
31978L0633[1]
A Bizottság nyolcadik irányelve (1978. június 15.) a takarmányok hatósági ellenőrzésére szolgáló közösségi analitikai módszerek meghatározásáról
A BIZOTTSÁG NYOLCADIK IRÁNYELVE
(1978. június 15.)
a takarmányok hatósági ellenőrzésére szolgáló közösségi analitikai módszerek meghatározásáról
(78/633/EGK)
AZ EURÓPAI KÖZÖSSÉGEK BIZOTTSÁGA,
tekintettel az Európai Gazdasági Közösséget létrehozó szerződésre,
tekintettel a legutóbb a csatlakozási okmánnyal módosított, a takarmányok hatósági ellenőrzésénél alkalmazandó közösségi mintavételi és analitikai módszerek bevezetéséről szóló, 1970. július 20-i 70/373/EGK bizottsági irányelvre ( 1 ) és különösen annak 2. cikkére,
mivel az említett irányelv előírja, hogy a takarmányok hatósági ellenőrzését, amelynek célja a takarmányok minőségére és összetételére vonatkozó törvényi, rendeleti és közigazgatási rendelkezésekből eredő követelmények betartásának ellenőrzése, közösségi mintavételi és analitikai módszerek alkalmazásával kell végrehajtani;
mivel az 1971. június 15-i 71/250/EGK ( 2 ), az 1971. november 18-i 71/393/EGK ( 3 ), az 1972. április 27-i 72/199/EGK ( 4 ), az 1972. december 5-i 73/46/EGK ( 5 ), az 1974. március 25-i 74/203/EGK ( 6 ), az 1974. december 20-i 75/84/EGK ( 7 ) és az 1976. március 1-i 76/372/EGK ( 8 ) bizottsági irányelvek már számos közösségi analitikai módszert meghatároztak; mivel az azóta eltelt időszakban, az adott területen mutatkozó előrehaladás miatt ajánlatos egy nyolcadik módszersorozat elfogadása;
mivel az ebben az irányelvben előírt intézkedések összhangban vannak a Takarmányok Állandó Bizottsága véleményével,
ELFOGADTA EZT AZ IRÁNYELVET:
1. cikk
(1) A tagállamok előírják, hogy a takarmányok hatósági ellenőrzésére szolgáló, azok cinkbacitracin-, flavofoszfolipol-, vas-, réz-, mangán- és cinktartalmára vonatkozó analitikai vizsgálatokat ezen irányelv mellékletében ismertetett módszerek szerint kell végrehajtani.
2. cikk
A tagállamok legkésőbb 1979. január 1-ig hatályba léptetik azon törvényi, rendeleti és közigazgatási rendelkezéseket, amelyek szükségesek ahhoz, hogy ennek az irányelvnek megfeleljenek. Erről haladéktalanul tájékoztatják a Bizottságot.
3. cikk
Ennek az irányelvnek a tagállamok a címzettjei.
MELLÉKLET
1. CINK BACITRACIN MEGHATÁROZÁSA
- agar táptalajon végzett diffúzióval -
1. CÉL ÉS ALKALMAZÁSI TERÜLET
A módszer a cink bacitracin takarmányokban és előkeverékekben történő meghatározására szolgál. A meghatározás alsó határa 5 mg/kg (5 ppm) ( 9 ).
2. VIZSGÁLATI ALAPELV
A pH 2-es mintát metil-alkohol/víz/sósav keverékével és nátrium-szulfid oldattal extraháljuk. A nátrium-szulfid hozzáadása a vizsgálat során esetleg interferenciát eredményező oldható réz-sók kicsapatására szolgál. A kivonatot beállítjuk pH 6,5-re, koncentráljuk (szükség esetén) és felhígítjuk. Antibiotikus aktivitását a cink bacitracin Micrococcus luteussal (flavussal) beoltott agar táptalajon végzett diffúziójának mérésével határozzuk meg. A diffúziót a mikroorganizmus gátlási zónáinak képződése mutatja. Ezeknek a zónáknak az átmérője egyenes arányban áll az antibiotikum-koncentrációnak az alkalmazott antibiotikum-koncentrációk tartománya feletti logaritmusával.
3. MIKROORGANIZMUS: MICROCOCCUS LUTEUS (FLAVUS) ATCC 10240
3.1. A törzstenyészet fenntartása
Oltsunk be ferde táptalajokat (4.1) tartalmazó csöveket Micrococcus luteussal (flavus), majd inkubáljuk 30 oC-on 24 órán át. A tenyészetet hűtőgépben tároljuk, 4 oC körüli hőmérsékleten. Kéthetenként oltsuk át.
3.2. A baktériumszuszpenzió elkészítése ( 10 )
2-3 ml nátrium-klorid oldat (4.3) segítségével gyűjtsük be egy nemrégiben elkészített ferde agarról (3.1) a növekményt. Ezzel a szuszpenzióval oltsunk be 250 ml, Roux-lombikban lévő táptalajt (4.1) és inkubáljuk 30 oC-on 18-20 órán át. Gyűjtsük be a növekményt 25 ml nátrium-klorid oldatban (4.3), majd keverjük össze. Hígítsuk fel a szuszpenziót nátrium-klorid oldattal (4.3) 1:10 arányban. A szuszpenziónak 650 nm-en, 1 cm-es cellában, a nátrium-klorid oldat (4.3) ellenében mért fényáteresztő képessége 75 % körüli kell, hogy legyen. Ez a szuszpenzió 4 oC körüli hőmérsékleten egy hétig tárolható.
4. TÁPTALAJOK ÉS REAGENSEK
4.1. Alaptáptalaj ( 11 )
| Hús pepton | 6,0 g |
| Tripton | 4,0 g |
| Élesztőkivonat | 3,0 g |
| Húskivonat | 1,5 g |
| Glükóz | 1,0 g |
| Agar | 10,0–20,0 g |
| Víz | 1 000 ml |
| pH 6,56,6 (sterilizálás után) |
4.2. Vizsgálati táptalaj (11)
| Tripton | 10,0 g |
| Élesztőkivonat | 3,0 g |
| Húskivonat | 1,5 g |
| Glükóz | 1,0 g |
| Agar | 10,0–20,0 g |
| Tween 80 | 1 ml |
| Víz | 1 000 ml |
| pH 6,5 (sterilizálás után) |
4.3. 0,8 %-os (w/v) nátrium-klorid oldat: oldjunk fel vízben 8 g nátrium-kloridot, majd hígítsuk 1 000 ml-re; sterilizáljuk.
4.4. Metil-alkohol/víz/sósav keveréke (4.6):
80/17,5/2,5 (v/v/v).
4.5. Foszfátpuffer, pH 6,5:
| Kálium–hidrogén-foszfát K2HPO4 | 22,15 g |
| Kálium-dihidrogén-foszfát KH2PO4 | 27,85 g |
| Vízzel kiegészítve | 1 000 ml-re |
4.6. Sósav (d: 1,18-1,19).
4.7. Sósav (0,1 M).
4.8. Nátrium-hidroxid 1 M oldat.
4.9. Nátrium-szulfid, körülbelül 0,5 M oldat.
4.10. Brómkrezol lila oldat 0,04 % (w/v): oldjunk fel 0,1 g brómkrezol lilát 18,5 ml 0,01 M nátrium-hidroxid oldatban. Töltsük fel vízzel 250 ml-re, majd keverjük össze.
4.11. Standard anyag: ismert aktivitású cink bacitracin (n.e.-ben).
5. STANDARD OLDATOK
Mérjünk ki (a jelzett aktivitás szerint) 1 050 n.e.-nek megfelelő standard cink bacitracint (4.11). Adjunk hozzá 5 ml 0,1 M sósavat (4.7) és 15 percig hagyjuk állni. Adjunk hozzá 30 ml vizet, foszfátpufferrel (4.5) (kb. 4 ml) állítsuk be pH 4,5-re, vízzel töltsük fel 50 ml-re, majd alaposan keverjük össze (1 ml = 21 n.e.).
Ebből a törzsoldatból, foszfátpufferrel (4.5) történő egymást követő hígítással készítsük el a következő oldatokat:
| s8 | 0,42 | n.e./ml |
| s4 | 0,21 | n.e./ml |
| s2 | 0,105 | n.e./ml |
| s1 | 0,0525 | n.e./ml |
6. A KIVONAT ÉS A VIZSGÁLATI OLDATOK ELKÉSZÍTÉSE
6.1. Extrakció
6.1.1. előkeverékek és ásványi takarmányok
Mérjünk ki 2,0- 5,0 g mintát, adjunk hozzá 29,0 ml-t a keverékből (4.4), valamint 1,0 ml nátrium-szulfid oldatot (4.9), majd rázassuk rövid ideig. Ellenőrizzük, hogy a pH érték 2 körül legyen. Rázzuk 10 percig, majd adjunk hozzá 30 ml foszfátpuffert (4.5), rázzuk 15 percig, majd centrifugáljuk. A felülúszó oldatból vegyünk egy megfelelő aliquot mennyiséget és pH-mérő vagy brómkrezol lila oldat (4.10) indikátor segítségével, 1 M nátrium-hidroxid oldattal (4.8) állítsuk be a pH-t 6,5-re. Hígítsuk fel foszfátpufferrel (4.5), hogy megközelítően 0,42 n.e./ml cink bacitracin tartalmat (= u8) kapjunk.
6.1.2. Fehérjekoncentrátumok
Mérjünk ki 10,0 g mintát, adjunk hozzá 49,0 ml-t a keverékből (4.4) valamint 1,0 ml nátrium-szulfid oldatot (4.9), majd rázassuk rövid ideig. Ellenőrizzük, hogy a pH-érték 2 körül legyen. Rázzuk 10 percig, majd adjunk hozzá 50 ml foszfátpuffert (4.5), rázzuk 15 percig, majd centrifugáljuk. A felülúszó oldatból vegyünk egy megfelelő mennyiséget és pH-mérő vagy brómkrezol lila oldat (4.10) indikátor segítségével, 1 M nátrium-hidroxid oldattal (4.8) állítsuk be a pH-t 6,5-re. Rotációs bepárlóban, 35 oC-ot meg nem haladó hőmérsékleten pároljuk be körülbelül a mennyiség felét.
Hígítsuk fel foszfátpufferrel (4.5), hogy megközelítőleg 0,42 n.e./ml cink bacitracin tartalmat (= u8) kapjunk.
6.1.3. Egyéb takarmányok
Mérjünk ki 10,0 g mintát (20 grammot 5 mg/kg-os várt cink bacitracin tartalom esetében). Adjunk hozzá 24,0 ml-t a keverékből (4.4), valamint 1,0 ml nátrium-szulfid oldatot (4.9), majd 10 percig homogenizáljuk. Adjunk hozzá 25 ml foszfátpuffert (4.5), rázzuk 15 percig, majd centrifugáljuk. A felülúszó oldatból vegyünk 20 ml-t és pH-mérő vagy brómkrezol lila oldat (4.10) indikátor segítségével, 1 M nátrium-hidroxid oldattal (4.8) állítsuk be a pH-t 6,5-re. Rotációs bepárlóban,35 oC-ot meg nem haladó hőmérsékleten pároljunk be körülbelül 4 ml-re. A maradék anyagot hígítsuk fel foszfátpufferrel (4.5), hogy 0,42 n.e./ml-es várt cink bacitracin tartalmat (= u8) kapjunk.
6.2. Vizsgálati oldatok
Az u8 oldatból a foszfátpufferrel (4.5) való egymást követő hígításokkal (1 + 1) készítsünk u4 (várt tartalom: 0,21 n.e./ml), u2 (várt tartalom: 0,105 n.e./ml) és u1 (várt tartalom: 0,0525 n.e./ml) oldatokat.
7. A VIZSGÁLATI MÓDJA
7.1. A vizsgálati táptalaj beoltása
Oltsuk be a vizsgálati táptalajt (4.2) a baktériumszuszpenzióval (3.2), körülbelül 50 oC-on. A vizsgálati táptalajjal (4.2) öntött lemezeken végzett előzetes vizsgálatokkal határozzuk meg azt a szükséges baktériumszuszpenzió mennyiséget, amely a különböző cink bacitracin koncentrációkkal a legnagyobb és legtisztább gátlási zónákat adja.
7.2. A lemezek elkészítése
Az agardiffúziót a négy standard oldat koncentrációjával (s8, s4, s2, s1) és a négy vizsgálati oldat koncentrációjával (u8, u4, u2, u1) öntött lemezeken végezzük. A standardnak és a kivonatnak ezt a négy-négy koncentrációját minden egyes lemezre fel kell vinnünk. Ezért megfelelő nagyságú lemezeket kell választanunk ahhoz, hogy az agar táptalajban legalább nyolc darab, 10-13 mm átmérőjű, egymástól legalább 30 mm távolságra lévő lyukat alakíthassunk ki. A vizsgálat olyan lemezeken végezhető el, amelyek egy üveglapból és egy annak tetejére helyezett 200 mm átmérőjű és 20 mm magas csiszolt alumínium vagy műanyag karimagyűrűből állnak.
A lemezekre öntsünk annyi, a 7.1 pont szerint beoltott táptalajt (4.2), hogy az egy körülbelül 2 mm vastagságú réteget alkosson (200 mm átmérőjű lemez esetében ez 60 ml). Hagyjuk egyenletesen eloszlani, fúrjuk ki a lyukakat, és helyezzük el bennük a vizsgálati és standard oldatok pontosan kimért mennyiségeit (az átmérő függvényében lyukanként 0,10-0,15 ml). Minden koncentrációt legalább négyszer alkalmazzunk, hogy minden meghatározás 32 gátlási zóna kiértékelése alapján történjen.
7.3. Inkubáció
Inkubáljuk a lemezeket 30 ± 2 oC-on, 16-18 órán át.
8. KIÉRTÉKELÉS
Mérjük meg a gátlási zónák átmérőjét 0,1 mm-es pontossággal. Fél-logaritmikus milliméterpapírra jegyezzük fel az egyes koncentrációknál mért eredmények középértékeit, jelezve a koncentrációknak a gátlási zónák átmérőihez vonatkoztatott logaritmusát. Szerkesszük meg mind a standard oldat, mind a kivonat regressziós vonalát, például az alábbiak szerint:
Határozzuk meg a standard legalacsonyabb szint (SA) regressziós pontját a következő képlet segítségével:
(a)
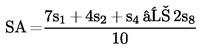
Határozzuk meg a standard legmagasabb szint (SM) regressziós pontját a következő képlet segítségével:
(b)
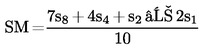
Hasonló módon számítsuk ki a regressziós pontokat a kivonat legalacsonyabb szintjére (UA) és a kivonat legmagasabb szintjére (UM), a fenti képletekben az s1, s2, s4 és s8 értékek helyett az u1, u2, u4 és u8 értékeket használva.
Jegyezzük fel a kiszámított SA és SM értékeket ugyanarra a milliméterpapírra és kössük össze őket, hogy megkapjuk a standard oldat regressziós vonalát. Hasonló módon jegyezzük fel az UA és UM értékeket és kössük össze őket, hogy megkapjuk a kivonat regressziós vonalát.
Ha interferencia nem lép fel, a vonalaknak párhuzamosnak kell lenniük. Gyakorlati megfontolásból a vonalakat párhuzamosnak tekinthetjük, ha az (SM - SA) és (UM - UA) érték középértékeiktől való eltérése nem haladja meg a 10 %-ot.
Ha a vonalak nem tekinthetőek párhuzamosnak, akkor akár az u1 és s1, akár az u8 és s8 elhagyható, és az SA, SM, UA és UM értékek alternatív képletekkel számolhatók ki, hogy megkapjuk az alternatív regressziós vonalakat:
| (a′) SA = | [Kép #1] | vagy | [Kép #2] |
| (b′) SM = | [Kép #3] | vagy | [Kép #4] |
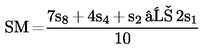
Kép #1
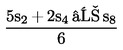
Kép #2
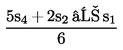
Kép #3
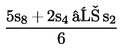
Kép #4
és hasonló módon az UA-ra és UM-ra vonatkozóan. Ugyanazt a párhuzamossági feltételt kell teljesíteni. Azt a tényt, hogy az eredmény három tényező alapján került kiszámításra, fel kell tüntetni a zárójelentésben.
Ha a vonalak párhuzamosnak tekinthetőek, akkor számoljuk ki a relatív aktivitás (A) logaritmusát (log A) a következő képletek egyikének felhasználásával, attól függően, hogy három vagy négy tényező alapján történt-e a párhuzamosság megállapítása.
Négy tényező esetén
(c)
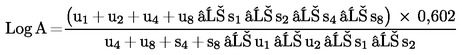
Három tényező esetén
(d)
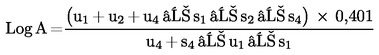
vagy
(d′)
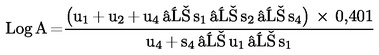
A mintakivonat aktivitása = a vonatkozó standard aktivitása x A
(u8 = s8 × A)
Ha a relatív aktivitás a 0,5 és 2,0 közötti tartományon kívül esik, akkor ismételjük meg a vizsgálatot a kivonat koncentrációin, vagy ahol ez nem lehetséges, ott a standard oldatokon végzett megfelelő módosításokkal. Ha a relatív aktivitást nem tudjuk az előírt tartományon belülre korrigálni, akkor bármely kapott eredményt közelítő eredménynek kell tekinteni, és ezt fel kell tüntetni a zárójelentésen.
Ha a vonalak nem tekinthetőek párhuzamosnak, meg kell ismételni a meghatározást. Ha a párhuzamosságot még ezután nem sikerült elérni, akkor a meghatározást nem kielégítőnek kell tekinteni.
Az eredményt milligrammban kifejezett cink bacitracinre és kilogrammban kifejezett takarmányra vonatkoztatva fejezzük ki.
9. ISMÉTELHETŐSÉG
Az ugyanazon mintán, ugyanazon analitikus által párhuzamosan végzett két meghatározás eredménye közötti különbség nem haladhatja meg:
- a 10 mg/kg alatti cink bacitracin tartalom esetén a 2 mg/kg-ot, abszolút értékben,
- a 10 és 25 mg/kg közötti tartalom esetén a legmagasabb érték 20 %-át,
- a 25 és 50 mg/kg közötti tartalom esetén az 5 mg/kg-ot, abszolút értékben,
- az 50 mg/kg feletti tartalom esetén a legmagasabb érték 10 %-át.
2. A FLAVOFOSZFOLIPOL AGAR TÁPTALAJON, DIFFÚZIÓ ÚTJÁN TÖRTÉNŐ MEGHATÁROZÁSA
1. CÉL ÉS ALKALMAZÁSI TERÜLET
A módszer a takarmányokban, koncentrátumokban és előkeverékekben található flavofoszfolipol meghatározására szolgál. A meghatározás alsó méréshatára 1 mg/kg (1 ppm).
2. VIZSGÁLATI ALAPELV
A mintát hígított metil-alkohollal, visszafolyós hűtő alatt hevítve extraháljuk. Centrifugálás után, a kivonatot ioncserélő gyantával történő kezeléssel derítjük (szükség esetén), majd hígítjuk. Antibiotikus aktivitását a flavofoszfolipol, Staphylococcus aureusszal beoltott agar táptalajon végbemenő diffúziójának mérésével határozzuk meg. A diffúziót a mikroorganizmus gátlási zónáinak képződése jelzi. E zónák átmérője egyenes arányos az antibiotikum-koncentrációnak az alkalmazott antibiotikum-koncentrációk tartománya feletti logaritmusával.
3. A MIKROORGANIZMUS: STAPHYLOCOCCUS AUREUS ATCC 6538 P
3.1. A törzstenyészet fenntartása
Oltsunk Staphylococcus aureusszal ferde agar táptalajt (4.1.). 37 °C-on, 24 órán keresztül inkubáljuk, tároljuk hűtőszekrényben kb. 4 °C-on, és havonta oltsuk át ferde agarra.
3.2. A baktérium-szuszpenzió elkészítése ( 12 )
Tegyünk félre két, törzstenyészetet tartalmazó (3.1.) csövet és hetente oltsuk át őket. 37 °C-on, 24 órán keresztül inkubáljuk, és tároljuk hűtőszekrényben kb. 4 °C-on.
A vizsgálat megkezdése előtt 24 órával, oltsunk be ezzel a szaporulattal kettő-négy, ferde agar táptalajt (4.1.) tartalmazó csövet. 37 °C-on, 16-18 órán keresztül inkubáljuk. Készítsük el a szaporulat szuszpenzióját nátrium-klorid oldatban (4.3.). A szuszpenziónak, 578 nm-en mérve, 1 cm-es mérőcellában, nátrium-klorid oldat ellenében (4.3.) körülbelül 40 %-os fényáteresztésűnek kell lennie.
4. TÁPTALAJOK ÉS REAGENSEK
4.1. Alaptáptalaj ( 13 )
| Húspepton | 6,0 g |
| Tripton | 4,0 g |
| Élesztőkivonat | 3,0 g |
| Húskivonat | 1,5 g |
| Glükóz | 1,0 g |
| Agar | 15,0 g |
| Víz | 1 000 ml |
| Sterilizálás utáni pH: 6,5 |
4.2. Vizsgálati táptalaj
4.2.1. Alapréteg ( 14 )
| Húspepton | 6,0 g |
| Élesztőkivonat | 3,0 g |
| Húskivonat | 1,5 g |
| Agar | 10,0 g |
| Víz | 1 000 ml |
| Sterilizálás utáni pH: 6,5 |
4.2.2. Oltási réteg
Mint a 4.1 pontban, 2,0 g szilikon habzásgátló emulzió ( 15 ) hozzáadásával.
4.3. 0,4 %-os (w/v) nátrium-klorid oldat: oldjunk fel 4 g nátrium-kloridot a.t. vízben, és hígítsuk fel 1 000 ml-re; sterilizáljuk.
4.4. Tiszta metil-alkohol.
4.5. 50 %-os (v/v) metil-alkohol: hígítsunk fel 500 ml metil-alkoholt (4.4.) 500 ml vízzel.
4.6. 80 %-os (v/v) metil-alkohol: hígítsunk fel 800 ml metil-alkoholt (4.4.) 200 ml vízzel.
4.7. Trisz-hidroximetil-aminometán, a.t.
4.8. Kálium-klorid 1,5 %-os (w/v) metil-alkoholos oldata: oldjunk fel 1,5 g a.t. kálium-kloridot 20 ml vízben, töltsük fel metil-alkohollal (4.4.) 100 ml-re.
4.9. Kationcserélő: Dowex 50 W × 8, 20-50 szemnagyságú, Na-típusú (Serva kat. 41600-as számú) vagy ezzel egyenértékű.
4.10. Anioncserélő: Dowex 1 × 2, 50-100 szemnagyságú, Cl-típusú (Serva kat. 41010-es számú) vagy ezzel egyenértékű. Használat előtt tartsa 12-14 óráig 80 %-os metil-alkoholban (4.6.).
4.11. Üvegvatta.
4.12. pH indikátorpapír (pH 6,6-8,1).
4.13. Aszkorbinsav.
4.14. Standard anyag: ismert aktivitású flavofoszfolipol.
5. ESZKÖZÖK
5.1. Kromatográfiás üvegcső, belső átmérője 9 mm, hossza 150-200 mm, szűkített alsó részénél elzárócsappal, felső részénél üvegcsiszolattal (ami összeköti a csepegtetőtölcsérrel [5.2.]) ellátott.
5.2. 250 ml-es csepegtetőtölcsér, elzárócsappal és üvegcsiszolattal.
5.3. 250 ml-es Erlenmeyer-lombik üvegcsiszolattal.
5.4. Visszafolyós hűtő üvegcsiszolattal.
6. STANDARD OLDATOK
Oldjunk fel egy pontosan kimért mennyiségű standard anyagot (4.14.) 50 %-os metil-alkoholban (4.5.) és hígítsuk fel úgy, hogy 100 μg/ml flavofoszfolipolt tartalmazó törzsoldatot kapjunk. Lezárt lombikokban, 4 °C-on tárolva az oldat két hónapig stabil marad.
Ezen törzsoldatból, 50 %-os metil-alkohollal (4.5.) történő többszöri hígítással készítsük el a következő oldatokat:
| S8 | 0,2 | μg/ml |
| S4 | 0,1 | μg/ml |
| S2 | 0,05 | μg/ml |
| S1 | 0,025 | μg/ml |
7. A KIVONAT ELKÉSZÍTÉSE
7.1. Extrahálás
7.1.1. Koncentrátumok, előkeverékek és ásványi takarmányok
Mérjünk ki 2,0-5,0 g mintát, és adjunk hozzá kb. 150 mg aszkorbinsavat (4.13.). Homogenizáljuk 150 ml 50 %-os metil-alkohollal (4.5.) egy Erlenmeyer-lombikban (5.3.), és kb. 400 mg trisz-hidroximetil-aminometán (4.7.) segítségével állítsuk be a pH-t 8,1-8,2-re. Ellenőrizzük a pH-t indikátorpapírral (4.12.). Hagyjuk állni 15 percig, azután újra állítsuk be a pH-t trisz-hidroximetil-aminometánnal (4.7.) 8,1-8,2-re, majd forraljuk 10 percig visszafolyós hűtő alatt (5.4.), állandó keveréssel. Hagyjuk lehűlni, centrifugáljuk a keveréket, és dekantáljuk a kivonatot.
7.1.2. Egyéb takarmányok
Mérjünk ki egy legalább 30 μg flavofoszfolipolt tartalmazó, 5,0-30,0 g tömegű mintát. Homogenizáljuk 150 ml 50 %-os metil-alkohollal (4.5.) egy Erlenmeyer-lombikban (5.3.), és kb. 400 mg trisz-hidroximetil-aminometán (4.7.) segítségével állítsuk be a pH-t 8,1-8,2-re. Ellenőrizzük a pH-t indikátorpapírral (4.12.). Hagyjuk állni 15 percig, azután újra állítsuk be a pH-t trisz-hidroximetil-aminometánnal (4.7.) 8,1-8,2-re, majd forraljuk 10 percig visszafolyós hűtő alatt (5.4.), állandó keveréssel. Hagyjuk kihűlni, centrifugáljuk a keveréket, és dekantáljuk a kivonatot.
7.2. Derítés (a vizsgálat ezen szakasza koncentrátumok, előkeverékek és ásványi takarmányok esetében elhagyható)
Keverjünk össze 110 ml kivonatot 11 g kationcserélővel (4.9.), forraljuk egy percig visszafolyós hűtő alatt (5.4.), állandó keveréssel. Válasszuk le a kationcserélőt centrifugálással vagy szűréssel. Keverjünk össze 100 ml kivonatot 150 ml metil-alkohollal (4.4.), majd tároljuk az oldatot 12-15 óráig 4 °C-on. Szűrjük le még hidegen a flokkulens anyagot.
Helyezzünk egy üvegvatta dugót (4.11.) az üvegcső aljára (5.1.), öntsünk a csőbe 5 ml anioncserélőt (4.10.) és mossuk az oszlopot 100 ml, 80 %-os metil-alkohollal (4.6.). A csepegtetőtölcsér (5.2.) segítségével öntsünk át az oszlopba legalább 100 ml szűrletet, amely várhatóan 16 μg flavofoszfolipolt tartalmaz (30 g-os takarmányminta esetében, 1 ppm-nél 200 ml-t). Adott esetben, mielőtt az oszlopba töltenénk, hígítsuk a szűrletet 80 %-os metil-alkohollal (4.6.), hogy a várt 16 μg/100 ml koncentrációjú flavofoszfolipol-tartalmat kapjuk. Állítsuk be az átfolyási sebességet kb. 2 ml/perc értékre. Öntsük el a szüredéket. Ezután mossuk az oszlopot 50 ml, 80 %-os metil-alkohollal (4.6.) és öntsük el a szüredéket.
Eluáljuk a flavofoszfolipolt kálium-klorid metil-alkoholos oldatával (4.8.), az átfolyási sebességet kb. 2 ml/perc értéken tartva. Vegyünk fel 50 ml eluátumot egy mérőlombikban, adjunk hozzá 30 ml vizet, és keverjük össze. Ezen oldat flavofoszfolipol-tartalmának 0,2 μg/ml-nek kell lennie (= U8).
7.3. Vizsgálati oldatok
Adott esetben (pl. a derítés kihagyása esetén), hígítsuk a 7.1.1. pontban kapott kivonatot 50 %-os metil-alkohollal (4.5.), hogy a várt 0,2 μg/ml koncentrációjú flavofoszfolipol-tartalmat kapjuk(= U8).
Az U8 oldatból, 50 %-os metil-alkohollal (4.5.) történő többszöri hígítás útján (1 + 1 arányban), készítsük el az U4 (várt tartalom: 0,1 μg/ml), az U2 (várt tartalom: 0,05 μg/ml) és az U1 (várt tartalom: 0,025 μg/ml) oldatokat.
8. A VIZSGÁLAT MÓDJA
8.1. A vizsgálati táptalaj beoltása
Oltsuk be baktérium-szuszpenzióval (3.2.) a vizsgálati táptalajt (4.2.2.) kb. 50 °C-on. A vizsgálati táptalajt (4.2.2.) tartalmazó tálcákon végzett előzetes vizsgálatokkal határozzuk meg a bakterium-szuszpenziónak azt a mennyiségét, amely a legnagyobb és legtisztábban látható gátlási zónákat adja a flavofoszfolipol különböző koncentrációival (kb. 30 ml/liter).
8. 2 A tálcák előkészítése
Az agaron végbemenő diffúziót a négyféle koncentrációjú standard oldatot (S8, S4, S2, S1) és a négyféle koncentrációjú vizsgálati oldatot (U8, U4, U2, U1) tartalmazó tálcákon végezzük. Ezt a négyféle koncentrációban meglévő kivonatot és standard oldatot minden egyes tálcán el kell helyezni. Ezért olyan tálcákat válasszunk, amelyek elég nagyok ahhoz, hogy az agar táptalajon legalább nyolc, egymástól minimum 30 mm távolságra lévő, egyenként 10-13 mm átmérőjű lyuk elférjen rajtuk. A vizsgálat elvégezhető olyan üveglemez tálcákon is, amelyeket egy 200 mm átmérőjű és 20 mm magas megmunkált alumínium vagy műanyag karimagyűrű szegélyez.
Öntsünk a tálcákra annyi táptalajt (4.2.1.), hogy az kb. 1,5 mm vastagságú réteget képezzen (45 ml-t egy 200 mm átmérőjű tálcára). Engedjük egyenletesen eloszlani, helyezzünk rá egy újabb, a 8.1. pontban leírt módon beoltott táptalajréteget (4.2.2.), hogy kb. 1 mm vastag réteget képezzen (30 ml-t egy 200 mm átmérőjű tálcán). Engedjük ismét egyenletesen eloszlani, fúrjunk lyukakat, és helyezzük el bennük a pontosan kimért mennyiségű vizsgálati és standard oldatokat (az átmérőtől függően 0,10 és 0,15 ml közötti mennyiséget lyukanként).
Minden egyes koncentrációt legalább négyszer vigye fel, így mindegyik meghatározás 32 gátlási zóna értékelésén alapulhat.
8.3. Inkubálás
Inkubálja a tálcákat 16-18 órán át 28-30 °C-on.
9. ÉRTÉKELÉS
Mérjük meg a gátlási zónák átmérőjét, 0,1 mm-es pontossággal. Az egyes koncentrációkon végzett mérések átlagát vigyük fel egy féllogaritmikus függvénydiagram-papírra, amely a gátlási zónák átmérőjével arányosan mutatja a koncentrációk logaritmusát. Szerkesszük meg mind a standard oldat, mind a kivonat regressziós egyenesét, például az alábbiak szerint.
A következő képlet segítségével határozzuk meg a standard legalacsonyabb értékéhez (SL) tartozó regressziós pontot:
(a)
(b)
Hasonló módon, határozzuk meg a kivonat legalacsonyabb (UL) és legmagasabb értékéhez (UH) tartozó regresz- sziós pontokat, úgy, hogy a fenti képletben az S1, S2, S4 és S8 jelöléseket az U1, U2, U4 és U8 jelölésekkel helyettesítjük.
Vigyük fel a kiszámított SL és SH értékeket ugyanarra a függvénydiagram-papírra, és kössük össze őket, hogy megkapjuk a standard oldathoz tartozó regressziós egyenest. Hasonló módon, vigyük fel az UL és UH értékeket és kössük össze őket, hogy megkapjuk a kivonathoz tartozó regressziós egyenest.
Amennyiben nem áll fenn semmilyen interferencia, a vonalaknak párhuzamosan kell futniuk. Gyakorlati szempontból a vonalak párhuzamosnak tekinthetőek, ha az (SH-SL) és (UH-UL) értékek legfeljebb 10 %-os mértékben térnek el átlagértéküktől.
Amennyiben a vonalak nem futnak párhuzamosan, akkor vagy az U1 és S1, vagy az U8 és S8 figyelmen kívül hagyható, az SL, SH, UL és UH pedig más képleteket alkalmazva számítható ki, amelyek alternatív regressziós egyeneseket adnak meg:
(a')
vagy
(b')
vagy
és ehhez hasonlóan az UL és UH esetében. Az alternatív regressziós egyenesek párhuzamosságát az előzőhöz hasonlóan kell ellenőrizni. A zárójelentésben meg kell említeni, hogy az eredmény három érték alapján lett kiszámítva.
Ha a vonalak párhuzamosnak tekinthetőek, a következő képlet segítségével számítsuk ki a relatív aktivitás logaritmusát (lg A).
Négy tényező esetén
(c)
Három tényező esetén
(d)
vagy
(d')
Tényleges aktivitás = feltételezett aktivitás × relatív aktivitás.
Ha a vonalak nem tekinthetőek párhuzamosnak, ismételjük meg a meghatározást. Ha a párhuzamosságot ezután sem sikerül elérni, számoljuk ki a relatív aktivitás logaritmusát (lg A) a c) képlet segítségével. A kapott eredmény azonban csak hozzávetőlegesnek tekinthető, és ezt fel kell tüntetni a zárójelentésben.
10. ISMÉTELHETŐSÉG
Ugyanazon analitikus által, ugyanazon a mintán végzett két meghatározás eredménye közötti különbség nem haladhatja meg:
a 0,5 mg/kg értéket, abszolút értékben, 1-2 mg/kg flavofoszfolipol-tartalmom esetén;
a 25 %-ot, 2-10 mg/kg közötti koncentráció legmagasabb értékeire vonatkoztatva;
a 20 %-ot, 10-25 mg/kg közötti koncentráció legmagasabb értékeire vonatkoztatva;
az 5 mg/kg értéket, abszolút értékben, 25-50 mg/kg közötti koncentráció esetén;
a 10 %-ot, 50 mg/kg feletti koncentráció legmagasabb értékeire vonatkoztatva.
3. A VAS, RÉZ, MANGÁN ÉS CINK NYOMELEMEK MEGHATÁROZÁSA
1. CÉL ÉS ALKALMAZÁSI TERÜLET
A módszer lehetővé teszi a vas, réz, mangán és cink nyomelemek takarmányokban történő meghatározását. A meghatározás alsó méréshatárai a következők:
vas (Fe): 20 mg/kg
réz (Cu): 10 mg/kg
mangán (Mn): 20 mg/kg
cink (Zn): 20 mg/kg
2. VIZSGÁLATI ALAPELV
Az esetleg jelen lévő szerves anyag roncsolása után, a mintát sósavban oldjuk. A vas, réz, mangán és cink nyomelemeket, megfelelő hígítás után, atomabszorpciós spektrometriával határozzuk meg.
3. REAGENSEK
Előzetes megjegyzések
A reagensek és az analitikai oldatok elkészítéséhez használt víznek a meghatározás alá eső kationoktól mentesnek kell lennie, amelyet vagy boroszilikát üveg, illetve kvarc lepárlókészülékben történő kétszeres desztillálással, vagy ioncserélő gyantával végzett kétszeres kezeléssel állíthatunk elő.
A reagenseknek legalább analitikai tisztaságúaknak (a.t.) kell lenniük. A meghatározandó elemektől való mentességet vakpróbával kell ellenőrizni. Szükség esetén, a reagenseket tovább kell deríteni.
Az alább leírásra kerülő standard oldatok helyett használhatunk olyan kereskedelmi forgalomban lévő standard oldatokat is, amelyek garantált minőségűek és ezt felhasználásuk előtt le is ellenőrizték.
3.1. Sósav, a.t. (d: 1,19).
3.2. Sósav, a.t. (6 N).
3.3. Sósav, a.t. (0,5 N).
3.4. 38-40 %-os (v/v) fluorsav, amelynek vastartalma kevesebb mint 1 mg Fe/liter és a bepárlás utáni maradék kevesebb mint 10 mg (mint szulfát)/liter.
3.5. Kénsav, a.t. (d: 1,84).
3.6. Hidrogén-peroxid, a.t. (megközelítőleg 100 térfogat oxigén [30 tömegszázalék]).
3.7. Standard vasoldat (1 000 μg Fe/ml) a következőképpen elkészítve: oldjunk fel 1 g a.t. vashuzalt 200 ml 6 N sósavban (3.2.), adjunk hozzá 16 ml hidrogén-peroxidot (3.6.), és töltsük fel vízzel 1 literre.
3.7.1. Standard vas-munkaoldat (100 μg Fe/ml), amelyet a standard oldat (3.7.) vízzel, 1 + 9 arányban történő hígításával készítünk el.
3.8. Standard rézoldat (1 000 μg Cu/ml) a következőképpen elkészítve: oldjunk fel 1 g (a.t.) porított rezet 25 ml 6 N sósavban (3.2.), adjunk hozzá 5 ml hidrogén-peroxidot (3.6.), és töltsük fel vízzel 1 literre.
3.8.1. Standard réz-munkaoldat (10 μg Cu/ml), amelyet a standard oldat (3.8.) vízzel, 1 + 9 arányban történő hígításával, majd az így kapott oldatnak vízzel, 1 + 9 arányban történő hígításával készítünk el.
3.9. Standard mangánoldat (1 000 μg Mn/ml) a következőképpen elkészítve: oldjunk fel 1 g (a.t.) porított mangánt 25 ml 6 N sósavban (3.2.), és töltsük fel vízzel 1 literre.
3.9.1. Standard mangán-munkaoldat (10 μg Mn/ml), amelyet a standard oldat (3.9.) vízzel, 1 + 9 arányban történő hígításával, majd az így kapott oldatnak vízzel, 1 + 9 arányban történő hígításával készítünk el.
3.10. Standard cinkoldat (1 000 μg Zn/ml) a következőképpen elkészítve: oldjunk fel 1 g (a.t.) cinkszalagot vagy -fóliát 25 ml 6 N sósavban (3.2.), és töltsük fel vízzel 1 literre.
3.10.1. Standard cink-munkaoldat (10 μg Zn/ml), amelyet a standard oldat (3.10.) vízzel, 1 + 9 arányban történő hígításával, majd az így kapott oldat vízzel, 1 + 9 arányban történő hígításával készítünk el.
3.11. Lantán-klorid oldat a következőképpen elkészítve: oldjunk fel 12 g lantán-oxidot 150 ml vízben, adjunk hozzá 100 ml 6 N sósavat (3.2.), és töltsük fel vízzel 1 literre.
4. ESZKÖZÖK
4.1. Hőmérséklet-szabályozóval és regisztrálókészülékkel ellátott tokos kemence.
4.2. Magas rezisztenciájú boroszilikát üvegeszközök. Ajánlatos kimondottan nyomelemek meghatározására szolgáló eszközöket használni.
4.3. Platinatégely és (választás szerint) kvarctégely.
4.4. Atomabszorpciós spektrofotométer, amely érzékenység és pontosság vonatkozásában a kívánt tartományban megfelel a módszer követelményeinek.
5. A VIZSGÁLAT MÓDJA
5.1. Szerves anyagot tartalmazó minták
5.1.1. Hamvasztás és az oldatok előkészítése az analízisre ( 16 )
i. Helyezzünk egy 0,2 mg pontossággal kimért, 5-10 g tömegű mintát kvarc- vagy platinatégelybe (4.3.) (lásd a b) megjegyzést), szárítsuk ki kemencében 105 °C-on, majd rakjuk be a tégelyt a hideg tokos kemencébe (4.1.). Zárjuk a kemencét (lásd a c) megjegyzést) és fokozatosan emeljük a hőmérsékletet kb. 90 perc alatt 450-475 °C-ra. Tartsuk ezt a hőmérsékletet 4-16 óráig (pl. egész éjszaka), hogy eltávozzanak a széntartalmú anyagok, majd nyissuk ki a kemencét, és hagyjuk kihűlni (lásd a d) megjegyzést).
Mossuk a tégelyt kb. 5 ml sósavval (3.1.), amit aztán lassan és óvatosan öntsünk a főzőpohárba (CO2-képződés miatt heves reakció léphet fel). Adjuk hozzá cseppenként a sósavat (3.1.) addig kevergetve, amíg a pezsgés meg nem szűnik. Párologtassuk el belőle a nedvességet, időnként üvegbottal megkeverve.
Ezután adjunk a maradékanyaghoz 15 ml, 6 N sósavat (3.2.), majd kb. 120 ml vizet. Keverjük meg az üvegbottal, amit a főzőpohárban hagyunk, és fedjük be a főzőpoharat egy kémlelőüveggel. Lassan forraljuk és tartsuk forrásponton addig, amíg a hamu feloldódása befejeződik. Szűrjük le hamumentes szűrőpapíron, és vegyük fel a szűrletet egy 250 ml-es mérőlombikban. Mossuk a főzőpoharat és a szűrőt 5 ml, forró 6 N sósavval (3.2) és kétszer forrásban lévő vízzel. Töltsük fel a mérőlombikot jelig vízzel (a HCl-koncentráció kb. 0,5 N).
ii. Ha a szűrőben lévő maradékanyag feketének látszik (szén), tegyük vissza a kemencébe, és hamvasszuk újra 450-475 °C-on. Ez a hamvasztás, amely csak pár órát vesz igénybe (kb. három-öt órát), akkor fejeződik be, amikor a hamu fehérnek vagy majdnem fehérnek tűnik. Oldjuk fel a maradékanyagot kb. 2 ml sósavval (3.1.), párologtassuk el belőle a nedvességet és adjunk hozzá 5 ml, 6 N sósavat (3.2.). Hevítsük, szűrjük az oldatot mérőlombikba, és töltsük fel a mérőlombikot jelig vízzel (a HCl-koncentráció kb. 0,5 N).
Megjegyzések:
a) A nyomelemek meghatározásakor fontos odafigyelni a - főleg a cink, réz és vas képezte - szennyeződés jelentette kockázatokra. Ezért a minták előkészítésére használt eszközöknek mentesnek kell lennie ezen fémektől.
A szennyeződés általános kockázatának csökkentése érdekében, dolgozzunk pormentes környezetben, gondosan tisztított eszközökkel és alaposan elmosott üvegedényekkel. Különösen a cink meghatározása érzékeny a szennyeződések számos típusára, pl. az üvegkészítményektől, a reagensektől, a portól, stb. származókra.
b) Az elhamvasztandó minta tömege a takarmány hozzávetőleges nyomelem-tartalma alapján számítható ki, az alkalmazott spektrofotométer érzékenységéhez viszonyítva. Egyes, kevés nyomelemet tartalmazó takarmányok esetén 10-20 g-os mintát kell venni, és a végső oldatot csak 100 ml-re kell feltölteni.
c) A hamvasztást zárt kemencében kell elvégezni, levegő vagy oxigén befúvatása nélkül.
d) A pirométer által jelzett hőmérséklet nem lépheti túl a 475 °C-ot.
5.1.2. Spektrofotometriás meghatározás
5.1.2.1. A kalibráló oldatok elkészítése
Minden egyes meghatározandó elemhez készítsünk a 3.7.1., 3.8.1., 3.9.1. és 3.10.1. pontban megadott standard munkaoldatokból egy kalibrálóoldat-sorozatot, úgy, hogy minden egyes kalibráló oldatnak a HCl-koncentrációja kb. 0,5 N és (vas, mangán és cink esetében) a lantán-klorid koncentrációja 0,1 %-os (w/v) lantánnal egyenértékű legyen. A kiválasztott nyomelem-koncentrációknak az alkalmazott spektrofotométer érzékenységi tartományán belül kell lenniük. Az alábbi táblázatok példákon keresztül mutatják a kalibráló oldat-sorozatok egy mintasorozatát; az alkalmazott spektrofotométer típusától és érzékenységi tartományától függően, adott esetben más koncentrációkat kell kiválasztani.
Vas
| μg Fe/ml | 0 | 0,5 | 1 | 2 | 3 | 4 | 5 |
| standard munkaoldat, ml-ben (3.7.1.) | 0 | 0,5 | 1 | 2 | 3 | 4 | 5 |
| (1 ml = 100 μg Fe) + ml 6 N HCl (3.2.) | 7 | 7 | 7 | 7 | 7 | 7 | 7 |
| + 10 ml lantán-klorid oldat (3.11.), töltsük fel vízzel 100 ml-re. | |||||||
Réz
| μg Cu/ml | 0 | 0,1 | 0,2 | 0,4 | 0,6 | 0,8 | 0,1 |
| standard munkaoldat, ml-ben (3.8.1.) (1 ml = 10 μg Cu) | 0 | 1 | 2 | 4 | 6 | 8 | 10 |
| + ml 6 N HCl (3.2.) | 8 | 8 | 8 | 8 | 8 | 8 | 8 |
| Töltsük fel vízzel 100 ml-re. | |||||||
Mangán
| μg Mn/ml | 0 | 0,1 | 0,2 | 0,4 | 0,6 | 0,8 | 1,0 |
| standard munkaoldat, ml-ben (3.9.1.) (1 ml = 10 μg Mn) | 0 | 1 | 2 | 4 | 6 | 8 | 10 |
| + ml 6 N HCl (3.2.) | 7 | 7 | 7 | 7 | 7 | 7 | 7 |
| + 10 ml lantán-klorid oldat (3.11.), töltsük fel vízzel 100 ml-re. | |||||||
Cink
| μg Zn/ml | 0 | 0,05 | 0,1 | 0,2 | 0,4 | 0,6 | 0,8 |
| standard munkaoldat, ml-ben (3.10.1.) (1 ml = 10 μg Zn) | 0 | 0,5 | 1 | 2 | 4 | 6 | 8 |
| + ml 6 N HCl (3.2.) | 7 | 7 | 7 | 7 | 7 | 7 | 7 |
| + 10 ml lantán-klorid oldat (3.11.), töltsük fel vízzel 100 ml-re. | |||||||
5.1.2.2. Az oldat előkészítése az analízisre
A réz meghatározásához az 5.1.1. pont szerint elkészített oldatot általában közvetlenül fel lehet használni. Ha mégis korrigálni kell koncentrációját a kalibrálóoldat-sorozaton belül, egy aliquot részt pipettázzunk egy 100 ml-es mérőlombikba, és töltsük fel jelig 0,5 N sósavval (3.3.).
A vas, mangán és cink meghatározásához pipettázzunk egy aliquot részt az 5.1.1. pont szerint elkészített oldatból egy 100 ml-es mérőlombikba, adjunk hozzá 10 ml lantán-klorid oldatot (3.11.), és töltsük fel jelig 0,5 N sósavval (3.3.) (lásd még az Észrevételek. 8. pontját).
5.1.2.3. Vakpróba
A vakpróba során a módszer összes meghatározott lépését el kell végezni, azzal a különbséggel, hogy a mintaanyagot elhagyjuk.
A "0" kalibráló oldatot nem szabad vakoldatként használni.
5.1.2.4. Az atomabszorpció mérése
Mérjük meg a kalibráló oldatok és az analizálandó oldat atomabszorpcióját oxidáló, levegő-acetilén láng alkalmazásával, a következő hullámhosszokon:
Fe : 248,3 nm
Cu : 324,8 nm
Mn : 279,5 nm
Zn : 213,8 nm
Minden mérést négyszer végezzünk el.
5.2. Ásványi takarmányok
Ha a minta nem tartalmaz szerves anyagot, az előzetes hamvasztás szükségtelen. Folytassuk a műveletet az 5.1.1.i. pont második bekezdésétől. A fluorsavval végzett bepárlás elhagyható.
6. AZ EREDMÉNYEK KISZÁMÍTÁSA
Egy kalibrációs görbe segítségével, számoljuk ki az analizálandó oldat nyomelem-koncentrációját, az eredményt a minta kilogrammonkénti nyomelemtartalmára vonatkoztatva, milligrammban adjuk meg (ppm).
7. ISMÉTELHETŐSÉG
Az ugyanazon analitikus által, ugyanazon a mintán végzett, két párhuzamos meghatározás eredménye közötti különbség nem haladhatja meg:
- az 5 mg/kg-ot, abszolút értékben, a vizsgált nyomelemtartalom legfeljebb 50 mg/kg értéke esetén,
- a legmagasabb érték 10 %-át a vizsgált nyomelemtartalom 50 és 100 mg/kg koncentráció közötti értéke esetén,
- a 10 mg/kg-ot, abszolút értékben, a vizsgált nyomelemtartalom 100 és 200 mg/kg közötti értéke esetén,
- a legmagasabb érték 5 %-át, a vizsgált nyomelemtartalom 200 mg/kg feletti értéke esetén.
8. ÉSZREVÉTEL
Jelentős mennyiségű foszfát jelenléte interferenciát okozhat a vas, a mangán és a cink meghatározásakor. Az ilyen interferenciát lantán-klorid oldat (3.11.) hozzáadásával korrigálhatjuk. Amennyiben a mintában a tömegarány
· 2, a lantán-klorid oldat (3.11.) az analizálandó oldathoz és a kalibrációs oldatokhoz való hozzáadása elhagyható.
( 1 ) HL L 170., 1970.8.3., 2. o.
( 2 ) HL L 155., 1971.7.12., 13. o.
( 3 ) HL L 279., 1971.12.20., 7. o.
( 4 ) HL L 123., 1972.5.29., 6. o.
( 5 ) HL L 83., 1973.3.30., 21. o.
( 6 ) HL L 108., 1974.4.22., 7. o.
( 7 ) HL L 32., 1975.2.5., 26. o.
( 8 ) HL L 102., 1976.4.15., 8. o.
( 9 ) 1 mg cink bacitracin takarmányszint 42 nemzetközi egységnek (n.e.) felel meg.
( 10 ) Más módszerek is alkalmazhatók feltéve, hogy megállapítást nyert, hogy azok hasonló baktériumszuszpenziókat eredményeznek.
( 11 ) Minden hasonló összetételű és ugyanezeket az eredményeket adó kereskedelmi táptalaj felhasználható.
( 12 ) Más módszerek is használhatók, feltéve, ha megállapították, hogy hasonló baktérium-szuszpenziókat lehet előállítani velük.
( 13 ) Bármely hasonló összetételű, kereskedelemben kapható táptalaj használható, amely hasonló eredményt ad, pl. oxoid antibiotikumos táptalaj 1 (CM 327) 3-as számú (L 13) oxoid agar hozzáadásával.
( 14 ) Bármely hasonló összetételű, kereskedelemben kapható táptalaj használható, amely hasonló eredményt ad, pl. oxoid antibiotikumos táptalaj 2 (CM 335) 3-as számú (L 13) oxoid agar hozzáadásával.
( 15 ) Pl. SE 2 a Wacker Chemie GmbH-tól, München.
( 16 ) A zöldtakarmányok (akár frissen, akár szárítva) nagy mennyiségű növényi eredetű kovasavat tartalmazhatnak, amely nyomelemeket tarthat vissza, ezért el kell távolítani. Az ilyen takarmányokból vett minták esetén ezért a következő, módosított módszert kell követni.
Végezzük el az 5.1.1.i. pontjában ismertetett műveletet a szűrésig. Az oldhatatlan maradékot tartalmazó szűrőpapírt mossuk át kétszer forrásban lévő vízzel, és helyezzük egy platinatégelybe (4.3.). Égessük el a tokos kemencében (4.1.) 550 °C alatti hőmérsékleten, amíg az összes széntartalmú anyag teljesen el nem tűnik. Hagyjuk kihűlni, adjunk hozzá pár csepp vizet, ezt követően 10-15 ml fluorsavat (3.4.), és párologtassuk el belőle a nedvességet kb. 150 °C-on. Ha valamennyi kovasav maradt a maradékanyagban, oldjuk fel újra pár milliliter fluorsavban (3.4.) és párologtassuk el belőle a nedvességet. Adjunk hozzá öt csepp kénsavat (3.5.) és melegítsük addig, amíg megszűnik a fehér füst képződése. Miután hozzáadtunk 5 ml 6 N sósavat (3.2.) és kb. 30 ml vizet, melegítsük meg és szűrjük le az oldatot egy 250 ml-es mérőlombikba, és töltsük fel vízzel jelig (a HCl-koncentráció kb. 0,5 N). Folytassuk a meghatározást az 5.1.3 ponttól.
Lábjegyzetek:
[1] A dokumentum eredetije megtekinthető CELEX: 31978L0633 - https://eur-lex.europa.eu/legal-content/HU/ALL/?uri=CELEX:31978L0633&locale=hu Utolsó elérhető, magyar nyelvű konszolidált változat CELEX: 01978L0633-19831220 - https://eur-lex.europa.eu/legal-content/HU/ALL/?uri=CELEX:01978L0633-19831220&locale=hu