipázegység definíciója a következő: az az enzimmennyiség, amely percenként 10 μ-savekvivalenst szabadít fel.
IX. MELLÉKLET
SPEKTROFOTOMETRIÁS VIZSGÁLAT ULTRAIBOLYA FÉNYBEN
ELŐSZÓ
Az ultraibolya fényben végzett spektrofotometriás vizsgálat segítségével a zsír minőségével, tartósítási állapotával és a technológiai folyamatok által benne okozott változásokkal kapcsolatos információk állapíthatók meg. A módszernél megadott hullámhosszok elnyelése az oxidációs folyamatok és/vagy finomítási eljárások következtében jelen lévő konjugált dién- és triénrendszereknek köszönhető. Ezeket az abszorpciókat fajlagos kioltásban
(azaz a zsír meghatározott oldószerrel készült, 1 %-os oldatában, 10 mm-es vastagságban mért kioltásban) fejezik ki, amelyet egyezményesen K-val (más néven "kioltási tényező") jelölnek.
1. ALKALMAZÁSI KÖR
A melléklet az olívaolajok ultraibolya fényben történő spektrofotometrikus vizsgálatának eljárását ismerteti.
2. A MÓDSZER ELVE
Egy mintát feloldanak a szükséges oldószerben, majd az adott hullámhossztartományban meghatározzák az oldat kioltását a tiszta oldathoz viszonyítva.
A fajlagos abszorbancia kiszámítása 232 nm-en és 268 nm-en izooktánban vagy 232 nm-en és 270 nm-en ciklohexánban, 1 százalékos koncentrációnál, 10 mm-es küvettában történik.
3. BERENDEZÉS
3.1. Egy, az ultraibolya hullámhosszon (220 és 360 mm között) történő mérésre alkalmas spektrofotométer, amely képes a nanométeregységek egyenkénti leolvasására. Az abszorbancia- és a hullámhosszskála, illetve a szórt fény pontosságát és reprodukálhatóságát rendszeresen ellenőrizni kell.
3.1.1. Hullámhosszskála: Ellenőrzése holmium-oxidot vagy holmium-oxid oldatot tartalmazó, jól elkülönülő abszorpciós sávokkal rendelkező (zárt vagy nem zárt) optikaiüveg-szűrőből álló referenciaanyag segítségével történhet. A referenciaanyagok alkalmasak a látható és ultraibolya tartományokban mérő, legfeljebb 5 nm névleges spektrális sávszélességgel rendelkező spektrofotométerek hullámhosszskáláinak ellenőrzésére és kalibrálására. A méréseket 640-240 nm hullámhossztartományban, levegős vakmintával szemben végzik a referenciaanyagokhoz mellékelt utasításoknak megfelelően. Az alapkorrekciót üres sugármenettel végzik minden egyes résszélesség-változásnál. A minta hullámhosszai a referenciaanyag tanúsítványában találhatóak.
3.1.2. Abszorbanciaskála: Ellenőrzése kereskedelmi forgalomba kapható, savas kálium-dikromát oldatokat tartalmazó, meghatározott koncentrációjú és a λmax mellett tanúsított abszorbancia értékű zárt referenciaanyagok segítségével történhet (kálium-dikromát négyféle, perklórsavas, négy UV-kvarcküvettába zárt oldata. A négy oldat a linearitás és fotometrikus pontosság UV-fényben történő mérésére szolgál). A kálium-dikromát oldatokat az alapkorrekciót követően a használt savból álló vakmintával szemben mérik a referenciaanyagokhoz mellékelt utasításoknak megfelelően. A minta abszorbanciaértékei a referenciaanyag tanúsítványában találhatóak.
Egy másik lehetőség a fotocella és a fénysokszorozó átvitelének ellenőrzéséhez a következő: mérjen ki 0,2000 g tiszta kálium-kromátot a spektrofotometriához és oldja fel 0,05 N kálium-hidroxid-oldatban egy 1 000 ml térfogatú mérőlombikban, majd a lombikot töltse fel a jelig. Az így kapott oldatból vegyen pontosan 25 ml-t, tegye át egy 500 ml térfogatú mérőlombikba, majd ugyanazzal a kálium-hidroxid-oldattal töltse fel a lombikot a jelig.
Mérje meg az így kapott oldat kioltását 275 nm hullámhosszon, referenciaként a kálium-hidroxid-oldatot használva. Az 1 cm-es küvettával mért kioltásnak 0,200 ± 0,005 értékűnek kell lennie.
3.2. Az ultraibolya-hullámhosszon (220 és 360 nm között) történő mérésre alkalmas, 10 mm-es optikai átviteli úttal rendelkező, kupakkal ellátott, szögletes kvarcküvetták. Vízzel vagy más megfelelő oldószerrel feltöltve a küvetták nem mutathatnak egymáshoz képest 0,01 kioltási egységnél nagyobb különbséget.
3.3. Egyjelű mérőlombikok, 25 ml térfogat, A osztály.
3.4. 0,0001 g pontossággal mérő analitikai mérleg.
4. REAGENSEK
Ellenkező értelmű utasítás hiányában az elemzés során csak elismert analitikai minőségű reagenseket és desztillált vagy ásványmentesített vizet, illetve hasonló tisztaságú vizet használjon.
Oldószer: Izooktán (2,2,4-trimetil-pentán) a 232 nm-en és 268 nm-en történő méréshez vagy ciklohexán a 232 nm-en és 270 nm-en történő méréshez, a desztillált vízhez viszonyítva 232 nm-en 0,12-nál, 270 nm-en pedig 0,05-nál kisebb abszorbanciával, 10 mm-es küvettában mérve.
5. ELJÁRÁS
5.1. A mintának teljesen homogénnek kell lennie, és nem lehetnek benne lebegő szennyeződések. Ellenkező esetben kb. 30 °C hőmérsékleten át kell szűrni papíron.
5.2. A fentiek szerint előkészített minta megközelítőleg 0,25 g-ját pontosan (1 mg pontossággal) be kell mérni egy 25 ml-es mérőlombikba, az előírt oldószerrel a jelig fel kell tölteni, majd homogenizálni kell. A kapott oldatnak teljesen átlátszónak kell lennie. Amennyiben opálosság vagy zavarosság lép fel, papíron gyorsan át kell szűrni.
MEGJEGYZÉS: Általánosságban 0,25-0,30 g tömeg elegendő a szűz és az extra szűz olívaolaj abszorbanciájának 268 nm-en és 270 nm-en történő méréséhez. A 232 nm-en történő mérésekhez általában 0,05 g tömegű minta szükséges, ezért általában két külön oldat készül. Az olívapogácsa-olajok, a finomított olívaolajok és a hamisított olívaolajok abszorbanciájának méréséhez általában kisebb mintaadag, pl. 0,1 g is elegendő magasabb abszorbanciájuknak köszönhetően.
5.3. Szükség esetén korrigálja az alapot (220-290 nm) mindkét kvarcküvettából (minta és referencia) vett oldattal, majd töltse fel a mintát tartalmazó kvarcküvettát a tesztoldattal, és a használt oldószert referenciaként használva mérje meg a kioltásokat 232, 268 vagy 270 nm-en.
A mért kioltási értékeknek 0,1 és 0,8 közé kell esniük, vagy a spektrofotométer linearitási tartományán belül kell lenniük, amelyet ellenőrizni kell. Ha nem így van, a mérést meg kell ismételni töményebb, illetve hígabb oldatokkal.
5.4. Az abszorbancia 268 vagy 270 nm-en történő mérését követően mérje meg az abszorbanciát λmax, λmax + 4 és λmax - 4 értékeken. Ezeket az abszorbanciaértékeket a fajlagos kioltásban bekövetkező változás (ΔΚ) meghatározására használják.
MEGJEGYZÉS: a λmax 268 nm izooktán oldószer esetében, illetve 270 nm ciklohexán esetében.
6. AZ EREDMÉNYEK KIFEJEZÉSE
6.1. Fel kell jegyezni a különböző hullámhosszokon a következő módon kiszámított fajlagos kioltásokat (kioltási tényezőket):
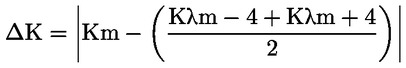
ahol:
Kλ = fajlagos kioltás λ hullámhosszon;
Eλ = a λ hullámhosszon mért kioltás;
C = az oldat koncentrációja g/100 ml-ben;
s = a kvarcküvetta átviteli úthossza cm-ben;
kerekítve 2 tizedesjegyre.
6.2. A fajlagos kioltás változása (ΔΚ)
A kioltás abszolút értékének változását (ΔΚ) az alábbi képlet adja meg:

ahol Km a maximális abszorpcióhoz tartozó, 270 nm és 268 nm (a használt oldattól függően) hullámhosszon mért fajlagos kioltás.
kerekítve 2 tizedesjegyre.
X. MELLÉKLET
ZSÍRSAV-METILÉSZTEREK GÁZKROMATOGRÁFIÁS MEGHATÁROZÁSA
1. ALKALMAZÁSI KÖR
Ez a melléklet útmutatást nyújt a növényi zsírokban és olajokban lévő szabad és kötött zsírsavak gázkromatográfiás meghatározásához azok zsírsav-metil-észterré alakítását követően.
A triacil-glicerinek kötött zsírsavai, valamint az észteresítési módszertől függően - a szabad zsírsavak zsírsav-metil-észterré alakulnak, amelyeket kapilláris gázkromatográfia segítségével lehet meghatározni.
Az ebben a mellékletben bemutatott módszer lehetővé teszi a zsírsav-metilészterek meghatározását C12-től C24-ig, beleértve a telített, a cisz- és a telítetlen transz-, valamint a cisz- és a többszörösen telítetlen transz zsírsav-metilésztereket.
2. ALAPELV
A zsírsav-metilészterek mennyiségi elemzésére gázkromatográfiát használnak. A zsírsav-metilésztert az A. résznek megfelelően készítik elő. Ezt követően injektorba fecskendezik és elpárologtatják azt. A zsírsav-metilészter elválasztását meghatározott polaritású és hosszúságú analitikai oszlopokon végzik. A zsírsav-metilészterek észlelésére lángionizációs detektort (FID) használnak. Az elemzés feltételei a B. részben találhatóak.
Vivőgázként (mozgó fázis) hidrogén vagy hélium használható a zsírsav-metilészterek FID-vel végzett gázkromatográfiája során. A hidrogén felgyorsítja az elválasztást és élesebb csúcsokat ad. A stacioner fázis egy szilícium-dioxidból készült közömbös, szilárd felületen lévő vékony folyadékfilm mikroszkopikus rétege.
Miközben áthaladnak a kapilláris oszlopokon, a vizsgált elpárologtatott vegyületek kölcsönhatásba lépnek az oszlop belső felületét borító stacioner fázissal. A különböző vegyületek az eltérő kölcsönhatás következtében eltérő időben (ezt nevezik a vegyület retenciós idejének egy adott elemzési paraméterkészleten) eluálnak. A retenciós idők összehasonlítása teszi lehetővé a különböző vegyületek azonosítását.
A. RÉSZ
ZSÍRSAV-METILÉSZTEREK ELŐÁLLÍTÁSA OLÍVAOLAJBÓL ÉS OLÍVAPOGÁCSA-OLAJBÓL
1. ALKALMAZÁSI KÖR
Ez a rész a zsírsav-metilészterek előállítását határozza meg. Zsírsav-metilészterek olívaolajból és olívapogácsa-olajból történő előállítására szolgáló módszereket foglal magában.
2. ALKALMAZÁSI KÖR
A zsírsav-metilészterek olívaolajból és olívapogácsa-olajból történő előállítását kálium-hidroxid metanolos oldatával történő átészterezéssel végzik szobahőmérsékleten. A mintának az átészteresítést megelőző tisztításának szükségessége a minta szabad zsírsavtartalmától és a meghatározandó analitikus paraméterétől függ; az alábbi táblázat alapján választható:
| Olajkategória | Módszer |
| ≤ 2,0 % savasságú szűz olívaolaj | 1. Zsírsav 2. Transz-zsírsavak 3. ΔECN42 (SPE szilikagéllel történő tisztítást követően) |
| Finomított olívaolaj | |
| Finomított olívaolajból és szűz olívaolajból álló olívaolaj | |
| Finomított olívapogácsa-olaj | |
| Olívapogácsa-olaj | |
| > 2,0 % savasságú szűz olívaolaj Nyers olívapogácsa-olaj | 1. Zsírsavak (SPE szilikagéllel történő tisztítást követően) 2. Transz-zsírsavak (SPE szilikagéllel történő tisztítást követően) 3. ΔECN42 (SPE szilikagéllel történő tisztítást követően) |
3. MÓDSZERTAN
3.1. Kálium-hidroxid metanolos oldatával szobahőmérsékleten történő átészterezés.
3.1.1. Alapelv
A metilészterek kálium-hidroxid metanolos oldatával történő átészterezéssel, az elszappanosítás bekövetkezését megelőző közbenső stádiumként képződnek.
3.1.2. Reagensek
3.1.2.1. Legfeljebb 0,5 tömegszázalék víztartalmú metanol.
3.1.2.2. Hexán, kromatográfiás minőség.
3.1.2.3. Heptán, kromatográfiás minőség.
3.1.2.4. Dietil-éter, elemzéshez stabilizált.
3.1.2.5. Aceton, kromatográfiás minőség.
3.1.2.6. Eluáló oldószer az olaj oszlop-/SPE-kromatográfiával történő tisztítására, hexán/dietil-éter 87:13 térfogatarányú elegye.
3.1.2.7. Kálium-hidroxid, kb. 2M metanololdat: oldjon fel 11,2 g kálium-hidroxidod 100 ml metanolban.
3.1.2.8. Szilikagél patronok, 1 g (6 ml), szilárd fázis kioldáshoz.
3.1.3. Készülékek
3.1.3.1. Lecsavarható fedelű kémcsövek (5 ml térfogatú) PTFE-tömítőgyűrűs kupakkal.
3.1.3.2. Kalibrált vagy automatikus pipetták, 2 ml-es és 0,2 ml-es.
3.1.4. Olajminták tisztítása
A mintákat szükség esetén úgy tisztítják, hogy az olajat szilikagél szilárd fázisú extrakciós patronon engedik át. A szilikagél patront (3.1.2.8.) egy vákuumos eluáló rendszerbe helyezik és6 ml hexánnal (3.1.2.2.) mossák át; a mosás vákuum nélkül történik. Ezt követően az olaj (hozzávetőlegesen 0,12 g) 0,5 ml hexánnal készült oldatát az oszlopra töltik. Az oldatot átfuttatják az oszlopon, majd 10 ml hexán/dietil-éterrel (87:13 térfogatarány) (3.1.2.6.) eluálják. A kombinált eluátumokat homogenizálják és két hasonló mennyiségre osztják szét. Rotációs bepárlóban csökkentett nyomás alatt, szobahőmérsékleten szárazra párolják az egyiket. A maradékot 1 ml heptánban feloldják, ezt követően az oldat készen áll a gázkromatográfiás zsírsavelemzésre. A másik részt elpárologtatják, majd a maradékot 1 ml acetonban feloldják a triglicerid szükség esetén elvégzendő folyadékkromatográfiás elemzéséhez.
3.1.5. Az eljárás
Egy 5 ml-es lecsavarható fedelű kémcsőben (3.1.3.1.) mérjen le kb. 0,1 g-ot az olajmintából. Adjon hozzá 2 ml heptánt (3.1.2.2.) és rázza össze. Adjon hozzá 0,2 ml metanolos kálium-hidroxid oldatot (3.1.2.7.), helyezze fel a PTFE-tömítőgyűrűs kupakot, szorítsa rá a kupakot, és rázza össze erőteljesen 30 másodpercig. Hagyja, hogy rétegek képződjenek, míg a felső oldat tiszta nem lesz. Dekantálja a metilésztert tartalmazó felső réteget. A heptános oldat ekkor injektálható a gázkromatográfba. Az elemzés megkezdéséig javasolt az oldatot hűtőszekrényben tárolni. Az oldat tárolása 12 óránál tovább nem javasolt.
B. RÉSZ
ZSÍRSAV-METILÉSZTEREK GÁZKROMATOGRÁFIÁS ELEMZÉSE
1. ALKALMAZÁSI KÖR
Ez a rész általános iránymutatót ad a kapilláris oszlopokat használó gázkromatográfiás vizsgálat alkalmazására, az A. részben megállapított módszer segítségével kapott zsírsav-metilészterekből álló keverék minőségi és mennyiségi jellemzőinek meghatározására.
Ez a rész nem alkalmazható polimerizált zsírsavakra.
2. REAGENSEK
2.1. Vivőgáz
Inert gáz (hélium vagy hidrogén), alaposan kiszárítva, kevesebb, mint 10 mg/kg oxigéntartalommal.
1. megjegyzés: A hidrogén megkétszerezi az elemzés sebességét, azonban veszélyes. Rendelkezésre állnak védőfelszerelések.
2.2. Segédgázok
2.2.1. Hidrogén (tisztaság ≥ 99,9 %), szerves szennyeződésektől mentes.
2.2.2. Levegő vagy oxigén, szerves szennyeződésektől mentes.
2.2.3. Nitrogén (tisztaság > 99 %).
2.3. Referenciaminta
Tiszta zsírsavak metil-észtereinek, illetve ismert összetételű zsírok metil-észtereinek keveréke, lehetőség szerint a vizsgált zsíros anyaghoz hasonló. Az oktadekán-, oktadekadién- és oktadekatrién-metilészterek cisz- és transz-izomerjei hasznosak a telítetlen savak transz-izomerjeinek azonosításához.
Meg kell akadályozni a többszörösen telítetlen zsírsavak oxidációját.
3. ESZKÖZÖK
A következő útmutató a kapilláris oszlopokat és láng-ionizációs detektort alkalmazó gázkromatográfiához használt szokásos felszerelésre vonatkozik.
3.1. Gázkromatográf
A gázkromatográf a következő elemekből áll:
3.1.1. Befecskendező rendszer
Használjon kapilláris oszlopokkal rendelkező befecskendező rendszert, amely esetben a befecskendező rendszernek kifejezetten az ilyen oszlopokkal történő használatra tervezettnek kell lennie. A befecskendező rendszer lehet osztott típusú vagy az oszlopon lévő osztatlan típusú.
3.1.2. Kemence
A kemencének képesnek kell lennie a kapilláris oszlop legalább 260 °C hőmérsékletre történő hevítésére, illetve a kívánt hőmérséklet tartására 0,1 °C-on belüli pontossággal. Az utóbbi követelmény különösen fontos olvasztott szilícium-dioxid csövek használata esetén.
A hőmérséklet-programozott fűtés használata minden esetben ajánlatos, különösen a 16-nál kevesebb szénatomot tartalmazó zsírsavak esetén.
3.1.3. Kapilláris oszlop
3.1.3.1. A vizsgált anyagok tekintetében semleges anyagból (általában üveg vagy szilícium-dioxid) készült cső. A belső átmérője 0,20-0,32 mm. A belső felületét a stacioner fázisból álló bevonat felvitele előtt megfelelően kezelni kell (például felületkikészítés, inaktiválás). A zsírsavakhoz és a zsírsavak cisz- és transz-izomerjeihez 60 m hosszúság elegendő.
3.1.3.2. Stacioner fázis, poláris polisziloxán (cianoszilikonok) kötött (keresztbe kapcsolt) oszlopok használhatók.
2. megjegyzés: Fennáll a veszélye, hogy a poláris polisziloxánok megnehezítik a linolénsav és a C20 savak azonosítását és leválasztását.
A bevonatoknak vékonyak, azaz 0,1-0,2 μm-eseknek kell lenniük.
3.1.3.3. Az oszlop összeszerelése és kondicionálása
Tartsa be a kapillárisoszlopok összeszerelésének normál szabályait, vagyis az oszlopok elrendezése a kemencén (talapzaton), szerelvények kiválasztása és összeszerelése (szivárgásmentesség), az oszlop végének elhelyezése a befecskendező rendszerben és a detektorban (a holtterek csökkentése). Helyezze az oszlopot vivőgáz árama alá (például 0,3 bar (30 kPa) 25 m hosszú és 0,3 mm belső átmérőjű oszlop esetén).
Kondicionálja az oszlopot a kemence környezeti hőmérsékletéről induló, a stacioner fázis lebomlási határa alatti 10 °C-ig terjedő, 3 °C/perc sebességű felfűtésével. Tartsa a kemencét ezen a hőmérsékleten egy órán keresztül, a nullapont stabilizálódásáig. Az izotermikus körülmények között történő munkához állítsa vissza 180 °C hőmérsékletre.
3. megjegyzés: A megfelelően kondicionált oszlopok kereskedelmi forgalomban kaphatók.
3.1.4. Lángionizációs detektor és konvertererősítő
3.2. Fecskendő
A fecskendő maximális térfogata 10 μl, 0,1 μl-es beosztásokkal.
3.3. Adatgyűjtő rendszer
Detektorokkal ellátott, online bekötött adatgyűjtő rendszerek, csúcsok összegzésére és normalizálásra képes szoftverrel.
4. AZ ELJÁRÁS
A 4.1-4.3. pontban leírt műveletek a láng-ionizációs detektor használatára vonatkoznak.
4.1. Vizsgálati körülmények
4.1.1. A kapilláris oszlopok optimális üzemi körülményeinek kiválasztása
A kapilláris oszlopok hatásfok- és permeabilitási jellemzőinek köszönhetően az alkotóelemek elválasztása és az elemzés időtartama nagymértékben függ a vivőgáz oszlopbeli sebességétől. Ezért az üzemi körülmények optimalizálására van szükség ennek a paraméternek (egyszerűbben az oszlop nyomásveszteségének) a változtatásával, attól függően, hogy az elválasztást szeretnénk növelni vagy gyorsabb analízist szeretnénk végezni.
Az alábbi körülmények bizonyultak megfelelőnek a zsírsav-metilészterek (C4-C26) elválasztására. A kromatogramokra a B. függelék tartalmaz példákat:
| Injektor hőmérséklete: | 250 °C |
| Detektor hőmérséklete: | 250 °C |
| Kemence hőmérséklete: | 165 °C (8 perc 210 °C-ra 2 °C/perc sebességgel |
| Hidrogén vivőgáz: | Oszlopfej nyomása: 179 kPa |
| Teljes átáramló mennyiség: | 154,0 mL/perc; |
| Megosztási arány: | 1:100 |
| Befecskendezett mennyiség: | 1 μl |
4.1.2. A feloldás meghatározása (lásd az A. mellékletet)
Az I. és II. szomszédos csúcsok R feloldása az alábbi képlettel számolható ki:
R = 2 × ((dr ( II ) - d r(I))/(ω(I) + ω(II))) vagy R = 2 × ((tr ( II ) - t r(I))/(ω(I) + ω(II))) (USP) (United States Pharmacopeia - Amerikai Gyógyszerkönyv),
vagy
R = 1,18 × ((tr ( II ) - tr(I))/(ω0,5(I) + ω0,5(II))) (EP, BP, JP, DAB), (JP (Japanese Pharmacopeia - Japán Gyógyszerkönyv), EP (Pharmacopée Européenne - Európai Gyógyszerkönyv), (BP (British Pharmacopeia - Brit Gyógyszerkönyv))
ahol:
d r(I) az I. csúcs retenciós távolsága;
d r(II) a II. csúcs retenciós távolsága;
tr(I) az I. csúcs retenciós ideje;
tr(II) a II. csúcs retenciós ideje;
ω(I) az I. csúcs alapjának szélessége;
ω(II) a II. csúcs alapjának szélessége;
ω0,5 az adott vegyület csúcsszélessége a csúcs középmagasságában;
Ha ω(I) ≈ ω(II), az R értékét az alábbi képlettel kell kiszámolni:
R = (dr ( II ) - d r(I))/ω = (dr ( II ) - d r(I))/4σ
ahol:
σ a standard eltérés (lásd A függelék, 1. ábra).
Ha a két csúcs közötti d r(II) - d r(I) dr távolság 4σ, az E feloldási tényező = 1.
Ha két csúcs nincs teljesen elválasztva, a két csúcs inflexiós pontjának tangensei a C pontban keresztezik egymást. A két csúcs teljes elválasztásához a két csúcs közötti távolságnak az alábbinak kell lennie:
d r(II) - d r(I) = 6 σ ahol R = 1,5 (lásd A. függelék, 3. ábra).
5. AZ EREDMÉNYEK KIFEJEZÉSE
5.1. Minőségi elemzés
Azonosítsa a B. függelék 1. ábrájának kromatogramjából a minta metil-észter csúcsait, amennyiben szükséges, interpolálás, vagy pedig a metil-észterekből álló referenciakeverékekhez (lásd a 2.3. pontot) tartozókkal való összehasonlítás útján.
5.2. Mennyiségi elemzés
5.2.1. Az összetétel meghatározása
Az alábbi módon számítsa ki az egyes zsírsav-metilészterek wi tömeghányadát (a metil-észter tömegszázalékában kifejezve):
5.2.2. Számítási módszer
5.2.2.1. Általános eset
Számítsa ki egy adott i összetevő esetében a mennyiséget a metil-észterek tömegének százalékában kifejezve úgy, hogy meghatározza az adott összetevő csúcsa alatti terület nagyságát az összes csúcs alatti területhez képest, a következő képlet segítségével:
wi = (Ai/ΣA) × 100
ahol:
Ai az egyes i zsírsav-metilészterek csúcsa alatti terület;
ΣA az egyes i zsírsav-metilészterek csúcsai alatti területek összege.
Az eredményeket két tizedesjegy pontossággal kell megadni.
4. megjegyzés: A zsírok és olajok esetében a zsírsav-metilészterek tömeghányada megegyezik a triacil-glicerinek tömeghányadával g/100 g-ban kifejezve. Azokat az eseteket, ahol ez a feltételezés nem érvényes, lásd az 5.2.2.2. pontban.
5.2.2.2. A korrekciós tényezők használata
Bizonyos esetekben, például a nyolcnál kevesebb szénatommal rendelkező zsírsavak vagy a másodlagos csoportokat tartalmazó savak jelenlétében, a területeket korrekciós tényezőkkel (Fci) kell korrigálni. Ezeket a tényezőket minden egyes eszközhöz meg kell határozni. Erre a célra a megfelelő tartományban lévő, hitelesített zsírsav-összetételű referenciaanyagokat kell használni.
5. megjegyzés: Ezek a korrekciós tényezők nem azonosak az A. mellékletben található elméleti FID korrekciós tényezőkkel, mivel magukban foglalják a befecskendező rendszer teljesítményét stb. is. A nagyobb eltérések esetén azonban a teljes rendszer teljesítményét ellenőrizni kell.
Ennél a referenciakeveréknél az i zsírsav-metilészter tömegszázaléka a következő képlet segítségével adható meg:
wi = (mi /Σm) × 100
ahol
mi az i zsírsav-metilészter tömege a referenciakeverékben;
Σm a referenciakeverékben lévő különböző zsírsav-metilészterek összes tömege.
A referenciakeverék kromatogramjából számítsa ki az i zsírsav-metilészter területre vetített százalékát a következő képlet segítségével:
wi = (Ai/ΣA) × 100
ahol:
Ai az i zsírsav-metilészter területe a referenciakeverékben;
ΣA a referenciakeverékben lévő különböző zsírsav-metilészterek összes területe.
Az Fc korrekciós tényező az alábbi:
Fc = (mi × ΣA)/(Ai/Σm)
Például az i egyes zsírsav-metilészterek tömegszázaléka a következő:
wi = (Fi × Ai)/Σ (Fi × Ai)
Az eredményeket két tizedesjegy pontossággal kell megadni.
6. megjegyzés: A számított érték megfelel az egyes zsírsav-metilészterek a triacil-glicerinként számolt tömegszázalékával g/100 g-ban kifejezve.
5.2.2.3. A belső standard használata
Bizonyos elemzések esetén (például amikor nem mindegyik zsírsav mennyiségét határozzuk meg, amikor négy és hat szénatomot tartalmazó savak vannak jelen a 16 és 18 szénatomos savak mellett, vagy ha egy mintában az egyik zsírsav pontos mennyiségét kell meghatározni) belső standardot kell használni. Leggyakrabban az 5, 15 vagy 17 szénatomos zsírsavakat használják. A belső standardhoz (amennyiben szükséges) meg kell határozni a korrekciós tényezőt.
Az i összetevő metil-észterekhez viszonyított tömegszázalékát a következő képlet segítségével lehet kiszámítani:
wi = (mIS × Fi × Ai)/(m × FIS × AIS)
ahol:
A i az i zsírsav-metilészter területe;
A IS a belső standard területe;
F i az i zsírsav zsírsav-metilészterben kifejezett korrekciós tényezője;
F IS a belső standard korrekciós tényezője;
m a vizsgált mennyiség tömege milligrammban;
m IS a belső standard tömege milligrammban.
Az eredményeket két tizedesjegy pontossággal kell megadni.
6. VIZSGÁLATI JEGYZŐKÖNYV
A vizsgálati jegyzőkönyvnek tartalmaznia kell a metil-észterek előkészítés és a gázkromatográfiás elemzés módszerét. Meg kell említeni benne minden, e standard módszerben nem szereplő vagy opcionálisnak tekinthető üzemi körülményt vagy az eredményekre esetlegesen hatással lévő bármilyen eseményt.
A vizsgálati jegyzőkönyvnek tartalmaznia kell a minta egyértelmű azonosításához szükséges valamennyi adatot.
7. A MÓDSZER PONTOSSÁGA
7.1. A körvizsgálatok eredményei
A módszer pontosságának körvizsgálatával kapcsolatos információk az IOC/T.20/Doc. No 33. szabvány C mellékletében találhatóak. Az ebből a körvizsgálatból származó értékek valószínűleg nem alkalmazhatók a megadottakon kívül más koncentrációtartományokra és mátrixokra.
7.2. Megismételhetőség
Az ugyanazon módszerrel ugyanazon tesztanyagon, ugyanazon laboratóriumban, ugyanazon kezelőszemély által, ugyanazon berendezésen, és a két teszt elvégzése között eltelt rövid idő alatt kapott két független egyedi teszteredmény közötti abszolút különbség az esetek nem több mint 5 %-ban lesz nagyobb az IOC/T.20/Doc. No. 33. szabvány C mellékletének 1-14. táblázatában megadott r értéknél.
7.3. Reprodukálhatóság
Az ugyanazon módszerrel ugyanazon tesztanyagon, különböző laboratóriumban, különböző kezelőszemély által, különböző berendezésen kapott két egyedi teszteredmény közötti abszolút különbség az esetek nem több mint 5 %-ában lesz nagyobb az IOC/T.20/Doc. No. 33. szabvány C mellékletének 1-14. táblázatában megadott R értéknél.
A. függelék
1. ábra

ω0,5 szélességgel az ABC háromszög magasságának felénél és b szélességgel az NPM háromszög magasságának felénél.
| 2. ábra | 3. ábra |
| [Kép #3] | [Kép #4] |

Kép #3

Kép #4
B. függelék
1. ábra
Olívapogácsa-olajból hideg metilezéssel nyert gázkromatográfiás profil.

Eltérő rendelkezés hiányában a kromatográfiás csúcsok a metil- és etil-észtereknek felelnek meg.
XI. MELLÉKLET
AZ OLÍVAOLAJ ILLÉKONY HALOGÉNEZETT OLDÓSZEREINEK MEGHATÁROZÁSA
1. MÓDSZER
A gőztér-mintavételezési technikán alapuló gázkromatográfiás elemzés.
2. BERENDEZÉS
2.1. Elektronbefogásos detektorral (ecd) felszerelt gázkromatográfiás berendezés.
2.2. Gőztér-mintavételező berendezés.
2.3. Üvegből készült 2 m hosszú, 2 mm átmérőjű gázkromatográfiás oszlop, stacioner fázis. OV101 10 % vagy avval egyenértékű a kalcinált, savazott, szilanizált és 80-100 szemcsenagyságú kovaföld impregnálásához.
2.4. Vivő- és segédgázok: az elektronbefogásos detektáláshoz alkalmas nitrogén a gázkromatográfiás eljáráshoz.
2.5. 10 ml és 15 ml térfogatú teflonborítású üveglombikok alumíniumdugóval, a fecskendő bevezetésére alkalmas kialakítással.
2.6. Hermetikus zárású bilincsek.
2.7. 0,5-2,0 ml térfogatú gázfecskendő.
3. REAGENSEK
Standard: a gázkromatográfiás eljáráshoz megfelelő tisztaságú halogénezett oldószerek.
4. ELJÁRÁS
4.1. Mérjen pontosan 3 g olajat egy üveglombikba (újból nem felhasználható); zárja le hermetikusan. Tegye egy termosztátban 70 °C hőmérsékletre egy óra hosszára. Egy fecskendő segítségével óvatosan szívjon ki a gőztérből 0,2-0,5 ml-t. A fecskendő tartalmát fecskendezze be a következő módon beállított gázkromatográfiás berendezés oszlopába:
- injektor hőmérséklete: 150 °C,
- oszlop hőmérséklete: 70-80 °C,
- detektor hőmérséklete: 200-250 °C.
Ettől eltérő hőmérsékletek is beállíthatók, amennyiben az eredmények egyenértékűek lesznek.
4.2. Referenciaoldatok: készítsen a mintában feltételezett tartalomnak megfelelő 0,05-1 ppm (mg/kg) töménységű standard oldatokat olyan finomított olívaolajból, amelyben oldószerek nyomokban sem találhatók meg. A halogénezett oldószerek pentán segítségével hígíthatók.
4.3. Mennyiségi becslés: hasonlítsa össze a minta, illetve a feltételezett töménységhez legközelebb álló standard oldat csúcsainak magasságát vagy területét. Amennyiben az eltérés 10 %-nál nagyobb mértékű, akkor az elemzést meg kell ismételni egy másik standard oldattal történő összehasonlítással, egészen addig, ameddig az eltérés 10 %-on belüli nem lesz. A tartalmazott mennyiség meghatározása az elemi befecskendezések átlaga alapján történik.
4.4. Az eredmények kifejezése: ppm-ben (mg/kg). Az eljárás érzékelési korlátja 0,01 mg/kg.
XII. MELLÉKLET
A NEMZETKÖZI OLÍVAOLAJ-TANÁCSNAK A SZŰZ OLÍVAOLAJOK ÉRZÉKSZERVI ÉRTÉKELÉSÉRE SZOLGÁLÓ MÓDSZERE
1. CÉL ÉS ALKALMAZÁSI TERÜLET
Az ebben a mellékletben bemutatott nemzetközi módszernek a célja, hogy megállapítsa az 1308/2013/EK európai parlamenti és tanácsi rendelet\ ( 4 ) VII. melléklete VIII. részének 1. pontja szerinti szűz olívaolaj érzékszervi jellemzőinek értékelési eljárását, és létrehozza az e jellemzők alapján történő osztályozásukra szolgáló módszert. A módszer a fakultatív címkézésre vonatkozó útmutatást is tartalmaz.
Az ismertetett módszer csak a szűz olívaolajokra alkalmazandó, valamint azok osztályozására vagy címkézésére az érzékelt hibák intenzitása és a gyümölcsösség szerint, egy válogatott, képzett és vizsgáztatott kóstolókból álló, értékelő bizottságként működő csoport által meghatározott módon.
Az IOC (Nemzetközi Olívaolaj-Tanács) e mellékletben említett szabványai a legutolsó rendelkezésre álló verziójukban kerültek alkalmazásra.
2. AZ ÉRZÉKSZERVI ELEMZÉS ÁLTALÁNOS ALAPSZÓKÉSZLETE
Lásd az IOC/T.20/Doc. No 4 "Sensory Analysis: General Basic Vocabulary" (Érzékeszervi elemzés: Általános alapszókészlet) szabványt.
3. SZAKKIFEJEZÉSEK
3.1. Negatív tulajdonságok
Dohos/seprős Az olyan olajbogyókból sajtolt olaj jellegzetes zamata, amelyek az egymásra halmozás, illetve a tárolási körülmények miatt a légmentes erjedés előrehaladott állapotában vannak, vagy olyan olajé, amely az olajtároló medencékben és kádakban a légmentes erjedés folyamatában lévő fejtési "üledékkel" érintkezett.
Penészes-nedves-földes A több napon keresztül nedves helyen történt tárolás következtében, a penész és élesztőgombák által megtámadott olajbogyóból nyert olaj jellegzetes zamata, vagy olyan olívabogyókból nyert olaj jellegzetes zamata, amelyeket földesen, sárosan takarítottak be, és lemosásuk elmaradt.
Boros-ecetes/savanyú-fanyar Némely olajnak a borra vagy ecetre emlékeztető jellegzetes zamata van. Ez a zamat alapvetően az olajbogyóknak, illetve a nem megfelelően lemosott sajtolószitán maradt olajbogyó-pépnek a levegőn való erjedési folyamatából származik, amelynek eredményeként ecetsav, etil-acetát és etanol képződik.
Avas Olyan olajok zamata, amelyek intenzív oxidációs folyamaton mentek keresztül.
Fagyott olíva (nedves fás) Az olajfán fagyásnak kitett olajbogyókból kivont olajok jellegzetes zamata.
3.1.1. Egyéb negatív tulajdonságok
| Sült vagy égett | Olyan olajok jellegzetes zamata, amelyek előállításuk során – és különösen az olajbogyópép termikus keverése alatt – túlzott mértékben és/vagy túl hosszú ideig lettek felmelegítve, ha ez nem megfelelő hőmérsékleti körülmények között történt. |
| Szénás-fás | A kiszáradt olajbogyóból kinyert bizonyos olajok jellegzetes zamata. |
| Nyers | Egyes régi olajok által keltett jellegzetes hatás, melyek kóstoláskor sűrű, nyúlós érzést okoznak. |
| Kenőzsíros | A gázolajra, gépzsírra vagy ásványi olajra emlékeztető zamat. |
| Vizes | Erjedt növényi nedvekkel hosszabb ideig érintkezésben maradt olajok által szerzett zamat. |
| Sós | A sós páclében tartósított olajbogyóból nyert olajok jellegzetes zamata. |
| Fémes | Fémekre emlékeztető aroma. Az olyan olaj jellegzetessége, amely a zúzás, a keverés, a sajtolás vagy a tárolás során hosszabb ideig fémfelületekkel érintkezett. |
| Eszpartó | Új eszpartófű-szitán kipréselt olívabogyókból származó olajok jellegzetes zamata. A zamat változhat attól függően, hogy a sajtolószítát zöld vagy száraz eszpartófűből készítették-e. |
| Férges | Az olajbogyó-fúrólégy (Bactrocera oleae) lárváitól erősen megtámadott olajbogyóból származó olaj zamata. |
| Uborkás | A hermetikusan lezárt állapotban, például bádogtartályban túl hosszú ideig tartott olaj jellegzetes zamata, amely a 2,6-nonadienal képződésének tulajdonítható. |
3.2. Pozitív tulajdonságok
| Gyümölcsös | Az olajra jellemző mindazon – olajbogyófajta szerint változó – szagérzetek, amelyek az egészséges és friss, zöld vagy érett gyümölcs közvetlen vagy retronazális úton való szaglásából származnak. |
| Keserű | A zöld vagy az érési folyamat kezdetén lévő olajbogyóból nyert olívaolaj jellegzetes alapíze, amelyet a nyelven V alakban elhelyezkedő kehelyformájú ízlelőbimbók érzékelnek. |
| Csípős | Az egész szájüregben, de különösen a torokban érzékelhető kaparó érzés, amely elsősorban a még éretlen olajbogyóból, a gazdasági év elején termelt olaj sajátossága. |
3.3. A címkézésnél alkalmazható fakultatív kifejezések
Az értékelő csoport elnöke kérelemre tanúsíthatja, hogy az értékelt olajok a tulajdonságok érzékelésének intenzitása tekintetében kizárólag a következő kifejezések és jelzők szerinti meghatározásoknak és fokozatoknak felelnek meg.
Pozitív tulajdonságok (gyümölcsös, kesernyés és csípős): Az érzékelés intenzitásának függvényében:
- erős, ha az érintett tulajdonság mediánja 6,0-nál nagyobb;
- közepes, ha az érintett tulajdonság mediánja 3,0 és 6,0 között van;
- enyhe, ha az érintett tulajdonság mediánja 3,0-nál kisebb.
| Gyümölcsösség | Az olajra jellemző mindazon – olajbogyófajta szerint változó – szagérzetek, amelyek olyan egészséges és friss olajbogyókból erednek, amelyekben sem a zöld, sem az érett gyümölcsösség nem domináns. E tulajdonság érzékelése közvetlen és/vagy retronazális úton történik. |
| Zöld gyümölcsösség | Az olajra jellemző mindazon – olajbogyófajta szerint változó – szagérzetek, amelyek a zöld gyümölcsre emlékeztetnek, és zöld, egészséges és friss olajbogyókból erednek. E tulajdonság érzékelése közvetlen és/vagy retronazális úton történik. |
| Érett gyümölcsösség | Az olajra jellemző mindazon – olajbogyófajta szerint változó – szagérzetek, amelyek az érett gyümölcsre emlékeztetnek, és egészséges és friss olajbogyókból erednek. E tulajdonság érzékelése közvetlen és/vagy retronazális úton történik. |
| Kiegyensúlyozott olaj | Olyan olaj, amely nem mutat kiegyensúlyozatlanságot; ezen azt a szaglási-ízlelési és érintési érzetet kell érteni, amikor a kesernyésség mediánja és a csípősség mediánja legfeljebb 2,0 ponttal van a gyümölcsösség mediánja fölött. |
| Lágy olaj | Olyan olaj, amelynél a kesernyésség és a csípősség mediánja 2,0 vagy annál kisebb. |
Kifejezések az érzékelés intenzitásának függvényében:
| Az érzékszervi vizsgálati jegyzőkönyvben szereplő kifejezések | Az érintett tulajdonság mediánja |
| Gyümölcsösség | — |
| Érett gyümölcsösség | — |
| Zöld gyümölcsösség | — |
| Enyhe gyümölcsösség | ≤ 3,0 |
| Közepes gyümölcsösség | 3,0 < Me ≤ 6,0 |
| Erős gyümölcsösség | > 6,0 |
| Enyhe érett gyümölcsösség | ≤ 3,0 |
| Közepes érett gyümölcsösség | 3,0 < Me ≤ 6,0 |
| Erős érett gyümölcsösség | > 6,0 |
| Enyhe zöld gyümölcsösség | ≤ 3,0 |
| Közepes zöld gyümölcsösség | 3,0 < Me ≤ 6,0 |
| Erős zöld gyümölcsösség | > 6,0 |
| Enyhe kesernyésség | ≤ 3,0 |
| Közepes kesernyésség | 3,0 < Me ≤ 6,0 |
| Erős kesernyésség | > 6,0 |
| Enyhe csípősség | ≤ 3,0 |
| Közepes csípősség | 3,0 < Me ≤ 6,0 |
| Erős csípősség | > 6,0 |
| Kiegyensúlyozott olaj | A kesernyésség mediánja és a csípősség mediánja legfeljebb 2,0 ponttal van a gyümölcsösség mediánja fölött. |
| Lágy olaj | A kesernyésség mediánja és a csípősség mediánja legfeljebb 2,0. |
4. AZ OLAJKÓSTOLÓ POHÁR
Lásd az IOC/T.20/Doc. No 5, "Glass for Oil Tasting" (Olajkóstoló pohár) szabványt.
5. VIZSGÁLÓTEREM
Lásd az IOC/T.20/Doc. No 6, "Guide for the Installation of a Test Room" (Segédlet a vizsgálóterem kialakításához) szabványt.
6. BERENDEZÉS
A kóstolók számára a feladatuk megfelelő ellátásához szükséges a következő felszerelést minden kóstolófülkében biztosítani és annak könnyen elérhetőnek kell lennie:
- a mintákat tartalmazó (azonos méretű) poharak, kódszámmal ellátva, óraüveggel lefedve és 28±2 °C-on tartva;
- értékelőlap (lásd 1. ábra) nyomtatva, vagy elektronikusan, feltéve, hogy megfelel az értékelőlap feltételeinek, szükség esetén a használatra vonatkozó utasításokkal;
- toll vagy kitörölhetetlen tinta;
- tálcán almaszeletek és/vagy víz, szénsavas víz és/vagy kétszersült;
- egy pohár környezeti hőmérsékletű víz;
- a 8.4. és 9.1.1. pontban felsorolt általános szabályokra emlékeztető tábla;
- köpőcsészék.
7. A CSOPORT ELNÖKE ÉS A KÓSTOLÓK
7.1. A csoport elnöke
A csoport elnökének alapos szakképzettséggel kell rendelkeznie, és a különféle típusú olívaolajok ismerőjének és tapasztalt szakértőjének kell lennie. Ez a személy a csoport kulcsembere, ő felelős a csoport munkájának megszervezéséért és lebonyolításáért.
A csoport vezetőjének feladata az érzékszervi vizsgálatokhoz használt eszközökkel, az érzékszervi képességekkel kapcsolatos továbbképzések megszervezése, az aprólékos előkészületek elvégzése, a tesztek megszervezése és lebonyolítása, valamint rendelkezik azokkal a képességekkel és türelemmel, amelyek szükségesek a tesztek tudományos alapossággal történő megtervezésévez és kivitelezéséhez.
Egy személyben felelős a kóstolók kiválasztásáért, a képzésükért, a képességük megfelelő szintentartása érdekében, és ellenőrzi teljesítményüket. Szintén az elnök felelős a kóstolók értékeléséért, amelynek mindig objektívnek kell lennie, és amelyhez speciális eljárásokat kell kifejleszteni. Az eljárások teszteken, valamint szigorú elfogadási és elutasítási kritériumokon kell alapulniuk. Lásd az IOC/T.20/Doc. No 14, "Guide for the selection, training and monitoring of skilled virgin olive oil tasters" (Útmutató a megfelelő képeségekkel rendelkező szűz olívaolaj kóstolók kiválasztásához, képzéséhez és ellenőrzéséhez) szabványt.
A csoport elnöke felel a csoport teljesítményéért, ennélfogva annak értékeléséért, melynek során mindig megbízhatónak és pártatlannak kell bizonyulnia. Minden esetben szemléltetnie kell, hogy a módszerek és a kóstolók az ellenőrzése alatt állnak. Javasolt a csoport időszakos felülvizsgálata (IOC/T.20/Doc. No 14, § 5).
Övé a végső felelősség a csoport feljegyzéseinek tárolásáért. Ezeknek a feljegyzéseknek bármikor nyomon követhetőknek kell lenniük. A feljegyzéseknek meg kell felelniük a nemzetközi érzékszervi vizsgálati szabványok által lefektetett biztosítékoknak és minőségi előírásoknak, és mindenkor biztosítaniuk kell a minták anonimitását.
Az elnök felelős a leltározásért, valamint annak biztosításáért, hogy a módszer előírásszerű betartásához szükséges berendezések és eszközök tisztítása és karbantartása megfelelő legyen, és az erről, valamint a megfelelő tesztkörülmények meglétéről szóló írott tanúsítványt megőrizze.
Az elnök felel a minták fogadásáért, valamint azok tárolásáért a laboratóriumba való beérkezésüktől kezdődően és a tesztelés után is. Ennek során végig biztosítania kell a minták anonimitását és megfelelő tárolását; ennek biztosítására írásos formában ki kell dolgoznia a folyamatot úgy, hogy az nyomonkövethető legyen és a megfelelő garanciákat biztosítsa.
Ezen felül, az elnök felel a minták előkészítéséért, kódolásáért és átadásáért a kóstolóknak, az előre rögzített protokollnak megfelelő kísérleti elrendezésben, valamint a kóstolók által szolgáltatott adatok összegyűjtéséért és statisztikai feldolgozásáért.
Az elnők felelőssége minden olyan eljárás kidolgozása vagy felvázolása, ami szükséges lehet ennek a szabványnak a teljesítéséért és a kóstolói csoport megfelelő működéséhez.
Annak érdekében, hogy ellenőrizze a csoport megfelelő működését, az elnöknek meg kell találnia a módját, hogy a csoportja adatait összhasonlíthassa más, szűz olívaolajokat elemző kóstolói csoportok adataival.
A csoport elnökének feladata a kóstolócsoport tagjainak ösztönzése, az érdeklődésük, kíváncsiságuk felkeltése a köztük kialakított versenyszellem révén. Ennek érdekében javasolt biztosítania a kétirányú információáramlást a csoport tagjaival, a tagokat folyamatosan informálnia az általuk elvégzett feladatokról és a kapott eredményekről. Biztosítja azt, hogy a véleményét ne ismerjék meg, illetve megakadályozza, hogy az erősebb személyiségek nézeteiket rákényszerítsék a többi kóstolóra.
Az elnök feladata még a kóstolók időben való értesítése, az esetleges kérdéseik megválaszolása a tesztek elvégzésével kapcsolatban, de tartózkodnia kell a javaslattételtől, illetve a mintákkal kapcsolatos véleménynyilvánítástól.
7.1.1. A csoport elnökhelyettese
A csoport elnökét indokolt esetben helyettesítheti bizottsági elnökhelyettes, aki a vizsgálatok elvégzésével kapcsolatos feladatokért fog felelni. A helyettesnek rendelkeznie kell a csoport elnökétől elvárt valamennyi készséggel.
7.2. Kóstolók
Az olívaolajok érzékszervi vizsgálatában részt vevő személyeknek önkéntesen kell jelentkezniük. Ezért javasolt a jelölteknek egy írásos kérelmet benyújtaniuk. A jelölteket a csoport elnöke választja ki, képzi és ellenőrzi a hasonló minták megkülönböztetésére irányuló képességeik szerint, szem előtt tartva, hogy a pontosságuk a képzések során egyre finomodni fog.
A kóstolóknak tényleges érzékszervi megfigyelőkként kell viselkedniük, félre kell tenniük a személyes véleményüket és csak az észlelt érzékszervi észleléseikről kell beszámolniuk. Ennek érdekében a kóstolóknak mindig csendben, nyugodt körülmények között kell dolgozniuk, sürgetés nélkül, úgy, hogy a lehető legnagyobb érzékszervi figyelmet szenteljék a vizsgált mintának.
Minden egyes teszthez 8 és 12 közötti számú kóstoló szükséges. Ugyanakkor célszerű ennél több kóstolót találni az esetleges hiányzók helyettesítésére.
8. TESZTKÖRÜLMÉNYEK
8.1. A minta tálalása
Az olajmintát a vizsgálathoz egy szabványos pohárban kell tálalni a következő szabvány szerint: IOC/T.20/Doc. No 5, "Glass for Oil Tasting" (Olajkóstoló pohár).
A pohárnak 14-16 ml vagy 12,8-14,6 g olajat kell tartalmaznia, ha a minták tömegének mérése szükséges, és azt minden esetben le kell fedni óraüveggel.
Minden egyes poharat egy kódszámmal kell ellátni, ami véletlenszerűen kiválasztott betűk vagy számok sorozatából áll. A kódokat szagtalan módszerrel kell jelölni.
8.2. A teszt és a minta hőmérséklete
A vizsgálatra szánt olajmintákat poharakban, 28 °C ± 2 °C-on kell tartani a teszt teljes időtartamára. Azért erre a hőmérsékletre esett a választás, mert ezen a hőmérsékleten könnyebb meghatározni az érzékszervi különbségeket, mint a környezeti hőmérsékleten, és mert az alacsonyabb hőmérsékleteken az aromaanyagok alig párolognak, illetve olyan illékony komponensek keletkeznek, amelyek a melegített olajokra jellemzőek. Lásd az IOC/T.20/Doc. No 5 "Glass for Oil Tasting" (Olajkóstoló pohár) szabványt azokra a módszerekre nézve, melyekkel a poharakban lévő minták melegíthetők.
A vizsgálóhelyiség hőmérsékletének 20 °C és 25 °C fok között kell lennie (lásd IOC/T.20/Doc. No 6).
8.3. Vizsgálati idő
Az olajok kóstolására a reggel a legalkalmasabb időszak. Bizonyított, hogy vannak a nap folyamán optimális íz- és illatérzékelési periódusok. Az étkezések előtt megfigyelhető egy időszak, amikor az illat- és ízlelési érzékenység megnövekszik, míg az étkezéseket követően csökken az érzékelő képesség.
Ezt a feltételt azonban nem szabad szélsőségesen kihasználni, mikor az éhség megzavarja a kóstolókat, és ezáltal csökkenti a megkülönböztetési képességüket, különösen a preferencia-, illetve elfogadási feltételeiket; ezért javasolt a kóstolás időpontját reggel 10:00 óra és dél (12:00) közöttre tenni.
8.4. Kóstolók: általános viselkedési szabályok
Ezek az ajánlások a kóstolók munkájuk során tanúsított viselkedésére vonatkoznak.
A kóstolóknak, amikor a csoport elnöke behívja őket egy érzékszervi vizsgálaton történő részvételre, a kitűzött időpont előtt meg kell jelenniük és be kell tartaniuk a következőket:
- A teszt meghatározott kezdési ideje előtt legalább 30 perccel nem dohányozhatnak, illetve nem fogyaszthatnak kávét.
- Nem használhatnak semmilyen illatosító szert, kozmetikai szert vagy szappant, amelynek az illata eltart a teszt kezdetéig. A szükséges gyakoriságú kézmosáshoz illatmentes szappant kell használniuk, amelyet leöblítenek, majd kezeiket megszárítják, az illat eltávolítása érdekében.
- A teszt végzése előtt legalább egy órával nem ehetnek.
- Fizikai rosszullét esetén, különösen ha ez érinti a szaglási vagy ízlelési képességet, vagy ha bármilyen olyan pszichológiai hatás alatt állnak, ami megakadályozza őket abban, hogy a munkájukra figyeljenek, a kóstolóknak tartózkodniuk kell a teszten való részvételtől, és erről a megfelelő módon értesíteniük kell a csoport vezetőjét.
- Amennyiben a fentieknek megfelelnek, a kóstolók a lehető legszabályosabb és legcsendesebb módon elfoglalják a számukra kijelölt fülkét.
- Figyelmesen elolvassák az értékelő lapon lévő utasításokat, és addig nem kezdik meg a minta vizsgálatát (nyugodtan és sürgetés nélkül), amíg teljesen biztosak nem lesznek az elvégzendő feladatban. Amennyiben valamilyen kétség merülne fel, személyesen megbeszélhetik a tapasztalt problémákat a csoport elnökével.
- A kóstolóknak a feladatuk elvégzése alatt csendben kell maradniuk.
- A mobiltelefonjukat végig kikapcsolva kell tartaniuk annak érdekében, hogy az ne vonja el sem a saját, sem a kollégáik figyelmét.
9. A SZŰZ OLÍVAOLAJ ÉRZÉKSZERVI ÉRTÉKELÉSÉNEK ÉS OSZTÁLYOZÁSÁNAK ELJÁRÁSA
9.1. Kóstolási technika
9.1.1. A kóstoló felveszi a poharat, ekkor még rajta tartja a poháron az óraüveget, és kissé megdönti; ezután ebben a helyzetben körbeforgatja a poharat, hogy a lehető legnagyobb mértékben benedvesítse a pohár belső felületét. Amikor eddig a fázisig eljutott, leveszi az óraüveget és lassú, mély lélegzetvételekkel megszagolja a mintát az olaj értékelése érdekében. A szaglás nem tarthat tovább 30 másodpercnél. Amennyiben ez alatt az idő alatt nem jut döntésre, akkor az újbóli próbálkozás előtt rövid pihenőt kell tartania.
A kóstoló a szaglási teszt befejezését követően az aromát (teljes retronazális, szaglási, ízlési és tapintási érzet) értékeli. Ennek elvégzéséhez kortyol egy kicsit (megközelítőleg 3 ml) az olajból. Nagyon fontos, hogy az olajat elossza a teljes szájüregben, a száj és a nyelv elejétől kezdődően végig az oldalak mentén a hátsó részig és a szájpadlásig, mivel ismert tény, hogy a négy alapíz, az édes, a sós, a savanyú és a keserű érzékelése a nyelv, a szájpadlás és a torok különböző területein változó intenzitású.
Nagyon fontos, hogy a kóstoló a nyelv hátsó részén megfelelő mennyiségű olajat lassan terítsen szét a szájpadlás és a torok irányába, mialatt arra figyel, hogy milyen sorrendben jelennek meg a kesernyés és a csípős ingerek. Amennyiben ez nem történik meg, egyes olajok esetén mindkét inger elkerülheti a figyelmet, vagy a kesernyés ingert elnyomhatja a csípős inger.
A szájon keresztül történő gyors, egymást követő belégzések nem csak a minta egész szájban történő szétterítését teszik lehetővé a kóstoló számára, hanem azt is, hogy az orr hátulján keresztül érzékelje az illékony aromás összetevőket.
Megjegyzés: ha a kóstolók egy minta estében nem érzékelnek gyümölcsösséget és a jellemző negatív tulajdonság intenzitása 3,5 vagy kevesebb, az értékelő csoport elnöke úgy határozhat, hogy a kóstolók szobahőmérsékleten ismételten végezzék el a minta vizsgálatát (COI/T.20/Doc. No 6/Rev. 1, 2007. szeptember, 3. szakasz - General specifications for installation of a test room - A vizsgálóterem kialakításának általános előírásai), meghatározva a szobahőmérséklet fogalmát és körülményeit. Amikor a minta eléri a szobahőmérsékletet, a kóstolóknak azt újra kell értékelniük kizárólag annak ellenőrzése céljából, hogy a gyümölcsösség érzékelhető-e. Amennyiben igen, annak intenzitását a skálán meg kell jelölniük.
A csípősség közvetlen érzékelését is figyelembe kell venni. Ennek érdekében javasolt az olajat le is nyelni.
9.1.2. Szűz olívaolajok érzékszervi vizsgálatánál javasolt egy sorozat alkalmával legfeljebb NÉGY MINTA vizsgálata, egy nap pedig három sorozat vizsgálata annak érdekében, hogy elkerülhető legyen a többi minta közvetlenül ezt követő kóstolása által okozott kontraszthatás.
Mivel az egymást követő kóstolások az érzékelő képesség kifáradását vagy elvesztését okozzák - amit az előző minták okoznak - olyan terméket kell használni, amellyel eltávolítható az előző kóstolás során a szájban maradó olaj.
Javasolt ehhez egy kis szelet almát összerágni, amely összerágás után egy köpőcsészébe köphető. Ezt követően a kóstoló öblítse ki a száját egy kevés szobahőmérsékletű vízzel. Legalább 15 percnek kell eltelnie egy kóstolás befejezése és egy újabb megkezdése előtt.
9.2. Hogyan használja a kóstoló az értékelőlapot?
A kóstolók által használandó értékelőlap ezen Melléklet 1. ábráján látható.
A csoportban részt vevő minden kóstolónak meg kell szagolnia, majd meg kell kóstolnia a megvizsgálandó ( 5 ) olajat.
Ezt követően a rendelkezésére álló értékelőlap 10 cm-es skáláján be kell jelölnie, hogy mekkora intenzitással érzékeli az egyes negatív és pozitív tulajdonságokat Abban az esetben, ha a kóstolók a 4. pontban fel nem tüntetett negatív tulajdonságot észlelnek, azokat - a meghatározással ellátott kifejezések közül a legpontosabban körülíró kifejezést, illetve kifejezéseket alkalmazva - feljegyzik az "egyéb" rovatba.
9.3. Hogyan dolgozza fel az értékelő csoport elnöke az adatokat?
Az értékelő csoport elnöke összegyűjti az egyes kóstolók által kitöltött értékelőlapokat; ellenőrzi a különféle tulajdonságokhoz rendelt intenzitási fokozatokat. Ha úgy véli, hogy rendellenességet észlel, felkéri a kóstolót az értékelőlap felülvizsgálatára, és - szükség esetén - a vizsgálat megismétlésére.
Az értékelő csoport elnöke beviheti az egyes kóstolók által megállapított adatokat az IOC/T.20/Doc. No 15 szabványban előírt számítógépes programba a vizsgálat eredményeinek statisztikai kiszámítása céljából, azok mediánjának kiszámítása alapján. Lásd a 9.4. pontot és ezen melléklet függelékét. Az egy mintára vonatkozó adatok feldolgozása egy mátrix segítségével történik, amely a 9 érzéki tulajdonságnak megfelelő 9 oszlopból és a csoport n számú kóstolójának megfelelő n számú sorból áll.
Ha egy, az értékelő csoportnak legalább 50 %-a által érzékelt tulajdonság az "egyéb" rovatba tartozik, ki kell számítani e hiba mediánját, és az olajat ennek megfelelően kell osztályozni.
A nagy jóságfokú variációs együttható (a legerősebb intenzitású és gyümölcsösségi tulajdonságú) értéke, ami meghatározza a besorolást, nem lehet nagyobb, mint 20 %.
Ha ennek az ellenkezője áll fenn, a csoport vezetőjének egy másik kóstolási időpontban meg kell ismételtetnie az adott minta értékelését.
Ha ez a helyzet gyakran előfordul, akkor a csoport elnökének javasolt külön képzésben részesítenie a kóstolókat (IOC/T.20/Doc. No 14, § 5) és alkalmaznia kell az ismételhetőségi indexet és az eltérési indexet a csoport teljesítményének ellenőrzésére (IOC/T.20/Doc. No 14, § 6).
9.4. Az olaj osztályozása
Az olaj a hibamedián és a gyümölcsösségi medián függvényében az alábbi osztályok valamelyikébe sorolandó. A hibamedián a legnagyobb intenzitással érzékelt hiba mediánja. A hibamediánt és a gyümölcsösségi mediánt egy tizedesjegy pontossággal kell megadni.
Az olaj osztályozása a hibamedián és a gyümölcsösségi medián értékeinek az alább bemutatott referenciatartományokkal való összehasonlítása alapján történik. E tartományok határai a módszerhiba figyelembevételével kerültek megállapításra, ezért abszolút értékeknek tekintendők. A számítógépes programok táblázatba foglalt statisztikai adatok, illetve grafikus ábrázolás útján lehetővé teszik az osztályozás vizuális megjelenítését.
a) Extra szűz olívaolaj: a hibamedián 0,0 és a gyümölcsösségi medián 0,0-nél nagyobb.
b) Szűz olívaolaj: a hibamedián 0,0-nél nagyobb, de legfeljebb 3,5, és a gyümölcsösségi medián 0,0-nél nagyobb.
c) Lampante szűz olívaolaj: a hibamedián 3,5-nél nagyobb, vagy a hibamedián legfeljebb 3,5 és a gyümölcsösségi medián 0,0.
1. megjegyzés: Ha a kesernyésség és/vagy a csípősség mediánja meghaladja az 5,0 értéket, a csoport elnöke ezt bejegyzi a vizsgálati jegyzőkönyvbe.
A megfelelőségi ellenőrzések keretében végzett elemzések esetében egy próbát végeznek. Egymásnak ellentmondó elemzések esetén külön kóstolások során két vizsgálatot kell elvégezni. A párhuzamos vizsgálatnak statisztikailag homogén eredményeket kell adnia (lásd a 9.5. pontot). Ellenkező esetben a minta vizsgálatát még kétszer el kell végezni. Az osztályozási tulajdonságok mediánjának végső értékét a két medián átlagaként kell kiszámolni.
9.5. A párhuzamos vizsgálat elfogadási és elutasítási kritériumai
A lejjebb meghatározott normalizált hiba szolgál annak meghatározására, hogy a párhuzamos vizsgálat eredményei homogének vagy statisztikailag elfogadhatók-e:

Ahol Me1 és Me2 a két vizsgálat mediánja (az első, illetve a második vizsgálaté) és U1 és U2 a két érték esetében kapott kiterjesztett bizonytalanság, amelyek számítása a függelékben meghatározottak szerint a következőképpen történik:
U 1 = c × s* and [Kép #5]

Kép #5
Kiterjesztett bizonytalanság, c = 1,96; ezért:
U1 = 0,0196 × CVr × Me1
ahol CVr a nagy jóságfokú variációs együttható.
Annak kijelentéséhez, hogy a két érték statisztikailag nem különböző, En -nek legfeljebb 1,0-nak kell lennie.
1. ábra
A SZŰZ OLÍVAOLAJ ÉRTÉKELŐ LAPJA
| A hibák érzékelésének intenzitása | ||||
| Dohos/seprős | ||||
| Penészes/nedves/földes | ||||
| Boros/ecetes savanyú/fanyar | ||||
| Fagyott olíva (nedves fás) | ||||
| Avas | ||||
| Egyéb negatív tulajdonságok: | ||||
| Leírás: | Fémes □ Szénás □ Férges □ Nyers□ Sós □ Sült vagy égett □ Vizes□ Eszpartó □ Uborkás □ Kenőzsíros□ | |||
| A pozitív tulajdonságok érzékelési intenzitása | ||||
| Gyümölcsös | ||||
| Zöld□ | Érett□ | |||
| Keserű | ||||
| Csípős | ||||
| A kóstoló neve: | A kóstoló azonosítója: | |||
| A minta kódja: | Aláírás: | |||
| Dátum: | ||||
| Megjegyzések: | ||||
Függelék
A medián és a konfidenciaintervallumok kiszámításására használt módszer
Medián
A medián az az Xm valós szám, amelyre egyszerre igaz, hogy az eloszlás értékei (X) 0,5 vagy annál kisebb valószínűséggel (p) kisebbek ennél a számnál (Xm), és hogy az eloszlás értékei 0,5 vagy annál nagyobb valószínűséggel (p) kisebbek vagy egyenlők ezzel a számmal Xm. Egy gyakorlatiasabb meghatározás szerint a medián a növekvő sorozatba rendezett számok 50. centilisének felel meg. Egyszerűbben kifejezve, a medián a páratlan számú sorrendbe rendezett sorozat középső értéke, vagy a páros számú sorrendbe rendezett sorozat két középső értékének átlaga.
Nagy jóságfokú szórás
A medián körüli szóródás megbízható becslése érdekében a Stuart és Kendall (4) szerinti nagy jóságfokú szórás becslésére vonatkozó módszert kell alkalmazni. A következő képlet az aszimptotikus szórást fejezi ki, azaz a megfigyelt adatok medián körüli szóródásának olyan, nagy jóságfokú becslését, ahol N a megfigyelések száma, az IQR az interkvartilis terjedelem, és amely a valószínű eloszlás eseteinek pontosan 50 %-át foglalja magában:

Az interkvartilis terjedelem kiszámításához meg kell határozni a 75. és a 25. centilis közötti eltérés nagyságát
A centilis az az Xpc érték, amelyre egyszerre igaz, hogy eloszlási értékei egy meghatározott századrésznek megfelelő vagy annál kisebb valószínűséggel (p) kisebbek Xpc-nél, és hogy eloszlási értékei egy meghatározott századrésznek megfelelő vagy annál nagyobb valószínűséggel (p) kisebbek vagy egyenlők mint Xpc. A századrész az eloszlás kiválasztott hányadát fejezi ki. A medián esetében ez 50/100-dal egyenlő.

A gyakorlatban a centilis az az eloszlási érték, amely egy, az eloszlási vagy sűrűségi görbe által behatárolt meghatározott területnek felel meg. Például a 25. centilis azt az elosztási értéket jelenti, amely megfelel egy 0,25 vagy 25/100 nagyságú területnek.
E szerint a módszer szerint a centilisek számítása a mátrix adataiban megjelenő valós értékek alapján történik (centilisszámítási-módszer).
Nagy jóságfokú variációs együttható %-ban kifejezve
A VEj% egy olyan tiszta, azaz mértékegység nélküli szám, amely a vizsgált számok sorozatának szóródási százalékát jelöli. Ez az együttható ezért igen alkalmas a csoporttagok megbízhatóságának ellenőrzésére.

A medián 95 %-os konfidenciaintervalluma
A 95 %-os konfidenciaintervallum (az elsőfajú hiba értéke 0,05, azaz 5 %) azt az értéktartományt jelenti, amelynek értékeit - feltételezve, hogy a vizsgálatot végtelen sokszor meg lehet ismételni - a medián felveheti. A gyakorlatban az említett intervallum a vizsgálatok szóródási tartományát adja meg az elfogadott működési körülmények között, feltételezve, hogy a vizsgálatokat többször el lehet végezni. Az intervallum, éppúgy, mint a VEj%, segít a vizsgálatok megbízhatóságának értékelésében.


ahol a C értéke 95%-os konfidenciaintervallum esetén 1,96-tal egyenlő.
A számítási lap egy példája megtalálható az IOC/T 20/Doc. No 15. szabvány 1. mellékletében.
Hivatkozások
(1) Wilkinson, L. 1990. Systat: The system for statistics. Evanston, IL.SYSTAT Inc.
(2) Cicchitelli, G. 1984. Probabilità e Statistica. Maggioli Editore, Rimini.
(3) Massart, D.L.; Vandeginste, B.G.M.; Deming, Y.; Michotte, L. 1988. Chemometrics. A textbook. Elsevier. Amsterdam.
(4) Kendall, M.G.; Stuart, A. 1967. The advanced theory of statistics. Vol. 1. Hafner Publishing Co.
(5) McGill, R.; Tukey, J.W.; Larsen, W.A. 1978. Variation of Box Plots. The American Statistician, 32, (2), 12-16.
(6) IOC/T.28/Doc. No 1 September 2007, Guidelines for the accreditation of sensory testing laboratories with particular reference to virgin olive oil according to standard ISO/IEC 17025:2005.
(7) IOC/T.20/Doc. No 14.
(8) IOC/T.20/Doc. No 15.
(9) ISO/IEC 17025:05.
XV. MELLÉKLET
1. AZ OLÍVAMARADÉK OLAJTARTALMA
1.1. Berendezés
- megfelelő extrakciós berendezés, 200-250 ml térfogatú gömbölyű aljú lombikkal felszerelve,
- elektromos fűtésű fürdő (például homokfürdő, vízfürdő) vagy főzőlap,
- analitikai mérleg,
- maximum 80 °C-ig szabályozható kemence,
- 103 ± 2 °C pontossággal szabályozható termosztáttal felszerelt elektromos fűtésű kemence, amely levegőárammal átjárható vagy csökkentett nyomáson üzemeltethető,
- mechanikai őrlő, amely könnyen tisztítható és lehetővé teszi az olíva-maradékanyagok hőmérséklet-emelkedés nélküli, illetve nedvességtartalmuk, illóanyag-tartalmuk vagy a hexánnal kivonható anyagtartalmuk érzékelhető változása nélküli zúzását,
- extraháló hüvely és pamutvatta vagy szűrőpapír, amelyekről a hexánnal kivonható anyagokat előzőleg már eltávolították,
- szárítóberendezés,
- 1 mm átmérőjű nyílásokkal rendelkező szita,
- előzőleg kiszárított apró habkődarabok.
1.2. Reagens
Műszaki fokozatú közönséges hexán, amely elpárolgását követően kevesebb, mint 0,002 g/100 ml maradékot hagy.
2. ELJÁRÁS
2.1. A tesztminta előkészítése
A minta megőrlésére szükség esetén használja az előzőleg megfelelően megtisztított mechanikus őrlőt, hogy olyan méretű részecskéket kapjon, amelyek maradék nélkül átjutnak a szitán.
Az őrlő tisztítására használja fel a minta megközelítőleg huszadrészét, ezt az őrleményt öntse ki, őrölje meg a fennmaradó mennyiséget, keverje össze óvatosan, majd késedelem nélkül elemezze.
2.2. A vizsgálati anyag mennyisége
Közvetlenül az őrlés befejezését követően a teszthez mérjen meg a mintából megközelítőleg 10 g mennyiséget 0,01 g pontossággal.
2.3. Az extraháló hüvely
Tegye a vizsgált mennyiséget a hüvelybe és pamutvattával dugózza le. Amennyiben szűrőpapírt használ, csomagolja bele az anyagot.
2.4. Előzetes szárítás
Amennyiben az olívamaradék nagyon nedves (azaz a nedvességtartalma és az illóanyagtartalma meghaladja a 10 %-ot), helyezze a betöltött hüvelyt (vagy szűrőpapírt) megfelelő időre egy legfeljebb 80 °C hőmérsékletre felfűtött kemencébe és végezzen előzetes szárítást, hogy a nedvességtartalmat és az illóanyag-tartalmat 10 % alá csökkentse.
2.5. A gömbölyű fenekű lombik előkészítése
Mérje meg a korábban egy 103 ± 2 °C hőmérsékletű kemencében kiszárított és szárítóban legalább 1 órán keresztül hűtött, 1-2 darab habkövet tartalmazó lombikot 1 mg pontossággal.
2.6. Előzetes extrahálás
Helyezze be a vizsgált anyagot tartalmazó hüvelyt (vagy szűrőpapírt) az extraháló berendezésbe. Öntse a lombikba a szükséges mennyiségű hexánt. Illessze a lombikot az extraháló berendezésbe, és helyezze ezt az összeállítást az elektromos fűtésű fürdőre. Állítsa be úgy a fűtési sebességet, hogy a visszaáramlás sebessége másodpercenként legalább 3 csepp legyen (mérsékelt, nem heves forrás). Négyórás extrahálást követően hagyja lehűlni. Vegye ki a hüvelyt az extraháló berendezésből és helyezze levegőáramba, hogy kihajtsa a beleivódott oldószert.
2.7. Második kivonás
Öntse a hüvely tartalmát a mikroőrlőbe és őrölje meg a lehető legfinomabbra. Öntse vissza a megőrölt keveréket veszteség nélkül a hüvelybe és helyezze vissza az extraháló berendezésbe.
Ugyanannak a kezdeti kivonatot tartalmazó lombiknak a segítségével folytassa az extrahálást további két órán keresztül.
A lombikban így kapott oldatnak tisztának kell lennie. Amennyiben mégse, szűrje át szűrőpapíron, és mossa át néhányszor az eredeti lombikot és a szűrőpapírt hexánnal. A szűrletet és a mosáshoz használt hexánt gyűjtse egy másik gömbölyű fenekű lombikba, amelyet előzőleg kiszárított és 1 mg pontossággal megmért.
2.8. Az oldószer eltávolítása és a kivonat mérése
Az oldószer nagyobbik részét desztillálással távolítsa el elektromos fűtésű fürdőn. Az oldószer többi részét a lombik 103 ± 2 °C hőmérsékletű kemencében történő 20 perces hevítésével távolítsa el. Az oldószer eltávolítását rendszeres légbefúvással vagy lehetőleg inertgáz-befúvással, illetve a nyomás csökkentésével segítse.
Hagyja a lombikot a szárítóberendezésben legalább egy órán keresztül lehűlni, majd mérje meg 1 mg pontossággal.
Melegítse ismét 10 percen keresztül a fenti módszerrel és hűtse le a szárítóberendezésben, majd mérje meg újra.
A két mérés közötti különbség nem haladhatja meg a 10 mg-ot. Amennyiben meghaladja, végezzen 10 perces melegítéseket és hűtéseket mindaddig, amíg az eltérés 10 mg vagy kevesebb. Jegyezze fel a lombik utoljára mért tömegét.
Végezzen ismételt meghatározást a vizsgálati mintán.
3. AZ EREDMÉNYEK KIFEJEZÉSE
3.1. A számítási módszer és képlet
a) A kivonat a kapott termék tömegszázalékában kifejezve:

,
| ahol: | S a kivonat a kapott termék tömegszázalékában kifejezve, m0 a vizsgált mennyiség tömege grammban, m1 a kivonat tömege a szárítást követően, grammban. |
Amennyiben a megismételhetőség feltétele teljesül, a két mérés számtani középértékét tekintse eredménynek.
Az eredményeket egy tizedesjegy pontossággal adja meg.
b) A kivonat olajtartalma szárazanyagtartalomban kifejezve, a következő képlet segítségével:

ahol
:
S = a kivonat a kapott termék tömegszázalékában kifejezve (lásd az (a) pontot),
U = annak nedvesség- és illóanyag-tartalma.
3.2. Megismételhetőség
Az egyidőben vagy az ugyanazon elemző által gyors egymásutánban megismételt meghatározással kapott két eredmény közötti különbség nem haladhatja meg a 0,2 g hexán kivonat/100 g mintaértéket.
Amennyiben ez a feltétel nem teljesül, ismételje meg az elemzést két újabb vizsgálati anyagon. Amennyiben a különbség ebben az esetben is meghaladja a 0,2 g-ot, akkor a négy meghatározás számtani középértékét tekintse eredménynek.
XVI. MELLÉKLET
A JÓDSZÁM MEGHATÁROZÁSA
1. CÉL
Ez a nemzetközi szabvány az állati és növényi zsírok és olajok, a továbbiakban: zsírok, jódszáma megállapításának módszerét írja le.
2. MEGHATÁROZÁS
E nemzetközi szabvány alkalmazásában a következő meghatározások érvényesek:
2.1. jódszám: a minta által e nemzetközi szabványban meghatározott üzemi körülmények mellett abszorbeált jód tömege.
A jódszámot g jód/100 g minta mértékegységben fejezik ki.
3. ALAPELV
A minta feloldása oldószerben, majd wijs reagens hozzáadása. egy meghatározott idő elteltével kálium-jodid oldat és víz hozzáadása, majd a felszabadított jód titrálása nátrium-tioszulfát oldattal.
4. REAGENSEK
Minden reagensnek elismert analitikai minőségűnek kell lennie:
4.1. víz, az ISO 3696 előírásainak megfelelő, 3. fokozat.
4.2. kálium-jodid, 100 g/l oldat, nem tartalmaz jódsavat vagy szabad jódot.
4.3. keményítő, oldat.
Keverjen el 5 g oldható keményítőt 30 ml vízben, adja ezt a keveréket 1 000 ml forrásban lévő vízhez, forralja három percig, majd hagyja lehűlni.
4.4. nátrium-tioszulfát, standard volumetrikus oldat, c (Na2S2O3.5H2O) = 0,1 mol/l, használat előtt hét napnál nem régebben standardizálva.
4.5. oldószer, azonos térfogatú ciklohexán és ecetsav összekeverésével készült,
4.6. Wijs reagens, jód-monoklorid ecetsavban. Használható a kereskedelmi forgalomban kapható Wijs reagens.
5. BERENDEZÉS
A szokásos laboratóriumi berendezés, valamint a következők:
5.1. üveg mérőkanalak, a vizsgált mennyiséghez és a lombikba történő benyúláshoz megfelelőek (6.2.),
5.2. kúpos fenekű lombikok, 500 ml térfogatúak, csiszolt üvegdugóval ellátva, teljesen szárazak.
6. A VIZSGÁLATI MINTA ELŐKÉSZÍTÉSE
Szárítsa ki a homogenizált mintát nátrium-szulfáton, majd szűrje le.
7. ELJÁRÁS
7.1. Vizsgált mennyiség
A vizsgált mennyiség tömege a várható jódszám függvényében változik az 1. táblázat szerint.
1. táblázat
| Várható jódszám | A vizsgált mennyiség tömege (g) |
| kevesebb, mint 5 | 3,00 |
| 5–20 | 1,00 |
| 21–50 | 0,40 |
| 51–100 | 0,20 |
| 101–150 | 0,13 |
| 151–200 | 0,10 |
Mérje meg a vizsgált mennyiséget egy üveg mérőkanállal (5.1.) 0,1 mg pontossággal.
7.2. Meghatározás
Helyezze a vizsgált mennyiséget az 500 ml térfogatú kémcsőbe (6.2.). Adjon hozzá 20 ml oldószert (4.5.), hogy feloldja a zsírt. Adjon hozzá pontosan 25 ml Wijs reagenst (4.6.), tegye be a dugót, forgassa össze a lombik tartalmát, és tegye a lombikot sötét helyre. A Wijs reagenshez ne használjon pipettát.
Készítsen vakpróbát hasonlóképpen az oldószerrel és reagenssel, de a vizsgált anyag kihagyásával.
A 150-nél alacsonyabb jódszámú minták esetén egy óráig, a 150-nél magasabb jódszámú minták, illetve a polimerizált termékek, és a jelentős mértékben oxidálódott termékek esetén két óráig hagyja a lombikot a sötétben.
A fenti idő elteltével adjon mindegyik kémcsőhöz 20 ml kálium-jodid oldatot (4.2.) és 150 ml vizet (4.1.).
Végezzen titrálást a standard volumetrikus nátrium-tioszulfát oldattal (4.4.), ameddig a jód miatt kialakult sárga szín majdnem teljesen eltűnik. Adjon hozzá néhány csepp keményítőoldatot (4.3.), és folytassa a titrálást, ameddig a kék szín heves rázás mellett el nem tűnik.
Megjegyzés: Megengedett a végpont potenciometrikus meghatározása.
7.3. A meghatározások száma
Végezzen a mintán két meghatározást.
8. AZ EREDMÉNYEK KIFEJEZÉSE
A jódszám a következő képlettel adható meg:

,
ahol:
c = a felhasznált standard volumetrikus nátrium-tioszulfát oldat (4.4.) pontos töménységének számértéke mól/l-ben;
V1 = a vakpróbához felhasznált standard volumetrikus nátrium-tioszulfát oldat (4.4.) térfogata ml-ben;
V2 = a meghatározáshoz felhasznált standard volumetrikus nátrium-tioszulfát oldat (4.4.) térfogata ml-ben;
m = a vizsgált anyag (7.1.) tömege g-ban.
A két meghatározás számtani középértéket tekintse eredményként, amennyiben teljesül a megismételhetőség követelménye (9.2.).
XVII. MELLÉKLET
A NÖVÉNYI OLAJOK SZTIGMASZTADIÉN-ÖSSZETÉTELÉNEK MEGHATÁROZÁSA
1. CÉL
A sztigmasztadiének meghatározása az e szénhidrogéneket alacsony koncentrációban tartalmazó növényi olajokban, különösen szűz olajban és nyers olívamaradék-olajban.
2. HATÁLY
Ez a szabvány minden növényi olajra alkalmazható, viszont a mérések csak abban az esetben megbízhatóak, ha ezeknek a szénhidrogéneknek a mennyisége 0,01-4,0 mg/kg között van. A módszer különösen alkalmas a finomított növényi olajok (olíva, olívamaradék, napraforgó, pálma stb.) jelenlétének szűz olívaolajban történő kimutatására, mivel a finomított olajok tartalmaznak sztigmasztadiéneket, a szűz olajok pedig nem.
3. ALAPELV
A nem szappanosítható anyag leválasztása. A szteroidos szénhidrogén frakció szilikagélen történő leválasztása oszlopkromatográfiás módszer segítségével és elemzése gázkromatográfiás módszer segítségével.
4. BERENDEZÉS
4.1. Visszafolyós hűtőben való felhasználásra alkalmas 250 ml térfogatú lombikok.
4.2. 500 ml térfogatú választótölcsérek.
4.3. 100 ml térfogatú kerek aljú lombikok.
4.4. Forgó bepárló.
4.5. Üveg kromatográfiás oszlop (1,5-2,0 cm belső átmérő, 50 cm hossz) tefloncsappal, üveggyapot dugóval vagy szinterelt üveglappal az alján. A szilikagél oszlop előkészítéséhez öntsön hexánt a kromatográfiás oszlopba megközelítőleg 5 cm mélységben, majd töltse fel hexánban eloszlatott szilikagélből (15 g szilikagél 40 ml hexánban) álló iszappal, további hexán hozzáadása mellett. Hagyja leülepedni, az ülepedés befejezését kismértékű rázással segítse. Adjon hozzá vízmentes nátrium-szulfátot megközelítőleg 0,5 cm magasságban, majd végül eluálja a feleslegben lévő hexánt.
4.6. Gázkromatográfiás berendezés lángionizációs detektorral, az oszlopra szerelt osztott vagy hideg befecskendezővel és ± 1 °C pontossággal programozható kemence.
4.7. Ömlesztett kovaföld kapillárisoszlop gázkromatográfiás eljáráshoz (0,25 vagy 0,32 mm belső átmérőjű, 25 m hosszú), 0,25 mm rétegvastagságban 5 % fenil-metil-szilikon fázis bevonatú.
1. megjegyzés:
Használhatók egyéb, azonos vagy alacsonyabb polaritású oszlopok is.
4.8. Integráló-rögzítő berendezés, lehetőleg völgy-völgy integrálási módszer elvégzésére képes.
4.9. 5-10 ml térfogatú mikrofecskendő a gázkromatográfiás eljáráshoz, cementált tűvel.
4.10. Elektromos fűtőpalást vagy főzőlap.
5. REAGENSEK
Minden reagensnek analitikai minőségűnek kell lennie, kivéve ha másként nincs előírva. A felhasznált víznek desztillált víznek vagy legalább hasonló tisztaságú víznek kell lennie.
5.1. Hexán vagy alkánok keveréke 65 és 70 °C közötti forrásponttal, rektifikáló oszlopon lepárolva. A hexán helyettesíthető izooktánnal (kromatográfia céljára alkalmas minőségű 2,2,4-trimetil-pentán), feltéve, hogy összehasonlítható pontossági értékek érhetők el. Az ellenőrzés történhet a 100 ml oldószer elpárologtatása után visszamaradó anyag vizsgálata útján. Az n-hexán forráspontjánál magasabb forráspontú oldószerek hosszabb idő alatt párolognak el. A hexán toxicitása miatt azonban előnyben kell részesíteni őket.
5.2. 96 v/v etanol.
5.3. Vízmentes nátrium-szulfát.
5.4. Kálium-hidroxid 10 %-os alkoholos oldata. Adjon 10 ml vizet 50 g kálium-hidroxidhoz, keverje el, majd öntse fel a keveréket etanollal 500 ml-re.
3. megjegyzés:
A kálium-hidroxid alkoholos oldatának színe állás közben barnává változik. Mindennap frissen kell készíteni, és jól lezárt sötét üvegpalackban kell tárolni.
5.5. Szilikagél 60 a kromatográfiás oszlophoz, 70-230 nyílásméretű (Merck, 7734 referenciaszámú vagy azzal megegyező).
4. megjegyzés:
A szilikagél rendszerint közvetlenül felhasználható, mindennemű kezelés nélkül. Néhány szilikagél adag azonban alacsony aktivitású lehet, ami rossz kromatográfiás szétválasztást eredményez. Ilyen körülmények mellett a szilikagélt az alábbi módon kell kezelni: aktiválja a szilikagélt legalább négy órán keresztül tartó, 550 °C hőmérsékleten történő hevítéssel. A felmelegítést követően tegye a szilikagélt szárítóberendezésbe, amíg a szilikagél le nem hűl, ekkor töltse be egy zárható lombikba. Adjon hozzá 2 % vizet, és rázza addig, amíg a por csomómentes nem lesz és szabadon folyik.
Amennyiben a szilikagéladagok a kromatogramokon zavaró csúcsokat eredményeznek, a szilikagélt a fenti módon kell kezelni. Egy másik lehetőség az extra tisztaságú szilikagél 60 használata (Merck, 7754 referenciaszám).
5.6. Koleszta-3, 5-dién (Sigma, 99 % tisztaságú) (200 ppm töménységű) törzsoldata hexánban (10 mg 50 ml-ben).
5.7. Koleszta-3, 5-dién 20 ppm töménységű standard oldata hexánban, a fenti oldat hígításából.
5. megjegyzés:
Az 5.6. és az 5.7. oldatok legalább 4 hónapon keresztül stabilak, amennyiben 4 °C-nál alacsonyabb hőmérsékleten tárolják.
5.8. n-nonakozán megközelítőleg 100 ppm töménységű hexánoldata.
5.9. Vivőgáz a kromatográfiás eljáráshoz: 99,9990 % tisztaságú hélium vagy hidrogén.
5.10. Segédgázok a lángionizációs detektorhoz: 99,9990 % tisztaságú hidrogén és tisztított levegő.
6. ELJÁRÁS
6.1. A nem szappanosítható anyag készítése
6.1.1. Mérjen 20 ± 0, 1 g olajat egy 250 ml térfogatú lombikba (4.1.), adjon hozzá 1 ml-t a koleszta-3, 5-dién standard oldatából (20 μg) és 75 ml-t a nátrium-hidroxid 10 %-os alkoholos oldatából, szerelje fel a visszafolyós hűtőt, és harminc percen keresztül forralja gyengén. Vegye le a fűtőeszközről a mintát tartalmazó lombikot, és hagyja kissé lehűlni az oldatot (ne hagyja, hogy teljesen lehűljön, mert a minta megköt). Adjon hozzá 100 ml vizet, majd 100 ml hexán felhasználásával öntse a mintát a választótölcsérbe (4.2.). Rázza a mintát hevesen 30 másodpercen keresztül, majd hagyja szétválni.
6. megjegyzés:
Amennyiben olyan emulzió keletkezik, amely nem tűnik el gyorsan, adjon hozzá kis mennyiségű etanolt.
6.1.2. Öntse az alsó vizes fázist egy másik választótölcsérbe, és 100 ml hexán segítségével végezzen ismét extrahálást. Engedje ki még egyszer az alsó fázist, és mossa át háromszor a hexánkivonatokat (másik választótölcsérben vegyítve) mindhárom alkalommal etanol-víz (1: 1) keverékével, amíg semleges pH-értéket nem ér el.
6.1.3. Vezesse át a hexánoldatot vízmentes nátrium-szulfáton (50 g), mossa át 20 ml hexánnal, és párolja be a forgó bepárlóban 30 °C hőmérsékleten csökkentett nyomás mellett, amíg ki nem szárad.
6.2. A szteroidos szénhidrogén-frakció leválasztása
6.2.1. A maradékot két 1 ml-es hexánadag segítségével öntse a frakcionáló oszlopba, engedje a mintát az oszlopba addig, ameddig az oldat szintje a nátrium-szulfátig süllyed, és kezdje meg a kromatográfiás elúciót a hexánnal megközelítőleg 1 ml/perc sebességgel. Az eluátum első 25-30 ml-jét öntse ki, majd gyűjtse össze a következő 40 ml-nyi frakciót. Az összegyűjtést követően öntse át ezt a frakciót egy 100 ml térfogatú kerek aljú lombikba (4.3.).
7. megjegyzés:
Az első frakció a telített szénhidrogéneket tartalmazza (lásd az 1a. ábrát), a második a szteroidosakat. A további elúció eredményeképpen szkvalén és rokonvegyületei keletkeznek. A telített és a szteroidos szénhidrogének jó szétválasztása érdekében szükség van a frakciók térfogatainak optimalizálására. Ehhez az első frakció térfogatát úgy kell beállítani, hogy a második frakció elemzésekor a telített szénhidrogéneket jelentő csúcsok alacsonyak legyenek (lásd az 1c. ábrát); amennyiben nem jelennek meg, de a standard csúcs intenzitása alacsony, csökkenteni kell a térfogatot. Az első és a második frakció összetevői közötti szétválasztásra máskülönben nincs szükség, mivel amennyiben a gázkromatográfiás körülmények a 6.3.1. pontnak megfelelően vannak beállítva, akkor a gázkromatográfiás elemzési eljárás során nem fordul elő a csúcsok átfedése. A második frakció térfogatának optimalizálására általában nincsen szükség, mivel a további komponensek szétválasztása megfelelő. A standard oldatnál 1,5 perccel alacsonyabb retenciós időnél megfigyelhető nagyméretű csúcs jelenléte azonban a szkvalénnek köszönhető és a rossz felbontás jele.
6.2.2. Párolja be a második fázist a forgó bepárlóban 30 °C hőmérsékleten csökkentett nyomás mellett, amíg ki nem szárad, majd ezután azonnal oldja fel a maradékot 0,2 ml hexánban. Az elemzés megkezdéséig tartsa az oldatot hűtőszekrényben.
8. megjegyzés:
A 6.1.3. és a 6.2.2. maradék nem tartható szárazon és szobahőmérsékleten. A keletkezésüket követően azonnal hozzá kell adni az oldószert, és hűtőszekrényben kell tárolni.
6.3. Gázkromatográfiás eljárás
6.3.1. Az osztott befecskendezés üzemi körülményei:
- injektor hőmérséklete: 300 °C,
- detektor hőmérséklete: 320 °C,
- integráló-rögzítő berendezés: az integrálás paramétereit úgy kell rögzíteni, hogy a területek pontos becslését eredményezze. Javasolható a völgy-völgy módszer használata,
- érzékenység: a minimális hígítás 16-szorosa,
- a befecskendezett oldat mennyisége: 1 μl,
- a kemence beprogramozott hőmérséklete: 235 °C kiindulási hőmérséklet 6 percen keresztül, majd percenként 2 °C-os hőmérséklet-emelkedés 285 °C hőmérsékletig,
- injektor 1:15 arányú áramlásosztóval,
- vivőgáz: hélium vagy hidrogén megközelítőleg 120 kPa nyomással.
Ezek a körülmények a kromatográfiás berendezés és az oszlop jellemzőinek megfelelően szabályozhatók be úgy, hogy a kromatogramok megfeleljenek az alábbi követelményeknek: belső standard csúcsa megközelítőleg a 6.3.2. pontban megadott időn belül öt perccel; a belső standard csúcsa a teljes skálának legalább 80 %-a.
A gázkromatográfiás rendszert a kolesztadién törzsoldatának (5.6.) és az n-nonakozán oldat (5.8.) befecskendezésével kell ellenőrizni. A koleszta-3, 5-dién csúcsnak az n-nonakozán csúcs előtt kell jelentkeznie (1c. ábra); amennyiben nem ez történik, két dolgot tehet: csökkenti a kemence hőmérsékletét és/vagy kevésbé poláris oszlopot használ.
6.3.2. A csúcsok azonosítása
A belső standard csúcsa megközelítőleg 19 percnél jelentkezik, és a 3,5-sztigmasztadiéné pedig ehhez képest megközelítőleg 1,29 perc retenciós időnél (lásd az 1b. ábrát). A 3,5-sztigmasztadién rendszerint egy kis mennyiségű izomerrel együtt fordul elő, és a kettő együtt eluálódik egyetlen kromatografikus csúcsként. Ha azonban az oszlop túlságosan poláris vagy magas felbontóképességet mutat, az izomer egy kis csúcsként jelentkezik a 3,5-sztigmasztadién csúcsa előtt és ahhoz közel (lásd 2. ábra). Annak biztosítására, hogy a sztigmasztadiének egyetlen csúcsként eluálódjanak, javasolható egy kevésbé poláris vagy nagyobb belső átmérőjű oszlop használata.
9. megjegyzés:
Referenciaként sztigmasztadiéneket finomított növényi olaj elemzéséből lehet nyerni, kisebb mennyiségű minta használatával (1-2 g). A sztigmasztadiének egy kimagasló és könnyen azonosítható csúcsot eredményeznek.
6.3.3. Mennyiségi elemzés
A sztigmasztadién-tartalom az alábbi képlet alapján határozható meg:
sztigmasztadiének mennyisége mg/kg-ban =

| ahol: | As = a sztigmasztadién-csúcs területe (amennyiben a csúcs két izomerre bomlik fel, a két csúcs területének összege), Ac = a belső standard területe (kolesztadién), Mc = a hozzáadott standard tömege, mikrogrammban, Mo = a felhasznált olaj tömege grammban. |
Érzékelési korlát: megközelítőleg 0,01 mg/kg.
10. megjegyzés: Amennyiben a sztigmasztadiének koncentrációja meghaladja a 4 mg/kg értéket, akkor szükség esetén a mennyiségi meghatározáshoz a Nemzetközi Olívatanács által a finomított olajok szterinjeinek meghatározása céljára elfogadott módszert kell alkalmazni.

[Kép #6] 1. ábra
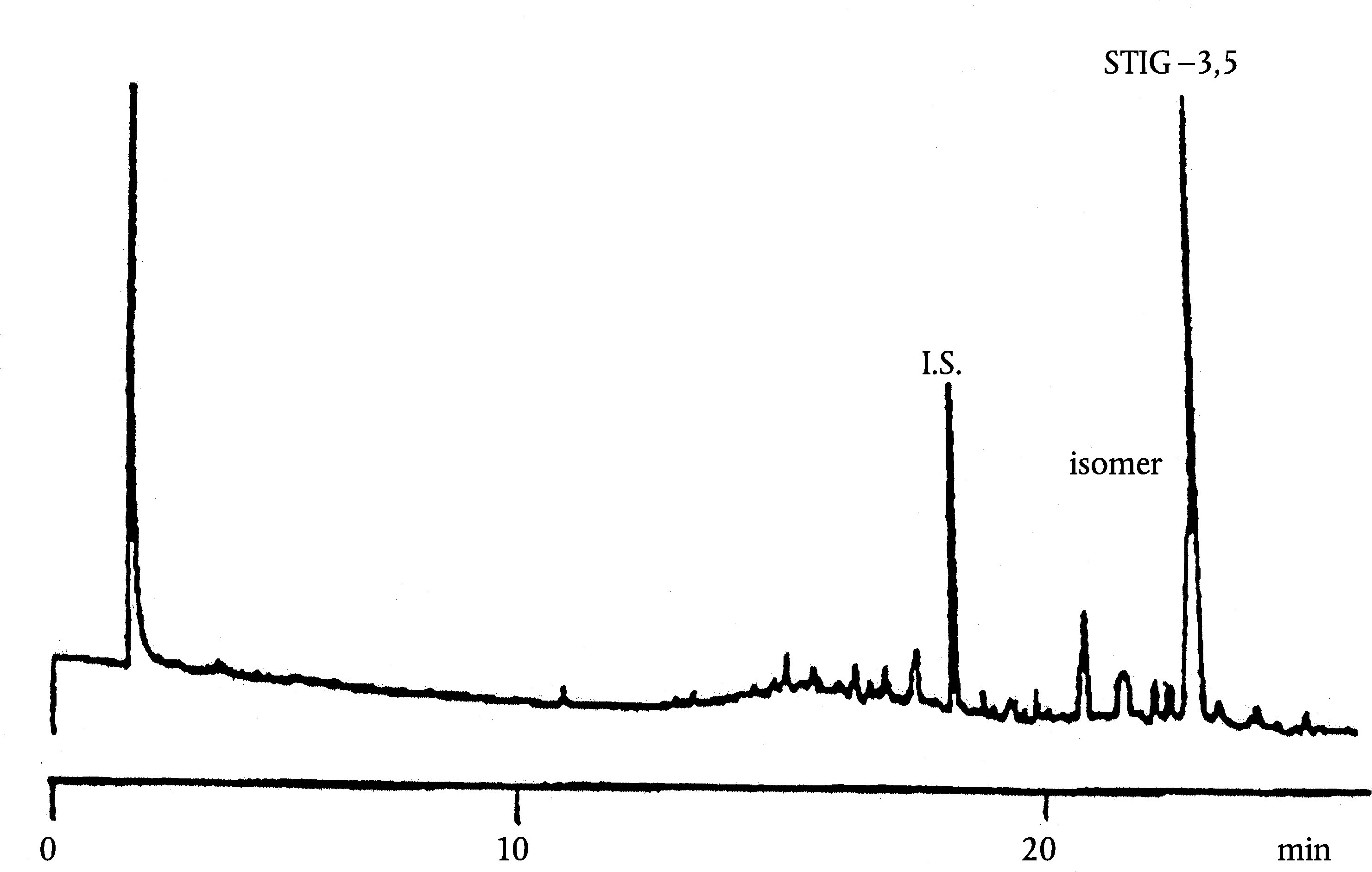
Kép #6
Az ömlesztett kovaföld kapilláris oszlopon (0,25 mm belső átmérő, 25 m, 5 % fenilmetil-szilikon bevonat 0,25 μm rétegvastagságban) elemzett olívaolaj-mintákból kapott gázkromatogramok.
a) Első frakció (30 ml) szűz olívaolajból, a standard csúcsaival.
b) Második frakció (40 ml) 0,10 mg/kg sztigmasztadién-tartalmú olívaolajból.
c) Második frakció (40 ml), amely tartalmaz egy kis mennyiséget az első frakcióból.
[Kép #7] 2. ábra

Kép #7
DB-5 oszlopon elemzett finomított olívaolaj gázkromatogramja, amelyen látszik a 3,5-sztigmasztadién izomerje.
XVIII. MELLÉKLET
AZ ECN-42-ES TRIACIL-GLICERINEK TÉNYLEGES ÉS ELMÉLETI MENNYISÉGE KÖZÖTTI KÜLÖNBSÉG MEGHATÁROZÁSA
1. HATÁSKÖR
A módszer a nagy teljesítményű folyadékkromatográfiás (HPLC) elemzés révén az olajban mért, 42-es ekvivalens szénszámú triacil-glicerinek kísérleti értékeinek (ECN-42HPLC) és a zsírsavösszetételből kiszámított, 42 ekvivalens szénszámú triacil-glicerinek elméleti értékének (ECN-42elméleti) abszolút különbségét határozza meg.
2. ALKALMAZÁSI KÖR
A módszer olívaolajokra alkalmazandó. A módszer a (linolsavban gazdag) magolajok kis mennyiségeinek jelenlétét hivatott kimutatni bármilyen kategóriájú olívaolajban.
3. A MÓDSZER ALAPELVE
Az ECN-42-es triacil-glicerinek nagy teljesítményű folyadékkromatográfiás elemzés révén meghatározott mennyisége és az ECN-42-es triacil-glicerinek (a zsírsavösszetétel gáz-folyadék-kromatográfiás módszer [GLC] szerint meghatározott] elméleti mennyisége a valódi olívaolajok esetében bizonyos határértéken belül megegyezik. Amennyiben a különbség meghaladja az egyes olajtípusokra elfogadott értékeket, megállapítható, hogy az olaj magolajokat tartalmaz.
4. MÓDSZER
Az ECN-42-es triacil-glicerinek elméleti mennyiségének megállapítására és a HPLC-adattal összevetve kapott különbség kiszámítására szolgáló módszer végrehajtása alapvetően más módszerek révén nyert analitikai adatok koordinációjával történik. A módszeren belül három fázis különíthető el: a zsírsavösszetétel meghatározása kapilláris gázkromatográfiával, az ECN-42-es triacil-glicerinek összetételének elméleti kiszámítása és az ECN-42-es triacil-glicerinek HPLC-vel történő meghatározása.
4.1. Eszközök
4.1.1. 250 és 500 ml-es gömblombikok.
4.1.2. 100 ml-es főzőpoharak.
4.1.3. Üveg kromatográfiás kolonna, 21 mm belső átmérővel, 450 mm hosszúsággal, csappal és felül normalizált kúpos furattal.
4.1.4. 250 ml-es elválasztó tölcsér, alul normalizált kúpos dugóval a kolonna tetejéhez való csatlakoztatás biztosítására.
4.1.5. 600 mm hosszú üvegpálca.
4.1.6. 80 mm átmérőjű üvegtölcsér.
4.1.7. 50 ml-es mérőlombikok.
4.1.8. 20 ml-es mérőlombikok.
4.1.9. Rotációs bepárló.
4.1.10. Nagy teljesítményű folyadékkromatográf, amely lehetővé teszi a kolonna hőmérsékletének termosztatikus szabályozását.
4.1.11. 10 μl befecskendezők.
4.1.12. Detektor: differenciál-refraktométer. A teljes skálán mért érzékenység legalább a refraktív index 10-4 egysége.
4.1.13. Kolonna: 250 mm hosszú és 4,5 mm belső átmérőjű rozsdamentes acél cső, megtöltve 5 μm átmérőjű, 22-23 %-ban oktadecil-szilán alakban lévő szenet tartalmazó szilikarészecskékkel.
4.1.14. Adatfeldolgozó szoftver.
4.1.15. Kb. 2 ml térfogatú ampullák, teflonbevonatú szeptummal és csavaros kupakkal.
4.2. Reagensek
A reagenseknek analitikai tisztaságúnak kell lenniük. Az eluáló oldószerek, melyeket gáztalanítani kell, többször újra felhasználhatók anélkül, hogy ez hatással lenne a szétválasztásra.
4.2.1. Kromatográfia céljára alkalmas minőségű petróleum-éter (40-60 °C) vagy hexán. A hexán helyettesíthető izooktánnal (kromatográfia céljára alkalmas minőségű 2,2,4-trimetil-pentán), feltéve, hogy összehasonlítható pontossági értékek érhetők el. Az n-hexán forráspontjánál magasabb forráspontú oldószerek hosszabb idő alatt párolognak el. A hexán toxicitása miatt azonban előnyben kell részesíteni őket.
4.2.2. Frissen desztillált, peroxidmentes etil-éter.
4.2.3. Eluáló oldószer az olaj oszlopkromatográfiával történő tisztítására: petróleum-éter és etil-éter 87:13 térfogatarányú elegye.
4.2.4. Szilikagél, 70-230 mesh, "Merck 7734" típus, 5 tömegszázalékon standardizált víztartalommal.
4.2.5. Üveggyapot.
4.2.6. Aceton HPLC-hez.
4.2.7. Acetonitril vagy propionitril HPLC-hez.
4.2.8. Eluáló oldószer HPLC-hez: acetonitril + aceton (az arányokat a kívánt elválasztásnak megfelelően kell beállítani, 50:50 arányú keverékkel kezdve) vagy propionitril.
4.2.9. Oldáskönnyítő oldószer: aceton.
4.2.10. Referenciatrigliceridek: használhatók a kereskedelmi forgalomban kapható trigliceridek (tripalmitin, triolein stb.), ekkor a retenciós időket az ekvivalens szénszám függvényében kell ábrázolni, vagy használhatók szójaolajról (szójaolaj, valamint olívaolaj és tiszta olívaolaj 30:70 arányú keverékével) készült referenciakromatogrammok (lásd az 1. és 2. megjegyzést, illetve az 1-4. ábrát).
4.2.11. Szilárd fázisú extrakciós oszlop szilikafázissal, 1 g, 6 ml.
4.2.12. Kromatográfia céljára alkalmas minőségű heptán. A heptán helyettesíthető izooktánnal (kromatográfia céljára alkalmas minőségű 2,2,4-trimetil-pentán).
4.3. A minták előkészítése
Mivel több interferáló anyag hamis pozitív eredményekhez vezethet, a mintát mindig tisztítani kell, a sütőolajokban található poláris vegyületek meghatározásához használt 2507-es IUPAC-módszernek megfelelően.
4.3.1. A kromatográfiás kolonna előkészítése
Töltse meg a kolonnát (4.1.3.) körülbelül 30 ml eluláló oldószerrel (4.2.3.), majd a kolonna belsejébe helyezzen üveggyapotot (4.2.5.), amelyet az üvegpálca segítségével (4.1.5.) nyomjon a kolonna aljára.
Egy 100 ml-es főzőpohárban szuszpendáljon 25 g szilikagélt (4.2.4.) 80 ml eluáló keverékben (4.2.3.), majd üvegtölcsér (4.1.6.) segítségével öntse a kolonnába.
Annak biztosításához, hogy a szilikagél teljes mennyisége átjusson a kolonnába, mossa ki a főzőpoharat az eluáló keverékkel, és a mosásra használt mennyiségeket is juttassa a kolonnába.
Nyissa ki a csapot, és annyi oldószert engedjen a kolonnából eluálni, hogy annak szintje kb. 1 cm-rel lépje túl a szilikagél szintjét.
4.3.2. Oszlopkromatográfia
Mérjen be 0,001 g pontossággal 2,5 ± 0,1 g, előzőleg szűrt, homogenizált és szükség esetén vízmentesített olajat egy 50 ml-es mérőlombikba (4.1.7).
Oldja fel körülbelül 20 ml eluáló oldószerben (4.2.3.). Az oldódás elősegítéséhez szükség esetén enyhén hevítse. Hűtse szobahőmérsékleten, és az eluáló oldószerrel állítsa be a volument.
Mérőpipetta segítségével tegyen 20 ml oldatot a 4.3.1. pont szerint előkészített kolonnába, nyissa meg a csapot, és engedje az oldószert a szilikagélréteg szintjéig eluálni.
Ezután eluáljon 150 ml eluáló oldószerrel (4.2.3.), az oldószer sebességét kb. 2 ml/percre beállítva (így kb. 60-70 percig tart, amíg 150 ml átmegy a kolonnán).
Az eluátumot egy előzőleg sütőben kitárált, 250 ml-es gömblombikban (4.1.1.) fogja fel, és tömegét pontosan mérje meg. Rotációs bepárló (4.1.9.) segítségével, csökkentett nyomáson távolítsa el az oldószert, és mérje meg a HPLC-elemzéshez és metil-észter készítéséhez használatos oldat elkészítéséhez szükséges maradékanyag tömegét.
A kolonnából az extra szűz, a szűz, a közönséges, a finomított és az olívaolaj kategóriák esetében legalább a minta 90 %-át, lampante és olívapogácsa-olajok esetében legalább 80 %-át kell visszanyerni.
4.3.3. Szilárd fázisú extrakcióval történő (SPE) tisztítás
A szilikatöltetű SPE-kolonnát 6 ml hexán (4.2.3) vákuumban - a kiszárítás elkerülése mellett - történő áteresztésével kell aktiválni.
Mérjen ki 0,001 g pontossággal 0,12 g-ot 2 ml-es ampullába (4.1.15), és oldja fel 0,5 ml hexánnal (4.2.3.).
Töltse meg az SPE-kolonnát az oldattal, és vákumban eluálja 10 ml (87:13 térfogatarányú) hexán-dietil-éterrel (4.2.3.).
Az összegyűjtött frakciót a rotációs bepárlóval (4.1.9.) csökkentett nyomás mellett, szobahőmérsékleten, szárazságig kell bepárolni. A maradékanyagot 2 ml, triacilglicerol elemzéséhez szolgáló acetonban (4.2.6.) fel kell oldani.
4.4. HPLC-elemzés
4.4.1. Minták előkészítése kromatográfiás elemzéshez
A vizsgálati mintából készítsen 5 %-os oldatot, úgy, hogy egy 10 ml-es mérőlombikba mérje ki a minta 0,5 ± 0,001 g-ját, és az oldáskönnyítő oldószerrel (4.2.9.) 10 ml-ig töltse fel.
4.4.2. Eljárás
Állítsa össze a kromatográfiás rendszert. A teljes rendszer kitisztításához szivattyúzza 1,5 ml/perc sebességgel az eluáló oldószert (4.2.8.). Várjon, ameddig kialakul egy stabil alapvonal.
Fecskendezzen be 10 μl mennyiségű, a 4.3. pont szerint előkészített mintát.
4.4.3. Az eredmények kiszámítása és kifejezése
Használja a területnormálási módszert, azaz tételezze fel, hogy az ECN-42-ECN-52-es triacil-glicerinekhez tartozó csúcsok alatti területek összege 100 %.
Számítsa ki az egyes trigliceridek relatív százalékát a következő képlet segítségével:
.
Az eredményeket legalább két tizedesjegyig kell megadni.
Lásd az 1-4. megjegyzést.
4.5. Triacil-glicerinek összetételének (mólszázalék) kiszámítása zsírsav-összetételi adatokból (területszázalék)
4.5.1. A zsírsavösszetétel meghatározása
A zsírsavösszetételt az ISO 5508 segítségével, kapilláris kolonna felhasználásával kell meghatározni. A metil-észtereket a COI/T.20/Doc. No 24 szerint kell elkészíteni.
4.5.2. A számításhoz szükséges zsírsavak
A glicerideket ekvivalens szénszámuk (ECN) alapján kell csoportosítani, figyelembe véve az ECN és a zsírsavak közötti alábbi ekvivalenciákat. Csak a 16 és 18 szénatomszámú zsírsavakat kellett figyelembe venni, az olívaolaj tekintetében ugyanis kizárólag ezek fontosak. A zsírsavakat 100 %-ra kell normálni.
| Zsírsav | Rövidítés | Molekulatömeg (MW) | ECN |
| Palmitinsav | P | 256,4 | 16 |
| Palmitoleinsav | Po | 254,4 | 14 |
| Sztearinsav | S | 284,5 | 18 |
| Olajsav | O | 282,5 | 16 |
| Linolsav | L | 280,4 | 14 |
| Linolénsav | Ln | 278,4 | 12 |
4.5.3. A területszázalék mólra való átszámítása valamennyi zsírsav esetében (1)
| [Kép #8] | [Kép #9] | [Kép #10] |
| [Kép #11] | [Kép #12] | [Kép #13] |
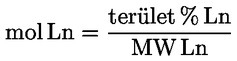
Kép #8
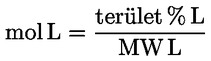
Kép #9
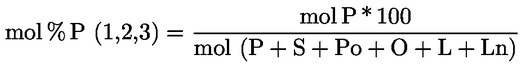
Kép #10

Kép #11
Kép #12
Kép #13
4.5.4. Zsírsavak 100 %-ra történő normálása (2)


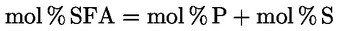
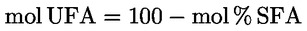

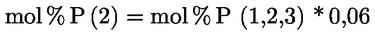
Az eredmény azt adja meg, hogy a triacil-glicerinekben az összes (1, 2, 3-) pozícióban egyes zsírsavak milyen mólszázalékos arányban vannak jelen.
Ezután a P és S telített zsírsavak (SFA) és a Po, O, L és Ln telítetlen zsírsavak (UFA) összegét kell kiszámítani, az alábbiak szerint (3):
4.5.5. A zsírsavösszetétel kiszámítása a triacil-glicerinek 2- és 1,3- pozícióiban
A zsírsavakat az alábbiak szerint három csoportra osztják: az egyik csoport a 2- pozícióban, két másik, azonos csoport pedig az 1- és a 3- pozícióban lévő zsírsavakra vonatkozik, ahol a telített (P és S) és telítetlen zsírsavakra (Po, O, L és Ln) különböző tényezőket alkalmaznak.
4.5.5.1. Telített zsírsavak a 2- pozícióban [P(2) és S(2)] (4)
4.5.5.2. Telítetlen zsírsavak a 2- pozícióban [Po(2), O(2), L(2) és Ln(2)] (5):
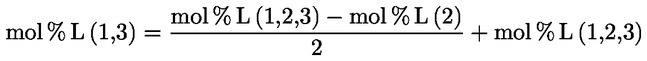
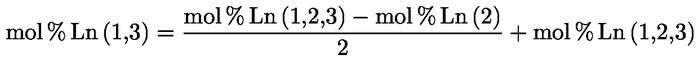
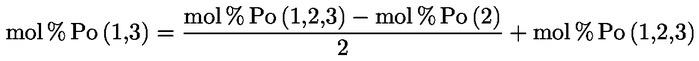
4.5.5.3. 1,3- pozícióban lévő zsírsavak [P(1,3), S(1,3), Po(1,3), O(1,3), L(1,3) és Ln(1,3)] (6):


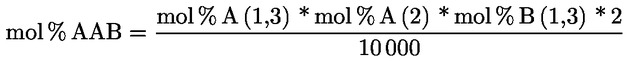
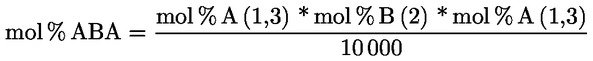

4.5.6. Triacil-glicerinek kiszámítása
4.5.6.1. Egy zsírsavval rendelkező triacil-glicerinek (AAA, itt LLL, PoPoPo) (7)

4.5.6.2. Két zsírsavval rendelkező triacil-glicerinek (AAB, itt PoPoL, PoLL) (8)

4.5.6.3. Három különböző zsírsavval rendelkező triacil-glicerinek (ABC, itt OLLn, PLLn, PoOLn, PPoLn) (9)



4.5.6.4. ECN-42-es triacil-glicerinek
Az ECN-42-es triacil-glicerineket a (7), (8) és (9) egyenleteknek megfelelően kell kiszámítani, és a HPLC-nél a várt elúció sorrendjében (általában csak három csúcs) megadni.
LLL
PoLL és LPoL pozíciós izomer
OLLn, valamint OLnL és LnOL pozíciós izomerek
PoPoL és PoLPo pozíciós izomer
PoOLn, valamint OPoLn and OLnPo pozíciós izomerek
PLLn, valamint LLnP és LnPL pozíciós izomerek
PoPoPo
SLnLn és LnSLn pozíciós izomer
PPoLn, valamint PLnPo és PoPLn pozíciós izomerek
Az ECN-42-es triacil-glicerineket a kilenc triacilglicerol - pozíciós izomerjeikkel együtt vett - összege révén, legalább két tizedesjegyig kell megadni.
5. AZ EREDMÉNYEK ÉRTÉKELÉSE
Össze kell hasonlítani a kiszámított elméleti tartalmat és a HPLC-elemzéssel meghatározott tartalmat. Ha a HPLC-adatból kivont elméleti adat különbsége abszolút értékben nagyobb, mint a megfelelő olajkategóriára a szabványban meghatározott értékek, a minta magolajat tartalmaz.
Az eredményeket két tizedesjegyig kell megadni.
6. PÉLDA (A SZÁMOK A MÓDSZERLEÍRÁSBAN SZEREPLŐ PONTOKRA UTALNAK)
- 4.5.1. Zsírsavak mólszázalékának kiszámítása GLC-adatokból (normált területszázalék)
A GLC-módszerrel a zsírsavösszetételre az alábbi adatokat kaptuk:
| Zsírsav | P | S | Po | O | L | Ln |
| MW | 256,4 | 284,5 | 254,4 | 282,5 | 280,4 | 278,4 |
| Terület % | 10,0 | 3,0 | 1,0 | 75,0 | 10,0 | 1,0 |
- 4.5.3. A területszázalék mólra való átszámítása valamennyi zsírsav esetében (lásd az (1) képletet)
mol P = [Kép #14]
Kép #14
mol S = [Kép #15]
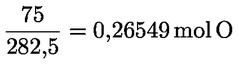
Kép #15
mol Po = [Kép #16]
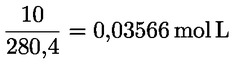
Kép #16
mol O = [Kép #17]
Kép #17
mol L = [Kép #18]

Kép #18
mol Ln = [Kép #19]

Kép #19
Összesen = 0,35821 mol triacilglicerol
- 4.5.4. Zsírsavak 100 %-ra történő normálása (lásd a (2) képletet)
mol % P(1,2,3) = [Kép #20]
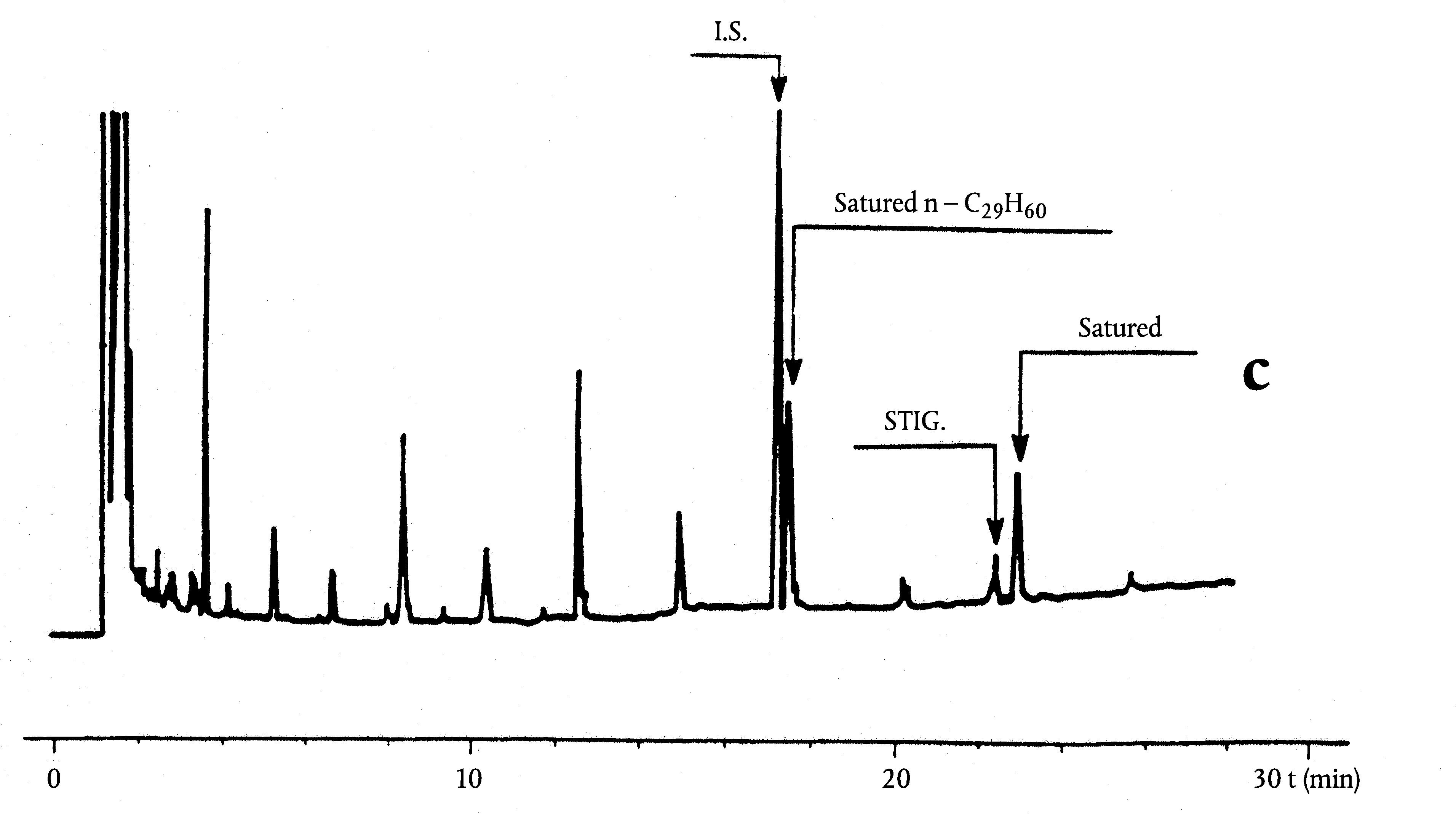
Kép #20
mol % S(1,2,3) = [Kép #21]
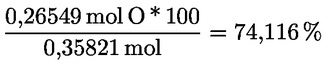
Kép #21
mol % Po(1,2,3) = [Kép #22]
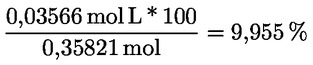
Kép #22
mol % O(1,2,3) = [Kép #23]

Kép #23
mol % L(1,2,3) = [Kép #24]

Kép #24
mol % Ln(1,2,3) = [Kép #25]

Kép #25
Összes mol % = 100 %
Triacilglicerolok 1,2,3- pozíciójában lévő telített és telítetlen zsírsavak összege (lásd a (3) képletet):
- 4.5.5 A zsírsavösszetétel kiszámítása a triacil-glicerinek 2- és 1,3- pozícióiban
- 4.5.5.1. Telített zsírsavak a 2- pozícióban [P(2) és S(2)] (lásd a (4) képletet)
- 4.5.5.2. Telítetlen zsírsavak a 2- pozícióban [Po(1,3), O(1,3), L(1,3) és Ln(1,3)] (lásd az (5) képletet)

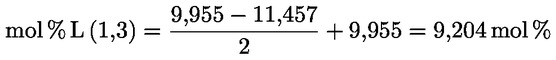
- 4.5.5.3 1,3-pozícióban lévő zsírsavak [P(1,3), S(1,3), Po(1,3), O(1,3), L(1,3) és Ln(1,3)] (lásd a (6) képletet)






- 4.5.6. Triacil-glicerinek kiszámítása
Az sn-2- és sn-3- pozíciókban kiszámított zsírsavösszetételből:
| Zsírsav | 1,3- poz. | 2- poz. |
| P | 16,004 % | 0,653 % |
| S | 4,325 % | 0,177 % |
| Po | 1,015 % | 1,262 % |
| O | 68,526 % | 85,296 % |
| L | 9,204 % | 11,457 % |
| Ln | 0,927 % | 1,153 % |
| Össz | 100,0 % | 100,0 % |
az alábbi triacil-glicerinek kiszámítására kerül sor:
LLL
PoPoPo
PoLL 1 pozíciós izomerrel
SLnLn 1 pozíciós izomerrel
PoPoL 1 pozíciós izomerrel
PPoLn 2 pozíciós izomerrel
OLLn 2 pozíciós izomerrel
PLLn 2 pozíciós izomerrel
PoOLn 2 pozíciós izomerrel
- 4.5.6.1. Egy zsírsavval rendelkező triacil-glicerinek (LLL, PoPoPo) (lásd a (7) képletet)
[Kép #26] = 0,09706 mol LLL
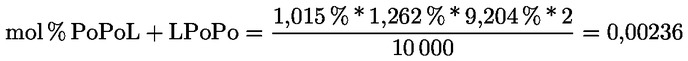
Kép #26
- 4.5.6.2. Két zsírsavval rendelkező triacil-glicerinek (PoLL, SLnLn, PoPoL) (lásd a (8) képletet)
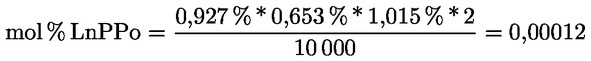
0,03210 mol PoLL

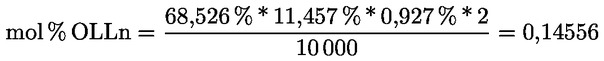
0,00094 mol SLnLn

0,00354 mol PoPoL
- 4.5.6.3. Három különböző zsírsavval rendelkező triacil-glicerinek (PoPLn, OLLn, PLLn, PoOLn) (lásd a (9) képletet)

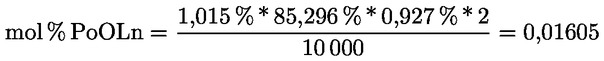
0,00761 mol PPoLn
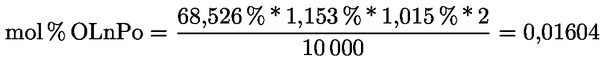

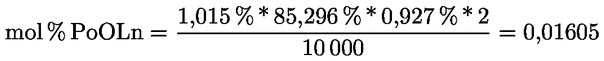
0,43655 mol OLLn
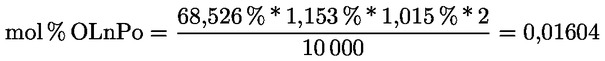
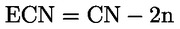

0,06907 mol PLLn

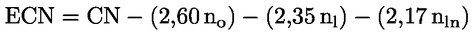

0,04812 mol PoOLn
ECN42=0,69512 mol triacil-glicerinek
1. megjegyzés: Az eluálási sorrend meghatározható az ekvivalens szénszámok kiszámításával, amelyet gyakran az [Kép #27] összefüggéssel adnak meg (ahol a CN a szénatomszám, az n pedig a kettős kötések száma), de a kettős kötések eredetének figyelembevételével sokkal pontosabban számítható. Amennyiben no, nl, illetve nln az olajsavra, linolsavra, illetve linolénsavra jellemző kettős kötések számát jelenti, az ekvivalens szénszám a következő képlettel számítható ki:
Kép #27
ahol a do, dl és dln együtthatókat a referenciatrigliceridek alapján lehet kiszámolni. Az e módszerben meghatározott feltételek mellett a kapott összefüggés megközelítőleg a következő:
2. megjegyzés: Több referenciatriglicerid segítségével ki lehet számítani a felbontást a triolein tekintetében is:
trioleina redukált retenciós idő felhasználásával:
A log α és az f (a kettős kötések száma) függvénye lehetővé teszi a retenciós értékek kiszámítását a referenciatrigliceridekben található zsírsavak minden trigliceridjére (lásd az 1. ábrát).
3. megjegyzés: Az oszlop hatásfoka tegye lehetővé a trilinolein csúcsának a szomszédos retenciós idejű trigliceridektől történő egyértelmű elkülönülését. Az elúciót az ECN-52-es csúcsig kell végezni.
4. megjegyzés: Ezen meghatározás szempontjából valamennyi fontos csúcs területének pontos mérése akkor biztosított, ha az ECN-50-hez tartozó második csúcs az adatrögzítő készülék teljes skálájának 50 %-ánál van.
1. ábra
A log α és f (kettős kötések száma) grafikonja

Kettős kötések száma
La: laurinsav My: mirisztinsav; P: palmitinsav; S: sztearinsav; O: olajsav; L: linolsav; Ln: linolénsav
2. ábra
Alacsony linolsavtartalmú olívaolaj
a)

Oldószer: aceton/acetonitril.
a) profil: A kromatográfiás csúcsok fő összetevői: ECN-42: (1) LLL + PoLL; (2) OLLn + PoOLn; (3) PLLn; ECN-44: (4) OLL + PoOL; (5) OOLn + PLL; (6) POLn + PPoPo; (7) OOL + PoOO; ECN-46: (8) OOL + LnPP; (9) PoOO; (10) SLL + PLO; (11) PoOP + SPoL + SOLn + SPoPo; (12) PLP; ECN-48: (13) OOO + PoPP; (14 + 15) SOL + POO; (16) POP; ECN-50: (17) SOO; (18) POS + SLS.
b)

Oldószer: propionitril.
b) profil: A kromatográfiás csúcsok fő összetevői: ECN-42: (1) LLL; (2) OLLn + PoLL; (3) PLLn; ECN-44: (4) OLL; (5) OOLn + PoOL; (6) PLL + PoPoO; (7) POLn + PPoPo + PPoL; ECN-46: (8) OOL + LnPP; (9) PoOO; (10) SLL + PLO; (11) PoOP + SPoL + SOLn + SPoPo; (12) PLP; ECN-48: (13) OOO + PoPP; (14) SOL; (15) POO; (16) POP; ECN-50: (17) SOO; (18) POS + SLS
3. ábra
Magas linolsavtartalmú olívaolaj
a)
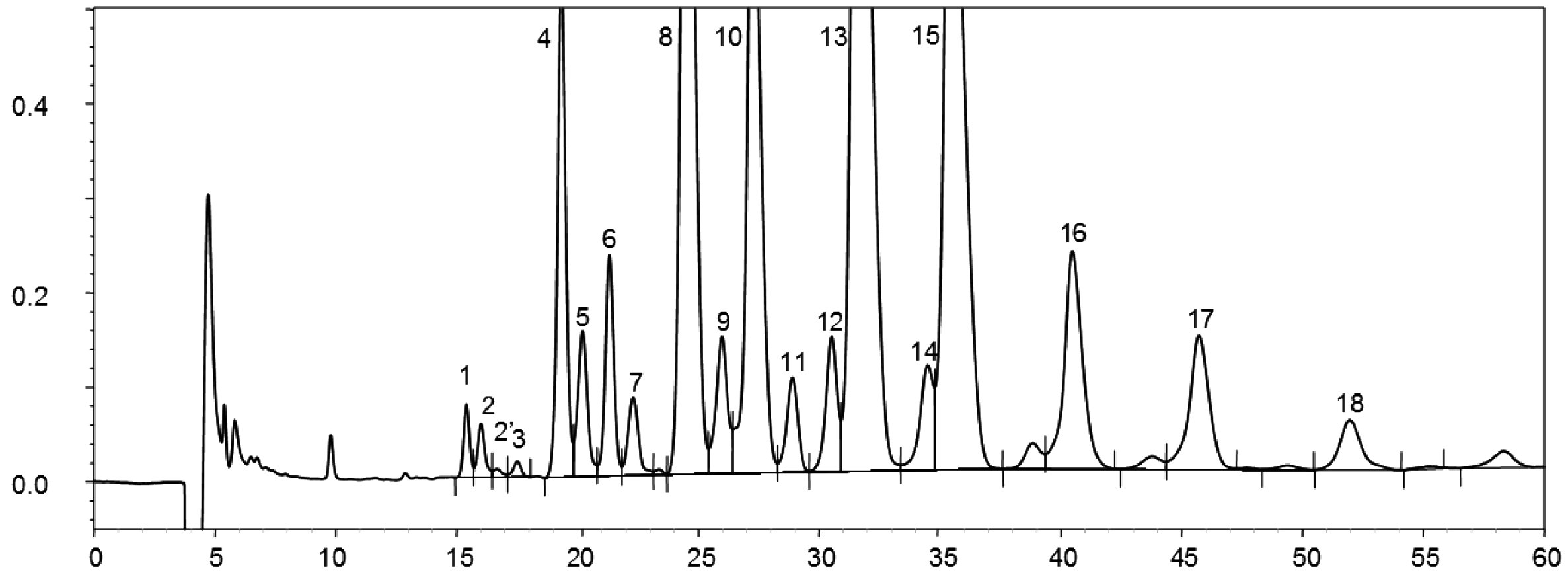
Oldószer: aceton/acetonitril (50:50).
a) profil: A kromatográfiás csúcsok fő összetevői: ECN-42: (1') LLL + PoLL; (2') OLLn + PoOLn; (3') PLLn; ECN-44: (4') OLL + PoOL; (5') OOLn + PLL; (6') POLn + PPoPo; ECN46: (7') OOL + PoOO; (8') PLO + SLL + PoOP; (9') PLP + PoPP; ECN48: (10') OOO; (11') POO + SLL + PPoO; (12') POP + PLS; ECN-50: (13') SOO; (14') POS + SLS
b)

Oldószer: propionitril.
b) profil: A kromatográfiás csúcsok fő összetevői: ECN-42: (1) LLL; (2 + 2') OLLn + PoLL; (3) PLLn; ECN-44: (4) OLL; (5) OOLn + PoOL; (6) PLL + PoPoO; (7) POLn + PPoPo + PPoL; ECN-46: (8) OOL + LnPP; (9) PoOO; (10) SLL + PLO; (11) PoOP + SPoL + SOLn + SPoPo; ECN-48: (12) PLP; (13) OOO + PoPP; (14) SOL; (15) POO; (16) POP; ECN-50: (17) SOO; (18) POS + SLS; ECN-52: (19) AOO.
XIX. MELLÉKLET
A SZTERINÖSSZETÉTEL ÉS -TARTALOM, VALAMINT AZ ALKOHOLOS VEGYÜLETEK MEGHATÁROZÁSA KAPILLÁRIS GÁZKROMATOGRÁFIÁVAL
1. ALKALMAZÁSI KÖR
A módszer az olívaolajokban és az olívapogácsa-olajokban, valamint ezek keverékeiben található egyes alkoholos vegyületek mennyiségének és az ilyen vegyületek összmennyiségének meghatározására szolgáló eljárást ismerteti.
Az olívaolajok és az olívapogácsa-olajok alkoholos vegyületei lehetnek alifás alkoholok, szterinek és triterpén-dialkoholok.
2. ALAPELV
Az olajakat, amelyekhez belső standardként α-kolesztanolt és 1-eikozanolt adnak, etanolos kálium-hidroxid-oldattal elszappanosítják, majd az el nem szappanosítható anyagot etiléterrel kivonják.
A különböző alkoholos vegyületek frakcióit vagy vékonyréteg-kromatográfiával, bázisos szilikagél lemezen (referencia-módszer), vagy nagy teljesítményű folyadékkromatográfiával (HPLC), szilikagél oszlop alkalmazásával elválasztják az el nem szappanosítható anyagtól. A szilikagél felhasználásával elvégzett elválasztás útján visszanyert frakciókat trimetilszilil-éterré alakítják és kapilláris oszlopos gázkromatográfia alkalmazásával elemzik.
1. RÉSZ
AZ EL NEM SZAPPANOSÍTHATÓ ANYAG ELŐÁLLÍTÁSA
1. ALKALMAZÁSI KÖR
Ez a rész az el nem szappanosítható anyag előállításának és kivonásának módját ismerteti. Az olíva- és olívapogácsa-olajokból származó, el nem szappanosítható anyag előállítását és kivonását foglalja magában.
2. ALAPELV
A vizsgált mennyiséget az etanolos kálium-hidroxid-oldattal, visszafolyatás mellett történő forralással elszappanosítják. Az el nem szappanosítható anyagot dietil-éterrel kivonják.
3. BERENDEZÉS
Hagyományos laboratóriumi felszerelés különös tekintettel a következőkre:
3.1. 250 ml-es, gömbölyű fenekű lombik visszafolyós hűtővel és csiszolt üvegcsatlakozókkal.
3.2. 500 ml-es elválasztó tölcsér.
3.3. 250 ml-es lombikok.
3.4. 100 μl-es és 500 μl-es mikrofecskendők.
3.5. Megközelítőleg 2 cm átmérőjű és megközelítőleg 5 cm magas, vákuum alatti szűrésre alkalmas hengeres szűrőtölcsér G3 porózus válaszfallal (15-40 μm-es porozitás), csiszolt üveg apacsatlakozóval.
3.6. A szűrőtölcsérhez (3.5.) csatlakoztatható, 50 ml-es Erlenmeyer-lombik csiszolt üveg anyacsatlakozóval.
3.7. 10 ml-es, kúpos fenekű kémcső csiszolt üvegdugóval.
3.8. Kalcium-kloriddal működő szárítóberendezés.
4. REAGENSEK
4.1. Kálium-hidroxid (minimális titer: 85 %).
4.2. Kálium-hidroxid megközelítőleg 2 M etanolos oldata.
130 g kálium-hidroxid (4.1.) hűtés közben feloldva 200 ml desztillált vízben, majd etanollal (4.7.) feltöltve egy liter mennyiségűre. Az oldatot jól lezárt, sötét színű üvegben kell tárolni, legfeljebb 2 napig.
4.3. Analízis céljára alkalmas minőségű etiléter.
4.4. Analízis céljára alkalmas minőségű, vízmentes nátrium-szulfát.
4.5. Kromatográfia céljára alkalmas minőségű aceton.
4.6. Kromatográfia céljára alkalmas minőségű etiléter.
4.7. Analízis céljára alkalmas minőségű etanol.
4.8. Analízis céljára alkalmas minőségű etil-acetát.
4.9. Belső standard, α-kolesztanol, 99 % feletti tisztaságú (a tisztaságot gázkromatográfiás elemzéssel kell ellenőrizni).
4.10. α-kolesztanol belső standard oldat, 0,2 % (m/V) oldat etil-acetátban (4.8.).
4.11. Fenolftaleinoldat, 10 g/l etanolban (4.7.).
4.12. 0,1 %-os (m/V) 1-eikozanol-oldat etil-acetátban (belső standard).
5. ELJÁRÁS
Egy 500 μl-es mikrofecskendő (3.4.) segítségével tegyen a 250 ml-es lombikba (3.1.) olyan mennyiségű α-kolesztanol belső standard oldatot (4.10.) és olyan mennyiségű 1-eikozanolt (4.12.), amelyben a kolesztanol, illetve az eikozanol mennyisége a minta szterin- és alkoholtartalmának megközelítőleg 10 %-a. Például 5 g olívaolaj-mintához adjon 500 μl α-kolesztanol-oldatot (4.10.) és 250 μl 1-eikozanol-oldatot (4.12.). Az olívapogácsa-olajok esetében az α-kolesztanol-oldatból (4.10.) és az 1-eikozanol-oldatból (4.12.) egyaránt 1 500 μl-t adjon a mintához. Enyhe nitrogénáramban való párologtatással, meleg vízfürdőben szárítsa ki. A lombik lehűtése után mérjen 5,00 ± 0,01 g-ot a száraz, szűrt mintából ugyanabba a lombikba.
1. megjegyzés: Az észlelhető mennyiségű koleszterint tartalmazó állati vagy növényi olajok és zsírok esetében olyan csúcsok jelenhetnek meg, amelyek retenciós ideje megegyezik a kolesztanoléval. Ennek előfordulása esetén a szterinfrakció esetében két elemzést kell végezni, az egyiket belső standard nélkül, a másikat pedig belső standarddal.
Adjon hozzá 50 ml 2 M etanolos kálium-hidroxid-oldatot (4.2.) és néhány forrkövet, csatlakoztassa a visszafolyós hűtőt, és vízfürdőben fokozatosan hevítse forrásig, amíg a szappanképződés meg nem indul (az oldat tisztává válik). Folytassa a melegítést további 20 percen keresztül, majd adjon hozzá 50 ml desztillált vizet a visszafolyós hűtő tetején keresztül, ezután szerelje le a hűtőt, és hűtse le a lombikot megközelítőleg 30 oC-ra.
A lombik tartalmát töltse át egy 500 ml-es elválasztó tölcsérbe (3.2.) több rész desztillált vízzel (50 ml) történő öblítés mellett. Adjon hozzá megközelítőleg 80 ml etilétert (4.6.) és körülbelül 60 másodpercen keresztül rázza erőteljesen; a nyomás kiengedéséhez időnként fordítsa át az elválasztó tölcsért és nyissa meg az elzárócsapot. Hagyja állni, amíg a két fázis teljesen szét nem válik (2. megjegyzés). Ezután válassza le a szappanos oldatot amennyire csak lehetséges, és helyezze át egy másik elválasztó tölcsérbe. Végezzen két további kivonást a vizes-alkoholos fázison hasonló módon, esetenként 60-70 ml etiléter (4.6.) felhasználásával.
2. megjegyzés: Az emulziók megszüntethetők kis mennyiségű etanol (4.7.) hozzáadásával.
A három éterkivonatot keverje össze egy közös elválasztó tölcsérben 50 ml vízzel. Folytassa az átmosást vízzel (50 ml-enként), amíg a mosóvíz már nem válik rózsaszínűvé egy csepp fenolftaleinoldat (4.11.) hozzáadására. A mosóvíz eltávolítását követően szűrje át vízmentes nátrium-szulfáton (4.4.) egy 250 ml-es lombikba, amelynek súlyát előzőleg lemérte; a tölcsért és a szűrőt mossa ki több adagban, kis mennyiségű etiléterrel (4.6.).
Desztillációval párologtassa el az oldószert rotációs bepárlóban, 30 °C-on, vákuumban. Adjon hozzá 5 ml acetont (4.5.), és az illékony oldószert teljesen távolítsa el enyhe nitrogénáramban. Kemencében 103 ± 2 °C-os hőmérsékleten szárítsa a maradékot 15 percen keresztül. Szárítóberendezésben hagyja kihűlni, és mérje meg a súlyát 0,1 mg pontossággal.
2. RÉSZ
AZ ALKOHOLOS VEGYÜLETEK FRAKCIÓINAK ELVÁLASZTÁSA
1. ALKALMAZÁSI KÖR
Az 1. részben foglaltak szerint előállított, el nem szappanosítható anyagnak a különböző alkoholos vegyületek - az alifás alkoholok, a szterinek és a triterpén-dialkoholok (eritrodiol és uvaol) - frakcióira bontása.
2. ALAPELV
Az el nem szappanosítható anyag bázisos vékonyréteg-kromatográfiával (referencia-módszer) frakcionálható, kimutatható, a megfelelő sávok pedig lekaparhatóak és kivonhatóak. Alternatív elválasztási módszerként HPLC is alkalmazható szilikagél oszlop és UV-detektor használatával, összegyűjtve a különböző frakciókat. Az alifás és a triterpén-alkoholok, valamint a szterin és a triterpén-dialkoholok együtt kerülnek elkülönítésre.
3. BERENDEZÉS
Hagyományos laboratóriumi felszerelés különös tekintettel a következőkre:
3.1. Komplett elemzőberendezés vékonyréteg-kromatográfiához, 20 × 20 cm-es nagyságú üveg tárgylemezekkel.
3.2. 366 vagy 254 nm hullámhosszúságú ultraibolya lámpa.
3.3. 100 μl-es és 500 μl-es mikrofecskendők.
3.4. Megközelítőleg 2 cm átmérőjű és megközelítőleg 5 cm magas, vákuum alatti szűrésre alkalmas hengeres szűrőtölcsér G3 porózus válaszfallal (15-40 μm-es porozitás), csiszolt üveg apacsatlakozóval.
3.5. A szűrőtölcsérhez (3.4.) csatlakoztatható, 50 ml-es Erlenmeyer-lombik csiszolt üveg anyacsatlakozóval.
3.6. 10 ml-es, kúpos fenekű kémcső csiszolt üvegdugóval.
3.7. Kalcium-kloriddal működő szárítóberendezés.
3.8. HPLC-rendszer, amely a következőkből áll:
3.8.1. Bináris pumpa.
3.8.2. 200 μl-es injektáló hurokkal felszerelt kézi vagy automatikus injektor.
3.8.3. Integrált gáztalanító.
3.8.4. UV-VIS vagy infravörös detektor.
3.9. HPLC-oszlop (25 cm × 4 mm belső átmérő), szilikagél 60 használatával (5 μm-es részecskeméret).
3.10. 0,45 μm-es fecskendőszűrő.
3.11. 25 ml-es Erlenmeyer-lombik.
4. REAGENSEK
4.1. Kálium-hidroxid (minimális titer: 85 %).
4.2. Kálium-hidroxid megközelítőleg 2 M etanolos oldata.
130 g kálium-hidroxid (4.1.) hűtés közben feloldva 200 ml desztillált vízben, majd etanollal (4.9.) feltöltve egy liter mennyiségűre. Az oldatot jól lezárt, sötét színű üvegben kell tárolni, legfeljebb 2 napig.
4.3. Analízis céljára alkalmas minőségű etiléter.
4.4. Kálium-hidroxid megközelítőleg 0,2 M etanolos oldata.
13 g kálium-hidroxid (4.1.) feloldva 20 ml desztillált vízben, majd etanollal (4.9.) feltöltve egy liter mennyiségűre.
4.5. Szilikagél bevonatú, 0,25 mm vastagságú, 20 x 20 cm-es üveg tárgylemezek, fluoreszcens indikátor nélkül (kereskedelmi fogalomban használatra készen kapható).
4.6. Kromatográfia céljára alkalmas minőségű aceton.
4.7. Kromatográfia céljára alkalmas minőségű n-hexán.
4.8. Kromatográfia céljára alkalmas minőségű etiléter.
4.9. Analízis céljára alkalmas minőségű etanol.
4.10. Analízis céljára alkalmas minőségű etil-acetát.
4.11. Referenciaoldat a vékonyréteg-kromatográfiához: koleszterin, fitoszterinek, alkoholok és eritrodiol 5 %-os oldata etil-acetátban (4.10.)
4.12. 2,7-diklór-fluoreszcein 0,2 %-os oldata etanolban. Enyhén meglúgosítva néhány csepp 2 M alkoholos kálium-hidroxid-oldat (4.2.) hozzáadásával.
4.13. n-hexán (4.7.) és etiléter (4.8.) 65:35 térfogatarányú (V/V) elegye.
4.14. A HPLC mozgófázisa: n-hexán (4.7.) és etiléter (4.8.) 1:1 (V/V) arányban.
5. REFERENCIA-MÓDSZER: AZ ALKOHOLOS VEGYÜLETEK ELVÁLASZTÁSA BÁZISOS VÉKONYRÉTEG-KROMATOGRÁFIÁS LEMEZ HASZNÁLATÁVAL
A bázisos vékonyréteg-kromatográfiás lemezek előkészítése. Merítse vagy mártsa be a szilikagél bevonatú lemezeket (4.5.) 4 cm mélységben 0,2 M etanolos kálium-hidroxid-oldatba (4.4.) 10 másodpercig, majd hagyja őket füstszekrényben száradni két órán át, végül helyezze őket 100 °C hőmérsékletű kemencébe egy órára.
Vegye ki a lemezeket a kemencéből, és felhasználásig tegye őket kalcium-kloriddal működő szárítóberendezésbe (3.7.) (az így kezelt lemezeket 15 napon belül fel kell használni).
A hexán és az etiléter elegyét (4.13.) (3. megjegyzés) vezesse egy futtatókádba megközelítőleg 1 cm mélységben. Zárja le a kádat megfelelő fedéllel, és hagyja hűvös helyen legalább egy fél órán keresztül annak érdekében, hogy beálljon az egyensúly a gőz és a folyadék között. A kád belső felületein az eulensbe merülő szűrőpapírcsíkok helyezhetők el. Ezzel megközelítőleg egyharmadával csökkenthető a futtatási idő, és egyenletesebb lesz az összetevők eluálódása.
3. megjegyzés: A futtatóelegyet a tökéletesen reprodukálható elúciós körülmények megteremtése érdekében minden vizsgálatnál cserélni kell. Alternatív megoldásként használható n-hexán és etiléter 50:50 térfogatarányú (V/V) oldata is.
Az 1. részben foglaltaknak megfelelően előállított, el nem szappanosítható anyagból készítsen megközelítőleg 5 %-os etil-acetátos (4.10.) oldatot, és a 100 μl-es mikrofecskendő (3.3.) segítségével ebből az oldatból 0,3 ml-t vigyen fel egyenletes és vékony csíkban egy kromatográfiás lemez (4.5.) alsó részére (2 cm-re a szélétől). A csíkhoz igazodva vigyen fel 2-3 μl-t a megadott anyagok referenciaoldatából (4.11.) annak érdekében, hogy a szterin-, triterpén-dialkohol- és alkoholsávok azonosíthatók legyenek a futtatás után.
Helyezze a lemezt a futtatókádba (3.1.). A környezeti hőmérsékletet 15-20 °C között kell tartani (4. megjegyzés). Azonnal zárja le a kádat a fedéllel, és hagyja a lemezt eluálódni, amíg az oldószerfront el nem éri a lemez felső szélétől számított 1 cm távolságot. Ekkor vegye ki a lemezt a futtatókádból, és meleglevegő-áram segítségével vagy a lemezt rövid időre elszívófülkébe helyezve párologtassa el az oldószert.
4. megjegyzés: Magasabb hőmérséklet hatására romlik az elválasztás hatásfoka.
Kismértékben és egyenletesen permetezze be a lemezt 2,7-diklór-fluoreszcein-oldattal (4.12.), és hagyja megszáradni. A lemez ultraibolya lámpa (3.2.) alatti vizsgálatakor a szterin-, triterpén-dialkohol- és alkoholsávokat a referenciaoldat (4.11.) által hagyott foltok segítségével lehet elkülöníteni. A fluoreszkáló terület mentén fekete ceruzával jelölje be a sávok határait (lásd az 1. ábrán látható vékonyréteg-kromatográfiás lemezt).
A kijelölt területen található szilikagélt kaparja le egy fémspatulával. Az eltávolított és apróra zúzott anyagot tegye egy szűrőtölcsérbe (3.4.). Adjon hozzá 10 ml forró etil-acetátot (4.10.), és alaposan keverje össze a fémspatula segítségével, majd (szükség esetén vákuumban) szűrje le, a szűrletet gyűjtse össze a szűrőtölcsérhez csatlakoztatott Erlenmeyer-lombikban (3.5.).
A lombikban maradó anyagot mossa ki háromszor etiléterrel (4.3.) (minden alkalommal megközelítőleg 10 ml-rel), a szűrletet gyűjtse ugyanabba a tölcsérhez csatlakoztatott lombikba. Párolja be a szűrletet megközelítőleg 4-5 ml térfogatúra, és a maradék oldatot töltse egy 10 ml-es kémcsőbe (3.6.), amelynek súlyát előzőleg lemérte; enyhe melegítéssel, gyenge nitrogénáramban párologtatva szárítsa ki az oldatot, pótolja ki újra néhány csepp acetonnal (4.6.), majd párologtassa el újra a kiszáradásig. A kémcsőben maradt anyag a szterin- és a triterpén-dialkohol, illetve az alkoholok és a triterpén-alkoholok frakcióiból áll
6. AZ ALKOHOLOS FRAKCIÓ ELVÁLASZTÁSA NAGY TELJESÍTMÉNYŰ FOLYADÉKKROMATOGRÁFIÁVAL (HPLC)
Az 1. részben foglaltaknak megfelelően előállított, el nem szappanosítható anyagot oldja fel 3 ml mozgófázisban (4.14.), majd szűrje át az oldatot egy fecskendőszűrővel (3.10.), és tegye félre.
Fecskendezzen be 200 μl-t az el nem szappanosított anyag leszűrt oldatából a HPLC-rendszerbe (3.8.)
Futtassa a HPLC-elválasztást 0,8 ml/perc sebességgel, öntse ki az első 5 percben átáramlott mennyiséget, majd gyűjtse 25 ml-es Erlenmeyer-lombikokba (3.11.) az 5 és 10 perc közötti idősávban átáramló oldatot az alifás és a triterpén-alkoholok, aztán a 11 és 25 perc közötti idősávban átáramló oldatot a szterinek, az eritrodiol és az uvaol elválasztásához (5. megjegyzés).
Az elválasztás 210 nm-es hullámhosszra beállított UV-detektorral vagy törésmutató detektorral követhető nyomon (lásd a 6. ábrát).
A frakciókat párologtatással ki kell szárítani és elő kell készíteni a kromatográfiás elemzéshez.
5. megjegyzés: Gondosan felügyelje a HPLC-pumpa nyomását: az etiléter növelheti a nyomást, ezért az áramlást a nyomás megfelelő szinten tartása érdekében szükség szerint ki kell igazítani.
3. RÉSZ
AZ ALKOHOLOS VEGYÜLETEK FRAKCIÓINAK GÁZKROMATOGRÁFIÁS ELEMZÉSE
1. ALKALMAZÁSI KÖR
Ez a rész általános útmutatást ad az e módszer 2. részében ismertetett eljárás szerint elkülönített alkoholos vegyületek minőségi és mennyiségi összetételének kapilláris oszlopos gázkromatográfia alkalmazásával történő meghatározásához.
2. ALAPELV
Az el nem szappanosítható anyagból vékonyréteg-kromatográfiával vagy nagy teljesítményű folyadékkromatográfiával kigyűjtött frakciókat származékképzés útján trimetilszilil-éterekké alakítják át, majd megosztó (split) injektor és lángionizációs detektor használatával végzett kapilláris oszlopos gázkromatográfia alkalmazásával elemzik.
3. BERENDEZÉS
Hagyományos laboratóriumi felszerelés különös tekintettel a következőkre:
3.1. 10 ml-es, kúpos fenekű kémcső csiszolt üvegdugóval.
3.2. Megosztó injektoros kapilláris oszloppal használható gázkromatográf, amely a következőkből áll:
3.2.1. termosztatikus kamra az oszlopok számára (oszlopkemence), amely alkalmas arra, hogy a kívánt hőmérsékletet ± 1 °C pontossággal fenntartsa;
3.2.2. szabályozható hőmérsékletű befecskendező egység perszilanizált üvegből készült gőzölögtető elemmel és megosztásos (split) rendszerrel;
3.2.3. lángionizációs detektor (FID);
3.2.4. a lángionizációs detektorral (3.10.3.) használható, manuális integrációra alkalmas adatgyűjtő rendszer.
3.3. Olvasztott szilícium-dioxidból készült, 20-30 m hosszúságú, 0,25-0,32 mm belső átmérőjű, egyenletes 0,10-0,30 μm vastagságú, 5 % difenil- és 95 % dimetil-polisziloxán összetételű bevonattal ellátott (SE-52 vagy SE-54 vagy ezekkel egyenértékű állófázist tartalmazó) kapilláris oszlop.
3.4. 10 μl térfogatú mikrofecskendő gázkromatográfiához, megosztó injektáláshoz alkalmas cementált tűvel.
4. REAGENSEK
4.1. Kromatográfia céljára alkalmas minőségű vízmentes piridin.
4.2. Analízis céljára alkalmas minőségű hexametil-diszilazán.
4.3. Analízis céljára alkalmas minőségű trimetil-klórszilán.
4.4. Szterin-trimetilszilil-éterek referenciaoldatai. A felhasználáskor kell előállítani az azokat tartalmazó olajokból származó szterinekből és eritrodiolból.
4.5. C20-C28 alifás alkoholok trimetilszilil-étereinek standard oldatai. A felhasználáshoz tiszta alkoholok elegyeiből frissen készíthetők el.
4.6. Vivőgáz: gázkromatográfiás tisztaságú hidrogén vagy hélium.
4.7. Segédgázok: gázkromatográfiás tisztaságú hidrogén, hélium, nitrogén és levegő.
4.8. Szililező reagens piridin, hexametil-diszilazán és trimetil-klórszilán 9:3:1 (V/V/V) térfogatarányú elegyéből.
4.9. Kromatográfia céljára alkalmas minőségű n-hexán.
5. A TRIMETILSZILIL-ÉTEREK ELŐÁLLÍTÁSA
Töltse az alkoholos vegyület frakcióját tartalmazó kémcsőbe (3.1.) a szililező reagenst (4.8.) (6. megjegyzés), az alkoholos vegyület minden milligrammjához 50 μl-nyi mennyiségben, úgy, hogy az közben ne vegyen fel nedvességet (7. megjegyzés).
6. megjegyzés: A használatra kész oldatok kereskedelmi forgalomban kaphatók. Egyéb szililező reagensek - például a bisz-trimetilszilil-trifluor-acetamid + 1 % trimetil-klórszilán, amelyet azonos térfogatú vízmentes piridinnel kell hígítani - szintén kaphatók. A piridin helyettesíthető azonos mennyiségű acetonitrillel.
7. megjegyzés: Kismértékű opálosság kialakulhat; ez normális jelenség, nem okoz problémát. A fehér pelyhek képződése vagy a rózsaszín elszíneződés megjelenése nedvesség jelenlétét, illetve a reagens elhasználódását jelzi. Ilyen esetben a tesztet meg kell ismételni (kizárólag hexametil-diszilazán és trimetil-klórszilán elegyének használata esetén).
Zárja le a kémcsövet (3.1.), és óvatosan (felfordítás nélkül) rázza addig, amíg az összetevők teljesen fel nem oldódnak. Hagyja az oldatot legalább 15 percig szobahőmérsékleten állni, majd ezt követően centrifugálja néhány percig. A tiszta oldat készen áll a gázkromatográfiás elemzésre.
6. GÁZKROMATOGRÁFIÁS ELEMZÉS
6.1. Előzetes műveletek és az oszlop tömítése
A kapilláris oszlopot (3.3.) illessze be a gázkromatográfba úgy, hogy az oszlop bemenetét csatlakoztassa a megosztó injektorhoz, az oszlop kimenetét pedig a detektorhoz.
Végezze el a gázkromatográfiás berendezés általános ellenőrzését (gázszerelvények szivárgása, a detektor hatásfoka, a megosztásos rendszer és a regisztráló rendszer hatásfoka stb.).
Amennyiben első alkalommal használja az oszlopot, célszerű azt kondicionálni: vezessen át egy enyhe gázáramot a kapilláris oszlopon, majd kapcsolja be a gázkromatográfiás berendezést, és kezdje el fokozatosan melegíteni addig, amíg a hőmérséklet legalább 20 °C-kal meghaladja az üzemi hőmérsékletet (8. megjegyzés). Ezt a hőmérsékletet tartsa legalább két órán keresztül, majd helyezze az egész berendezést üzemi körülmények közé (a gázáramok és a megosztásos folyamat szabályozása, a láng begyújtása, csatlakoztatás az elektronikus íróhoz, az oszlop beszabályozása, a detektor és az injektor hőmérsékletének beállítása stb.), és rögzítse a jelet az analízis során tervezett legmagasabb szintnél legalább kétszer nagyobb érzékenységgel. Az alapvonalnak lineárisnak kell lennie, mindennemű csúcs és ingadozás nélkül. A negatív irányú, egyenes vonalú elmozdulás az oszlop csatlakozásainak tökéletlen tömítettségét jelzi, míg a pozitív irányú elmozdulás azt mutatja, hogy az oszlop kondicionálása nem megfelelő.
8. megjegyzés: A kondicionálás hőmérsékletének mindig legalább 20 °C-kal alacsonyabbnak kell lennie az alkalmazott állófázis esetében várható maximális hőmérsékletnél.
6.2. Üzemi körülmények
Optimalizálja a hőmérséklet programozását és a vivőgáz áramlását annak érdekében, hogy a 3-6. ábrán láthatóhoz hasonló kromatogramokat kapjon.
A következő paraméterek vizsgálat tárgyát képezték és hasznosnak bizonyultak:
6.2.1. Alifás alkoholok
| A kemence programozása | 180 °C (8 perc) → 260 °C (5 °C/perc) → 260 °C (15 perc) |
| Az injektor hőmérséklete | 280 °C |
| A detektor hőmérséklete | 290 °C |
| A vivőgáz lineáris sebessége | hélium (20–30 cm/s); hidrogén (30–50 cm/s) |
| Megosztási arány | 1:50 – 1:100 |
| Befecskendezett mennyiség | 0,5–1 μl TMSE-oldat |
6.2.2. Szterin és triterpén-dialkoholok
| A kemence programozása | 260 ± 5 °C, izotermikus |
| Az injektor hőmérséklete | 280–300 °C |
| A detektor hőmérséklete | 280–300 °C |
| A vivőgáz lineáris sebessége | hélium (20–30 cm/s); hidrogén (30–50 cm/s) |
| Megosztási arány | 1:50 – 1:100 |
| Befecskendezett mennyiség | 0,5–1 μl TMSE-oldat |
A fenti feltételek az oszlop és a gázkromatográf jellemzőinek megfelelően módosíthatók, hogy a kapott kromatogramok megfeleljenek a következő feltételeknek:
- A C26 alkohol retenciós idejének 18 ± 5 percnek kell lennie.
- A C22 alkoholcsúcsnak a teljes skálára vonatkozó érték 80 ± 20 %-ának kell lennie az olívaolaj esetében, és a teljes skálára vonatkozó érték 40 ± 20 %-ának kell lennie az olívapogácsa-olaj esetében.
- A β-szitoszterin-csúcs retenciós idejének 20 ± 5 percnek kell lennie.
- A kampeszterin-csúcsnak olívaolaj esetében (3 %-os átlagos tartalom mellett) a teljes skála 20 ± 5 %-ának kell lennie.
- Minden jelen lévő szterint el kell választani. A szétválasztáson túl a csúcsokat teljesen fel kell bontani, azaz a csúcs vonalának a következő csúcs kezdete előtt vissza kell térnie az alapvonalhoz. Azonban a nem teljes felbontás elfogadható abban az esetben, ha az 1,02 értékű relatív retenciós időhöz tartozó csúcs (szitosztanol) a függőleges segítségével kiszámítható.
6.3. Az elemzési eljárás
A 10 μl-es mikrofecskendővel (3.4.) szívjon fel 1 μl hexánt, majd 0,5 μl levegőt, ezt követően pedig 0,5-1 μl mintaoldatot. A mikrofecskendő dugattyúját felfelé mozgatva ürítse ki a tűt. A tűt vezesse be az injektor válaszfalán, majd 1-2 másodperc elmúltával fecskendezze be gyorsan az oldatot, és körülbelül 5 másodperc múlva lassan húzza ki a tűt. Automatikus injektor is alkalmazható.
Folytassa a rögzítést mindaddig, amíg a jelen lévő alkoholos vegyületek TMSE-elegyének elúciója teljesen végbe nem ment. Az alapvonalnak továbbra is meg kell felelnie a vonatkozó üzemi körülmények (6.2.1. vagy 6.2.2.) követelményeinek.
6.4. A csúcsok azonosítása
Az egyes csúcsok azonosítását a retenciós idők alapján, valamint az alifás és a triterpén-alkoholok, illetve a szterin és a triterpén-dialkoholok azonos körülmények között elemzett TMSE-elegyével összehasonlítva végezze. A 3. ábrán az alifás és a triterpén-alkoholok frakciójának kromatogramja látható, míg a szterinekre és a triterpén-dialkoholokra vonatkozó kromatogramokat a 2. ábra illusztrálja.
Az alifás alkoholok a következő sorrendben eluálódnak: C20-ol (belső standard), C22-ol, C23-ol, C24-ol, C25-ol, C26-ol, C27-ol és C28-ol.
A szterinek és a triterpén-dialkoholok a következő sorrendben eluálódnak: koleszterin, brasszikaszterin, ergoszterin, 24-metilén-koleszterin, kampeszterin, kampesztanol, sztigmaszterin, Δ7-kampeszterin, Δ5,23-sztigmasztadienol, kleroszterin, β-szitoszterin, szitosztanol, Δ5-avenaszterin, Δ5,24-sztigmasztadienol, Δ7-sztigmasztenol, Δ7-avenaszterin, eritrodiol és uvaol.
6.5. Mennyiségi értékelés
Az 1-eikozanol és a C22, C24, C26 és C28 alifás alkoholok csúcsterületeit adatgyűjtő rendszer használatával kell kiszámítani. Az 1-eikozanol választényezőjének értékét vegye 1-nek.
Az α-kolesztanol, valamint a szterin és a triterpén-dialkoholok csúcsainak területét az integrátor segítségével számítsa ki. Hagyja figyelmen kívül az 1. táblázat felsorolásában nem szereplő vegyületekhez tartozó csúcsokat (az ergoszterin esetében nem kell elvégezni a számítást). Az α-kolesztanol választényezőjének értékét vegye 1-nek.
Az egyes alkoholos vegyületek zsírban mért, mg/kg mértékegységben kifejezett koncentrációja a következő módon számítandó ki:

ahol:
Ax = az x alkoholos vegyület csúcsának területe a rendszer által használt mértékegységben.
As = az 1-eikozanol/α-kolesztanol csúcsának területe a rendszer által használt mértékegységben.
ms = a hozzáadott 1-eikozanol/α-kolesztanol tömege milligrammban.
m = a meghatározás céljára használt minta tömege grammban.
7. AZ EREDMÉNYEK KIFEJEZÉSE
Jegyezze fel az egyes alifás és triterpén-alkoholok zsírban mért koncentrációját mg/kg mértékegységben, illetve ezen értékek összegét mint "teljes alifásalkohol-tartalmat". A teljes tartalom a C22, C24, C26 és C28 alkoholok mennyiségének összege.
Az egyes alkoholos vegyületek összetételét egy tizedes pontosságig kell megadni.
A teljes szterinkoncentrációt tizedes nélkül kell megadni.
Az egyes szterinek koncentrációját a vonatkozó csúcs területének és az összes szterincsúcs területének hányadosából határozza meg:
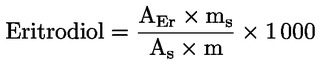
ahol:
Ax = az x szterin csúcsának területe.
ΣA = az összes szterincsúcs területe.
Látható β-szitoszterin: Δ5,23-sztigmasztadienol + kleroszterin + β-szitoszterin + szitosztanol + Δ5-avenaszterin + Δ5,24-sztigmasztadienol.
Számítsa ki az eritrodiol és az uvaol százalékos arányát:
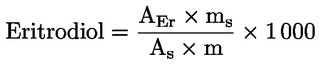
ahol:
AEr = az eritrodiol területe a rendszer által használt mértékegységben.
AUv = az uvaol területe a rendszer által használt mértékegységben.
Σ AT = a szterin + az eritrodiol + az uvaol összterülete a rendszer által használt mértékegységben.
Az egyes szterinek és triterpén-dialkoholok relatív százalékos arányának és a szterinek összkoncentrációjának a kiszámításán túlmenően az eritrodiol és az uvaol koncentrációját és ezek összegét is ki kell számítani (zsírban mért koncentráció mg/kg mértékegységben), a következőképpen fejezve ki az eredményeket:


ahol:
AEr = az eritrodiol csúcsának területe a rendszer által használt mértékegységben.
AUv = az uvaol területe a rendszer által használt mértékegységben.
As = az α-kolesztanol csúcsának területe a rendszer által használt mértékegységben.
ms = a hozzáadott α-kolesztanol tömege milligrammban.
m = a meghatározás céljára használt minta tömege grammban.
Függelék
| [Kép #28] | 1. Szénhidrogének 2. α-Tokoferol 3. Prenolok 4. Triterpén-alkoholok 5. Alifás alkoholok 6. Metil-szterinek 7. Szterinek 8. Triterpén-dialkoholok |
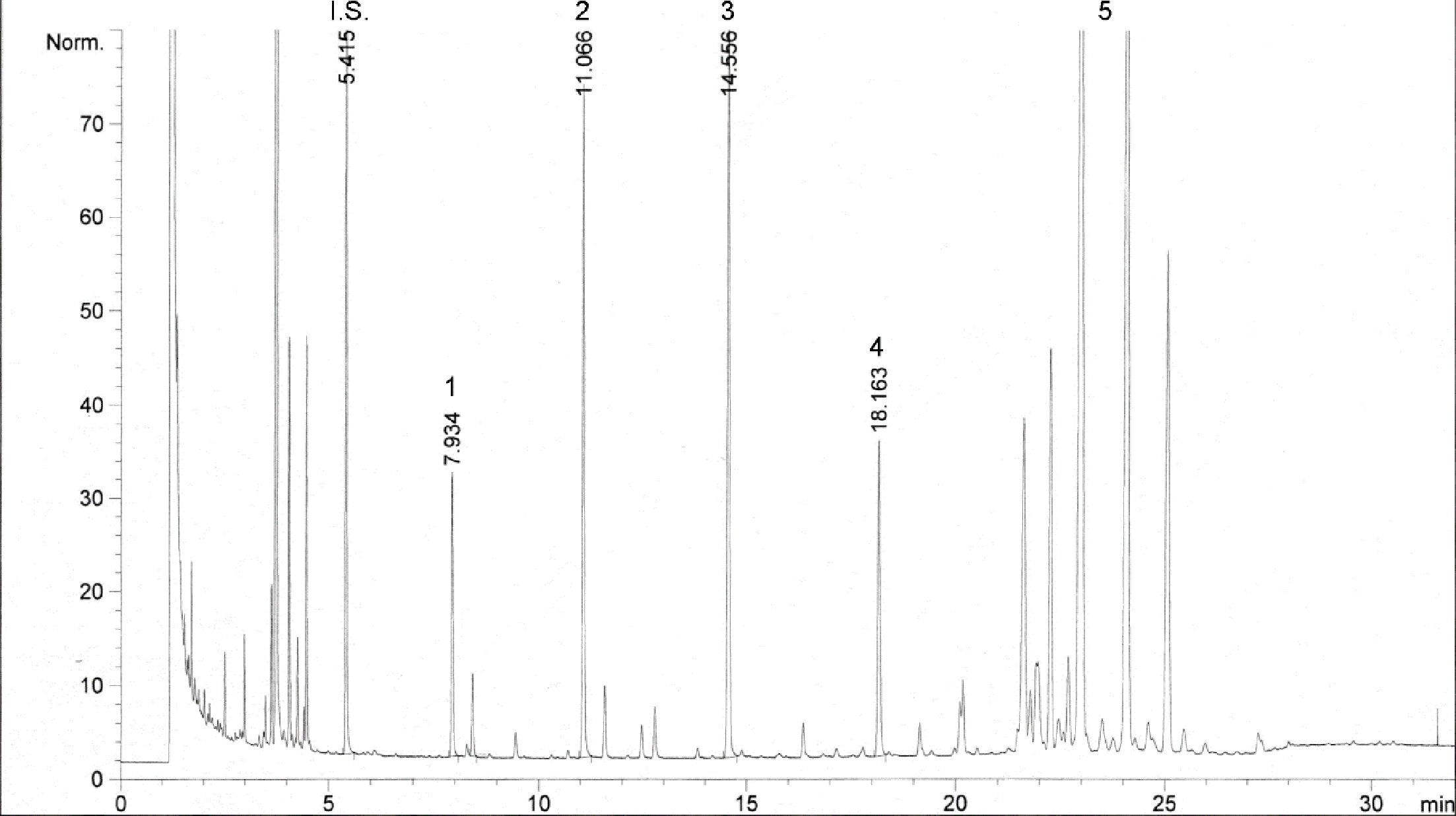
Kép #28
1. ábra - Az olívapogácsa-olaj hexán és dietil-éter (65:35 térfogatarányú) elegyével kétszer eluált, SO4H2 (50 %) alkalmazásával futtatott és melegített, el nem szappanosítható frakciójának vékonyréteg-kromatográfiája A lekaparandó sávok azok, amelyek a téglalapokon belül találhatók; az 1. téglalap az alifás alkoholokhoz tartozó sávokat, a 2. téglalap a szterinekhez és a triterpén-dialkoholokhoz tartozó sávokat tartalmazza.
I. táblázat - A szterinek relatív retenciós ideje
| Csúcs | Megnevezés | Relatív retenciós idő | ||
| SE 54 oszlop | SE 52 oszlop | |||
| 1. | Koleszterin | Δ-5-kolesztén-3ß-ol | 0,67 | 0,63 |
| 2. | Kolesztanol | 5α-kolesztán-3ß-ol | 0,68 | 0,64 |
| 3. | Brasszikaszterin | [24S]-24-metil-Δ-5,22-kolesztadién-3β-ol | 0,73 | 0,71 |
| * | Ergoszterin | [24S]-24-metil-Δ-5,7,22-kolesztatrién-3β-ol | 0,78 | 0,76 |
| 4. | 24-metilén-koleszterin | 24-metilén-Δ-5,24-kolesztadién-3ß-ol | 0,82 | 0,80 |
| 5. | Kampeszterin | (24R)-24-metil-Δ-5-kolesztén-3ß-ol | 0,83 | 0,81 |
| 6. | Kampesztanol | (24R)-24-metil-kolesztán-3ß-ol | 0,85 | 0,82 |
| 7. | Sztigmaszterin | (24S)-24-etil-Δ-5,22-kolesztadién-3ß-ol | 0,88 | 0,87 |
| 8. | Δ-7-kampeszterin | (24R)-24-metil-Δ-7-kolesztén-3ß-ol | 0,93 | 0,92 |
| 9. | Δ-5,23-sztigmasztadienol | (24R,S)-24-etil-Δ-5,23-kolesztadién-3ß-ol | 0,95 | 0,95 |
| 10. | Kleroszterin | (24S)-24-etil-Δ-5,25-kolesztadién-3ß-ol | 0,96 | 0,96 |
| 11. | ß-szitoszterin | (24R)-24-etil-Δ-5-kolesztén-3ß-ol | 1,00 | 1,00 |
| 12. | Szitosztanol | 24-etil-kolesztán-3ß-ol | 1,02 | 1,02 |
| 13. | Δ-5-avenaszterin | (24Z)-24-etilidén-Δ-kolesztén-3ß-ol | 1,03 | 1,03 |
| 14. | Δ-5,24-sztigmasztadienol | (24R,S)-24-etil-Δ-5,24-kolesztadién-3ß-ol | 1,08 | 1,08 |
| 15. | Δ-7-sztigmasztenol | (24R,S)-24-etil-Δ-7-kolesztén-3ß-ol | 1,12 | 1,12 |
| 16. | Δ-7-avenaszterin | (24Z)-24-etilidén-Δ-7-kolesztén-3ß-ol | 1,16 | 1,16 |
| 17. | Eritrodiol | 5α-oleán-12-én-3β,28-diol | 1,41 | 1,41 |
| 18. | Uvaol | Δ12-urszén-3β,28-diol | 1,52 | 1,52 |
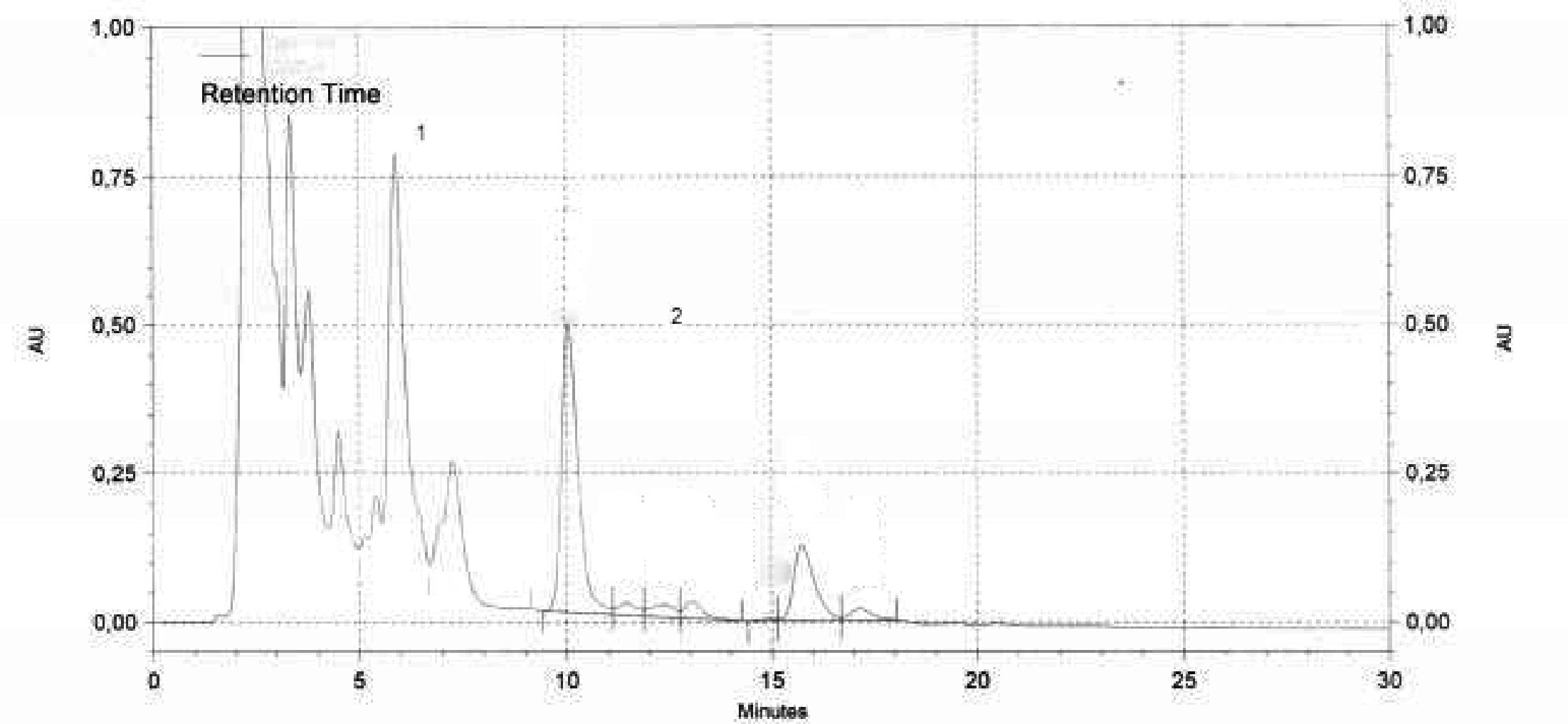
2. ábra - Finomított olívaolajból nyert szterin és triterpén-dialkoholok lángionizációs detektorral végzett gázkromatográfia szerinti profilja (a továbbiakban: GC-FID kromatográfiás profil). 1. koleszterin, 2. α-kolesztanol (belső standard), 3. 24-metilén-koleszterin, 4. kampeszterin, 5. kampesztanol, 6. sztigmaszterin, 7. Δ5,23-sztigmasztadienol, 8. kleroszterin, 9. β-szitoszterin, 10. szitosztanol, 11. Δ5-avenaszterin, 12. Δ5,24-sztigmasztadienol, 13. Δ7-sztigmasztenol, 14. Δ7-avenaszterin, 15. eritrodiol, 16. uvaol.

3. ábra - Lampante olívaolajból nyert szterin és triterpén-dialkoholok GC-FID kromatográfiás profilja. 1. koleszterin, 2. α-kolesztanol, 3. brasszikaszterin, 4. 24-metilén-koleszterin, 5. kampeszterin, 6. kampesztanol, 7. sztigmaszterin, 8. Δ7-kampeszterin, 9. Δ5,23-sztigmasztadienol, 10. kleroszterin, 11. β-szitoszterin, 12. szitosztanol, 13. Δ5-avenaszterin, 14. Δ5,24-sztigmasztadienol, 15. Δ7-sztigmasztenol, 16. Δ7-avenaszterin, 17. eritrodiol, 18. uvaol.

4. ábra - Az olívaolaj alifás alkoholjainak és triterpén-alkoholjainak GC-FID kromatográfiás profilja. (Belső standard) C20-ol, 1. C22-ol, 2. C24-ol, 3. C26-ol, 4. C28-ol, 5. triterpén-alkoholok.

5. ábra - Finomított olívaolaj és egy második centrifugáláson átesett olívaolaj alifás alkoholjainak és triterpén-alkoholjainak GC-FID kromatográfiás profilja. (Belső standard) C20-ol, 1. C22-ol, 2. C24-ol, 3. C26-ol, 4. C28-ol, 5. triterpén-alkoholok.

6. ábra - El nem szappanosítható, UV-detektor alkalmazásával végzett HPLC-vel szétválasztott olívaolaj HPLC-kromatogramja. 1. alifás és triterpén-alkoholok; 2. szterinek és triterpén-dialkoholok.
XX. MELLÉKLET
A viasz-, valamint a zsírsav-metilészter-és zsírsav-etilészter-tartalom kapilláris gázkromatográfiával történő meghatározásának módszere
1. A MÓDSZER CÉLJA
A módszer célja az olívaolaj viasz-, zsírsav-metilészter- és zsírsav-etilészter-tartalmának meghatározása. Az egyes viaszok és alkilészterek elválasztása a szénatomszám alapján történik. Javasolt a módszer alkalmazása az olívaolaj és az olívapogácsa-olaj megkülönböztetésére, valamint az extra szűz olívaolaj minőségének meghatározására, mivel alkalmas az extra szűz olívaolajból és alacsonyabb minőségű olajból - legyen az szűz, lampante vagy valamely szagtalanított olaj - megtévesztő szándékkal létrehozott keverékek azonosítására.
2. ALAPELV
Megfelelő belső standardok hozzáadása az olajhoz, majd frakcionálás kromatográfiás eljárás segítségével hidratáltszilikagél-oszlopon. A vizsgálati körülmények között eluált (a triacilglicerineknél kisebb polaritású) frakció leválasztása, majd kapilláris gázkromatográfia segítségével történő közvetlen elemzése.
3. ESZKÖZÖK
3.1. 25 ml-es térfogatú Erlenmeyer-lombik,
3.2. 15,0 mm belső átmérőjű, 30-40 cm hosszú folyadékkromatográfiás üvegoszlop megfelelő csappal,
3.3. kapillárisoszloppal történő használatra alkalmas gázkromatográf, közvetlenül az oszlopra történő injektálásra alkalmas rendszerrel, amely a következő részekből áll:
3.3.1. termosztátvezérlésű kemence hőmérséklet-programozással,
3.3.2. közvetlenül az oszlopra történő injektálásra alkalmas hideg befecskendező rendszer,
3.3.3. lángionizációs detektor és konverter-erősítő,
3.3.4. regisztráló-integráló berendezés (1. megjegyzés) a konverter-erősítővel (3.3.3. pont) történő használatra, melynek válaszideje nem haladja meg az egy másodpercet, és változtatható papírsebességű,
1. megjegyzés: Lehetséges olyan informatizált rendszerek használata is, amelyek alkalmasak a gázkromatográfiai adatok számítógépes feldolgozására.
3.3.5. ömlesztett szilícium-dioxidból készült, 8-12 m hosszúságú, 0,25-0,32 mm belső átmérőjű, belülről egyenletesen 0,10-0,30 μm rétegvastagságú folyadékfázissal (2. megjegyzés) borított (a viaszok, zsírsav-metilészterek és zsírsav-etilészterek elemzésére szolgáló) kapillárisoszlop,
2. megjegyzés: E célra használhatók a kereskedelmi forgalomban lévő megfelelő folyadékfázisok, mint az SE52, SE54 stb.
3.4. 10 μl térfogatú mikrofecskendő közvetlenül az oszlopra történő injektáláshoz, keményített tűvel,
3.5. elektromos rázóberendezés,
3.6. rotációs bepárló,
3.7. tokos kemence,
3.8. analitikai mérleg, ± 0,1 mg-os mérési pontossággal,
3.9. szokványos laboratóriumi üvegeszközök.
4. REAGENSEK
4.1. szilikagél, 60-200 μm mesh. Helyezze a szilikagélt az 500 °C hőmérsékletű tokos kemencébe legalább négy óra hosszára. Hagyja lehűlni, majd adjon hozzá a felhasznált szilikagél mennyisége 2 %-ának megfelelő vízmennyiséget. Alaposan rázza fel a sűrű szuszpenziót, hogy homogénné váljék, majd használat előtt legalább 12 órán keresztül tartsa a szárítóberendezésben.
4.2. n-hexán, kromatográfia vagy maradékvizsgálat céljára alkalmas. A hexán helyettesíthető izooktánnal (kromatográfia céljára alkalmas minőségű 2,2,4-trimetil-pentán), feltéve, hogy összehasonlítható pontossági értékek érhetők el. Az n-hexán forráspontjánál magasabb forráspontú oldószerek hosszabb idő alatt párolognak el. A hexán toxicitása miatt azonban előnyben kell részesíteni őket. A tisztaságot ellenőrizni kell, például a 100 ml oldószer elpárologtatása után visszamaradó anyag vizsgálata útján.
FIGYELEM! Gőzei a levegővel keveredve gyúlékonyak! A vizsgálathoz távolítson el minden gyújtóforrást (hőforrás, szikra, nyílt láng)! Gondoskodjon róla, hogy az üvegek mindig jól le legyenek zárva! Használat során gondoskodjon megfelelő szellőzésről! Előzze meg a gőzfelhalmozódást, és távolítson el minden tűzveszélyes tárgyat, például a nem éghetetlen anyagokból készült fűtőberendezéseket és elektromos készülékeket! Az anyag veszélyes, belélegezve idegsejt-károsodást okozhat. Ne lélegezze be a gőzt! Szükség esetén használjon megfelelő lélegeztető berendezést! Szembe és bőrre ne kerüljön!
Az izooktán gyúlékony, tűzveszélyes folyadék. Robbanási határértékei a levegőben 1,1 % és 6,0 % között változnak (térfogatszázalék). Lenyelve és belélegzés útján mérgező. Ezen oldószer alkalmazása során használjon jó üzemállapotban lévő elszívófülkét!
4.3. Etil-éter, kromatográfia céljára alkalmas. FIGYELEM! Kiemelten tűzveszélyes és közepesen toxikus anyag! Bőrrel érintkezve irritációt okoz. Belégzése káros. Szemkárosodást okozhat. Késleltetett káros hatásokat okozhat. Robbanó peroxidokat képezhet. Gőzei a levegővel keveredve gyúlékony elegyet alkothatnak. A vizsgálathoz távolítson el minden gyújtóforrást (hőforrás, szikra, nyílt láng)! Győződjön meg róla, hogy az üvegek mindig jól le legyenek zárva! Használat során gondoskodjon megfelelő szellőzésről! Előzze meg a gőzfelhalmozódást és távolítson el minden tűzveszélyes tárgyat, pl. a nem éghetetlen anyagokból készült fűtőberendezéseket és elektromos készülékeket! Ne párolja szárazra, illetve majdnem teljesen szárazra! Víz vagy más megfelelő redukálószer hozzáadásával mérsékelhető a peroxidképződés. Ne igya meg az anyagot! Ne lélegezze be a gőzt! Kerülje a hosszan tartó vagy ismételt bőrkontaktust!
4.4. n-heptán, kromatográfia céljára alkalmas, vagy izo-oktán.
FIGYELEM! Tűzveszélyes anyag! Belégzése káros. A vizsgálathoz távolítson el minden gyújtóforrást (hőforrás, szikra, nyílt láng)! Győződjön meg róla, hogy az üvegek mindig jól le legyenek zárva! Használat során gondoskodjon megfelelő szellőzésről! Ne lélegezze be a gőzt! Kerülje a hosszan tartó vagy ismételt bőrkontaktust!
4.5. Lauril-arachidát 0,05 % (m/v) standard oldata (3. megjegyzés) heptánban (belső standard viaszhoz).
3. megjegyzés: Palmitil-palmitát, mirisztil-sztearát és arachidil-laureát egyaránt használható.
4.6. Metil-heptadekanoát 0,02 % (m/v) standard oldata heptánban (belső standard metil- és etilészterekhez).
4.7. Szudán 1 (1-fenilazo-2-naftol).
4.8. Vivőgáz: hidrogén vagy hélium, gázkromatográfia céljára alkalmas tisztaságban.
FIGYELMEZTETÉS
Hidrogén. Nyomás alatt kiemelten tűzveszélyes. A vizsgálathoz távolítson el minden gyújtóforrást (hőforrás, szikra, nyílt láng) és a nem éghetetlen anyagokból készült elektromos készülékeket! Győződjön meg róla, hogy a használaton kívüli palack szelepe zárva legyen! Mindig használjon nyomáscsökkentőt! Mielőtt kinyitná a szelepet, csökkentse a nyomáscsökkentő rugójának feszességét! A szelep nyitásakor ne álljon a kieresztőnyílás elé! Használat során gondoskodjon megfelelő szellőzésről! Ne vezessen hidrogént egyik palackból a másikba! Ne keverje a palackban lévő gázt! Gondoskodjon róla, hogy a palack ne borulhasson fel! Ne tegye ki a palackot napfénynek vagy egyéb hőforrásnak! Tartsa a palackot rozsdamentes környezetben! Ne használjon sérült vagy címke nélküli palackot!
Hélium. Nagy nyomáson sűrített gáz. Csökkenti a belélegezhető oxigén koncentrációját. Tartsa zárva a palackot! Használat során gondoskodjon megfelelő szellőzésről! Nem megfelelő szellőzés esetén ne menjen a tárolótérbe! Mindig használjon nyomáscsökkentőt! Mielőtt kinyitná a szelepet, csökkentse a nyomáscsökkentő rugójának feszességét! Ne vezessen gázt egyik palackból a másikba! Gondoskodjon róla, hogy a palack ne borulhasson fel! A szelep nyitásakor ne álljon a kieresztőnyílás elé! Ne tegye ki a palackot napfénynek vagy egyéb hőforrásnak! Tartsa a palackot rozsdamentes környezetben! Ne használjon sérült vagy címke nélküli palackot! Ne lélegezze be a gázt! Kizárólag ipari céllal használható fel.
4.9. Segédgázok
:
- Hidrogén, gázkromatográfia céljára alkalmas tisztaságban.
- Levegő, gázkromatográfia céljára alkalmas tisztaságban.
FIGYELMEZTETÉS
Levegő. Nagy nyomáson sűrített gáz. Éghető anyagok jelenlétében óvatosan kezelendő, mivel nagy nyomáson a levegőben található legtöbb szerves vegyület öngyulladási hőmérséklete jelentősen csökken. Győződjön meg róla, hogy a használaton kívüli palack szelepe zárva legyen! Mindig használjon nyomáscsökkentőt! Mielőtt kinyitná a szelepet, csökkentse a nyomáscsökkentő rugójának feszességét! A szelep nyitásakor ne álljon a kieresztőnyílás elé! Ne vezessen gázt egyik palackból a másikba! Ne keverje a palackban lévő gázt! Gondoskodjon róla, hogy a palack ne borulhasson fel! Ne tegye ki a palackot napfénynek vagy egyéb hőforrásnak! Tartsa a palackot rozsdamentes környezetben! Ne használjon sérült vagy címke nélküli palackot! Az ipari felhasználásra szánt levegőt nem szabad belélegezni és lélegeztető berendezésekben alkalmazni.
5. ELJÁRÁS
5.1. A kromatográfiás oszlop előkészítése
Készítsen szuszpenziót 15 g szilikagélből (4.1. pont) és n-hexánból (4.2. pont), és töltse az oszlopba (3.2. pont). Hagyja magától leülepedni. Teljes ülepedés után elektromos rázóberendezéssel tömörítse, hogy homogénebb kromatográfiás réteget kapjon. Az esetleges szennyeződések eltávolítása érdekében 30 ml n-hexánnal mossa át az oszlopot. Az analitikai mérleg (3.8. pont) segítségével mérjen pontosan 500 mg mintát a 25 ml térfogatú lombikba (3.1. pont), majd a feltételezett viasztartalom függvényében adjon hozzá megfelelő mennyiségű belső standardot (4.5. pont), azaz adjon hozzá olívaolaj esetén 0,1 mg, olívapogácsa-olaj esetén 0,25-0,5 mg lauril-arachidátot, az olívaolajokhoz pedig 0,05 mg metil-heptadekanoátot (4.6. pont).
Az ily módon előkészített mintát 2 x 2 ml n-hexán (4.2. pont) segítségével vigye fel a kromatográfiás oszlopra.
Hagyja lefutni az oldatot 1 mm-rel az abszorbens felszíne fölé. Ismét mossa át az oszlopot 99:1 arányú n-hexán/etil-éter eleggyel, és gyűjtsön össze 220 ml-t úgy, hogy 10 másodpercenként megközelítőleg 15 csepp folyjon át. (Az így kapott frakció tartalmazza a metil- és etilésztereket és viaszokat.) (4. megjegyzés) (5. megjegyzés).
4. megjegyzés: A 99:1 arányú n-hexán/etil-éter keveréket minden nap frissen kell elkészíteni.
5. megjegyzés: A viaszok megfelelő eluálódásának vizuális ellenőrzése érdekében a mintaoldathoz hozzáadhat 100 μl szudán 1 színezőanyagot, az eluáló elegy 1 %-a arányában.
A színezőanyag retenciós ideje a viaszoké és a triacilglicerineké között helyezkedik el. Ezért amikor a színezőanyag eléri a kromatográfiás oszlop alját, függessze fel az eluálást, ekkorra ugyanis valamennyi viasz eluálódott.
Az így kapott frakciókat a rotációs bepárlóban párolja szárazra, amíg az oldószer majdnem teljesen eltűnik belőle. Az oldószer utolsó 2 ml-ét gyenge nitrogénárammal távolítsa el. Gyűjtse össze a metil- és etilésztereket tartalmazó frakciót 2-4 ml n-heptánnal vagy izo-oktánnal hígítva.
5.2. Gázkromatográfiás elemzés
5.2.1. Előzetes műveletek
Az oszlopot illessze be a gázkromatográfba (3.3. pont) úgy, hogy az oszlop bemenetét az oszlopra szerelt ("on-column") rendszerhez, az oszlop kimenetét pedig a detektorhoz csatlakoztatja. Ellenőrizze a gázkromatográfiás berendezést (gázszerelvények szorossága, a detektor hatékonysága, a regisztráló rendszer hatékonysága stb.).
Az első alkalommal használt kapillárisoszlopokat ajánlatos kondicionálni. Fúvasson át egy kevés gázt az oszlopon, majd kapcsolja be a gázkromatográfiás berendezést. Melegítse fokozatosan addig, amíg körülbelül 4 óra elteltével el nem éri a 350 °C hőmérsékletet.
Ezt a hőmérsékletet tartsa legalább két órán keresztül, majd hozza a berendezést üzemi körülmények közé (gázáram szabályozása, bontóláng begyújtása, csatlakoztatás az elektronikus regisztráló berendezéshez [3.3.4 pont], az oszlophőmérséklet szabályozása a kemencén, a detektor szabályozása stb.). Rögzítse a jelet az elemzés elvégzéséhez szükségesnél legalább kétszer nagyobb érzékenység mellett. Az alapvonalnak lineárisnak, mindennemű csúcstól és drifttől mentesnek kell lennie.
A negatív egyenes vonalú drift az oszlop illesztékeinek tökéletlenségét, míg a pozitív drift az oszlop nem megfelelő kondicionálását jelzi.
5.2.2. Az üzemi körülmények megválasztása a viaszok, valamint a metil- és etilészterek esetében (6. megjegyzés)
Az üzemi körülmények általában az alábbiak:
- Oszlophőmérséklet :
20 °C/perc 5 °C/perc
kiindulási hőmérséklet 80 °C (1′) [Kép #29] 140 °C [Kép #30] 335 °C (20)

Kép #29
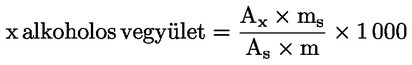
Kép #30
- Detektor hőmérséklete : 350 °C.
- Befecskendezett anyag mennyisége : 1 μl n-heptán-oldat (2-4 ml).
- Vivőgáz : hélium vagy hidrogén, a kiválasztott gáz számára optimális lineáris sebességgel (lásd az A. függeléket).
- Berendezés érzékenysége : a fenti körülményeknek megfelelően.
6. megjegyzés: Tekintettel a magas véghőmérsékletre, pozitív drift előfordulása megengedhető, amely azonban nem haladhatja meg a teljes skálaérték 10 %-át.
A fenti feltételek az oszlop és a gázkromatográf jellemzőinek megfelelően módosíthatók, hogy a kapott kromatogramok lehetővé tegyék valamennyi viasz, illetve zsírsav-metilészter és zsírsav-etilészter leválasztását, valamint hogy a csúcsok kielégítően elkülöníthetőek legyenek (lásd a 2., 3. és 4. ábrát), miközben a lauril-arachidát belső standard retenciós ideje 18 ± 3 perc. A viaszok legreprezentatívabb csúcsának a teljes skálaérték 60 %-ánál nagyobbnak kell lennie, míg a metil- és etilészterek esetében a metil-heptadekanoát belső standardnak el kell érnie a teljes skálaértéket.
A csúcsok integrálási paramétereit úgy kell meghatározni, hogy lehetővé váljon az érintett csúcsok alatti területek pontos becslése.
5.3. Az elemzés menete
A 10 μl térfogatú mikrofecskendő segítségével szívjon fel 10 μl oldatot, majd a mikrofecskendő dugattyúját felfelé mozgatva ürítse ki a tűt. A tűt vezesse be a befecskendező rendszerbe, majd egy-két másodperc elmúltával gyorsan fecskendezze be az oldatot. Körülbelül öt másodperc elteltével finoman húzza ki a tűt.
Addig folytassa a rögzítést, ameddig a viaszok vagy sztigmasztadiének teljesen eluálódtak, attól függően, hogy melyik frakció analízisét végzi.
Az alapvonalnak minden esetben meg kell felelnie az előírt feltételeknek.
5.4. A csúcsok azonosítása
Az egyes csúcsok azonosítását a retenciós idő alapján, az azonos körülmények között analizált, ismert retenciós idejű viaszkeverékekkel összehasonlítva kell végezni. Az alkilészterek azonosítása az olívaolajokban található főbb zsírsavak (palmitinsav és olajsav) metil- és etilésztereinek keverékei alapján történik.
Az 1. ábrán egy szűz olívaolaj viaszkromatogramja látható. A 2. és a 3. ábrán két, kiskereskedelmi forgalomban kapható extra szűz olívaolaj kromatogramja látható, metil- és etilészterekkel, illetve azok nélkül. A 4. ábra egy csúcsminőségű extra szűz olívaolaj és egy 20 %-ban szagtalanított olajjal kevert ugyanilyen olaj kromatogramját mutatja.
5.5. A viasztartalom mennyiségi értékelése
Az integráló berendezés segítségével határozza meg a lauril-arachidát belső standardnak és a nyílt szénláncú C40-C46-os észtereknek megfelelő csúcsok alatti területet.
A teljes viasztartalom mg/kg zsír alakban történő meghatározásához adja össze az egyes viaszok mennyiségét, az alábbiak szerint:
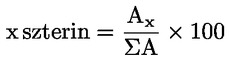
ahol
Ax = az egyes észterek csúcsa alatti terület, a számítógép által használt mértékegységben
As = a lauril-arachidát belső standard csúcsa alatti terület, a számítógép által használt mértékegységben
ms = a hozzáadott lauril-arachidát belső standard tömege milligrammban
m = a meghatározni kívánt minta tömege grammban.
5.5.1. A metil- és etilészterek mennyiségi értékelése
Az integráló berendezés segítségével határozza meg a metil-heptadekanoát belső standardnak, a C16-os és a C18-os zsírsavak metilésztereinek és etilésztereinek megfelelő csúcsok alatti területet.
Határozza meg az egyes alkilészterek mennyiségét mg/kg zsír alakban, az alábbiak szerint:

ahol
Ax = az egyes C16-os és C18-as észterek csúcsa alatti terület, a számítógép által használt mértékegységben
As = a metil-heptadekanoát belső standard csúcsa alatti terület, a számítógép által használt mértékegységben
ms = a hozzáadott metil-heptadekanoát belső standard tömege milligrammban
m = a meghatározni kívánt minta tömege grammban.
6. AZ EREDMÉNYEK KIFEJEZÉSE
A különböző C40-C46-os viasztartalmak összegét (7. megjegyzés) mg/kg zsír alakban tüntesse fel.
Tüntesse fel a teljes C16-C18-as metilészter- és etilészter-tartalmat, valamint a kettő összegét.
Az eredményeket a közelebbi mg/kg értékre kell kerekíteni.
7. megjegyzés: A meghatározandó mennyiségek a páros szénatomszámú C40-C46-os észtereknek megfelelő csúcsoknál leolvasható értékek, ahogy azt a mellékelt ábrán látható kromatogramminta (az olívaolaj viasztartalma) mutatja. Amennyiben a C46-észter duplán jelenik meg, javasolt helyette egy olívapogácsa-olaj viasztartalmát elemezni, ahol a C46 túlsúlya miatt a megfelelő csúcs könnyen azonosítható.
Tüntesse fel a jelen lévő etil- és metilészterek arányát.
1. ábra
Gázkromatogram-minta egy olívaolaj viaszfrakciójáról ( 6 )
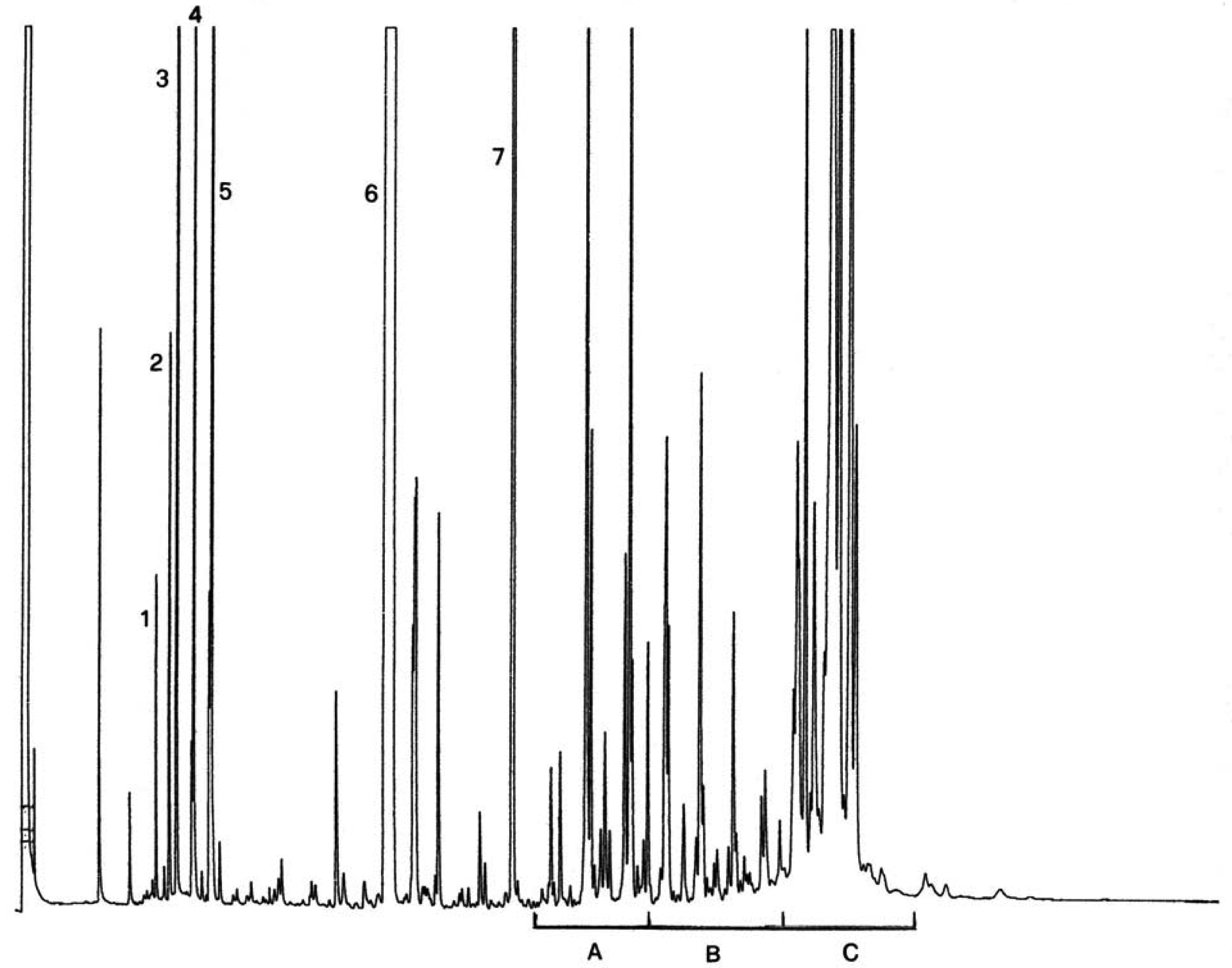
Jelmagyarázat:
Az 5-8 perc retenciós idejű zsírsav-metilészterek és zsírsav-etilészterek csúcsai
I. S. = lauril-arachidát
1 = diterpén észterek
2+2′ = C40-es észterek
3+3′ = C42-es észterek
4+4′ = C44-es észterek
5 = C46-os észterek
6 = szterinészterek és triterpén alkoholok
2. ábra
Egy szűz olívaolajban található metilészterek, etilészterek és viaszok

Jelmagyarázat:
1 - metil C16
2 - etil C16
3 - metil-heptadekanoát
4 - metil C18
5 - etil C18
6 - szkvalén
7 - lauril-arachidát I.S.
A - diterpén észterek
B - viaszok
C - szterinészterek és triterpén észterek
3. ábra
Egy extra szűz olívaolajban található metilészterek, etilészterek és viaszok

Jelmagyarázat:
1 - metil-heptadekanoát
2 - metil C18
3 - etil C18
4 - szkvalén
5 - lauril-arachidát I.S.
A - diterpén észterek
B - viaszok
C - szterinészterek és triterpén észterek
4. ábra
Egy extra szűz olívaolaj és egy szagtalanított olajjal kevert ugyanilyen olaj kromatogramjának részlete

Jelmagyarázat:
1 - metil-mirisztát I.S.
2 - metil-palmitát
3 - etil-palmitát
4 - metil-heptadekanoát I.S.
5 - metil-linoleát
6 - metil-oleát
7 - metil-sztearát
8 - etil-linoleát
9 - etil-oleát
10 - etil-sztearát
A. függelék
A gáz lineáris sebességének meghatározása
Fecskendezzen 1-3 μl metánt (vagy propánt) a normál üzemi körülményekre beállított gázkromatográfiás berendezésbe. Mérje meg a gáz oszlopon történő átáramlásának idejét a befecskendezés pillanatától a csúcs kiemelkedésének pillanatáig (tM).
A lineáris sebességet (cm/mp) az L/tM képlettel kell meghatározni, ahol L az oszlop cm-ben megadott hosszúsága, tM pedig a másodpercben mért idő.
XXI. MELLÉKLET
A 8. cikk (2) bekezdésében említett, olívaolajokon végzett megfelelőségi ellenőrzések eredményei
| Címkézés | Kémiai paraméterek | Érzékszervekkel meghatározható jellemzők (4) | Végkövetkeztetések | ||||||||||||||
| Minta | Kategória | Származási ország | Az ellenőrzés helyszíne (1) | Hivatalos név | Eredetmegjelölés | Tárolási feltételek | Hibás információ | Olvashatóság | M/NM (3) | Határértékeken kívül eső paraméterek I/N | Ha igen, melyek ezek? (2) | M/NM (3) | Hibamedián | Gyümölcsösségi medián | M/NM (3) | Szükséges intézkedés | Szankció |
| (1) Belső piac (sajtoló, palackozók, kiskereskedelmi fázis), export, import. (2) Az olívaolaj I. mellékletben felsorolt jellemzőinek mindegyikéhez tartozik egy kód. (3) Megfelelő/nem megfelelő. (4) Olívaolaj és pogácsaolaj esetében nem szükséges megadni. | |||||||||||||||||
( 1 ) HL L 228., 2009.9.1., 3. o.
( 2 ) Az Európai Parlament és a Tanács 2011/91/EU irányelve (2011. december 13.) azon árutételt azonosító jelzésekről és jelölésekről, amelyekhez az adott élelmiszer tartozik (HL L 334., 2011.12.16., 1. o.).
( 3 ) A szterinészterek eluálódása után a kromatográfiás vonalon nem mutatkozhatnak jelentős csúcsok (trigliceridek).
( 4 ) Az Európai Parlament és a Tanács 2013. december 17-i 1308/2013/EU rendelete a mezőgazdasági termékpiacok közös szervezésének létrehozásáról és a 922/72/EGK, a 234/79/EK, az 1037/2001/EK és az 1234/2007/EK tanácsi rendelet hatályon kívül helyezéséről (HL L 347., 2013.12.20., 671. o.).
( 5 ) Visszautasíthatják az olaj megkóstolását, ha rendkívül intenzíven érzékelnek egy vagy több negatív tulajdonságot; ebben az esetben ezt a rendkívüli körülményt fel kell jegyezniük az értékelőlapra.
( 6 ) A szterinészterek eluálódása után a kromatográfiás vonalon nem mutatkozhatnak jelentős csúcsok (triacilglicerinek).
Lábjegyzetek:
[1] A dokumentum eredetije megtekinthető CELEX: 31991R2568 - https://eur-lex.europa.eu/legal-content/HU/ALL/?uri=CELEX:31991R2568&locale=hu Utolsó elérhető, magyar nyelvű konszolidált változat CELEX: 01991R2568-20191020 - https://eur-lex.europa.eu/legal-content/HU/ALL/?uri=CELEX:01991R2568-20191020&locale=hu