
32021R0808[1]
A Bizottság (EU) 2021/808 végrehajtási rendelete (2021. március 22.) az élelmiszer-termelő állatokon alkalmazott gyógyszerhatóanyagok maradékanyagaira vonatkozó analitikai módszerek elvégzéséről és az eredmények értelmezéséről, valamint az alkalmazandó mintavételi módszerekről és a 2002/657/EK és 98/179/EK határozatok hatályon kívül helyezéséről
A BIZOTTSÁG (EU) 2021/808 VÉGREHAJTÁSI RENDELETE
(2021. március 22.)
az élelmiszer-termelő állatokon alkalmazott gyógyszerhatóanyagok maradékanyagaira vonatkozó analitikai módszerek elvégzéséről és az eredmények értelmezéséről, valamint az alkalmazandó mintavételi módszerekről és a 2002/657/EK és 98/179/EK határozatok hatályon kívül helyezéséről
(EGT-vonatkozású szöveg)
1. cikk
Tárgy és hatály
Ez a rendelet az (EU) 2022/1646 bizottsági végrehajtási rendelet ( 1 ) 3. cikkében meghatározott nemzeti tervek keretében szabályokat állapít meg a gyógyszerhatóanyagok maradékanyagaival kapcsolatos mintavételhez és laboratóriumi elemzésekhez használt elemzési módszereket illetően. Szabályokat állapít meg továbbá a laboratóriumi elemzések analitikai eredményeinek értelmezéséhez.
E rendelet a gyógyszerhatóanyagok maradékanyagainak előfordulásával kapcsolatos követelményeknek való megfelelés vizsgálatát célzó hatósági ellenőrzésekre alkalmazandó.
2. cikk
Fogalommeghatározások
E rendelet alkalmazásában az (EU) 2019/2090 felhatalmazáson alapuló bizottsági rendelet ( 2 ) 2. cikkében, az (EU) 2019/1871 bizottsági rendeletben ( 3 ), a 470/2009/EK európai és parlamenti és tanácsi rendelet ( 4 ) 2. cikkében és a 315/93/EGK tanácsi rendeletben ( 5 ) szereplő fogalommeghatározások érvényesek.
Ezenkívül a következő fogalommeghatározásokat is alkalmazni kell:
1. "abszolút visszanyerés": egy analitra vonatkozóan az analitikai folyamat utolsó szakaszának hozama az analit eredeti minta szerinti mennyiségével osztva és százalékban kifejezve;
2. "pontosság": egy vizsgálati eredmény és az elfogadott valódi referenciaérték közötti egyezés közelisége, a valódiság és precizitás becslésével meghatározva ( 6 );
3. "alfa (α) hiba": annak valószínűsége, hogy a vizsgált minta valójában megfelelt, annak ellenére, hogy nem megfelelő mérési eredmény született;
4. "analit": valamely rendszer elemzendő komponense;
5. "engedélyezett anyag": a 2001/82/EK európai parlamenti és tanácsi irányelvvel ( 7 ) összhangban az élelmiszer-termelő állatokon való alkalmazásra engedélyezett gyógyszerhatóanyag;
6. "béta (β) hiba": annak valószínűsége, hogy a vizsgált minta valójában nem megfelelő, annak ellenére, hogy megfelelő mérési eredmény született;
7. "torzítás": a vizsgálati eredmény becsült értéke és egy elfogadott referenciaérték különbözete;
8. "kalibráló standard": visszavezethető mérési referencia, amely egy referenciabázishoz kötött módon jeleníti meg a kérdéses anyag mennyiségének értékét;
9. "hiteles anyagminta (CRM)": egy érvényes eljárásokat alkalmazó felhatalmazott szerv által kiadott dokumentációval kísért és bizonytalanságokkal és nyomonkövethetőséggel társuló egy vagy több meghatározott tulajdonságot mutató referenciaanyag ( 8 );
10. "párhuzamos kromatográfia": technika, amelynek során egy vagy több ismert vegyület mellett egy ismeretlen anyagot visznek fel a kromatográfiás hordozóra azt feltételezve, hogy az ismert és ismeretlen anyagok egymáshoz viszonyított viselkedése segíti majd az ismeretlen anyag azonosítását;
11. "kollaboratív vizsgálat": azonos minta/minták azonos módszerrel való elemzése a módszer teljesítményjellemzőinek megállapítása céljából különböző laboratóriumokban, ahol a vizsgálat alapján kiszámítható az adott módszer véletlenszerű mérési hibája és laboratóriumi torzítása;
12. "megerősítő módszer": teljes vagy kiegészítő információt szolgáltató módszer ahhoz, hogy elvégezhető legyen az anyag egyértelmű azonosítása és - szükség esetén - számszerűsítése az alábbi módok egyike szerint:
a) az engedélyezett anyagok maradékanyag-határértékén vagy maximális szintjén;
b) intézkedési referenciapontok (RPA) azon tiltott vagy nem engedélyezett anyagoknál, amelyekhez intézkedési referenciapont van megállapítva;
c) az észszerűen elérhető legalacsonyabb koncentráció szinten azon tiltott vagy nem engedélyezett anyagoknál, amelyekhez nincs intézkedési referenciapont megállapítva;
13. "kiterjesztési tényező (k)": a kívánt konfidenciaszintet kifejező és a kiterjesztett mérési bizonytalansághoz társuló szám;
14. "megerősítési döntési határérték (CCα)": az a határérték, amelynél és amely felett α hibavalószínűséggel állapítható meg egy minta nem-megfelelt jellege, és az 1 - α érték az engedélyezett határérték túllépésének százalékos statisztikai bizonyosságát jelenti;
15. "a szűrés kimutatási képessége (CCβ)": az a legkisebb analittartalom, amely egy mintából β hibavalószínűséggel kimutatható vagy számszerűsíthető:
a) tiltott vagy nem engedélyezett gyógyszerhatóanyagoknál a CCβ az a legkisebb koncentráció, ahol egy módszer 1 - β statisztikai bizonyossággal képes kimutatni vagy számszerűsíteni a tiltott vagy nem engedélyezett anyagok maradékanyagait tartalmazó mintákat;
b) engedélyezett anyagoknál a CCβ az a koncentráció, ahol egy módszer 1 - β statisztikai bizonyossággal képes kimutatni az engedélyezett határérték alatti koncentrációkat;
16. "megerősített mintaanyag": a kimutatandó vagy számszerűsítendő analit ismert mennyiségével feldúsított minta;
17. "körvizsgálat": ugyanazon minta/minták vizsgálatainak előre meghatározott feltételek szerinti megszervezése, elvégzése és kiértékelése két vagy több laboratórium által a vizsgálati teljesítmény kiértékelése céljából, akár kollaboratív vizsgálatként, akár jártassági vizsgálatként;
18. "belső standard (IS)": a mintából hiányzó, de az azonosítandó vagy számszerűsítendő analit fizikai-kémiai tulajdonságaihoz a lehető legjobban hasonlító fizikai-kémiai tulajdonságokkal rendelkező anyag;
19. "kérdéses szint": egy anyag vagy analit azon szignifikáns koncentrációja a mintában, amely meghatározza a jogszabályoknak való megfelelést az alábbiakat illetően:
a) maradékanyag-határérték vagy az engedélyezett anyagok maximális szintje a 124/2009/EK bizottsági rendelettel ( 9 ) és a 37/2010/EU bizottsági rendelettel ( 10 ) összhangban;
b) intézkedési referenciapontok azon tiltott vagy nem engedélyezett anyagoknál, amelyekhez intézkedési referenciapont van megállapítva az (EU) 2019/1871 rendeletnek megfelelően;
c) az analitikailag elérhető legalacsonyabb koncentráció azon tiltott vagy nem engedélyezett anyagoknál, amelyekhez nincs intézkedési referenciapont megállapítva;
20. "legalacsonyabb kalibrált szint (LCL)": az a legalacsonyabb koncentráció, amelyre a mérési rendszert kalibrálták;
21. "mátrix": az anyag, amelyből mintát vesznek;
22. "mátrixhatás": az analitikai válasz különbözősége egy oldószerben feloldott standard és egy mátrix-illesztett standard között, vagy belső standarddal történő korrekció nélkül vagy belső standarddal történő korrekcióval;
23. "mátrix-illesztett standard": analitot nem tartalmazó vakmátrix, amelyhez a minta feldolgozását követően adják hozzá különböző koncentrációkban az analitot;
24. "mátrix-erősített standard": analitot nem tartalmazó vakmátrix, amelyet az oldószeres extrakció és a minta feldolgozása előtt különböző koncentrációkban dúsítanak az analittal;
25. "mérendő mennyiség": a mérés tárgyát képező adott mennyiség;
26. "mérési bizonytalanság": a mérés eredményéhez társított nem negatív paraméter, amely a felhasznált információk alapján jellemzi a mérendő mennyiséghez észszerűen hozzárendelhető értékek szóródását;
27. "teljesítménykritériumok": egy teljesítményjellemzőre vonatkozó követelmények, amelyek alapján megítélhető, hogy az analitikai módszer alkalmas-e a tervezett felhasználásra és megbízható eredményeket produkál-e;
28. "precizitás": a megszabott feltételek mellett kapott független vizsgálati eredmények közötti egyezés közelisége a vizsgálati eredmények szórásaként vagy relatív szórásaként kifejezve;
29. "kvalitatív módszer": olyan analitikai módszer, amely egy anyagot vagy anyagcsoportot annak kémiai, biológiai vagy fizikai tulajdonságai alapján mutat ki vagy azonosít;
30. "kvantitatív módszer": olyan analitikai módszer, amely meghatározza egy anyag mennyiségét vagy tömegszázalékát, hogy azután azt megfelelő mértékegységek számértékeként lehessen kifejezni;
31. "visszanyerés": egy analit visszanyeréssel korrigált mennyisége a mátrixmintában található erősített analit mennyiségével osztva, százalékban kifejezve;
32. "visszanyerési korrekció": a belső standardok használata, a mátrix kalibráló görbe használata és a visszanyerési korrekciós tényező használata, illetve ezek kombinációja;
33. "referenciaanyag": olyan egy vagy több tulajdonságát tekintve kellően homogén és stabil anyag, amelyről megállapították, hogy alkalmas egy mérési folyamatban vagy névleges tulajdonságok vizsgálatához való tervezett felhasználására ( 11 );
34. "relatív mátrixhatás": az analitikai válasz különbözősége egy oldószerben feloldott standard és egy mátrix-illesztett standard között, korrekció mellett használt belső standard esetén;
35. "ismételhetőség": precizitás olyan körülmények között, ahol a független vizsgálati eredményeket ugyanazzal a módszerrel, azonos vizsgálati tételeken, azonos laboratóriumban, azonos vizsgáló személy, ugyanazokkal a berendezésekkel, kis időintervallumú eltéréssel kapja;
36. "reprodukálhatóság": precizitás olyan körülmények között, ahol a vizsgálati eredményeket ugyanazzal a módszerrel, azonos vizsgálati tételeken, különböző laboratóriumokban, különböző vizsgáló személyek, különböző berendezésekkel kapják ( 12 );
37. "zavartűrés": egy analitikai módszer érzékenysége azon kísérleti körülmények változásaira, amelyek mellett a módszer az előírás szerint vagy meghatározott apróbb módosításokkal elvégezhető;
38. "szűrési módszer": az a módszer, amelyet a kérdéses szinten egy anyag vagy anyagcsoport szűréséhez alkalmaznak;
39. "szűrési célkoncentráció (STC)": a CCβ alatti vagy azzal egyenlő koncentráció, amelynél a szűrési mérés a potenciálisan nem-megfelelt "szűrési pozitív" kategóriába sorolja a mintát és megerősítő vizsgálatot indít el;
40. "szelektivitás": egy módszer azon képessége, hogy különbséget tud tenni az éppen mért analit és más anyagok között;
41. "egylaboros vizsgálat" vagy "házon belüli validálás": olyan analitikai vizsgálat, amelybe egyetlen laboratórium van bevonva, ahol egyetlen módszert alkalmazva elemzik ugyanazon vagy különböző vizsgálati anyagokat különböző körülmények és indokoltan nagy időintervallumok mellett;
42. "standard addíció": olyan eljárás, amelynél a minta egyik részét változtatás nélkül elemzik, míg a többi vizsgálati részhez az elemzést megelőzően ismert mennyiségű standard analitet adnak;
43. "standard analit": ismert és hitelesített tartalmú és tisztaságú analit, amelyet referenciaként használnak az elemzésben;
44. "anyag": állandó összetételű anyag, amelyet az azt alkotó összetevők és bizonyos fizikai tulajdonságok jellemeznek;
45. "vizsgálati rész": a mintából kivett azon anyagmennyiség, amelyen a vizsgálatot vagy megfigyelést végzik;
46. "valódiság": a vizsgálati eredmények nagy sorozatából nyert átlagérték és egy elfogadott referenciaérték közötti egyezés közelisége;
47. "mértékegységek": az ISO 80000-1:2022 ( 13 ) és a 80/181/EGK tanácsi irányelv ( 14 ) szerinti mértékegységek;
48. "validálás": vizsgálattal való megerősítése és tényleges bizonyítása annak, hogy valamilyen konkrét, tervezett felhasználás meghatározott követelményei teljesülnek ( 15 ), egylaboros vizsgálat vagy kollaboratív vizsgálat útján;
49. "laboratóriumon belüli reprodukálhatóság" vagy "közbenső precizitás/házon belüli reprodukálhatóság": mérési precizitás egy konkrét laboratórium laboratóriumon belüli körülményei között.
3. cikk
Analitikai módszerek
A tagállamok gondoskodnak arról, hogy az (EU) 2017/625 rendelet 34. cikkével összhangban vett minták elemzése az alábbi követelményeknek megfelelő módszerek alkalmazásával történjen:
(1) vizsgálati utasításokban dokumentáltak, lehetőség szerint az ISO 78-2:1999 Kémia - A szabványok elrendezése - 2. rész: Kémiai elemzési módszerek ( 16 ) mellékletei szerint;
(2) megfelelnek az analitikai módszerekhez az e rendelet I. mellékletének 1. fejezetében megállapított teljesítménykritériumoknak és egyéb követelményeknek;
(3) validálásukat az e rendelet I. mellékletének 2. és 4. fejezetében megállapított követelményekkel összhangban végezték;
(4) lehetővé teszik az (EU) 2019/1871 rendeletben megállapított intézkedési referenciapontok érvényesítését, a tiltott és nem engedélyezett anyagok jelenlétének azonosítását, a 315/93/EGK rendelet és a 124/2009/EK rendelet alapján rögzített legmagasabb szintek (ML-ek) érvényesítését, valamint az 1831/2003/EK és 470/2009/EK rendeletek alapján rögzített maximális maradékanyag-határértékek (MRL-ek) érvényesítését.
Ha a validálás során az I. melléklet 1. és 2. táblázatában meghatározott kritériumoktól való eltérések figyelhetők meg, az említett eltéréseknek a validálás eredményére gyakorolt hatását dokumentált és visszakereshető módon elemezni kell.
4. cikk
Minőségellenőrzés
A tagállamok biztosítják az (EU) 2017/625 rendelet szerint végzett elemzések eredményeinek minőségét, különösen a vizsgálatok vagy kalibrálási eredmények figyelemmel kísérése útján, az ISO/IEC 17025:2017: Vizsgáló- és kalibrálólaboratóriumok felkészültségének általános követelményeivel, továbbá a rutinelemzések alatti minőség-ellenőrzés e rendelet I. mellékletének 3. fejezetében megállapított követelményeivel összhangban.
5. cikk
Az eredmények értelmezése
(1) Egy elemzés eredménye akkor tekintendő nem megfelelőnek, ha egyenlő vagy nagyobb mint a megerősítési döntési határérték (CCα).
(2) Azoknál az engedélyezett anyagoknál, amelyekhez nincs megállapítva MRL vagy ML, a megerősítési döntési határértéket (CCα) az a koncentráció jelenti, amelynél és amely felett 1 - α statisztikai bizonyossággal dönthető el, hogy megtörtént-e az engedélyezett határérték túllépése.
(3) A nem engedélyezett vagy tiltott anyagoknál, illetve azoknál az engedélyezett anyagoknál, amelyekhez nincs megállapítva MRL vagy ML egy adott faj vagy termék esetében, a megerősítési döntési határértéket (CCα) az a legalacsonyabb koncentráció jelenti, amelynél 1 - α statisztikai bizonyossággal dönthető el az adott analit jelenléte.
(4) A nem engedélyezett vagy tiltott gyógyszerhatóanyagoknál az α hiba legfeljebb 1 % lehet. Minden egyéb anyag esetében az α hiba legfeljebb 5 % lehet.
6. cikk
Mintavételi módszerek
A tagállamok gondoskodnak arról, hogy a minták vétele, kezelése és címkézése az e rendelet II. mellékletében megállapított részletes mintavételi módszerek szerint történjen.
7. cikk
Hatályon kívül helyezés és átmeneti intézkedések
A 2002/657/EK és a 98/179/EK határozat e rendelet hatálybalépésének napjától hatályát veszti.
Az e rendelet hatálybalépése előtt validált módszerekre azonban 2026. június 10-ig továbbra is a 2002/657/EK határozat I. mellékletének 2. és 3. pontjában meghatározott követelmények alkalmazandók.
8. cikk
Hatálybalépés
Ez a rendelet az Európai Unió Hivatalos Lapjában való kihirdetését követő huszadik napon lép hatályba.
Ez a rendelet teljes egészében kötelező és közvetlenül alkalmazandó valamennyi tagállamban.
I. MELLÉKLET
1. FEJEZET:
TELJESÍTMÉNYKRITÉRIUMOK ÉS EGYÉB KÖVETELMÉNYEK ANALITIKAI MÓDSZEREKHEZ
1.1. A szűrési módszerek követelményei
1.1.1. A megfelelő szűrési módszerek kategóriái
A kvalitatív, félkvantitatív vagy kvantitatív módszerek jelentik a megfelelő szűrési módszereket.
1.1.2. A biológiai, biokémiai vagy fizikai-kémiai szűrési módszerek követelményei
Tiltott vagy nem engedélyezett anyagok esetében a CCβ értékének az észszerűen elérhető legalacsonyabbnak kell lennie, illetve mindenképpen alacsonyabbnak kell lennie az olyan anyagok intézkedési referenciapontjánál (RPA), amelyekhez RPA kerül megállapításra az (EU) 2019/1871 rendelet értelmében.
Az engedélyezett gyógyszerhatóanyagok esetében a CCβ értékének alacsonyabbnak kell lennie mint az MRL vagy ML értékének.
Szűrési célokra csak azok az analitikai módszerek használhatók, amelyeknél dokumentált és visszakereshető módon bizonyítható, hogy megtörtént a validálásuk és a hamis megfelelt rátájuk legfeljebb 5 % (β hiba). A gyaníthatóan nem-megfelelt eredmények esetén az adott eredményt megerősítő módszerrel kell megerősíteni.
A szűréshez és megerősítéshez egyaránt használt kvantitatív szűrési módszereknek az 1.2.2.1. és 1.2.2.2. alatt leírtakkal azonos követelményeket kell teljesíteniük a pontosság, tartomány és precizitás terén.
1.2. A megerősítő módszerek követelményei
1.2.1. A megerősítő módszerek általános követelményei
Tiltott vagy nem engedélyezett anyagok esetében a CCα értékének az észszerűen elérhető legalacsonyabbnak kell lennie. Tiltott vagy nem engedélyezett anyagok esetében, amelyekhez RPA kerül megállapításra az (EU) 2019/1871 rendelet értelmében, a CCα értéke nem lehet nagyobb az intézkedési referenciapontnál.
Engedélyezett anyagok esetében a CCα értékének az MRL vagy ML értékénél nagyobbnak, de ahhoz a lehető legközelebbinek kell lennie.
Megerősítési célokra csak azok az analitikai módszerek használhatók, amelyeknél dokumentált és visszakereshető módon bizonyítható, hogy megtörtént a validálásuk, és a hamis nem-megfelelt rátájuk (α hiba) legfeljebb 1 % a tiltott vagy nem engedélyezett anyagok esetében, illetve legfeljebb 5 % az engedélyezett anyagok esetében.
A megerősítő módszereknek az analit kémiai szerkezetére vonatkozóan információkkal kell szolgálniuk. Ebből következően a csupán kromatográfiás elemzésen alapuló - tömegspektrometriás kimutatás alkalmazása nélküli - megerősítő módszerek önmagukban nem alkalmasak arra, hogy a tiltott vagy nem engedélyezett gyógyszerhatóanyagok megerősítő módszerei legyenek. Amennyiben a tömegspektrometria nem alkalmas az engedélyezett anyagokhoz, egyéb módszerek, például HPLC-DAD, HPLC-FLD vagy ezek kombinációja alkalmazható.
Amennyiben a megerősítő módszer szerint szükséges, úgy egy megfelelő belső standardot kell hozzáadni a vizsgálati részhez az extrakciós eljárás kezdetén. Rendelkezésre állástól függően vagy az analit stabil, izotóppal megjelölt - tömegspektrometriás kimutatásra különösen alkalmas - formái, vagy az analittel szoros szerkezeti rokonságot mutató vegyületek alkalmazandók erre a célra.
1.2.1a. A párhuzamos kromatográfia konkrét alkalmazása, ha nem áll rendelkezésre belső standard
Amikor nem használható megfelelő belső standard, akkor az analit azonosítását lehetőleg párhuzamos kromatográfiával ( 17 ) kell megerősíteni. Ilyen esetben csak egy csúcs jelentkezhet, amelynél a megnövekedett csúcsmagasság (vagy -terület) a hozzáadott analit mennyiségének felel meg. Ha ez nem megvalósítható, akkor mátrix-illesztett vagy mátrix-erősített standardok használandók.
1.2.2. A megerősítő módszerek általános teljesítménykritériumai
1.2.2.1. Valódiság visszanyerés útján
Valamely hiteles anyagminta ismételt elemzései esetén a kísérleti úton meghatározott, visszanyeréssel korrigált közepes tömegszázaléknak a hitelesített értéktől való eltérésének meg kell felelnie meg az 1. táblázat szerinti minimális valódiság-tartományoknak.
1. táblázat
A kvantitatív módszerek minimális valódisága
| Tömegszázalék | Tartomány |
| ≤ 1 μg/kg | –50 % és +20 % között |
| > 1 μg/kg és 10 μg/kg között | –30 % és +20 % között |
| ≥ 10 μg/kg | –20 % és +20 % között |
Ha nem áll rendelkezésre hiteles anyagminta, a mérések valódisága egyéb módokon is értékelhető, például körvizsgálatokból származó hozzárendelt értékű anyagok alkalmazásával vagy ismert analitmennyiség(ek) vakmátrixhoz való hozzáadásain keresztül.
1.2.2.2. Precizitás
Egy referencia- vagy megerősített anyag - laboratóriumon belüli reprodukálhatósági körülmények közötti - ismételt elemzésének relatív szórása (CV) nem haladhatja meg a Horwitz-egyenlettel kalkulált szintet. Az egyenlet:
CV = 2(1 - 0,5 log C)
ahol C a 10 hatványaként (kitevőjeként) kifejezett tömegszázalék (pl. 1 mg/g = 10-3). 120 μg/kg alatti tömegszázalékoknál a Horwitz-egyenlet alkalmazása elfogadhatatlanul magas értékeket ad. A megengedett maximális relatív szórás ezért nem lehet nagyobb a 2. táblázatban látható értékeknél.
2. táblázat
Elfogadható relatív szórás
| Tömegszázalék | Reprodukálhatósági CV (%) |
| > 1 000 μg/kg | 16 (a Horwitz-egyenlet alapján) |
| > 120 μg/kg és 1 000 μg/kg között | 22 (a Horwitz-egyenlet alapján) |
| 10–120 μg/kg | 25 () |
| < 10 μg/kg | 30 () |
| (1) A bemutatott CV (%) iránymutatás és az észszerűen elérhető legalacsonyabb legyen. | |
Az ismételhetőségi körülmények között elvégzett elemzéseknél az ismételhetőségi körülmények közötti relatív szórás általában a 2. táblázatban látható értékek kétharmada alatt van, és nem haladhatja meg a reprodukálhatósági körülmények közötti relatív szórást.
1.2.3. A kromatográfiás elválasztásra vonatkozó követelmények
1.2.3.1. Elfogadható minimum retenciós idő
Folyadék- (LC) vagy gázkromatográfia (GC) esetén a vizsgálat tárgyát képező analit(ok) elfogadható minimum retenciós ideje kétszerese az oszlop holt térfogatához tartozó retenciós időnek.
1.2.3.2. Az extraktumban lévő analit retenciós ideje
Az extraktumban lévő analit retenciós idejének ± 0,1 perc tűréshatár mellett meg kell felelnie a kalibráló standard, a mátrix-illesztett standard vagy a mátrix-erősített standard retenciós idejének. Gyors kromatográfia esetén, ahol a retenciós idő 2 percnél kisebb, a retenciós idő legfeljebb 5 %-os eltérése elfogadható.
1.2.3.3. Retenciós idő belső standard használata esetén
Belső standard használatakor az analit kromatográfiás retenciós idejének a belső standard retenciós idejéhez viszonyított arányának (azaz az analit relatív retenciós ideje) meg kell felelnie a kalibráló standard, a mátrix-illesztett standard vagy a mátrix-erősített standard retenciós idejének, gázkromatográfia esetén 0,5 % és folyadékkromatográfia esetén 1 % maximális eltérés mellett, az e rendelet hatálybalépése óta validált módszerekre vonatkozóan.
1.2.4. A tömegspektrometria specifikus teljesítménykritériumai
1.2.4.1. Tömegspektrometriás kimutatás
A tömegspektrometriás kimutatást az alábbi lehetőségek valamelyikével kell végezni:
1. teljes letapogatású (FS) tömegspektrumok rögzítése;
2. szelektált ion monitoring (SIM);
3. szekvenciális tömegspektrometria (MSn) technikák, mint például a kiválasztott reakció monitoring (SRM);
4. a tömegspektrometria (MS) vagy szekvenciális tömegspektrometria (MSn) technikák kombinálása megfelelő ionizálási módokkal.
Egyaránt megfelelő a kisfelbontású tömegspektrometria (LRMS, egységnyi tömegfelbontás) és a nagyfelbontású tömegspektrometria (HRMS), például kettős fókuszálású szektorok, Time of Flight (TOF) és Orbitrap műszerek.
Nagyfelbontású tömegspektrometria (HRMS) esetén egy analit azonosításának megerősítésekor az összes diagnosztikai ion tömegeltérésének 5 ppm (vagy m/z < 200 esetén 1 mDa) alatt kell lennie. Ez alapján a célnak megfelelő tényleges felbontást kell választani, és a felbontásnak a teljes tömegtartományra vonatkozóan jellemzően 10 %-os völgynél 10 000 vagy félértékszélességnél (FWHM) 20 000 felett kell lennie.
Amikor a tömegspektrometriás meghatározás teljes letapogatású (LRMS és HRMS) spektrumok rögzítésével történik, akkor csak a kalibráló standard, mátrix-illesztett standard vagy mátrix-erősített standard referenciaspektrumában 10 % feletti relatív intenzitással előforduló diagnosztikai ionok megfelelőek. A diagnosztikai ionok közé tartoznak a molekulaionok (ha a báziscsúcs legalább 10 %-os relatív intenzitása mellett fordulnak elő) és a jellemző fragmens- vagy termékionok.
Prekurzorion kiválasztás: Amikor a tömegspektrometriás meghatározást a prekurzorion kiválasztása után fragmentálással végzik, akkor a prekurzorion kiválasztás egységnyi vagy nagyobb tömegfelbontás mellett történik. A kiválasztott prekurzoriont a molekulaion, a molekulaion jellemző adduktjai, a jellemző termékionok vagy azok egyik izotópionja jelentik. Amennyiben a prekurzor kiválasztás tömegablaka egy Dalton feletti méretű (pl. Data Independent Acquisition esetén), úgy a technika teljes letapogatású megerősítő elemzésnek tekintendő.
Fragmens- és termékionok: A kiválasztott fragmens- vagy termékionok a mért analit/termék diagnosztikai fragmensét jelentik. A nem szelektív átmenetek (pl. tropilium kationok vagy vízvesztés) lehetőség szerint elhagyandók. A diagnosztikai ionok abundanciáját az integrált extrahált ionkromatogramok csúcsterületéből vagy magasságából kell meghatározni. Ez vonatkozik arra az esetre is, amikor azonosításhoz használnak teljes letapogatású méréseket. A jel/zaj viszony minden diagnosztikai ion esetében legalább három az egyben (3:1) legyen.
Relatív intenzitások: A diagnosztikai ionok relatív intenzitásainak (ionarány) kifejezése a legnagyobb arányban előforduló ion vagy átmenet intenzitásának százalékaként történik. Az ionarányt a spektrumok összehasonlítása vagy az extrahált ion tömegnyomok jeleinek integrálása útján kell meghatározni. A megerősítendő analit ionarány feleljen meg a mátrix-illesztett standardok, a mátrix-erősített standardok vagy a standard oldatok ionarányának összehasonlítható koncentrációk mellett, azonos körülmények között mérve, ±40 % relatív eltérésen belül.
Valamennyi tömegspektrometriás elemzéshez legalább egy ionarányt kell meghatározni. Ezek lehetőség szerint az egyetlen pásztázással elért ionok, de az ionok származhatnak ugyanazon injektálás különböző pásztázásaiból is (teljes pásztázás és fragmentációs pásztázás).
1.2.4.2. Azonosítás
Azonosítási pontrendszer segítségével kell kiválasztani a megfelelő adatgyűjtési módot és értékelési kritériumokat. Az azonosítás megerősítéséhez legalább 4 azonosítási pont szükséges egy mátrix azon anyagainál, amelyekhez MRL van megállapítva (engedélyezett felhasználás). A nem engedélyezett vagy tiltott anyagok esetében 5 azonosítási pontra van szükség. A kromatográfiás elválasztásból egy pont származhat. A 3. táblázatban látható, hogy az egyes technikák mennyi azonosítási pontszámot kaphatnak. A megerősítéshez szükséges azonosítási pontszámok összegéhez más technikákból szerzett azonosítási pontszámok is hozzáadhatók.
1. Valamennyi tömegspektrometriás elemzést olyan elválasztástechnikával kell kombinálni, amely az adott alkalmazáshoz elegendő elválasztási erőt és szelektivitást mutat. A megfelelő elválasztástechnikák közé tartozik például a folyadék- és gázkromatográfia, a kapilláris elektroforézis (CE) és a szuperkritikus folyadékkromatográfia (SFC). Az izobár vagy izomer vegyületet tartalmazó analiteknél az azonosítás megerősítéséhez kötelező a retenciós idő elfogadhatósága (± 0,5 % GC és ± 1 % LC és SFC esetén).
2. A minimális számú azonosítási pontszám megszerzéséhez maximum három különálló technika kombinálható.
3. A különböző ionizációs módok (pl. elektron ionizáció és kémiai ionizáció) különböző technikáknak tekintendők.
3. táblázat
Azonosítási pontszám technikánként
| Technika | Azonosítási pontszám |
| Elválasztás (GC, LC, SFC, CE mód) | 1 |
| LR-MS ion | 1 |
| Prekurzorion kiválasztás <±0,5 D tömegtartománynál | 1 (közvetett) |
| LR-MSn termékion | 1,5 |
| HR-MS ion | 1,5 |
| HR-MSn termékion | 2,5 |
4. táblázat
Példák az azonosítási pontszámokra, az egyes technikákra és azok kombinálására (n = egész szám)
| Technika/technikák | Elválasztás | Ionok száma | Azonosítási pontszám |
| GC-MS (EI vagy CI) | GC | n | 1 + n |
| GC-MS (EI és CI) | GC | 2 (EI) + 2 (CI) | 1 + 4 = 5 |
| GC-MS (EI vagy CI) 2 származék | GC | 2 (A származék) + 2 (B származék) | 1 + 4 = 5 |
| LC-MS | LC | n (MS) | 1 + n |
| GC- vagy LC-MS/MS | GC vagy LC | 1 prekurzor + 2 termék | 1 + 1 + 2 × 1,5 = 5 |
| GC- vagy LC-MS/MS | GC vagy LC | 2 prekurzor + 2 termék | 1 + 2 + 2 × 1,5 = 6 |
| GC- vagy LC-MS3 | GC vagy LC | 1 prekurzor + 1 MS2 termék + 1 MS3 termék | 1 + 1 + 1,5 + 1,5 = 5 |
| GC- vagy LC-HRMS | GC vagy LC | n | 1 + n × 1,5 |
| GC- vagy LC-HRMS/MS | GC vagy LC | 1 prekurzor (<±0,5 Da tömegtartomány) + 1 termék | 1 + 1 + 2,5 = 4,5 |
| GC- vagy LC-HRMS és HRMS/MS | GC vagy LC | 1 teljes letapogatású ion + 1 HRMS termékion () | 1 + 1,5 + 2,5 = 5 |
| GC- és LC-MS | GC és LC | 2 ion (GCMS) + 1 ion (LCMS) | 1 + 1 + 2 + 1 + 1 = 6 |
| (1) a A prekurzorion kiválasztásáért nem szerezhető további azonosítási pontszám, ha ez a prekurzorion ugyanaz az ion (vagy addukt vagy izotóp), mint a teljes pásztázáson figyelt HRMS ion. | |||
1.2.5. Specifikus teljesítménykritériumok az analit tömegspektrometriától eltérő kimutatási technikákkal végzett folyadékkromatográfiás meghatározásához
Csak az engedélyezett anyagok esetében használhatók az alábbi technikák a tömegspektrometria alapú módszerek alternatívájaként, amennyiben teljesülnek az e technikákra vonatkozó kritériumok:
1. teljes letapogatású diódasoros detektálású spektrofotometria (DAD) HPLC melletti használat esetén;
2. fluoreszcenciás detektálású spektrofotometria (FLD) HPLC melletti használat esetén.
Az UV/VIS detektálás (egyetlen hullámhossz) melletti folyadékkromatográfia önmagában nem alkalmas arra, hogy megerősítő módszerként használják.
1.2.5.1. Teljesítménykritériumok teljes letapogatású diódasoros spektrofotometriához
A kromatográfiás elválasztás 1.2.3. fejezetben foglalt teljesítménykritériumainak teljesülniük kell.
Az UV-spektrum abszorpciós maximumainak ugyanazokon a hullámhosszokon kell jelentkezniük, mint a mátrixban lévő kalibráló standardéinak, a kimutatási rendszer felbontásától függő maximális határon belül. A diódasoros kimutatásnál ez a maximális határ jellemzően ± 2 nm értéken belül van. Az analit 220 nm feletti spektruma - a két spektrum azon részeit illetően, amelyeknek legalább 10 % a relatív abszorbanciája - nem mutathat szemmel észlelhető különbséget a kalibráló standard spektrumához képest. Ez a kritérium akkor teljesül, amikor - először - ugyanazok a maximumok jelentkeznek és - másodszor - amikor a két spektrum között egy ponton sem figyelhető meg nagyobb különbség, mint a kalibráló standard abszorbanciájának 10 %-a. Számítógéppel segített könyvtári kutatás és megfeleltetés alkalmazása esetén a hatósági mintákból származó spektrometriás adatoknak a kalibráló oldat ugyanezen adataival való összehasonlítása meg kell, hogy haladjon egy kritikus megfeleltetési tényezőt. Ezt a tényezőt a validálási folyamat során kell meghatározni minden egyes analitre vonatkozóan, mégpedig a fenti kritériumoknak megfelelő spektrumok alapján. A mintamátrix és a detektorteljesítmény által a spektrumokban előidézett variabilitást ellenőrizni kell.
1.2.5.2. Teljesítménykritériumok fluoreszcenciás detektálású spektrofotometriához
A kromatográfiás elválasztás 1.2.3. fejezetben foglalt teljesítménykritériumainak teljesülniük kell.
A gerjesztési és emissziós hullámhosszoknak a kromatográfiás körülményekkel kombinált megválasztását úgy kell végezni, hogy a vakminta extraktumokban minimális legyen a zavaró komponensek hatása. A gerjesztési és emissziós hullámhosszok között legalább 50 nanométer távolság legyen.
A kromatogram legközelebbi csúcsmaximumának a kijelölt analit-csúcstól legalább egy teljes csúcsszélességgel kell elkülönülnie, az analit-csúcs maximum magasságának 10 %-ánál.
Ez csak a natív fluoreszcenciát mutató molekulákra, valamint a transzformálás vagy derivatizálás után fluoreszcenciát mutató molekulákra érvényes.
2. FEJEZET
VALIDÁLÁS
2.1. Analitikai módszerekhez meghatározandó teljesítményjellemzők
A módszer validálásán keresztül azt kell bizonyítani, hogy az analitikai módszer megfelel az adott teljesítményjellemzőkre vonatkozó kritériumoknak. Az eltérő kontrollcélok eltérő módszercsoportokat igényelnek. Az 5. táblázat azt mutatja, hogy az egyes módszertípusoknál milyen teljesítményjellemzőket kell igazolni, minden paraméter további magyarázata e fejezetben található.
5. táblázat
Az analitikai módszerek osztályozása a meghatározandó teljesítményjellemzők szerint
| Módszer | Megerősítés | Szűrés | |||
| Kvalitatív | Kvantitatív | Kvalitatív | Félkvantitatív () | Kvantitatív | |
| Anyagok | A | A, B | A, B | A, B | A, B |
| 1.2. szerinti azonosítás | x | x | |||
| CCα | x | x | |||
| CCβ | – | x | x | x | |
| Valódiság | x | x | |||
| Precizitás | x | (x) | x | ||
| Relatív mátrixhatás/abszolút visszanyerés (*1) | x | x | |||
| Szelektivitás/specificitás | x | x | x | x | |
| Stabilitás () | x | x | x | x | |
| Zavartűrés | x | x | x | x | |
| (1) A félkvantitatív szűrési módszer egy kvantitatív eredményeket produkáló szűrési módszer, amely azonban nem felel meg az e rendelet I. mellékletének 2. táblázata szerinti precizitási követelményeknek. (*1) Az MS módszerek tekintetében validálással kell bizonyítani a teljesítményjellemzőkre vonatkozó követelmények teljesülését. A módszer relatív mátrixhatását akkor kell meghatározni, ha annak értékelése nem történt meg a validálási eljárás során. A módszer abszolút visszanyerését akkor kell meghatározni, ha nem kerül sor belső standard vagy mátrix-erősített kalibrálás alkalmazására. (2) Ha egy mátrixban lévő analit stabilitási adatai elérhetők a szakirodalomból vagy egy másik laboratóriumtól, ezeket az adatokat nem kell újra meghatároznia az érintett laboratóriumnak. Az oldatban lévő analitok elérhető stabilitási adataira való hivatkozás azonban csak azonos feltételek alkalmazása esetén fogadható el. x: A validálással bizonyítani kell a teljesítményjellemzőkre vonatkozó követelmények teljesülését. x) Félkvantitatív szűrési módszerek esetén nem kell teljesíteni az 1.2.2.2. fejezet szerinti precizitási követelményeket. A hamis megfelelt analitikai eredmények elkerülése érdekében azonban meg kell határozni a precizitást a módszer stabilitásának bizonyításához. A: tiltott vagy nem engedélyezett anyagok B: engedélyezett anyagok | |||||
2.2. Valódiság, ismételhetőség és laboratóriumon belüli reprodukálhatóság
Ez a fejezet a validálási eljárásokra ad példákat és hivatkozásokat. A módszer teljesítménykritériumoknak való megfelelése egyéb megközelítésekkel is bizonyítható, amennyiben azok ugyanazt az információs szintet és minőséget érik el.
2.2.1. Hagyományos validálás
A paraméterek hagyományos módszerekkel történő kiszámítása több egyedi kísérlet elvégzését igényli (lásd e melléklet 5. táblázatát). Nagyobb változások esetében ellenőrizni kell a teljesítményjellemzők állandó érvényességét. Sokanalites módszerek esetén több analit is elemezhető egyidejűleg, amennyiben sikerült kizárni az esetleg releváns interferenciákat. Több teljesítményjellemző határozható meg hasonló módon. A munkamennyiség minimalizálása érdekében ezért ajánlatos a kísérleteket minél inkább összevonni (pl. ismételhetőség és laboratóriumon belüli reprodukálhatóság a specificitással, a vakmintáknak a megerősítési döntési határérték meghatározására szolgáló elemzésével és a specificitás vizsgálatával együtt).
2.2.1.1. Valódiság hiteles anyagminta alapján
Előnyösebb egy analitikai módszer valódiságát hiteles anyagminta (CRM) útján meghatározni. Ennek eljárását az ISO 5725-4:1994 ( 18 ) írja le.
Erre vonatkozik a következő példa:
1. Végezze el a CRM hat ismétlésének elemzését a módszerre vonatkozó vizsgálati utasítások szerint;
2. Határozza meg az egyes ismétlések mintáiban található analit koncentrációját;
3. Számítsa ki erre a hat ismétlésre az átlagot, a szórást és a relatív szórást (%);
4. Számítsa ki a valódiságot úgy, hogy a kimutatott átlagkoncentrációt elosztja a (koncentrációként mért) hitelesített értékkel és megszorozza 100-zal, hogy az eredmény százalékos formájú legyen.
Valódiság (%) = visszanyeréssel korrigált kimutatott átlagkoncentráció) × 100/hitelesített érték
2.2.1.2. Valódiság megerősített minták alapján
Ha nem áll rendelkezésre hiteles anyagminta, a módszer valódiságát kísérletek útján kell meghatározni erősített vakmátrix használatával, legalább az alábbi sémának megfelelően:
1. Az e rendelet hatálybalépése óta validált módszerek esetében válasszon vakanyagot és erősítse meg az alábbi koncentrációszinteken:
a) az RPA 0,5-szöröse ( 19 ), 1,0-szorosa és 1,5-szöröse; vagy
b) az MRL vagy ML 0,1-szerese ( 20 ), 1,0-szorosa és 1,5-szöröse engedélyezett anyagok esetén; vagy
c) az LCL 1,0-szorosa, 2,0-szorosa és 3,0-szorosa nem engedélyezett anyagok esetén (amelyekhez nincs megállapítva RPA).
2. Az elemzést minden szinten hat ismétléssel kell végezni.
3. Végezze el a minták elemzését.
4. Számítsa ki az egyes mintákban kimutatott koncentrációt.
5. Az alábbi egyenlet segítségével számítsa ki minden mintához a valódiságot, majd a hat eredményhez minden koncentrációszinten számítsa ki az átlagvalódiságot és a relatív szórást.
Valódiság (%) = visszanyeréssel korrigált kimutatott átlagkoncentráció) × 100/erősítési szint
Az e rendelet hatálybalépése előtt validált engedélyezett anyagokra vonatkozó módszereknél elegendő meghatározni a módszer valódiságát 6 erősített alikvot használatával, az MRL vagy ML 0,5-szörösét, 1,0-szorosát és 1,5-szörösét kitevő koncentráció mellett.
2.2.1.3. Ismételhetőség
1. Az e rendelet hatálybalépése óta validált módszerek esetében ugyanazon faj azonos vakmátrixaiból kell mintacsoportot készíteni. Ezeket az analittal kell megerősíteni az alábbiaknak megfelelő koncentrációk eléréséhez:
a) az RPA 0,5-szöröse ( 21 ), 1,0-szorosa és 1,5-szöröse; vagy
b) az MRL vagy ML 0,1-szerese ( 22 ), 1,0-szorosa és 1,5-szöröse engedélyezett anyagok esetén, vagy
c) az LCL 1,0-szorosa, 2,0-szorosa és 3,0-szorosa nem engedélyezett vagy tiltott anyagok esetén, ha nincs RPA.
2. Az elemzést minden szinten legalább hat ismétléssel kell végezni.
3. Végezze el a minták elemzését.
4. Számítsa ki az egyes mintákban kimutatott koncentrációt.
5. Számítsa ki a megerősített minták átlagkoncentrációját, szórását és relatív szórását (%).
6. Legalább két másik alkalommal ismételje meg a fenti lépéseket, szintenként összesen legalább 18 ismétlést végezve.
7. Számítsa ki a megerősített minták átlagkoncentrációját, szórását (az egyes alkalmak négyzetre emelésével kapott szórás átlagolásával és abból négyzetgyök vonásásával) és relatív szórását.
Az e rendelet hatálybalépése előtt validált engedélyezett anyagokra vonatkozó módszereknél elegendő meghatározni az ismételhetőséget a megerősített mátrixok használatával, az MRL vagy ML 0,5-szörösét, 1,0-szorosát és 1,5-szörösét kitevő koncentráció mellett.
Az ismételhetőség kiszámítása az ISO 5725-2:2019 ( 23 ) szerint is elvégezhető.
2.2.1.4. Laboratóriumon belüli reprodukálhatóság
1. Az e rendelet hatálybalépése után végzett validálásoknál az alábbiaknak megfelelő koncentrációk eléréséhez analit(ok) útján megerősített meghatározott vizsgálati anyagból (azonos vagy különböző mátrixokból) kell mintacsoportot készíteni:
a) az RPA 0,5-szöröse ( 24 ), 1,0-szorosa és 1,5-szöröse; vagy
b) az MRL vagy ML 0,1-szerese(6), 1,0-szorosa és 1,5-szöröse engedélyezett anyagok esetén, vagy
c) az LCL 1,0-szorosa, 2,0-szorosa és 3,0-szorosa nem engedélyezett vagy tiltott anyagok esetén, ha nincs RPA.
2. Az elemzést minden koncentrációszinten a vakanyag legalább hat ismétlésével kell végezni.
3. Végezze el a minták elemzését.
4. Számítsa ki az egyes mintákban kimutatott koncentrációt.
5. Legalább két másik alkalommal ismételje meg a fenti lépéseket (szintenként összesen legalább 18 ismétlést végezve), lehetőség szerint más vakanyag-tételekkel, más vizsgáló személyekkel és a lehető legtöbb eltérő környezeti körülmény között, pl. más reagens- és oldószer-tételek, más szobahőmérsékletek, más műszerek vagy egyéb módosított paraméterek alkalmazásával.
6. Határozza meg a megerősített minták átlagkoncentrációját, szórását és relatív szórását (%).
Az e rendelet hatálybalépése előtt validált engedélyezett anyagokra vonatkozó módszereknél elegendő meghatározni a laboratóriumon belüli reprodukálhatóságot a megerősített mátrixok használatával, az MRL vagy ML 0,5-szörösét, 1,0-szorosát és 1,5-szörösét kitevő koncentráció mellett.
A laboratóriumon belüli reprodukálhatóság/közbenső precizitás kiszámítása az ISO 5725-2:2019, ISO 11843-1:1997 ( 25 ), Codex CAC/GL 59-2006 ( 26 ) szerint is elvégezhető.
2.2.2. Validálás alternatív modellek szerint
A paraméterek alternatív modellekkel összhangban történő kiszámítása egy kísérleti terv elvégzését igényli. A vizsgálat tárgyát képező különböző fajok és különböző faktorok számától függően kísérleti tervet kell összeállítani. A teljes validálási eljárás első lépéseként tehát mérlegelni kell a jövőben a laboratóriumban elemzésre kerülő mintapopulációkat azon legfontosabb fajok és tényezők meghatározása érdekében, amelyek befolyásolhatják a mérési eredményeket. A faktoriális megközelítéssel kiértékelhető az adott laboratóriumban sokféle vizsgálati körülmény - például más elemzők, más műszerek, más reagenstételek, más mátrixok, más vizsgálati időtartamok és más vizsgálati hőmérsékletek - mellett kapott vizsgálati eredmények mérési bizonytalansága. Ezt követően a koncentrációtartományt célhoz szabottan kell megválasztani, engedélyezett anyagoknál az MRL vagy ML szerint, tiltott vagy nem engedélyezett anyagoknál pedig az RPA vagy LCL szerint.
A faktoriális megközelítés célja a kiválasztott faktorok egyidejű ellenőrzött variációjával megbízható precizitási adatokat és mérési adatokat biztosítani. Lehetővé teszi a faktoriális hatások és véletlenszerű hatások együttes hatásának értékelését. A kísérleti kialakítás lehetővé teszi továbbá az analitikai módszer zavartűrésének ( 27 ) vizsgálatát és a házon belüli reprodukálhatóság mátrixok közötti szórásának meghatározását.
Az alábbiakban egy ortogonális kísérleti tervet alkalmazó alternatív megközelítés példája szerepel.
Legfeljebb hét faktor (zajfaktor) vizsgálható. A vizsgálat úgy van kialakítva, hogy a precízió, valódiság (megerősített minták alapján), érzékenység, mérési bizonytalanság és kritikus koncentrációk a kísérleti terv végrehajtásával egyidejűleg határozhatók meg.
6. táblázat
Egy validálási vizsgálat keretén belül nyolc sorozat (faktorszintű kombináció), 7 faktor és két változó (A/B) alkalmazásával végzett ortogonális kísérleti terv példája
| Faktor | I | II | III | IV | V | VI | VII |
| 01 sorozat | A | A | A | A | A | A | A |
| 02 sorozat | A | A | B | A | B | B | B |
| 03 sorozat | A | B | A | B | A | B | B |
| 04 sorozat | A | B | B | B | B | A | A |
| 05 sorozat | B | A | A | B | B | A | B |
| 06 sorozat | B | A | B | B | A | B | A |
| 07 sorozat | B | B | A | A | B | B | A |
| 08 sorozat | B | B | B | A | A | A | B |
A módszer jellemzőinek kiszámítását Jülicher et al. ( 28 ) leírása vagy az ISO/TS 23471:2022 ( 29 ) szerint kell végezni.
2.2.3. Egyéb validálási megközelítések
A módszer teljesítményjellemzőkre vonatkozó teljesítménykritériumoknak való megfelelése egyéb megközelítésekkel is bizonyítható, amennyiben azok ugyanazt az információs szintet és minőséget érik el. Validálás végzése történhet a Codex Alimentarius, az ISO vagy az IUPAC ( 30 ) által megállapított körvizsgálat lefolytatásával vagy olyan alternatív módszerek szerint is, mint az egylaboros vizsgálatok vagy a házon belüli validálás ( 31 ). Alternatív validálási eljárások alkalmazása esetén az alapmodellt és stratégiát - a vonatkozó feltételekkel, feltételezésekkel és formulákkal együtt - rögzíteni kell a validálási protokollban, vagy legalább hivatkozni kell azok rendelkezésre állására.
2.3. Szelektivitás/specificitás
Az analit és a vele szoros rokonságot mutató anyagok közötti különbségtétel képességét a lehető legnagyobb mértékben meg kell határozni. Meg kell határozni a kérdéses maradvány, a mátrixvegyületek vagy bármely más, potenciálisan zavaró anyag homológjainak, izomerjeinek, bomlástermékeinek, endogén anyagainak, analógjainak és metabolitjainak interferenciáját, és az azonosított interferenciák elkerülése érdekében szükség esetén módosítani kell a módszert. A módszer specificitásának meghatározásához az alábbi megközelítés alkalmazandó:
1. Válasszon ki egy sor kémiailag rokon vegyületet vagy a kérdéses vegyülettel valószínűleg együtt előforduló olyan egyéb anyagokat, amelyek jelen lehetnek a mintákban, és ellenőrizze, hogy azok zavarhatják-e a célzott analit(ok) elemzését.
2. Elemezzen megfelelő számú reprezentatív vakmintát pl. különböző tételeket vagy sok különböző állatfajt (n ≥ 20) és ellenőrizze a jelek, csúcsok vagy ionnyomok interferenciáit a kérdéses tartományban, ahol a célzott analit elúciója várható.
3. Erősítse meg a reprezentatív vakmintákat a vonatkozó koncentrációnál olyan anyagokkal, amelyek várhatóan interferálnak az analit azonosításával és/vagy számszerűsítésével, és vizsgálja meg, hogy a hozzáadott anyag:
a) vezethet-e hamis azonosításhoz;
b) akadályozza-e a célzott analit azonosítását;
c) jelentősen befolyásolja-e a számszerűsítést.
2.4. Zavartűrés
Az analitikai módszer folyamatos teljesítményét különböző kísérleti körülmények között kell tesztelni, ideértve például a különböző mintavételi körülményeket és a rutinvizsgálatok során lehetséges kisebb változásokat. A módszer zavartűrésének tesztelésekor a kísérleti körülmények kisebb változásaira van szükség. E változások jelentőségét kell kiértékelni. Minden teljesítményjellemzőt azon kisebb változásokra kell meghatározni, amelyek kimutatottan jelentős hatást gyakorolnak a vizsgálati módszer teljesítményére.
2.5. Stabilitás
Meg kell határozni a kalibráló standard, mátrix-illesztett standard és/vagy mátrix-erősített standard és a mintában lévő analit vagy mátrixkomponensek tárolás vagy elemzés alatti stabilitását, mivel az instabilitás befolyásolhatja a vizsgálati eredmény alakulását.
Az analit különböző tárolási körülmények közötti stabilitása rendszerint jól jellemzett. A standardok és minták tárolási körülményeinek a szokásos laboratóriumi akkreditációs és minőségellenőrzési rendszer részét képező figyelemmel kísérése biztosíthatja a szükséges információkat. Ha egy mátrixban lévő analitek stabilitási adatai elérhetők (pl. uniós referencialaboratóriumok információi, közzétett adatok stb. alapján), ezen adatokat nem kell meghatároznia minden egyes laboratóriumnak. Az oldatban és mátrixban lévő analitek elérhető stabilitási adataira való hivatkozás azonban csak azonos feltételek alkalmazása esetén fogadható el.
Az alábbi megközelítések alkalmazandók, ha a szükséges stabilitási adatok nem érhetők el. Emellett az e melléklet 7. táblázatában szereplőhöz hasonló tárolási hőmérsékleti sémával alkalmazott izokron megközelítés ( 32 ) is lehetővé teszi az analitok potenciális instabilitásának megállapítását és a megfelelő tárolási időszakok becslését, és szintén alkalmazható.
2.5.1. Oldatban lévő analit stabilitásának meghatározása
1. Készítsen az analit(ek)ből friss törzsoldatokat és a vizsgálati utasítások szerint végezzen hígítást, hogy minden kiválasztott koncentrációból elegendő alikvot (pl. 40) álljon rendelkezésre. Az alábbiakból kell mintákat készíteni:
a) Az erősítéshez használt analitoldatok;
b) A végső elemzéshez használt analitoldatok;
c) Bármely egyéb szükséges oldat (pl. derivatizált standardok).
2. A vizsgálati utasítások szerint mérje meg az analit-tartalmat a frissen készített oldatban.
3. Öntsön megfelelő mennyiségeket alkalmas tárolóedényekbe, majd címkézze fel és tárolja azokat a 7. táblázatban foglalt séma fény- és hőviszonyai szerint. Az alkalmazott analitikai gyakorlat alapján kell megválasztani a tárolási időt, ami ideális esetben addig tart amíg az azonosítás és/vagy számszerűsítés során nem észlelhetők az első bomlási jelek. Ha a stabilitási vizsgálat során nem észlelhető bomlás, a stabilitási vizsgálat tárolási ideje egyezzen meg az oldat maximális tárolási időtartamával.
4. Az alábbi képlettel számítsa ki az egyes alikvotokban található analit(ok) koncentrációját a frissen készített oldatban lévő analit koncentrációjához viszonyítva:
Fennmaradó analit (%) = Ci × 100/Cfriss
Ci = az i időpontbeli koncentráció
Cfriss = a friss oldat koncentrációja
A tárolt öt párhuzamos oldat középértéke legfeljebb 15 %-kal térhet el a frissen készített öt párhuzamos oldat középértékétől. A frissen készített öt párhuzamos oldat középértékét kell alapul venni a százalékos eltérés kiszámításához.
7. táblázat
Séma az oldatban lévő analit stabilitásának meghatározásához
| –20 °C | +4 °C | +20 °C | |
| Sötét | 10 alikvot | 10 alikvot | 10 alikvot |
| Világos | 10 alikvot |
2.5.2. Mátrixban lévő analit(ok) stabilitásának meghatározása
1. Lehetőség szerint már előfordult mintákat kell használni. Ha nem áll rendelkezésre már előfordult mátrix, akkor analittel erősített vakmátrix használandó.
2. Amennyiben rendelkezésre áll megfelelő, már előfordult mátrix, akkor homogenizálja (lehetőleg addig, amíg az még friss). Ossza fel a mátrixot öt részre, és mindegyik részből elemezzen egy alikvotot.
3. Ha nem áll rendelkezésre már előfordult mátrix, akkor vegyen némi vakmátrixot, majd homogenizálja. Ossza fel a mátrixot öt részre. Erősítse meg mindegyik részt a kérdéses szint körüli analittel, amit lehetőség szerint kis mennyiségű vizes oldatban kell elkészíteni. Azonnal elemezzen minden részből egy alikvotot.
4. A már előfordult homogenizált mátrix vagy az erősített dúsított vakmátrix részeit (részmintáit) tárolja olyan hőmérsékleten, amely megfelel a laboratóriumban az adott analit/mátrix-kombináció tekintetében elfogadott tárolási körülményeknek, és határozza meg az analit koncentrációját a rövid távú tárolást, a középtávú tárolást és a hosszú távú tárolást követően (legalább annyi ideig, amíg a mintát rendszerint a laboratóriumban tartják).
5. A tárolt részből vett öt alikvot középértéke legfeljebb a módszer laboratóriumon belüli reprodukálhatóságával térhet el a frissen készített öt alikvot középértékétől. A frissen készített öt alikvot középértékét kell alapul venni a százalékos eltérés kiszámításához.
6. Jegyezze fel az elfogadható maximális tárolási időt és az optimális tárolási körülményeket.
2.6. Megerősítési döntési határérték (CCα)
A megerősítő módszerekhez szükség van a CCα meghatározására. A CCα megállapítását az azonosítás vagy az azonosítás plusz számszerűsítés 1. fejezetben foglalt "Teljesítménykritériumok és egyéb követelmények analitikai módszerekhez" követelményeinek megfelelő körülmények között kell végezni.
A minták megfelelőségének ellenőrzéséhez a kombinált standard mérési bizonytalanságát már figyelembe vették a CCα értékénél (megerősítési döntési határérték).
1. A nem engedélyezett vagy tiltott gyógyszerhatóanyagoknál a CCα számítása a következő:
a)
1. módszer: az ISO 11843-1:1997 ( 33 ) szerinti kalibráló görbe eljárással (itteni hivatkozása a nettó állapotváltozó kritikus értéke). Ebben az esetben vakanyagot kell használni, amely ekvidisztáns osztásokban van megerősítve az RPA vagy LCL értékén és a felett. Végezze el a minták elemzését. Az azonosítás után lehetőség szerint ábrázolja grafikusan a jel görbéjét vagy az újraszámított koncentrációt a hozzáadott koncentrációval szemben. Az y-metszékhez tartozó koncentráció plusz a metszék laboratóriumon belüli reprodukálhatósága szórásának 2,33-szorosa adja a döntési határértéket. E módszer csak kvantitatív próbákra érvényes. Az e megközelítéssel kapott döntési határértékeket a kalkulált döntési határértéknél megerősített vakmátrix elemzésével kell ellenőrizni.
b)
2. módszer: mátrixonként legalább 20 reprezentatív vakanyag elemzésével annak érdekében, hogy kiszámítható legyen a jel/zaj viszony az analit várható időablakánál. Döntési határértékként a jel/zaj viszony háromszorosa használható. Ez kvantitatív és kvalitatív próbákra érvényes. Az e megközelítéssel kapott döntési határértékeket a kalkulált döntési határértéknél megerősített vakmátrix elemzésével kell ellenőrizni.
c)
3. módszer: CCα = LCL + k (egyoldalú, 99 %) × (kombinált) standard mérési bizonytalanság LCL értéknél
A nem engedélyezett vagy tiltott gyógyszerhatóanyagoknál, a validálási kísérlettől (és annak adott szabadságfokától) függően, észszerűen alkalmazható a t-eloszlás vagy - a Gauss-eloszlás (egyoldalú, n=∞) alapul vétele esetén - 2,33 értékű k-faktor alkalmazandó.
A laboratóriumon belüli reprodukálhatóság és a valódiság alkalmas a (kombinált) standard mérési bizonytalanság meghatározására, ha annál minden releváns befolyásoló faktort figyelembe vesznek.
Az e rendelet hatálybalépése előtt validált módszerek esetében csak 2026. január 1-jéig használható a CCα számítás 2. módszere. Az e rendelet hatálybalépése után validált módszerek esetében csak az 1. vagy 3. módszer használható.
2. Az engedélyezett anyagoknál a CCα számítása a következő:
a) Mátrix/fajkombinációkan lévő engedélyezett anyagok esetén, amelyekhez nincs rögzítve MRL vagy ML:
i.
1. módszer: az ISO 11843-1:1997 szerinti kalibráló görbe eljárással (itteni hivatkozása a nettó állapotváltozó kritikus értéke). Ebben az esetben vakanyagot kell használni, amely ekvidisztáns osztásokban van megerősítve az MRL vagy ML értékén és a felett. Végezze el a minták elemzését. Az azonosítás után lehetőség szerint ábrázolja grafikusan a jel görbéjét vagy az újraszámított koncentrációt a hozzáadott koncentrációval szemben. Az MRL vagy ML értékhez tartozó koncentráció plusz a laboratóriumon belüli reprodukálhatóság megengedett határérték melletti szórásának 1,64-szorosa adja a döntési határértéket (α = 5 %).
ii.
2. módszer: CCα = MRL (vagy ML) + k (egyoldalú, 95 %) × (kombinált) standard mérési bizonytalanság MRL vagy ML értéknél.
Az engedélyezett anyagoknál, a validálási kísérlettől (és annak adott szabadságfokától) függően, észszerűen alkalmazható a t-eloszlás vagy - a Gauss-eloszlás (egyoldalú, n=∞) alapul vétele esetén - 1,64 értékű k-faktor alkalmazandó.
A laboratóriumon belüli reprodukálhatóság és a valódiság alkalmas a (kombinált) standard mérési bizonytalanság meghatározására, ha annál minden releváns befolyásoló faktort figyelembe vesznek.
Azon gyógyszerhatóanyagok esetében, amelyeknél MRL van megállapítva a különböző anyagok összegéhez, a mintában lévő legmagasabb koncentrációjú anyag CCα értékét kell alkalmazni a mért mintában lévő anyagok összegének felmérési CCα értékeként.
b) Mátrix/fajkombinációkan lévő engedélyezett anyagok esetén, amelyekhez nincs rögzítve MRL, nem fordulhat elő maradékanyag, hacsak nem történt meg a 2001/82/EK irányelv 11. cikke szerinti engedélyezett kezelés. Engedélyezett anyagok esetén, amelyekhez nincs rögzítve MRL, a Bizottság (EU) 2018/470 végrehajtási rendelete ( 34 ) alapján megállapított lépcsőzetes MRL alkalmazandó a CCα számításához. A fenti bekezdés szerinti 1. vagy 2. módszer alkalmazandó, de az "MRL" jelentése "a lépcsőzetes MRL 0,5-szöröse, a lépcsőzetes MRL 0,1-szeresét célozva, amennyiben észszerűen megvalósítható".
2.7. Kimutatási képesség szűréshez (CCβ)
A szűrési módszerekhez szükség van a CCβ meghatározására. A CCβ megállapítását az e melléklet 1. fejezetében foglalt "Teljesítménykritériumok és egyéb követelmények analitikai módszerekhez" alapján és az 5. táblázatban foglalt követelmények szerint kell végezni. A szűrési módszerekhez azonban nem kell alkalmazni az azonosítás összes követelményét (vö. 1.2.3., 1.2.4., 1.2.5.).
1. A nem engedélyezett vagy tiltott gyógyszerhatóanyagoknál maximum 5 % β hiba biztosítandó. A CCβ számítása a következők szerint történik:
a)
1. módszer: Az ISO 11843-1:1997 szerinti kalibráló görbe eljárás (itteni hivatkozása a nettó állapotváltozó minimálisan kimutatható értéke). Ebben az esetben reprezentatív vakanyagot kell használni, amely ekvidisztáns osztásokban van megerősítve az RPA értékén és az alatt vagy - RPA hiányában - az STC körüli értéken. Végezze el a minták elemzését. Ábrázolja grafikusan a jel görbéjét a hozzáadott koncentrációval szemben. Az STC értékhez tartozó koncentráció plusz az STC értéknél mért átlagtartalom laboratóriumon belüli reprodukálhatósága szórásának 1,64-szerese adja a kimutatási képességet. A legalacsonyabb erősítési szinttől jóval elmaradó (a legalacsonyabb erősítési szintnél 50 %-kal kisebb) extrapolálást kísérleti adatokkal kell megerősíteni a validálási lépésnél.
b)
2. módszer: Megerősített vakanyag vizsgálata az eredetileg választott STC koncentrációszintjén. Ezen a koncentrációszinten - a meghatározás megbízható alapjának biztosítására - 20 megerősített vakanyagot kell elemezni. Ha ezen a koncentrációszinten a hamis megfelelt eredmények aránya továbbra is ≤ 5 %, akkor ez a szint jelenti a módszer kimutatási képességét. Ha a kapott hamis megfelelt eredmények aránya > 5 %, a választott STC-t meg kell emelni, és a vizsgálatot meg kell ismételni a legfeljebb 5 % hamis megfelelt eredményre vonatkozó követelménynek való megfelelés ellenőrzése érdekében.
c)
3. módszer: CCβ = STC + k (egyoldalú, 95 %) × (kombinált) standard mérési bizonytalanság STC értéknél vagy a felett.
A nem engedélyezett vagy tiltott gyógyszerhatóanyagoknál, a validálási kísérlettől (és annak adott szabadságfokától) függően, észszerűen alkalmazható a t-eloszlás vagy - a Gauss-eloszlás (egyoldalú, n=∞) alapul vétele esetén - 1,64 értékű k-faktor alkalmazandó.
A laboratóriumon belüli reprodukálhatóság és a valódiság alkalmas a (kombinált) standard mérési bizonytalanság meghatározására, ha annál minden releváns befolyásoló faktort figyelembe vesznek.
2. Az engedélyezett anyagoknál maximum 5 % β hiba biztosítandó. A CCβ számítása a következők szerint történik:
a)
1. módszer: az ISO 11843-1:1997 szerinti kalibráló görbe eljárással (itteni hivatkozása a nettó állapotváltozó minimálisan kimutatható értéke). Ebben az esetben reprezentatív vakanyagot kell használni, amely ekvidisztáns osztásokban van megerősítve a megengedett határértéken és az alatt, az STC értékről indulva. Végezze el a minták elemzését és az analit(ek) azonosítását. Számítsa ki az STC értéknél mért átlagtartalom szórását.
Az STC értékhez tartozó koncentráció plusz az STC értéknél mért átlagtartalom laboratóriumon belüli reprodukálhatósága szórásának 1,64-szerese adja a kimutatási képességet.
b)
2. módszer: megerősített vakanyag vizsgálatával a megengedett határérték alatti koncentrációszinteken. A meghatározás megbízható alapjának biztosításához minden koncentrációszinthez 20 megerősített vakanyagot kell elemezni. Az a koncentrációszint jelenti a módszer kimutatási képességét, ahol legfeljebb csupán 5 % hamis megfelelt eredmények maradnak.
c)
3. módszer: CCβ = STC + k (egyoldalú, 95 %) × (kombinált) standard mérési bizonytalanság STC értéknél vagy a felett.
Az engedélyezett anyagoknál, a validálási kísérlettől (és annak adott szabadságfokától) függően, észszerűen alkalmazható a t-eloszlás vagy - a Gauss-eloszlás (egyoldalú, n=∞) alapul vétele esetén - 1,64 értékű k-faktor alkalmazandó (lépcsőzetes alkalmazás vagy szokásos MRL alkalmazás esetén).
A laboratóriumon belüli reprodukálhatóság és a valódiság alkalmas a (kombinált) standard mérési bizonytalanság meghatározására, ha annál minden releváns befolyásoló faktort figyelembe vesznek.
2.8. Kalibráló görbék
Ha kalibráló görbékkel történik a számszerűsítés:
1. lehetőség szerint legalább öt ekvidisztáns szintet (a nulla szintet is ideértve) kell használni a görbe felépítéséhez;
2. le kell írni a görbe munkatartományát;
3. le kell írni a görbe matematikai képletét és az adatok görbéhez illeszkedési jóságát (R2 determinációs együttható);
4. le kell írni a görbeparaméterek elfogadhatósági tartományait.
Standard oldaton, mátrix-illesztett standardokon vagy mátrix-erősített standardokon alapuló kalibráló görbéknél jelezni kell a kalibráló görbe paramétereinek elfogadhatósági tartományait, amelyek sorozatonként változhatnak.
2.9. Abszolút visszanyerés
A módszer abszolút visszanyerését nem szükséges meghatározni, ha rendelkezésre áll belső standard, mátrix-erősített kalibrálás vagy mindkettő. Minden más esetben meg kell határozni a módszer abszolút visszanyerését.
Fix korrekciós tényező alkalmazható, ha teljesülnek az 1. táblázat szerinti valódisági követelmények. Egyéb esetben az adott tételre kapott visszanyerési tényező alkalmazandó. A visszanyerési korrekciós tényező helyett a standard hozzáadás ( 35 ) eljárása vagy belső standard is alkalmazható.
Az abszolút visszanyerést a mátrix legalább hat reprezentatív tételére ki kell számítani.
Az extrakció előtt egy alikvot vakmátrixot meg kell erősíteni az analittel, a minta elkészítése után pedig egy második alikvot vakmátrixot kell megerősíteni a releváns koncentrációszinten, majd meg kell határozni az analit koncentrációját.
A visszanyerés számítása a következők szerint történik:
Visszanyerés (analit) = (mátrix-erősített standard)/(mátrix-illesztett standard) × 100
2.10. Relatív mátrixhatások
A relatív mátrixhatást minden esetben meg kell határozni. Ez a validálás részeként vagy külön kísérletekben végezhető. A relatív mátrixhatás számítását a mátrixok/fajok legalább 20 különböző vaktételére kell elvégezni a módszer hatókörétől pl. a bevonandó különböző fajoktól függően.
Az extrakció után a vakmátrixot meg kell erősíteni az analittel az RPA, MRL vagy ML értékén, és az analit tiszta oldatával együtt kell elemezni.
A relatív mátrixhatás vagy mátrixfaktor (MF) számítása a következők szerint történik:
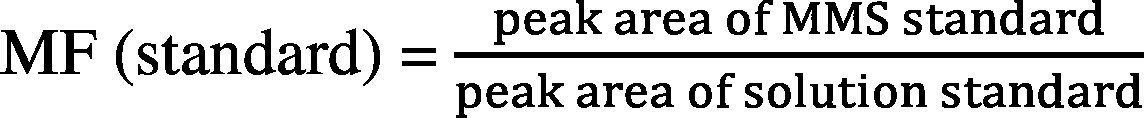
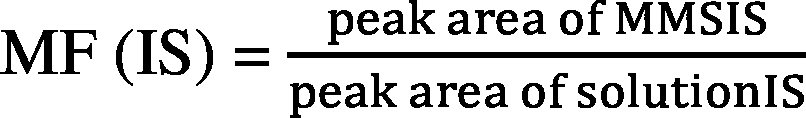
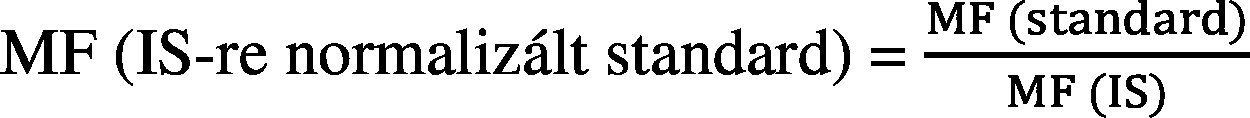
IS : belső standard
MMS : mátrix-illesztett standard
Az MF (IS-re normalizált standard) esetében a relatív szórás nem haladhatja meg az e melléklet 2. táblázatában megadott értékeket.
3. FEJEZET
RUTINELEMZÉS ALATTI MINŐSÉG-ELLENŐRZÉS - A MÓDSZER TELJESÍTMÉNYÉNEK FOLYAMATOS ELLENŐRZÉSE
Az ISO/IEC 17025:2017 ( 36 ) 7.7. fejezete szerinti analitikai eredmények minőségének biztosítására vonatkozó követelményeknek teljesülniük kell.
Rutinelemzés során a hiteles anyagminták (CRM-ek) elemzése jelenti a megfelelő opciót a módszer teljesítményének bizonyításához. Mivel az adott analiteket a szükséges koncentrációszinten tartalmazó CRM-ek ritkán állnak rendelkezésre, helyettük olyan referenciaanyagok is használhatók, amelyeket uniós referencialaboratóriumok vagy ISO/IEC 17043:2010 ( 37 ) akkreditációval rendelkező laboratóriumok biztosítanak és jellemeznek. További lehetőségként rendszeresen ellenőrzött házon belüli referenciaanyagok is alkalmazhatók.
Rutinelemzés során a hiteles anyagminták (CRM-ek) elemzése jelenti a megfelelő opciót a módszer teljesítményének bizonyításához. Mivel az adott analitokat a szükséges koncentrációszinten tartalmazó CRM-ek ritkán állnak rendelkezésre, helyettük olyan referenciaanyagok is használhatók, amelyeket uniós referencialaboratóriumok vagy ISO/IEC 17043:2023 ( 38 ) szerinti akkreditációval rendelkező laboratóriumok biztosítanak és jellemeznek. További lehetőségként rendszeresen ellenőrzött, házon belüli referenciaanyagok is alkalmazhatók.
1. A szűrési lépéshez:
Az elvégzett elemzések minden sorozata (tétele) esetében a következő minőségellenőrzési mintákat kell egyidejűleg elemezni:
a) kontrollminta a műszer rendszeralkalmasságához, ideális esetben módszerspecifikus;
b) az STC értékéhez közeli és ideális esetben a CCβ értéke szerinti koncentrációnál megerősített minőségellenőrzési minták az engedélyezett gyógyszerhatóanyagok, valamint a tiltott vagy nem engedélyezett anyagok szűréséhez;
c) megfelelt kontrollminták (vakminták) és adott esetben vakreagensek.
2. A megerősítő lépéshez:
Az elvégzett elemzések minden sorozata (tétele) esetében a következő minőségellenőrzési mintákat kell egyidejűleg elemezni:
a) kontrollminta a műszer rendszeralkalmasságához, ideális esetben módszerspecifikus;
b) engedélyezett gyógyszerhatóanyagoknál az MRL vagy ML értékéhez közeli, illetve tiltott vagy nem engedélyezett anyagoknál az RPA vagy LCL értékéhez közeli koncentrációnál megerősített minőségellenőrzési minták (nem-megfelelt kontrollminták);
c) megfelelt kontrollminták (vakminták) és adott esetben vakreagensek.
Az alábbi sorrend ajánlott a minőségellenőrzési mintákhoz: kontrollminta a műszer rendszeralkalmasságához, megfelelt kontrollminta, megerősítendő minta/minták, megerősítendő kontrollminta ismét, majd megerősített minőségellenőrzési minta (nem-megfelelt kontrollminta).
A kvantitatív módszereknél a hatósági minták minden tétele esetében a fenti minták előtt vagy után el kell végezni a kalibráló görbe elemzését és mérését.
Amennyiben megvalósítható, a nem-megfelelt kontrollmintákban lévő összes célzott analit (megerősített mintákon alapuló) valódiságát minőségellenőrzési grafikonok útján kell értékelni az ISO/IEC 17025:2017 7.7. fejezete szerint. Ha ez aránytalanul nagy számú valódiság meghatározást igényel, akkor az analitek száma a reprezentatív analitek számára csökkenthető.
4. FEJEZET
EGY ELŐZETESEN VALIDÁLT MÓDSZER VALIDÁLÁSI HATÓKÖRÉNEK KITERJESZTÉSE
Néha előfordul, hogy egy előzetesen átfogó módon validált módszer hatókörét ki kell terjeszteni. Ilyenkor a hatókör kiterjesztését hatékonyan és analitikai szempontból megbízhatóan kell elvégezni. Erre a teljes validáláshoz viszonyítva egy kisebb mintaszámon (pl. a minták felén) végzett validálás útján kerülhet sor.
Mindazonáltal a csökkentett validálás során validálandó módosítások típusát és számát mindig szakismeretek és korábbi tapasztalatok alapján kell megállapítani, pl. a detektálási technika módosítása bármely esetben teljes validálást tesz szükségessé.
Állandó érvényességének biztosításához a módszert általában folyamatosan figyelni kell és össze kell vetni a kezdetben kapott validálási paraméterekkel. A módszer teljesítményének folyamatos ellenőrzése ideális esetben úgy van kialakítva, hogy a teljes validáláshoz hiányzó adatok idővel összegyűjthetők (pl. az egyes analitikai sorozatok minőségellenőrzési mintáinak néhány adatpontjával).
4.1. Módszerek kiterjesztése a koncentrációtartományok tekintetében
Az MRL, ML és RPA értékek változásai miatt az adott módszer validálásához tartozó koncentrációtartomány kiigazítására. Ilyen esetben elfogadott a csökkentett validálás alkalmazása.
A módosított tartomány kalibráló görbéit a validált eljárás szerint kell elkészíteni. Különböző koncentrációszinteken megerősített tételeket (vö. 2.2.1., 2.2.2.) kell elemezni. A valódiság, ismételhetőség és laboratóriumon belüli reprodukálhatóság/közbenső precizitás elfogadható tartományon belül kell, hogy legyen a módszer eredeti validálásához viszonyítva. Amennyiben szükséges, úgy el kell végezni a CCβ (szűrési módszerek) és CCα (megerősítő módszerek) újraszámolását.
4.2. Módszerek kiterjesztése a további anyagok tekintetében
A módszerek további anyagokra való kiterjesztése általában csak olyan analiteknél lehetséges, amelyek az analitikai módszerbe már bevontakéhoz hasonló szerkezettel és jellemzőkkel rendelkeznek. Ilyen esetben elfogadott a csökkentett validálás alkalmazása. Emellett a módszer leírásától semmilyen eltérés nem lehetséges.
A további anyagok kalibráló görbéit a validált eljárás szerint kell elkészíteni. Különböző koncentrációszinteken megerősített mátrixanyag tételeket (vö. 2.2.1., 2.2.2.) kell elemezni. A valódiság, ismételhetőség és laboratóriumon belüli reprodukálhatóság/közbenső precizitás a módszer eredeti validálásához tartozó többi analitéhez képest hasonló tartományon belül és az 1.2.2. szerinti követelményeknek megfelelő kell, hogy legyen. Az új analitekhez el kell végezni a CCβ (szűrési módszerek) és CCα (megerősítő módszerek) kiszámolását.
4.3. Módszerek kiterjesztése a mátrixok/fajok tekintetében
Új mátrixok vagy fajok bevonása egy már validált analitikai módszerbe mindig csak eseti döntéssel történhet, a módszer kapcsán addig szerzett ismeretek és tapasztalatok, valamint a potenciális mátrixhatásokat és interferenciákat felmérő előzetes tapasztalatok alapján. Ez általában csak a hasonló tulajdonságokkal rendelkező mátrixok és a nem kritikus analitek esetében lehetséges (stabilitás, kimutathatóság).
A kalibráló görbéket (standard vagy mátrix) a validált eljárás szerint kell elkészíteni. Különböző koncentrációszinteken megerősített mátrixanyag tételeket (vö. 2.2.1., 2.2.2.) kell elemezni. A valódiság, ismételhetőség és laboratóriumon belüli reprodukálhatóság/közbenső precizitás a módszer eredeti validálásához képest elfogadható tartományon belül és az 1.2.2. szerinti követelményeknek megfelelő kell, hogy legyen. A validálási megközelítéstől függően a CCβ (szűrési módszerek) vagy CCα (megerősítő módszerek) újraszámolására lehet szükség.
Ha az eredeti mátrix értékeihez képest az eredmények nincsenek elfogadható tartományban, akkor egy további teljes validálásra lesz szükség a mátrix/fajok egyedi teljesítményparamétereinek meghatározása érdekében.
Ha egy adott anyag MRL értékei bizonyos mátrixoknál eltérést mutatnak, akkor valószínűleg nehéz lesz a módszer hatókörét a további mátrixhoz/fajhoz igazítani, mivel ebben az esetben két módosítást kell figyelembe venni. Ilyen esetben teljes validálás elvégzése ajánlott.
II. MELLÉKLET
MINTAVÉTELI ELJÁRÁSOK ÉS A HATÓSÁGI MINTÁK KEZELÉSE
1. Mintamennyiség
A minimális mintamennyiségeket az országos maradékanyag-ellenőrzési programban kell meghatározni. A minimális mintamennyiségnek elegendőnek kell lennie ahhoz, hogy a jóváhagyott laboratóriumok elvégezzék a szűrési és megerősítő elemzésekhez szükséges analitikai eljárásokat. Konkrétan a baromfi, akvakultúra, nyúl, tenyésztett vad, hüllők és rovarok esetében egy mintát egy vagy több állat alkot, az analitikai módszerek követelményeitől függően. Tojások esetében a minta mérete legalább 12 vagy több tojás, az alkalmazott elemzési módszerektől függően. Ha egyetlen mintán belül több anyagkategóriát kell különböző analitikai módszerekkel elemezni, akkor a mintaméretet ennek megfelelően kell növelni.
2. Almintákra történő felosztás
Hacsak technikailag nem megoldhatatlan, vagy a nemzeti jogszabályok nem írják ezt elő, minden mintát legalább két egyenértékű, az analitikai eljárás elvégzésére alkalmas almintára kell felosztani. A felosztás történhet a mintavételi helyen vagy a laboratóriumban.
3. Visszavezethetőség
Minden mintát úgy kell venni, hogy az visszavezethető legyen a származási gazdasághoz és az állatcsoporthoz vagy adott esetben a konkrét állathoz. A tagállam választása szerint különösen tej esetében lehet a mintavételt az alábbi helyek valamelyikén végezni:
1. a gazdaságban a gyűjtőtartályból;
2. tejipari szinten, a tej leengedése előtt.
4. Mintatároló edények
A mintákat az épség és a visszavezethetőség megőrzésére alkalmas tárolóedényekben kell gyűjteni. A tárolóedényeknek különösen a helyettesítést, a keresztszennyeződést és a lebomlását kell megakadályozniuk. A tárolóedényeket hatósági plombával kell lezárni.
5. Mintavételi jelentés
Minden mintavételi eljárás után jelentést kell készíteni.
Az ellenőr legalább a következő adatokat gyűjti be a mintavételi jelentésbe:
1. az illetékes hatóságok címe;
2. az ellenőr neve vagy azonosító kódja;
3. a minta hatósági kódszáma;
4. a mintavétel dátuma;
5. az állatok vagy az állati eredetű termékek tulajdonosának, illetve a kezelésükkel megbízott személynek a neve és a címe;
6. a gazdaság neve és címe, ahonnan az állat származik (gazdaságban történő mintavétel esetén);
7. a létesítmény nyilvántartási száma/vágóhíd száma;
8. az állat vagy a termék azonosítása;
9. az állat faja;
10. a mintamátrix;
11. adott esetben, a mintavételt megelőző négy hétben történt gyógyszeres kezelés (gazdaságban történő mintavétel esetén;
12. a vizsgálandó anyag vagy anyagcsoportok;
13. különleges megjegyzések.
A mintavételi eljárástól függően a jelentést nyomtatott vagy elektronikus formában kell benyújtani. A mintavételi jelentést és annak példányait a hitelességet és jogi érvényességet biztosító módon kell kitölteni, amelyhez szükség lehet e dokumentumok ellenőr általi aláírására. Gazdaságban történő mintavétel esetén a gazdálkodó vagy helyettese kérhető az eredeti mintavételi jelentés aláírására.
A mintavételi jelentés eredeti példánya az illetékes hatóságnál marad, amely garantálja, hogy jogosulatlan személyek nem juthatnak hozzá az eredeti jelentéshez.
Szükség esetén a gazdálkodó vagy a létesítmény tulajdonosa tájékoztatható az elvégzett mintavételről.
6. Laboratóriumi mintavételi jelentés
Az illetékes hatóságok által létrehozott laboratóriumi mintavételi jelentés megfelel az ISO/IEC 17025:2017 ( 39 ) 7. fejezetében foglalt követelményeknek és legalább az alábbi információkat tartalmazza:
1. az illetékes hatóságok vagy kijelölt szervek címe;
2. az ellenőr neve vagy azonosító kódja;
3. a minta hatósági kódszáma;
4. a mintavétel dátuma;
5. az állat faja;
6. a mintamátrix;
7. a vizsgálandó anyagok vagy anyagcsoportok;
8. különleges megjegyzések.
A laboratóriumba küldött mintát a laboratóriumi mintavételi jelentés kíséri.
7. Szállítás és tárolás
A maradékanyag-ellenőrzési programok meghatározzák a megfelelő tárolási és szállítási feltételeket minden egyes analit/mátrix kombinációnál az analit stabilitásának és a minta épségének biztosításához. A szállítási idő a lehető legrövidebb és a szállítási hőmérséklet megfelelő legyen az analit stabilitásának biztosításához.
Különös figyelmet kell fordítani a szállítódobozokra, a hőmérsékletre és a felelős laboratóriumba történő szállítás idejére.
Az ellenőrzési program követelményeinek be nem tartása esetén a laboratórium késedelem nélkül tájékoztatja az illetékes hatóságot.
( 1 ) A Bizottság (EU) 2022/1646 végrehajtási rendelete (2022. szeptember 23.) az állatgyógyászati készítményként vagy takarmány-adalékanyagként engedélyezett gyógyszerhatóanyagok, valamint a tiltott vagy nem engedélyezett gyógyszerhatóanyagok és azok maradékanyagai felhasználása tekintetében végzett hatósági ellenőrzésekre, a többéves nemzeti ellenőrzési tervek meghatározott tartalmára és az elkészítésükkel kapcsolatos különös intézkedésekre vonatkozó egységes gyakorlati szabályokról (HL L 248., 2022.9.26., 32. o., ELI: http://data.europa.eu/eli/reg_impl/2022/1646/oj).
( 2 ) A Bizottság (EU) 2019/2090 felhatalmazáson alapuló rendelete (2019. június 19.) az (EU) 2017/625 európai parlamenti és tanácsi rendeletnek az állatgyógyászati készítményekben vagy takarmány-adalékanyagként történő felhasználásra engedélyezett gyógyszerhatóanyagok, illetve a tiltott vagy nem engedélyezett gyógyszerhatóanyagok használatára vagy maradékanyagaira vonatkozó uniós szabályoknak való feltételezett vagy megállapított meg nem felelés esetei tekintetében történő kiegészítéséről (HL L 317., 2019.12.9., 28. o.).
( 3 ) A Bizottság (EU) 2019/1871 rendelete (2019. november 7.) az állati eredetű élelmiszerekben előforduló nem engedélyezett farmakológiai hatóanyagokra vonatkozó intézkedési referenciapontokról és a 2005/34/EK határozat hatályon kívül helyezéséről (HL L 289., 2019.11.8., 41. o.).
( 4 ) Az Európai Parlament és a Tanács 470/2009/EK rendelete (2009. május 6.) az állati eredetű élelmiszerekben előforduló farmakológiai hatóanyagok maradékanyag-határértékeinek meghatározására irányuló közösségi eljárásokról, a 2377/90/EGK tanácsi rendelet hatályon kívül helyezéséről, és a 2001/82/EK európai parlamenti és tanácsi irányelv, valamint a 726/2004/EK európai parlamenti és tanácsi rendelet módosításáról (HL L 152., 2009.6.16., 11. o.).
( 5 ) A Tanács 315/93/EGK rendelete (1993. február 8.) az élelmiszerekben előforduló szennyező anyagok ellenőrzésére vonatkozó közösségi eljárások megállapításáról (HL L 037., 1993.2.13., 1. o.).
( 6 ) ISO 3534-1: 2006 Statisztika - Szójegyzék és jelölések - 1. rész: Általános statisztikai és valószínűségi fogalmak (1. fejezet).
( 7 ) Az Európai Parlament és a Tanács 2001/82/EK irányelve (2001. november 6.) az állatgyógyászati készítmények közösségi kódexéről (HL L 311., 2001.11.28., 1. o.).
( 8 ) JCGM 200:2008, Nemzetközi metrológiai szótár - Alapvető és általános fogalmak és kapcsolódó kifejezések (VIM), harmadik kiadás 2008: https://www.iso.org/sites/JCGM/VIM-JCGM200.htm (5. fejezet Mérési szabványok (etalonok)).
( 9 ) A Bizottság 124/2009/EK rendelete (2009. február 10.) a nem céltakarmányokban elkerülhetetlen átvitel következtében jelen lévő kokcidiosztatikumoknak vagy hisztomonosztatikumoknak az élelmiszerekben megengedhető legnagyobb szintjeinek meghatározásáról (HL L 040., 2009.2.11., 7. o.).
( 10 ) A Bizottság 37/2010/EU rendelete (2009. december 22.) a farmakológiai hatóanyagokról és az állati eredetű élelmiszerekben előforduló maximális maradékanyag-határértékek szerinti osztályozásukról (HL L 015., 2010.1.20., 1. o.).
( 11 ) Codex Alimentarius Bizottság, ENSZ Élelmezési és Mezőgazdasági Szervezet/Egészségügyi Világszervezet, Analitikai terminológia iránymutatások (CAC/GL 72-2009).
( 12 ) ISO 5725-1:2023 Mérési módszerek és eredmények pontossága (valódiság és precizitás) - 1. rész: Általános elvek és meghatározások (3. fejezet).
( 13 ) ISO 80000-1:2022 Mennyiségek és mértékegységek - 1. rész: Általános (bevezetés).
( 14 ) A Tanács 80/181/EGK irányelve (1979. december 20.) a mértékegységekre vonatkozó tagállami jogszabályok közelítéséről és a 71/354/EGK irányelv hatályon kívül helyezéséről (HL L 39., 1980.2.15., 40. o., ELI: http://data.europa.eu/eli/dir/1980/181/oj).
( 15 ) ISO/IEC 17025:2017: Vizsgáló- és kalibrálólaboratóriumok felkészültségének általános követelményei. (3. fejezet).
( 16 ) ISO 78-2: 1999 Kémia - A szabványok elrendezése - 2. rész: Kémiai elemzési módszerek (Mellékletek).
( 17 ) A párhuzamos kromatográfia olyan eljárás, amelyben a kromatográfiás lépés(ek) előtt a minta extraktumot két részre osztják. Az egyik részt a szokásos módon kromatografálják. A másik részt összekeverik a mérendő standard analittel. Ezt a keveréket azután szintén kromatografálják. A hozzáadott standard analit mennyiségének és az extraktumban lévő analit becsült mennyiségének egymáshoz hasonlónak kell lennie. A párhuzamos kromatográfia rendeltetése az, hogy kromatográfiás módszerek alkalmazása esetén javítsa az analit azonosítását, különösen ha nem használható megfelelő belső standard.
( 18 ) ISO 5725-4:2020 Mérési módszerek és eredmények pontossága (valódiság és precizitás) - 4. rész: Alapmódszerek egy mértékadó mérési módszer valódiságának meghatározására (3. szakasz).
( 19 ) Amennyiben egy nem engedélyezett gyógyszerhatóanyag validálásához az RPA 0,5-szörösét kitevő koncentráció észszerűen nem érhető el, az RPA 0,5-szörösét kitevő koncentráció helyettesíthető az RPA 0,5-szöröse és 1,0-szorosa közötti azon legalacsonyabb koncentrációval, amely észszerűen elérhető, vagy pedig az LCL értékkel, amennyiben az alacsonyabb, mint az RPA 0,5-szöröse.
( 20 ) Amennyiben egy konkrét gyógyszerhatóanyag validálásához az MRL 0,1-szeresét kitevő koncentráció észszerűen nem érhető el, az MRL 0,1-szeresét kitevő koncentráció helyettesíthető az MRL 0,1-szerese és 0,5-szöröse közötti legalacsonyabb koncentrációval, amely észszerűen elérhető.
( 21 ) Amennyiben egy nem engedélyezett gyógyszerhatóanyag validálásához az RPA 0,5-szörösét kitevő koncentráció észszerűen nem érhető el, az RPA 0,5-szörösét kitevő koncentráció helyettesíthető az RPA 0,5-szöröse és 1,0-szorosa közötti azon legalacsonyabb koncentrációval, amely észszerűen elérhető, vagy pedig az LCL értékkel, amennyiben az alacsonyabb, mint az RPA 0,5-szöröse.
( 22 ) Amennyiben egy konkrét gyógyszerhatóanyag validálásához az MRL 0,1-szeresét kitevő koncentráció észszerűen nem érhető el, az MRL 0,1-szeresét kitevő koncentráció helyettesíthető az MRL 0,1-szerese és 0,5-szöröse közötti legalacsonyabb koncentrációval, amely észszerűen elérhető.
( 23 ) ISO 5725-2:2019 Mérési módszerek és eredmények pontossága (valódiság és precizitás) - 2. rész: Alapmódszer egy mértékadó mérési módszer ismételhetőségének és reprodukálhatóságának meghatározására (3. szakasz).
( 24 ) Amennyiben egy nem engedélyezett gyógyszerhatóanyag validálásához az RPA 0,5-szörösét kitevő koncentráció észszerűen nem érhető el, az RPA 0,5-szörösét kitevő koncentráció helyettesíthető az RPA 0,5-szöröse és 1,0-szorosa közötti azon legalacsonyabb koncentrációval, amely észszerűen elérhető, vagy pedig az LCL értékkel, amennyiben az alacsonyabb, mint az RPA 0,5-szöröse.
( 25 ) ISO 11843-1:1997 Kimutatási képesség - 1. rész: Kifejezések és fogalommeghatározások.
( 26 ) Codex Alimentarius Bizottság, ENSZ Élelmezési és Mezőgazdasági Szervezet, Világszervezet, Az eredmények bizonytalansági becslésére vonatkozó iránymutatások (CAC/GL 59-2006).
( 27 ) A mintaanyagok, analitek, tárolási körülmények, környezeti és/vagy mintakészítési viszonyok jelenthetik a kísérleti körülményekben bekövetkező hivatkozott változásokat. A gyakorlatban ingadozásnak kitett összes kísérleti körülmény (pl. a reagensek stabilitása, a minta összetétele, pH, hőmérséklet) esetében jelezni kell mindazokat az eltéréseket, amelyek befolyásolhatják az elemzési eredményt.
( 28 ) Jülicher, B., Gowik, P. and Uhlig, S. (1998) Assessment of detection methods in trace analysis by means of a statistically based in-house validation concept (Nyomelemzési kimutatási módszerek kiértékelése egy statisztikai alapú, házon belüli validálási koncepció útján). Analyst, 123, 173.
( 29 ) ISO/TS 23471:2022 - Kísérleti tervek a bizonytalanság értékeléséhez. Faktoriális tervezés alkalmazása a bizonytalansági függvények meghatározásánál.
( 30 ) IUPAC (1995), Protocol for the design, conduct and interpretation of method-performance studies (Protokoll a módszer teljesítményvizsgálatok tervezéséhez, lefolytatásához és értelmezéséhez), Pure & Applied Chem, 67, 331.
( 31 ) Gowik, P., Jülicher, B. and Uhlig, S. (1998) Multi-residue method for non-steroidal anti-inflammatory drugs in plasma using high performance liquid chromatography-photodiode-array detection (Fotodiódasoros kimutatást alkalmazó nagy teljesítményű folyadékkromatográfiával végzett multi-szermaradék módszer plazmában előforduló nem szteroid gyulladáscsökkentő gyógyszerekhez). Method description and comprehensive in-house validation (Módszerleírás és átfogó házon belüli validálás). J. Chromatogr., 716, 221.
( 32 ) Lamberty, A., Schimmel, H. and Pauwels, J. (1998) The study of the stability of reference materials by isochronous measurements (Referenciaanyagok stabilitásának vizsgálata izokron mérésekkel). Fres. J. Anal. Chem. 360, 359.
( 33 ) ISO 11843-1:1997 Kimutatási képesség - 1. rész: Kifejezések és fogalommeghatározások.
( 34 ) A Bizottság (EU) 2018/470 végrehajtási rendelete (2018. március 21.) a 2001/82/EK irányelv 11. cikkének rendelkezései szerint az EU-ban kezelt állatokból származó élelmiszerek ellenőrzése céljára alkalmazandó maradékanyag-határértékekre vonatkozó részletes szabályokról (HL L 79., 2018.3.22., 16. o.).
( 35 ) A hozzáadott standard analit mennyisége a mintában lévő analit becsült mennyiségének kétszerese-ötszöröse lehet. Az eljárás célja a mintában levő analit-tartalom meghatározása, figyelembe véve az analitikai eljárás visszanyerését.
( 36 ) ISO/IEC 17025: 2017 Vizsgáló- és kalibrálólaboratóriumok felkészültségének általános követelményei (7.7. fejezet).
( 37 ) ISO/IEC 17043:2010 Megfelelőségértékelés - Jártassági vizsgálatok általános követelményei.
( 38 ) ISO/IEC 17043:2023 Megfelelőségértékelés - Jártassági vizsgálatok általános követelményei.
( 39 ) ISO/IEC 17025: 2017 Vizsgáló- és kalibrálólaboratóriumok felkészültségének általános követelményei (7.7. fejezet).
Lábjegyzetek:
[1] A dokumentum eredetije megtekinthető CELEX: 32021R0808 - https://eur-lex.europa.eu/legal-content/HU/ALL/?uri=CELEX:32021R0808&locale=hu Utolsó elérhető, magyar nyelvű konszolidált változat CELEX: 02021R0808-20250217 - https://eur-lex.europa.eu/legal-content/HU/ALL/?uri=CELEX:02021R0808-20250217&locale=hu