
32008R0273[1]
A Bizottság 273/2008/EK rendelete ( 2008. március 5. ) a tej és tejtermékek elemzési és minőségértékelési módszerei tekintetében az 1255/1999/EK tanácsi rendelet alkalmazására vonatkozó részletes szabályok megállapításáról
A BIZOTTSÁG 273/2008/EK RENDELETE
(2008. március 5.)
a tej és tejtermékek elemzési és minőségértékelési módszerei tekintetében az 1255/1999/EK tanácsi rendelet alkalmazására vonatkozó részletes szabályok megállapításáról
AZ EURÓPAI KÖZÖSSÉGEK BIZOTTSÁGA,
tekintettel az Európai Közösséget létrehozó szerződésre,
tekintettel a tej- és tejtermékpiac közös szervezéséről szóló, 1999. május 17-i 1255/1999/EK tanácsi rendeletre ( 1 ) és különösen annak 10. és 15. cikkére, 26. cikkének (3) bekezdésére, 29. cikkének (1) bekezdésére és 31. cikkének (4) bekezdésére,
mivel:
(1) A Bizottság 213/2001/EK rendelete ( 2 ) a tej és tejtermékek elemzése és minőségértékelése tekintetében az 1255/1999/EK tanácsi rendelet alkalmazására vonatkozó különleges részletes szabályokat állapít meg. Az analitikai módszertan területén bekövetkezett műszaki fejlődés fényében további lényeges módosításokra van szükség. Az egyértelműség és a hatékonyság érdekében és a módosítások száma és műszaki jellege miatt a 213/2001/EK rendeletet hatályon kívül kell helyezni, és helyébe új rendeletet kell léptetni.
(2) Az 1255/1999/EK rendeletben megállapított intézkedések alapján a tejre és tejtermékekre vonatkozó összetételi és minőségi követelményeket ellenőrizni kell annak érdekében, hogy biztosítsák ezen követelmények szigorú betartását.
(3) Az ilyen ellenőrzésekre vonatkozó referencia-módszerek gyakran olyan nemzetközi szervezetek által közzétett módszerek, mint például az Európai Szabványügyi Bizottság (CEN), a Nemzetközi Tejipari Szövetség (IDF), a Nemzetközi Szabványügyi Szervezet (ISO) és az Analitikai Kémikusok Nemzetközi Szervezete (AOAC International), amelyek rendszeresen frissítik a módszereket. Egyes esetekben sor kerül közösségi referencia-módszer megállapítására, míg más esetekben a közösségi szabályokban referencia-módszert nem határoztak meg. A referencia-módszerek egységes alkalmazásának biztosítása érdekében össze kell állítani a referencia-módszerek listáját, és rendelkezni kell arról, hogy a Bizottság szükség esetén kiigazítsa a listát.
(4) A rutinmódszerek alkalmazását nem szabad kizárni. Ennek érdekében azok használatának minimális feltételeit meg kell határozni.
(5) Közös eljárásokat kell megállapítani annak érdekében is, hogy az elemzések eredményeinek értékelését, az érintett termékek érzékszervi értékelését, és a vitatott eredmények ismételt vizsgálatát egyöntetű gyakorlat alapján lehessen elvégezni.
(6) Egyes elemzések esetében jelenleg nincs nemzetközi szinten elfogadott és validált referencia-módszer, és ezért az elemzési eredményeknek a laboratóriumok közötti szórásáról információ nem áll rendelkezésre. Ezért közösségi módszerek megállapítására van szükség, amelyeket nemzetközileg elfogadott szabályok alapján validáltak, és amelyeket referencia-módszerként kell alkalmazni.
(7) A Bizottság 1898/2005/EK rendelete ( 3 ) megállapítja a tejszín, vaj és vajkoncentrátum közösségi piacon történő értékesítésére vonatkozó intézkedések tekintetében az 1255/1999/EK tanácsi rendelet végrehajtása részletes szabályait és rendelkezik a tejszín, vaj és vajkoncentrátum egyes körülmények között történő megjelöléséről, annak érdekében, hogy biztosítsák az ilyen termékek helyes végfelhasználását. A megjelölés fontos a rendszer megfelelő működéséhez. A részt vevő piaci szereplők egyenlő bánásmódja érdekében közös módszereket kell kialakítani egyes ilyen jelölőanyagok meghatározására.
(8) Az 1255/1999/EK rendelet 9. cikke értelmében a juhtejből készült sajtok magántárolására támogatás nyújtható. Ugyanezen termékekre különleges visszatérítés is adható az említett rendelet 31. cikkének értelmében. A juhtejből, kecsketejből vagy bivalytejből, illetve a juh-, kecske- és bivalytej elegyéből készült sajtok a Közösség területére egyes harmadik országokból kedvezményes feltételek mellett hozhatók be. A fentiekre tekintettel megfelelő ellenőrző vizsgálatokra van szükség annak biztosítására, hogy az érintett termékek nem tartalmaznak tehéntejet. Ezért meg kell határozni egy közösségi referencia-módszert a tehéntej kimutatására a rutinmódszerek használatának sérelme nélkül, feltéve hogy az ilyen rutinmódszerek megfelelnek egyes kritériumoknak.
(9) A fölözött tejből származó kazein és kazeinátok előállítására nyújtott támogatásról szóló, 1990. október 10-i 2921/90/EGK bizottsági rendelet ( 4 ) értelmében ki kell mutatni a kólibaktériumoktól való mentességet. A tejben és tejtermékekben található kólibaktériumok kimutatására a nemzetközileg elfogadott referencia-módszer az ISO 4831. E szabvány alapján közösségi referencia-módszert alakítottak ki.
(10) A vám- és a statisztikai nómenklatúráról, valamint a Közös Vámtarifáról szóló, 1987. július 23-i 2658/87/EGK tanácsi rendelet ( 5 ) a 2309 vámtarifaszám alá tartozó összetett takarmányok esetében a tejterméktartalom függvényében különböző vámtételekről rendelkezik. Annak biztosítása érdekében, hogy a szóban forgó szabályokat egységesen alkalmazzák, a laktóztartalom elemzésére a tagállamokban kötelezően alkalmazandó, általánosan elismert módszert kell meghatározni.
(11) Az 1255/1999/EK rendelet értelmében az intervencióra szánt vajnak és sovány tejpornak, valamint az állati takarmányozásra szánt sovány tejpornak meg kell felelnie bizonyos minőségi követelményeknek. Referencia-módszereket kell meghatározni a követelmények teljesítésének igazolására.
(12) Néhány módszert először vezet be ez a rendelet. A rendelet hatálybalépését követően elegendő időt kell hagyni annak érdekében, hogy a laboratóriumok megfelelően bevezethessék és alkalmazhassák ezeket az új módszereket. Amikor egy szabványügyi szervezet felülvizsgál és közzétesz az I. mellékletben említett valamely referencia-módszert, a laboratóriumoknak hat hónapot kell engedélyezni, hogy az új szabványnak megfelelően frissítsék analitikai eljárásaikat.
(13) Az e rendeletben előírt intézkedések összhangban vannak a Tej- és Tejtermékpiaci Irányítóbizottság véleményével,
ELFOGADTA EZT A RENDELETET:
I. FEJEZET
ÁLTALÁNOS RENDELKEZÉSEK
1. cikk
Tárgy és hatály
(1) E rendelet megállapít egyes, a tej- és tejtermékpiac közös szervezéséről szóló, 1255/1999/EK rendeletben foglalt előírások alapján a tej és tejtermékek kémiai, fizikai, mikrobiológiai elemzéséhez, illetve érzékszervi vizsgálatához szükséges referencia-módszereket és alkalmazásuk szabályait.
(2) Ennek a rendeletnek az I. melléklete határozza meg az (1) bekezdésben említett elemzésekhez alkalmazandó referencia-módszerek felsorolását.
(3) A Bizottság a felsorolást az 1255/1999/EK rendelet 42. cikkében meghatározott eljárásnak megfelelően frissíti.
2. cikk
Rutinmódszerek
A közösségi szabályok által megkövetelt elemzések rutinmódszerekkel is elvégezhetők, feltéve hogy ezeket kellően kalibrálják és a megfelelő referencia-módszerrel rendszeresen ellenőrzik. Az eredmények összehasonlítását a rendszeres hiba, az ismételhetőség és a reprodukálhatóság figyelembevételével kell végezni.
Vitás esetekben a referencia-módszerrel nyert eredmény lesz az irányadó.
A tagállamok tájékoztatják a Bizottságot a rutinmódszerek használatáról az 1. cikkben említett elemzés során.
II. FEJEZET
ELEMZÉSI MÓDSZEREK
3. cikk
Szállítmányok értékelése a jogszabály által előírt határértéknek való megfelelés szempontjából
A jelölőanyagok elemzésének kivételével e rendelet II. melléklete alkalmazandó az összetételre vonatkozó jogszabályi követelményeknek való megfelelés meghatározása céljából.
4. cikk
Érzékszervi értékelés
(1) A tej és tejtermékek esetében - az intervenciós raktározásra szánt vaj kivételével - tagállamok által az érzékszervi értékeléshez használandó referencia-módszer vagy a 99C:1997 IDF-szabvány, vagy egyéb, ezzel egybevethető módszer, amelyről értesítik a Bizottságot.
Az érzékszervi vizsgálatok során a III. mellékletben meghatározott eljárásokat kell alkalmazni a bírálók teljesítményének, valamint a kapott eredmények megbízhatóságának ellenőrzésére.
(2) Az intervenciós raktározásra szánt vaj esetében az érzékszervi vizsgálatok során a III. mellékletben meghatározott eljárásokat kell alkalmazni a bírálók teljesítményének, valamint a kapott eredmények megbízhatóságának ellenőrzésére.
Az érzékszervi értékelés referencia-módszereként a IV. mellékletben meghatározott eljárást kell alkalmazni.
5. cikk
Jelölőanyagok
(1) Az V. mellékletben meghatározott elemzési módszert kell a vaj, a vajolaj és a tejszín önantsav-triglicerid-tartalmának meghatározására szolgáló referencia-módszerként alkalmazni.
(2) A VI. mellékletben meghatározott elemzési módszert kell a vajkoncentrátum, a vaj és a tejszín vanillintartalmának meghatározására szolgáló referencia-módszerként alkalmazni.
(3) A VII. mellékletben meghatározott elemzési módszert kell a vajkoncentrátum és a vaj béta-apo-8'karotinsav etil-észter-tartalmának meghatározására szolgáló referencia-módszerként alkalmazni.
(4) A VIII. mellékletben meghatározott elemzési módszert kell a vajkoncentrátum és a vaj β-szitoszterin- vagy sztigmaszterintartalmának meghatározására szolgáló referencia-módszerként alkalmazni.
(5) A vajkoncentrátumnak, a vajnak és a tejszínnek a megjelölése akkor tekintendő a közösségi szabályokkal összhangban lévőnek, ha a kapott eredmények megfelelnek az V. melléklet 10. és 11. pontjában és a VI., VII. és VIII. melléklet 8. pontjában foglalt meghatározásoknak.
6. cikk
A tehéntejkazein kimutatása
(1) A IX. mellékletben meghatározott elemzési referencia-módszert kell használni annak biztosítására, hogy a csak és kizárólag juhtejből, kecsketejből, bivalytejből, illetve juhtej, kecsketej és bivalytej keverékéből készülő sajtok ne tartalmazzanak tehéntejkazeint.
A tehéntejkazein akkor minősül a mintában jelen lévőnek, ha az elemzett minta tehéntejkazein-tartalma egyenlő vagy nagyobb, mint a IX. mellékletben meghatározott, 1 % tehéntejet tartalmazó referenciaminta tehéntejkazein-tartalma.
(2) A tehéntejkazeinnek az (1) bekezdésben említett sajtokban történő kimutatására rutinmódszert is lehet alkalmazni feltéve, hogy:
a) a kimutatási határ legfeljebb 0,5 %; és
b) nem ad hamis pozitív eredményt; és
c) a tehéntejkazein megfelelő érzékenységgel még hosszabb érlelési időszakot követően is kimutatható, úgy ahogy a szokásos kereskedelmi körülmények mellett előfordulna.
Ha a fent említett követelmények bármelyike nem teljesül, a IX. mellékletben meghatározott referencia-módszereket kell alkalmazni.
7. cikk
Kólibaktériumok kimutatása
A vajban, sovány tejporban, kazeinben és kazeinátokban található kólibaktériumokat a X. mellékletben meghatározott referencia-módszerrel összhangban kell kimutatni.
8. cikk
A laktóztartalom meghatározása
A 2309 KN-kód alá tartozó termékek tejcukortartalmát a XI. mellékletben meghatározott referencia-módszer szerint kell meghatározni.
9. cikk
Oltós savó kimutatása
(1) Az intervenciós raktározásra szánt sovány tejporban található oltós savót a XII. mellékletben meghatározott referencia-módszerrel összhangban kell kimutatni.
(2) Az állati takarmányozásra szánt sovány tejporban és keverékekben található oltós savót a XII. mellékletben meghatározott referencia-módszerrel összhangban kell kimutatni. Az oltós savó kimutatása esetén a XIII. melléklet alkalmazandó.
10. cikk
Író kimutatása
A sovány tejporban található írót a XIV. mellékletben meghatározott referencia-módszerrel összhangban kell kimutatni.
11. cikk
Antimikrobiális maradványanyagok kimutatása
A sovány tejporban található antimikrobiális maradványanyagokat a XV. mellékletben meghatározott referencia-módszerrel összhangban kell kimutatni.
12. cikk
Soványtejpor-tartalom meghatározása
Az összetett takarmányok soványtejpor-tartalmát a XVI. mellékletben meghatározott referencia-módszerrel összhangban kell meghatározni.
13. cikk
Keményítő kimutatása
A sovány tejporban, denaturált tejporban és összetett takarmányokban található keményítőt a XVII. mellékletben meghatározott referencia-módszerrel összhangban kell kimutatni.
14. cikk
A szárított tejszín nedvességtartalmának meghatározása
A szárított tejszín nedvességtartalmát a XVIII. mellékletben meghatározott referencia-módszerrel összhangban kell meghatározni.
15. cikk
Savanyú írópor nedvességtartalmának meghatározása
A takarmányokban való használatra szánt savanyú írópor nedvességtartalmát a XIX. mellékletben meghatározott referencia-módszerrel összhangban kell meghatározni.
16. cikk
A tejzsírtisztaság meghatározása
A tejzsírtisztaságot a XX. mellékletben meghatározott referencia-módszerrel összhangban kell meghatározni.
III. FEJEZET
ÁLTALÁNOS ÉS ZÁRÓ RENDELKEZÉSEK
17. cikk
Minőségbiztosítás
Az elemzéseket belső minőségellenőrzési eljárásokat magában foglaló analitikai minőségbiztosítási rendszerrel rendelkező laboratóriumokban kell végezni. A nem akkreditált laboratóriumoknak legalább évente egyszer szakmai alkalmassági vizsgálatokon kell részt venniük és eredményeik nem térhetnek el 2σR-nél (a referencia-módszer reprodukálhatósági szórása) nagyobb mértékben a konszenzusos értéknél. Az alkalmazott rendszerek részletes leírásának - betekintés céljából - a laboratóriumban rendelkezésre kell állnia.
A takarmány- és élelmiszerjog, valamint az állat-egészségügyi és az állatok kíméletére vonatkozó szabályok követelményeinek történő megfelelés ellenőrzésének biztosítása céljából végrehajtott hatósági ellenőrzésekről szóló, 2004. április 29-i 882/2004/EK európai parlamenti és tanácsi rendelet ( 6 ) 12. cikkében említett szabványokkal összhangban akkreditált laboratóriumok mentesülnek a szakmai alkalmassági vizsgálaton való részvétel kötelezettsége alól.
18. cikk
A mintavétel és az elemzési eredmények vitathatósága
(1) A mintavételt az érintett termékre vonatkozó szabályozással összhangban kell végrehajtani. Amennyiben nincsenek mintavételi rendelkezések, az ISO 707 IDF 50, Tej és tejtermékek - Mintavételi útmutatóban megadott rendelkezések alkalmazandók.
(2) Az elemzési eredmények laboratóriumi jegyzőkönyveinek elegendő információt kell tartalmazniuk az eredményeknek a II. és XXI. melléklet szerinti értékeléséhez.
(3) A közösségi szabályok szerinti kötelező elemzésekhez ellenmintát kell venni.
(4) A XXI. mellékletben meghatározott módszert kell alkalmazni olyan esetekben, ahol az elemzés eredményét a piaci szereplő nem fogadja el.
(5) Amennyiben a gyártó a mintavételtől számított öt munkanapon belül bizonyítani tudja, hogy a mintavételi eljárást nem végezték el helyesen, a mintavételt - ha lehetséges - meg kell ismételni. Amennyiben a mintavétel nem ismételhető meg, a szállítmányt el kell fogadni.
19a. cikk
Értesítések
A 2. cikkben, a 4. cikk (1) bekezdésében és a III. C. mellékletében előírt értesítéseket a 792/2009/EK bizottsági rendeletnek ( 7 ) megfelelően kell megküldeni.
20. cikk
Hatályon kívül helyezés
A 213/2001/EK rendelet hatályát veszti.
A hatályon kívül helyezett rendeletre történő utalásokat úgy kell tekinteni, mintha erre a rendeletre vonatkoznának, és a XXII. mellékletben foglalt megfelelési táblázattal összhangban kell alkalmazni.
21. cikk
Hatálybalépés
Ez a rendelet az Európai Unió Hivatalos Lapjában való kihirdetését követő harmadik napon lép hatályba.
Ezt a rendeletet 2008. március 31-jétől kell alkalmazni.
Ez a rendelet teljes egészében kötelező és közvetlenül alkalmazandó valamennyi tagállamban.
I. MELLÉKLET
(1. cikk)
REFERENCIA-MÓDSZEREK FELSOROLÁSA
Index Min. = minimum, Max. = maximum, Melléklet = az idézett rendelet melléklete, SNF = zsírmentes szárazanyag, PV = peroxidérték, A = külső, F = íz, zamat, C = állomány/állag, TBC = összes csíraszám, Therm = termofil csíraszám, MS = tagállam, IDF = Nemzetközi Tejipari Szövetség, ISO = Nemzetközi Szabványügyi Szervezet, IUPAC = Elméleti és Alkalmazott Kémia Nemzetközi Uniója, ADPI = Amerikai Tejtermék Intézet, SCM = cukrozott sűrített tej, EMC = sűrített tej vagy tejszín.
A. RÉSZ
| Bizottsági rendelet | Termék | Paraméter | Határérték (1) | Referencia-módszer | Megjegyzés |
| 2771/1999/EK rendelet – Intervenciós raktározás | Sózatlan vaj | Zsír | Min. 82 % m/m | ISO 17189:2003IDF 194:2003 | |
| Víz | Legfeljebb 16 % m/m | ISO 3727–1:2001IDF 80–1:2001 | |||
| SNF | Legfeljebb 2 % m/m | ISO 3727–2:2001IDF 80–2:2001 | |||
| Zsírsavtartalom | 1,2 mmol/100g zsír | ISO 1740:2004IDF 6:2004 | |||
| PV (max.) | 0,3 meq. oxigén/1 000 g zsír | ISO 3976:2006IDF 74:2006 | 1. megjegyzés | ||
| Kólibaktériumok | Nem mutatható ki 1 g-ban | X. melléklet | 3. megjegyzés | ||
| Nem tejzsír | Nem mutatható ki triglicerid-analízissel | XX. melléklet | |||
| Szterin jelölőanyagok | Nem mutatható ki, β-szitoszterin ≤ 40 mg/kg | VIII. melléklet | |||
| Egyéb jelölőanyagok | |||||
| — vanillin | Nem mutatható ki | VI. melléklet | |||
| — karotinsav etil-észtere | ≤ 6 mg/kg | VII. melléklet | |||
| — önantsav trigliceridjei | Nem mutatható ki | V. melléklet | |||
| Érzékszervi jellemzők | A, F és C esetében legalább 5-ből 4 pont | IV. melléklet | |||
| Vízeloszlás | Legalább 4 pont | ISO 7586:1985IDF 112A:1989 | |||
| 2771/1999/EK rendelet – Magántárolás | Sózatlan vaj | Zsír | Min. 82 % m/m | ISO 17189:2003IDF 194:2003 | |
| Víz | Legfeljebb 16 % m/m | ISO 3727–1:2001IDF 80–1:2001 | |||
| SNF | Legfeljebb 2 % m/m | ISO 3727–2:2001IDF 80–2:2001 | |||
| 2771/1999/EK rendelet – Magántárolás | Sózott vaj | Zsír | Min. 80 % m/m | ISO 17189:2003IDF 194:2003 | |
| Víz | Legfeljebb 16 % m/m | ISO 3727–1:2001IDF 80–1:2001 | |||
| SNF (só kivételével) | Legfeljebb 2 % m/m | ISO 3727–2:2001IDF 80–2:2001 | |||
| Só | Legfeljebb 2 % m/m | ISO 15648:2004IDF 179:2004 | |||
| 1898/2005/EK rendelet II. fejezet | Sózatlan vaj | Zsír | Min. 82 % m/m | ISO 17189:2003IDF 194:2003 | |
| Nem tejzsír | XX. melléklet | ||||
| Víz | Legfeljebb 16 % m/m | ISO 3727–1 2001IDF 80–1:2001 | |||
| SNF | Legfeljebb 2 % m/m | ISO 3727–2:2001IDF 80–2:2001 | |||
| Jelölőanyagok: | |||||
| — szterinek | Lásd a VIII. mellékletet | VIII. melléklet | |||
| — vanillin | Lásd a VI. mellékletet | VI. melléklet | |||
| — karotinsav etil-észtere | Lásd a VII. mellékletet | VII. melléklet | |||
| — önantsav trigliceridjei | Lásd az V. mellékletet | V. melléklet | |||
| 1898/2005/EK rendelet II. fejezet | Sózott vaj | Zsír | Min. 80 % m/m | ISO 17189:2003IDF 194:2003 | |
| Nem tejzsír | XX. melléklet | ||||
| Víz | Legfeljebb 16 % m/m | ISO 3727–1:2001IDF 80–1:2001 | |||
| SNF (só kivételével) | Legfeljebb 2 % m/m | ISO 3727–2:2001IDF 80–2:2001 | |||
| Só | Legfeljebb 2 % m/m | ISO 15648:2004IDF 179:2004 | |||
| Jelölőanyagok: | |||||
| — szterinek | Lásd a VIII. mellékletet | VIII. melléklet | |||
| — vanillin | Lásd a VI. mellékletet | VI. melléklet | |||
| — karotinsav etil-észtere | Lásd a VII. mellékletet | VII. melléklet | |||
| — önantsav trigliceridjei | Lásd az V. mellékletet | V. melléklet | |||
| 1898/2005/EK rendelet II. fejezet | Vajkoncentrátum | Zsír | Min. 99,8 % m/m | IDF 24:1964 | |
| Víz és SNF | Legfeljebb 0,2 % m/m | ISO 5536:2002IDF 23:2002 (nedvesség) IDF 24:1964 (SNF) | |||
| Zsírsavtartalom | 1,2 mmol/100g zsír | ISO 1740:2004IDF 6:2004 | |||
| PV (max.) | 0,5 meq. oxigén/1 000 g zsír | ISO 3976:2006IDF 74:2006 | 1. megjegyzés | ||
| Nem tejzsír | Nincs | XX. melléklet | |||
| Íz | Tiszta | ||||
| Szag | Idegen szagoktól mentes | ||||
| Egyéb | Semlegesítőszerektől, antioxidánsoktól és tartósítószerektől mentes | ||||
| Jelölőanyagok: | |||||
| — szterinek | Lásd a VIII. mellékletet | VIII. melléklet | |||
| — vanillin | Lásd a VI. mellékletet | VI. melléklet | |||
| — karotinsav etil-észtere | Lásd a VII. mellékletet | VII. melléklet | |||
| — önantsav trigliceridjei | Lásd az V. mellékletet | V. melléklet | |||
| 1898/2005/EK rendelet II. fejezet | Tejszín | Zsír | Minimum 35 % m/m | ISO 2450:1999IDF 16 C:1987 | |
| Nem tejzsír | XX. melléklet | ||||
| Jelölőanyagok: | |||||
| — szterinek | Lásd a VIII. mellékletet | 2. megjegyzés | |||
| — vanillin | Lásd a VI. mellékletet | VI. melléklet | |||
| — karotinsav etil-észtere | Lásd a VII. mellékletet | 2. megjegyzés | |||
| — önantsav trigliceridjei | Lásd az V. mellékletet | V. melléklet | |||
| 1898/2005/EK rendelet III. fejezet | Vajkoncentrátum | Zsír | Min. 96 % m/m | 2. megjegyzés | |
| Nem tejzsír | XX. melléklet | ||||
| SNF | Legfeljebb 2 % m/m | 2. megjegyzés | |||
| Jelölőanyagok: | |||||
| — sztigmaszterin (95 % m/m) | 15 g/100 kg vajkoncentrátum | VIII. melléklet | |||
| — sztigmaszterin (85 % m/m) | 17 g/100 kg vajkoncentrátum | VIII. melléklet | |||
| — önantsav trigliceridjei | 10,34 kg/t vajkoncentrátum | V. melléklet | |||
| — vajsav és sztigmaszterin etil-észtere | — vajsav etil-észtere — sztigmaszterin: VIII. melléklet | 2. megjegyzés | |||
| — vajsav etil-észtere és önantsav trigliceridjei | — vajsav etil-észtere — önantsav trigliceridjei: V. melléklet | 2. megjegyzés | |||
| lecitin (E322) | Legfeljebb 0,5 % m/m | 2. megjegyzés | |||
| NaCl | Legfeljebb 0,75 % m/m | ISO 15648:2004IDF 179:2004 | |||
| Zsírsavtartalom | 1,2 mmol/100g zsír | ISO 1740:2004IDF 6:2004 | |||
| PV (max.) | Legfeljebb 0,5 meq. oxigén/1 000 g zsír | ISO 3976:2006IDF 74:2006 | 1. megjegyzés | ||
| Íz | Tiszta | ||||
| Szag | Idegen szagoktól mentes | ||||
| Egyéb | Semlegesítőszerektől, antioxidánsoktól és tartósítószerektől mentes | ||||
| 1898/2005/EK rendelet IV. fejezet | Sózatlan vaj | Zsír | Min. 82 % m/m | ISO 17189:2003IDF 194:2003 | |
| Víz | Legfeljebb 16 % m/m | ISO 3727–1:2001IDF 80–1:2001 | |||
| SNF | Legfeljebb 2 % m/m | ISO 3727–2:2001IDF 80–2:2001 | |||
| 1898/2005/EK rendelet IV. fejezet | Sózott vaj | Zsír | Min. 80 % m/m | ISO 17189:2003IDF 194:2003 | |
| Víz | Legfeljebb 16 % m/m | ISO 3727–1:2001IDF 80–1:2001 | |||
| SNF (só kivételével) | Legfeljebb 2 % m/m | ISO 3727–2:2001IDF 80–2:2001 | |||
| Só | Legfeljebb 2 % m/m | ISO 15648:2004IDF 179:2004 | |||
| 1255/1999/EK rendelet 9. cikke és II. címe | Juhtejből és/vagy kecsketejből készült sajt | Tehéntej | < 1 % m/m | IX. melléklet | |
| 2921/90/EGK rendelet | I. melléklet – savkazein | Víz | Legfeljebb 12,00 % m/m | ISO 5550:2006IDF 78:2006 | |
| Zsír | Legfeljebb 1,75 % m/m | ISO 5543:2004IDF127:2004 | |||
| Szabad savtartalom | Legfeljebb 0,30 ml 0,1 N NaOH-oldat/g | ISO 5547:1978IDF 91:1979 | |||
| 2921/90/EGK rendelet | I. melléklet – oltós kazein | Víz | Legfeljebb 12,00 % m/m | ISO 5550:2006IDF 78:2006 | |
| Zsír | Legfeljebb 1,00 % m/m | ISO 5543:2004IDF 127:2004 | |||
| Hamu | Min. 7,50 % m/m | ISO 5545:1978IDF 90:1979 | |||
| 2921/90/EGK rendelet | I. melléklet – kazeinátok | Víz | Legfeljebb 6,00 % m/m | ISO 5550:2006IDF 78:2006 | |
| Tejfehérje | Min. 88,00 % m/m | ISO 5549:1978IDF 92:1979 | |||
| Zsír és hamu | Legfeljebb 6,00 % m/m | ISO 5543:2004IDF 127:2004 | |||
| Kötött hamu | ISO 5544:1978IDF 89:1979 | ||||
| Hamu | ISO 5545:1978IDF 90:1979 | ||||
| 2921/90/EGK rendelet | II. melléklet – savkazein | Víz | Legfeljebb 10,00 % m/m | ISO 5550:2006IDF 78:2006 | |
| Zsír | Legfeljebb 1,50 % m/m | ISO 5543:2004IDF 127:2004 | |||
| Szabad savtartalom | Legfeljebb 0,20 ml 0,1 N NaOH-oldat/g | ISO 5547:1978IDF 91:1979 | |||
| TBC (max.) | 30 000/ g | ISO 4833:2003 | 3. megjegyzés | ||
| Kólibaktériumok | 0,1 g-ban nem mutatható ki | X. melléklet | 3. megjegyzés | ||
| Therm. (max.) | 5 000/ g | ISO 4833:2003 | 3. és 4. megjegyzés | ||
| 2921/90/EGK rendelet | II. melléklet – oltós kazein | Víz | Legfeljebb 8,00 % m/m | ISO 5550:2006IDF 78:2006 | |
| Zsír | Legfeljebb 1,00 % m/m | ISO 5543:2004IDF 127:2004 | |||
| Hamu | Min. 7,50 % m/m | ISO 5545:1978IDF 90:1979 | |||
| TBC (max.) | 30 000/ g | ISO 4833:2003 | 3. megjegyzés | ||
| Kólibaktériumok | 0,1 g-ban nem mutatható ki | X. melléklet | 3. megjegyzés | ||
| Therm. (max.) | 5 000/ g | ISO 4833:2003 | 3. és 4. megjegyzés | ||
| 2921/90/EGK rendelet | II. melléklet – kazeinátok | Víz | Legfeljebb 6,00 % m/m | ISO 5550:2006IDF 78:2006 | |
| Tejfehérje | Min. 88,00 % m/m | ISO 5549:1978IDF 92:1979 | |||
| Zsír és hamu | Legfeljebb 6,00 % m/m | ISO 5543:2004IDF 127:2004 ISO 5544:1978IDF 89:1979 vagy ISO 5545:1978IDF 90:1979 | |||
| TBC (max.) | 30 000/ g | ISO 4833:2003 | 3. megjegyzés | ||
| Kólibaktériumok | 0,1 g-ban nem mutatható ki | X. melléklet | 3. megjegyzés | ||
| Therm. (max.) | 5 000/g | ISO 4833:2003 | 3. és 4. megjegyzés | ||
| 2921/90/EGK rendelet | III. melléklet – kazeinátok | Víz | Legfeljebb 6,00 % m/m | ISO 5550:2006IDF 78:2006 | |
| Tejfehérje | Min. 85,00 % m/m | ISO 5549:1978IDF 92:1979 | |||
| Zsír | Legfeljebb 1,50 % m/m | ISO 5543:2004IDF 127:2004 | |||
| Laktóz | Legfeljebb 1,00 % m/m | ISO 5548:2004IDF 106:2004 | |||
| Hamu | Legfeljebb 6,50 % m/m | ISO 5544:1978IDF 89:1979 vagy ISO 5545:1978IDF 90:1979 | |||
| TBC (max.) | 30 000/g | ISO 4833:2003 | 3. megjegyzés | ||
| Kólibaktériumok | 0,1 g-ban nem mutatható ki | X. melléklet | 3. megjegyzés | ||
| Therm. (max.) | 5 000/ g | ISO 4833:2003 | 3. és 4. megjegyzés | ||
| 2799/1999/EK rendelet | Összetett takarmányok és sovány tejpor (SMP) (takarmányozásra) | Víz (savanyú írópor) | Legfeljebb 5 % m/m | XIX. melléklet | |
| Fehérje | 31,4 % m/m (min.) a zsírmentes szárazanyag-tartalomban | ISO 8968–123:2001IDF 20–123:2001 | |||
| Víz (SMP) | Legfeljebb 5 % m/m | ISO 5537:2004IDF 26:2004 | |||
| Zsírok (SMP) | Legfeljebb 11 % m/m | ISO 1736:2000IDF 9C:1987 | |||
| Oltós savó (SMP) | Nincs | XIII. melléklet | 6. megjegyzés | ||
| Keményítő (SMP) | Nincs | XVII. melléklet | |||
| Víz (keverékek) | Legfeljebb 5 % m/m a zsírmentes szárazanyag-tartalomban | ISO 5537:2004IDF 26:2004 | |||
| Zsír (keverékek) | A 84/4/EGK bizottsági irányelv (HL L 15., 1984.1.18., 29. o.) | ||||
| Oltós savó (keverékek) | Nincs | XIII. melléklet | |||
| (a végtermék) SMP-tartalma | Min. 50 % m/m | XVI. melléklet | |||
| Zsír (a végtermékben) | Min. 2,5 % m/m vagy 5 % m/m | A 84/4/EGK bizottsági irányelv (HL L 15., 1984.1.18., 29. o.) | 7. megjegyzés | ||
| Keményítő (a végtermékben) | Min. 2 % m/m | XVII. melléklet | 8. megjegyzés | ||
| Réz (a végtermékben) | 25 ppm | A 78/633/EGK bizottsági irányelv (HL L 206., 1987.7.26., 43. o.) | |||
| 214/2001/EK rendelet | Sovány tejpor (SMP) (porlasztva szárított) | Zsír | Legfeljebb 1,0 % m/m | ISO 1736:2000IDF 9C:1987 | |
| Fehérje | 31,4 % (2)m/m (min.) a zsírmentes szárazanyag-tartalomban | ISO 8968–1/2:2001IDF 20–1/2:2001 | |||
| Víz | Legfeljebb 3,5 % m/m | ISO 5537:2004IDF 26:2004 | |||
| Savasság | Legfeljebb 19,5 ml, 0,1 N NaOH, 10 g zsírmentes szárazanyag | ISO 6091:1980IDF 86:1981 | |||
| Laktátok | Legfeljebb 150 mg/100 g zsírmentes szárazanyag | ISO 8069:2005IDF 69:2005 | |||
| Foszfatáz | Negatív | ISO 11816–1:2006IDF 155–1:2006 | |||
| Oldhatósági index | Legfeljebb 0,5 ml 24 °C-on | ISO 8156:2005IDF 129:2005 | |||
| Égett szemcsék | A vagy B korong (15,0 mg) | ADPI (1990) | |||
| TBC | 40 000/ g | ISO 4833:2003 | 3. megjegyzés | ||
| Kólibaktériumok | Negatív/0,1 g | X. melléklet | 3. megjegyzés | ||
| Író | Negatív | XIV. melléklet | |||
| Oltós savó | Negatív | XII. melléklet | |||
| Savanyú savó | Negatív | 2. megjegyzés | |||
| Antimikrobiális szerek | XV. melléklet | ||||
| (1) Az adott rendelet követelményeinek sérelme nélkül. (2) A minimális fehérjetartalom 34 % lenne 2009. szeptember 1-jével. | |||||
B. RÉSZ
A B. részben felsorolt referencia-módszereket olyan termékek elemzésére lehet alkalmazni, amelyekre az 1. oszlopban említett rendeletek bármelyike vonatkozik.
| Bizottsági rendelet | Termék | KN-kód | Paraméter | Határérték | Referencia-módszer | Megjegyzés |
| 2658/87/EGK rendelet 2535/2001/EK rendelet 1282/2006/EK rendelet | Tej és tejszín nem sűrítve, cukor vagy más édesítőanyag hozzáadása nélkül | 0401 | Zsír (≤ 6 % m/m) | A határértékek az adott termék KN-kódjának leírásában megadottak, adott esetben a 3846/87/EGK bizottsági rendeletben (HL L 366., 1987.12.24., 1. o.) az export nómenklatúra 9. részében vagy a 2535/2001/EK rendeletben (HL L 341., 2001.12.22., 29. o.) pontosítva | ISO 1211:2001IDF 1D:1996 | |
| Zsír (> 6 % m/m) | ISO 2450:1999IDF 16C:1987 | |||||
| Tej és tejszín sűrítve vagy cukor vagy más édesítőanyag hozzáadásával | 0402 | Zsír (folyékony forma) | ISO 1737:1999IDF 13C:1987 | |||
| Zsír (szilárd forma) | ISO 1736:2000IDF 9C:1987 | |||||
| Fehérje | ISO 8968–123:2001IDF 20–123:2001 | |||||
| Szacharóz (normál tartalommal) | ISO 2911:2004 IDF 35:2004 | |||||
| Szacharóz (alacsony tartalommal) | 2. megjegyzés | |||||
| Szárazanyag (SCM) | ISO 6734:1989IDF 15B:1991 | |||||
| Szárazanyag (EMC) | ISO 6731:1989IDF 21B:1987 | |||||
| Víz (tejpor) | ISO 5537:2004IDF 26:2004 | |||||
| Víz (tejszínpor) | XVIII. melléklet | |||||
| Író, erjesztett vagy savanyított tej és tejszín, sűrítve vagy nem sűrítve, cukrozva vagy más édesítőanyag hozzáadásával | 0403 | Zsír | ISO 1211:2001IDF 1D:1996 ISO 1736:2000IDF 9C:1987 ISO 2450:1999IDF 16 C:1987 ISO 7208:1999IDF 22B:1987 ISO 8262–3:2005IDF 124–3:2005 | |||
| Fehérje | ISO 8968–123:2001IDF 20–123:2001 | |||||
| Szacharóz (normál tartalommal) | ISO 2911:2004IDF 35:2004 | |||||
| Szacharóz (alacsony tartalommal) | 2. megjegyzés | |||||
| Víz (savanyú írópor) | XIX. melléklet | |||||
| Víz (édes írópor) | ISO 5537:2004IDF26:2004 | |||||
| Szárazanyag (egyéb termékek) | Az illetékes hatóság által jóváhagyott módszerek. | |||||
| Savó, sűrítve vagy cukrozva vagy más édesítőszer hozzáadásával is; természetes tejalkotórészeket tartalmazó készítmény | 0404 | Zsír | ISO 1736:2000IDF 9C:1987 ISO 2450:1999IDF 16C:1987 ISO 7208:1999IDF 22B:1987 | |||
| Fehérje | ISO 8968–123:2001IDF 20–123:2001 | |||||
| Szacharóz (normál tartalommal) | ISO 2911:2004IDF 35:2004 | |||||
| Szacharóz (alacsony tartalommal) | 2. megjegyzés | |||||
| 0404 90 | Fehérje | ISO 8968 1/2 2001IDF 20–1/2:2001 | ||||
| Víz | IDF 21B:1987 | |||||
| Szárazanyag | ISO 6734:1989IDF 15B:1991 | |||||
| (Sűrített termékek) | ISO 6731:1989IDF 21B:1987 | |||||
| Vaj és tejből nyert más zsír; kenhető tejkészítmények | 0405 | Zsír (ha ≤ 85 % m/m) | ISO 17189:2003IDF 194:2003 | |||
| Vaj | Víz | ISO 3727–1:2001IDF 80–1:2001 | ||||
| SNF | ISO 3727–2:2001IDF 80–2:2001 | |||||
| NaCl | ISO 15648:2004IDF 179:2004 | |||||
| Zsír (ha > 99 % m/m) | IDF 24:1964 | |||||
| Vajolaj | Víz (ha zsír < 99 % m/m) | ISO 5536:2002IDF 23:2002 | ||||
| Sajt és túró | 0406 | Zsír | ISO 1735:2004IDF 5:2004 | |||
| Szárazanyag | ISO 5534:2004IDF 4:2004 | |||||
| Szárazanyag (Ricotta) | ISO 2920:2004IDF 58:2004 | |||||
| NaCl | ISO 5943:2006IDF 88:2006 | |||||
| Laktóz | ISO 5765–1/2:2002IDF 79–1/2:2002 | |||||
| 2658/87/EGK rendelet | Összetett takarmányok | 2309 | Laktóz | XI. melléklet |
Az Európai Unió referencia-módszerei felsorolásához tartozó megjegyzések
1. megjegyzés: az ISO 1740:1991 szabvány szerinti tejzsírizolálás (fény elleni védelem).
2. megjegyzés: nincs még elfogadott referencia-módszer. Az illetékes hatóság által jóváhagyott módszerek.
3. megjegyzés: az ISO 8261:2001IDF 122:2001 szabvány szerinti minta-előkészítés.
4. megjegyzés: inkubálás 48 órán keresztül 55 °C-os hőmérsékleten, ügyelni kell rá, hogy a táptalaj ne száradjon ki.
5. megjegyzés: % m/m SNF = % m/m szárazanyag - % m/m zsír.
6. megjegyzés: a Bizottság 84/4/EGK irányelve.
7. megjegyzés: a Bizottság 2799/1999/EK rendelete (HL L 340., 1999.12.31., 3-3-27. o.)
8. megjegyzés: a Bizottság 78/633/EGK irányelve.
II. MELLÉKLET
(3. cikk)
SZÁLLÍTMÁNYOK ÉRTÉKELÉSE A JOGSZABÁLY ÁLTAL ELŐÍRT HATÁRÉRTÉKNEK VALÓ MEGFELELÉS SZEMPONTJÁBÓL
1. ALAPELV
Amennyiben a vonatkozó jogszabályok részletes mintavételi eljárásokról rendelkeznek, ezeket az eljárásokat kell követni. Minden egyéb esetben az ellenőrzésnek alávetett szállítmányból véletlenszerűen vett legalább három mintaegységből álló mintát kell használni. Készíthető összetett minta. A kapott eredményeket össze kell hasonlítani a jogszabályban előírt határértékekkel 95 %-os megbízhatósági intervallum (kétszeres szórás) számításával, ahol a vonatkozó szórás attól függ, hogy (1) a módszert nemzetközi együttműködés során a σr -re és σR -re vonatkozó értékekkel validálták-e, vagy (2) házon belüli validálás esetén számítottak-e belső reprodukálhatóságot. Ez a megbízhatósági intervallum egyenlő lesz az eredmény mérési bizonytalanságával.
2. A MÓDSZERT NEMZETKÖZI EGYÜTTMŰKÖDÉS SORÁN VALIDÁLJÁK
Ebben az esetben a σr ismételhetőségi szórást és a σR reprodukálhatósági szórást megállapították, és a laboratórium bizonyítani tudja a validált módszer teljesítményjellemzőinek való megfelelést.
Számítsuk ki az n ismételt mérés
A következőképpen számítjuk ki
k
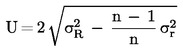
Ha az x mérési végeredményt az x = y 1 + y 2, x = y 1 - y 2, x = y 1 · y 2 vagy x = y 1 / y 2 formájú képletek alkalmazásával számítjuk ki, a szórások kombinálásának szokásos eljárását kell ilyen esetekben követni.
A szállítmány akkor tekintendő az UL előírt felső határértéknek nem megfelelőnek, ha
egyéb esetben az UL-nek megfelelőnek minősül.
A szállítmány akkor tekintendő az LL előírt alsó határértéknek nem megfelelőnek, ha
egyéb esetben az LL-nek megfelelőnek minősül.
3. HÁZON BELÜLI VALIDÁLÁS BELSŐ REPRODUKÁLHATÓSÁGI SZÓRÁS SZÁMÍTÁSÁVAL
Azokban az esetekben, amikor ebben a rendeletben nem szereplő módszereket használnak és nem folytatnak pontossági méréseket, házon belüli validálást kell végrehajtani. Az U kiterjesztett bizonytalanság számításának képletében σr és σ R helyett az sir belső ismételhetőségi szórást és a siR belső reprodukálhatósági szórást kell használni.
Az (1) bekezdésben említett döntési szabályok alkalmazandók. Amennyiben azonban a szállítmányt a jogszabályban előírt határértéknek nem megfelelőnek ítélik, a méréseket meg kell ismételni az e rendeletben meghatározott módszerrel, és döntést kell hozni az (1) bekezdés szerint.
III. MELLÉKLET
(4. cikk)
A BÍRÁLÓK ÉRTÉKELÉSE ÉS AZ EREDMÉNYEK MEGBÍZHATÓSÁGA AZ ÉRZÉKSZERVI ELEMZÉSEKNÉL
A következő eljárások alkalmazandók, ha pontozásos módszereket használunk (99C:1997 IDF-szabvány).
A. AZ "ISMÉTELHETŐSÉGI INDEX" MEGHATÁROZÁSA
Egy 12 hónapos időszakon belül a bíráló személynek legalább tíz mintát kell azonosítatlan mintaként elemeznie. Ez rendszerint több alkalommal történik. Az egyes termékjellemzők eredményeit a következő képlettel értékelhetjük:
ahol:
wI : az ismételhetőségi index
xi1 : az xi minta első értékelésére adott pontszám
xi2 : az xi minta második értékelésére adott pontszám
n : a minták száma
Az értékelendő mintáknak tág minőségi határokat kell átfogniuk. wI nem haladhatja meg az 1,5-ös értéket (5-pontos skála).
B. AZ "ELTÉRÉSI INDEX" MEGHATÁROZÁSA
Ezt az indexet annak ellenőrzésére használjuk, hogy valamely bíráló ugyanazt a skálát alkalmazza-e a minőség értékelésére, mint egy tapasztalt bírálókból álló csoport. A bíráló által adott pontszámokat összehasonlítjuk a bírálókból álló csoport által adott pontszámokból számított középértékkel.
A következő képlet alkalmazandó az eredmények értékelésére:
ahol:
xi1; xi2 : lásd az a) pontot
;
:
a bírálókból álló csoport által az xi mintára az első, illetve a második értékelés alkalmával adott átlagos pontszám
n : a minták száma (12 havonta legalább tíz).
Az értékelendő mintáknak tág minőségi határokat kell átfogniuk. A DI nem haladhatja meg az 1,5-ös értéket (5-pontos skála).
A tagállamoknak jelenteniük kell minden nehézséget, amely ennek az eljárásnak az alkalmazása során merül fel.
Ha az egyéni bírálók túllépik az eltérési vagy az ismételhetőségi index 1,5-ös határértékét, az illetékes hatóság szakértői az elkövetkező néhány hétben egy vagy több véletlenszerű "újbóli" ellenőrzést hajtanak végre a bírálók által osztályozott mintákon, vagy egy vagy több "kísért" ellenőrzést végeznek az érintett bírálókkal. Szoros megfigyelésre van szükség annak eldöntéséhez, hogy továbbra is igény van-e a szolgáltatásaikra. Az eredményeket dokumentálni kell, és a további intézkedések bizonyítékaként meg kell őrizni.
C. EGY TAGÁLLAM KÜLÖNBÖZŐ RÉGIÓIBAN, ILLETVE A KÜLÖNBÖZŐ TAGÁLLAMOKBAN NYERT EREDMÉNYEK KÖZÖTTI KÜLÖNBSÉGEK EGYBEVETÉSE
Amennyiben alkalmazható, évente legalább egyszer ellenőrző vizsgálatot kell szervezni a különböző régiókban működő bíráló személyek által nyert eredmények összehasonlítására. Amennyiben jelentős eltérések tapasztalhatók, meg kell tenni a szükséges lépéseket az okok azonosítására és egymással egybevethető eredmények kialakítására.
A tagállamok megszervezhetik a saját bírálóik által kapott eredmények és a szomszédos tagországok bírálói által kapott eredmények összevetésére alkalmas ellenőrző vizsgálatokat. Amennyiben jelentős eltérés tapasztalható, mélyreható nyomozást kell végezni annak érdekében, hogy egymással egybevethető eredmények szülessenek.
A tagállamoknak értesíteniük kell a Bizottságot az ilyen összehasonlító vizsgálatok eredményéről.
IV. MELLÉKLET
(4. cikk)
VAJ ÉRZÉKSZERVI ÉRTÉKELÉSE
1. TÁRGY
A vaj érzékszervi értékelésére vonatkozó ezen eljárás célja az összes tagállamban alkalmazandó egységes módszer kialakítása.
További részletekért lásd a tej és tejtermékekre vonatkozó hatályos nemzetközi IDF-szabványt, IDF 99-1., 2. és 3. rész az érzékszervi értékelésről.
2. FOGALOMMEGHATÁROZÁSOK
"Érzékszervi értékelés" (bírálat): egy termék jellemzőinek érzékszervekkel való vizsgálata.
"Bizottság": a kiválasztott bírálók egy csoportja, akik a bírálat során kölcsönös érintkezés és egymás befolyásolása nélkül járnak el.
"Bíráló": érzékszervi vizsgálat végzésére való alkalmassága miatt kiválasztott személy. A bírálók ezen típusa rendelkezhet korlátozott tapasztalattal.
"Szakértő bíráló": nagyfokú érzékszervi érzékenységgel és érzékszervi módszertani tapasztalattal rendelkező személy, aki képes a különféle termékek következetes és megbízható érzékszervi bírálatára. Az ilyen típusú bíráló jó hosszú távú érzékszervi memóriával rendelkezik.
"Pontozás": a bizottság érzékszervi vizsgálatainak értékelése egy számskála segítségével. Ehhez a hibajegyzéket kell használni.
"Osztályozás": minőségi osztályba sorolás, amelyet a pontozás alapján végeznek.
"Ellenőrző dokumentumok": azok a dokumentumok, amelyeket az egyes jellemzők pontjainak és a termék végső osztályozásának jegyzőkönyvezésére használnak. (Ez a dokumentum használható a kémiai összetétel nyilvántartásba vételére is.)
3. VIZSGÁLÓHELYISÉG
Részletekért lásd: ISO 8589 és ISO/DIS 22935-2 IDF 99-2, 7. bekezdés.
Gondoskodni kell arról, hogy a bírálókat a vizsgálóhelyiségben külső tényezők ne befolyásolják.
A vizsgálóhelyiségnek idegen szagoktól mentesnek és könnyen tisztíthatónak kell lennie. A falak világos színűek legyenek és ne tükröződjenek.
A vizsgálóhelyiség és annak megvilágítása olyan legyen, hogy azok a pontozni kívánt termék jellemzőit ne befolyásolják.
A szobát megfelelő termosztatikus szabályozással kell felszerelni, hogy a vajat állandó hőmérsékleten lehessen tartani. Az osztályozás idején a vaj hőmérséklete 12 °C (±2 °C) legyen.
4. A BÍRÁLÓK KIVÁLASZTÁSA
A bírálóknak ismerniük kell a vajtermékeket, és alkalmasnak kell lenniük az érzékszervi osztályozás elvégzéséhez. Az alkalmasságot rendszeres időközönként (évente legalább egyszer) az illetékes hatóságnak ellenőrizni kell.
4.1. Az ISO/DIS 22935-1 IDF 99-1 szabvány 4. bekezdése (toborzás) és 5.1. bekezdése részletezi az új bírálók hivatalos alkalmazását megelőzően alkalmazható általános követelményeket és szűrővizsgálatokat.
Alapvető fontosságú a folyamatos képzés, és rendszeresen tartani kell általános összejöveteleket. Lásd az ISO 8586-1 szabványt a bizottság képzésére vonatkozó információért.
4.2. A kezdeti képzés a következőkre terjed ki:
- az érzékszervi értékelés általános elmélete és gyakorlati fontossága,
- módszerek, skálák és az érzékszervi benyomások leírása,
- érzékszervi jellemzők észlelése és felismerése és specifikus érzékelési fogalmak,
- általános képzés a vaj gyártásáról,
- validált referenciák és minták, amelyek segítségével a bíráló azonosítani tud adott ízeket és ízintenzitásokat a terméken belül.
5. A BIZOTTSÁGGAL KAPCSOLATOS KÖVETELMÉNYEK
A bizottságban lévő bírálók számának páratlannak kell lennie, minimum három fő. A bírálók többsége az illetékes hatóság alkalmazottja vagy olyan felhatalmazott személy, aki nem a tejipar alkalmazottja.
A bizottság elnöke felelős a teljes eljárásért, és részt vehet a bizottság munkájában.
Mielőtt az értékelésre sor kerülne, a következő tényezőket kell figyelembe venni ahhoz, hogy a bírálóktól optimális teljesítményt várhassunk el:
- a bírálók nem szenvedhetnek olyan betegségben, amely teljesítményükre káros hatást gyakorolna. Ilyen esetben az érintett bírálót a bizottságban egy másik személlyel kell helyettesíteni,
- a bírálóknak időben kell megjelenniük a bírálatban való részvételre, és biztosítaniuk kell, hogy elegendő idő álljon rendelkezésükre a bírálat elvégzéséhez,
- a bírálók nem használhatnak erős illatú anyagokat, úgymint parfüm, arcszesz, dezodor, stb., valamint kerülniük kell az erősen ízesített (például nagyon fűszeres) ételeket,
- a bírálók nem dohányozhatnak, nem ehetnek és vízen kívül más folyadékot nem fogyaszthatnak a bírálatot megelőző fél órában.
6. TELJESÍTMÉNY
Képessége fenntartása érdekében minden bírálónak rendszeresen részt kell vennie érzékszervi értékelő bizottságokban. A gyakoriság a vaj mennyiségétől és az átbocsátási képességtől függ, és ha lehetséges, havonta legalább egyszer bizottságot kell összehívni.
A tapasztalt bírálóknak is évente számos bizottságban részt kell venniük, ha lehetséges legalább negyedévenként egyszer.
7. MINTAVÉTEL ÉS A MINTA ELŐKÉSZÍTÉSE
Alapvető, hogy a minták eredete rejtve maradjon a bírálat során az esetleges részrehajlás elkerülése érdekében. A mintákat kóddal kell ellátni.
Ezt az értékelés előtt meg kell szervezni. Meg kell határozni, hogy a vajat milyen hőmérsékleten kell tartani a vizsgálóhelyiségbe történő szállítás alatt (6 °C ±2 °C).
Amikor az érzékszervi vizsgálatra hűtőraktárban kerül sor, a mintát vajmintavevővel kell venni. Amennyiben az érzékszervi vizsgálatra nem hűtőraktárban kerül sor, hanem valamely más helyszínen, legalább 500 gramm mintát kell venni. A bírálat során a vaj hőmérséklete 12 °C (±2 °C) legyen (lásd: az ISO/DIS 22935-2 IDF 99-2 szabványban a vaj értékelési hőmérséklete 14 °C ±2 °C). Az ettől való nagy eltéréseket feltétlenül kerülni kell.
8. AZ EGYES JELLEMZŐK ÉRTÉKÉNEK ELBÍRÁLÁSA
8.1. Az érzékszervi vizsgálatokat mindig a következő három jellemzőre kell elvégezni: külső, állomány és íz, zamat.
A külső a következő jellemzőket foglalja magában: szín, látható tisztaság, fizikai szennyeződés hiánya, penészfoltok hiánya és egyenletes vízeloszlás. A víz eloszlását a 112A/1989 IDF-szabvány szerint kell ellenőrizni.
Az állomány a következő jellemzőket foglalja magában: szerkezet, textúra és szilárdság. A kenhetőség fizikai eszközökkel megfigyelhető, ha egy tagállam az ügyfelek elvárásainak teljesítése érdekében szükségesnek látja. A Bizottság dönthet a módszertan harmonizálásáról a jövőben.
A szerkezet a termék fogyasztás közbeni egységességére utaló kifejezés. Általában a szilárdsággal és a kenhetőséggel hozzák összefüggésbe, és egyenletesnek kell lennie az egész termékben. Szorosan kapcsolódik a textúrához, és ez a termék azon képessége, hogy megtartsa saját súlyát. Ezt jelzi a vágással szembeni ellenállás, és mechanikusan, valamint szájban történő érzékelés és tapintás útján mérhető.
Az íz, zamat a szájban, főként a nyelv ízlelőbimbói által érzékelt jellemző.
Az aroma az orr és a szaglás által érzékelt jellemző.
Az ajánlott hőmérséklettől való jelentős eltérés megakadályozza az állomány és zamat megbízható értékelését. A hőmérséklet kiemelkedő jelentőségű.
A vaj osztályozását el kell halasztani, ha a hőmérséklet az ajánlott tartományon kívül esik.
8.2. Minden egyes jellemzőt külön kell érzékszervi értékelésnek alávetni. A pontozást az 1. táblázat szerint kell végezni.
8.3. Az egységes megítélés elérése érdekében a bírálók számára kívánatos, hogy a bírálat előtt egy vagy több referenciamintán közösen végezzenek pontozást a külső, az állomány és a zamat vonatkozásában.
8.4. Az elfogadhatóság tekintetében a pontozás a következő:
Lásd a 7. részt - Nómenklatúra, és a pontozás során a pontokra alkalmazandó kritériumok leírása.
| Maximum | Szükséges | |
| Külső | 5 | 4 |
| Állomány | 5 | 4 |
| Zamat/aroma | 5 | 4 |
- Ahol a termék nem éri el a kívánt pontszámot, a hiba leírását meg kell adni.
- Az ellenőrző dokumentumban jegyzőkönyvezni kell az egyes bírálók által az egyes jellemzőkre adott pontokat.
- A terméket többségi döntés alapján fogadják el vagy utasítják el.
- Lehetőleg el kell kerülni az olyan eseteket, ahol az egyes tulajdonságokra adott egyedi pontszámok eltérése nagyobb mint a szomszédos pontszámok különbsége (legfeljebb 20 mintánként egyszer). Különben a bizottság vezetőjének ellenőriznie kell a bizottság alkalmasságát.
9. FELÜGYELET
Rendszerint a bizottság elnöke, aki az illetékes hatóság alkalmazottja, és aki tagja lehet a bizottságnak, felelős az eljárás egészéért. Az ellenőrző dokumentumban jegyzőkönyveznie kell az egyes jellemzőkre adott egyes pontokat, és igazolnia kell, hogy a terméket elfogadták vagy elutasították.
10. NÓMENKLATÚRA
Lásd a mellékelt 2. táblázatot.
11. HIVATKOZÁS
FIL-IDF 99C:1997 Sensory evaluation of dairy products by scoring - Reference method (Tejtermékek pontozásos érzékszervi értékelése - referencia-módszer)
ISO/DIS 22935IDF 99 International Standard for Milk and Milk Products - Sensory analysis - Parts 1-3 (A tejre és a tejtermékekre vonatkozó nemzetközi szabvány - Érzékszervi vizsgálat - 1-3. rész)
ISO 8586-1 Sensory analysis - General guidance for selection, training and monitoring of assessors - Part 1 (Érzékszervi vizsgálat - Általános útmutató a bírálók kiválasztásához, képzéséhez és folyamatos ellenőrzéséhez - 1. rész)
ISO 8589 sensory analysis - General guidance for the design of test rooms (Érzékszervi vizsgálat - Általános útmutató a vizsgálóhelyiségek kialakításához)
FIL-IDF 112A:1989 Butter - Determination of water dispersion value (Vaj - A vízeloszlási érték meghatározása)
1. táblázat
Vaj pontozása
| Külső | Állomány | Zamat + aroma | ||||||
| Pont | Szám (1) | Megjegyzés | Pont (minőségi osztály) | Szám (1) | Megjegyzés | Pont (minőségi osztály) | Szám (1) | Megjegyzés |
| 5 | Nagyon jó ideális típus legjobb minőség (egyenletesen száraz) | 5 | Nagyon jó ideális típus legjobb minőség (jól kenhető) | 5 | Nagyon jó ideális típus legjobb minőség (teljesen tiszta, finom aroma) | |||
| 4 | Jó (2) Nincs nyilvánvaló hibája | 4 | 17 18 | Jó (2) kemény lágy | 4 | Jó (2) Nincs nyilvánvaló hibája | ||
| 3 | 1 2 3 4 5 6 7 8 | Megfelelő (kisebb hibák) kivált (szabad) nedvesség nem egységes, kétszínű csíkos pettyes, márványos foltos olajkiválás túlszínezett laza, nyílt szerkezet | 3 | 14 15 16 17 18 | Megfelelő (kisebb hibák) tömör, törékeny, morzsálódó pépes, tésztás, zsíros ragadós kemény lágy | 3 | 21 22 25 27 33 34 35 | Megfelelő (kisebb hibák) tisztátalan idegen íz savanyú főtt íz, égett íz takarmányíz nyers, keserű túlsózott |
| 2 | 1 3 4 5 6 10 11 12 | Gyenge (nyilvánvaló hibák) kivált (szabad) nedvesség csíkos pettyes, márványos foltos olajkiválás idegen anyag penészes oldatlan só | 2 | 14 15 16 17 18 | Gyenge (nyilvánvaló hibák) tömör, törékeny, morzsálódó pépes, tésztás, zsíros ragadós kemény lágy | 2 | 21 22 23 25 32 33 34 35 36 38 | Gyenge (nyilvánvaló hibák) tisztátalan idegen íz állott savanyú oxidált íz, fémes íz takarmányíz nyers, keserű túlsózott dohos, bűzös vegyszerízű |
| 1 | 1 3 4 5 6 7 9 10 11 12 | Igen gyenge (súlyos hibák) kivált (szabad) nedvesség csíkos pettyes, márványos foltos olajkiválás túlszínezett szemcsés idegen anyag penészes oldatlan só | 1 | 14 15 16 17 18 | Igen gyenge (súlyos hibák) tömör, törékeny, morzsálódó pépes, tésztás, zsíros ragadós kemény lágy | 1 | 22 24 25 26 28 29 30 31 32 34 35 36 37 38 | Igen gyenge (súlyos hibák) idegen íz sajtos, savanyú sajt íz savanyú élesztős penészes íz avas olajos, halízű faggyú íz oxidált íz, fémes íz nyers, keserű túlsózott dohos, bűzös malátaízű vegyszerízű |
| (1) 2. táblázat. (2) A „jó” minősítésnél említett hibák csak igen kis mértékű eltérések az ideális típustól. | ||||||||
2. táblázat
A vaj hibáinak jegyzéke
| I. Külső |
| 1. kivált (szabad) nedvesség |
| 2. nem egységes, kétszínű |
| 3. csíkos |
| 4. pettyes, márványos |
| 5. foltos |
| 6. olajkiválás |
| 7. túlszínezett |
| 8. laza (nyílt szerkezet) |
| 9. szemcsés |
| 10. idegen anyag |
| 11. penészes |
| 12. oldatlan só |
| II. Állomány |
| 14. tömör, törékeny, morzsálódó |
| 15. pépes, tésztás, zsíros |
| 16. ragadós |
| 17. kemény |
| 18. lágy |
| III. Zamat és aroma |
| 20. íztelen |
| 21. tisztátlan (1) |
| 22. idegen íz |
| 23. állott |
| 24. sajtos, savanyú sajt íz |
| 25. savanyú |
| 26. élesztős |
| 27. a) főtt íz |
| b) égett íz |
| 28. penészes íz |
| 29. avas |
| 30. olajos, halízű |
| 31. faggyú íz |
| 32. a) oxidált íz |
| b) fémes íz |
| 33. takarmányíz |
| 34. nyers, keserű |
| 35. túlsózott |
| 36. dohos, bűzös |
| 37. malátaízű |
| 38. vegyszerízű |
| (1) Ezt az elnevezést minél ritkábban alkalmazzuk, és csak olyan esetekben, amikor a hibát más módon ennél pontosabban nem lehet leírni. |
V. MELLÉKLET
(5. cikk)
A VAJ, A VAJOLAJ ÉS A TEJSZÍN ÖNANTSAV-TRIGLICERID-TARTALMÁNAK MEGHATÁROZÁSA TRIGLICERIDEK GÁZKROMATOGRÁFIÁS ELEMZÉSÉVEL
1. TÁRGY
Ez a módszer a vaj, a vajolaj és a tejszín önantsav-triglicerid-tartalma meghatározásának módszerét ismerteti.
2. KIFEJEZÉSEK ÉS FOGALOMMEGHATÁROZÁSOK
Önantsavtartalom: az ebben a módszerben leírt eljárással meghatározott önantsav-triglicerid-tartalom.
Megjegyzés: Az önantsav-tartalmat a vajolaj és a vaj esetében kg/terméktonnában, tejszín esetében kg/tejzsírtonnában kell kifejezni.
3. A MÓDSZER ELVE
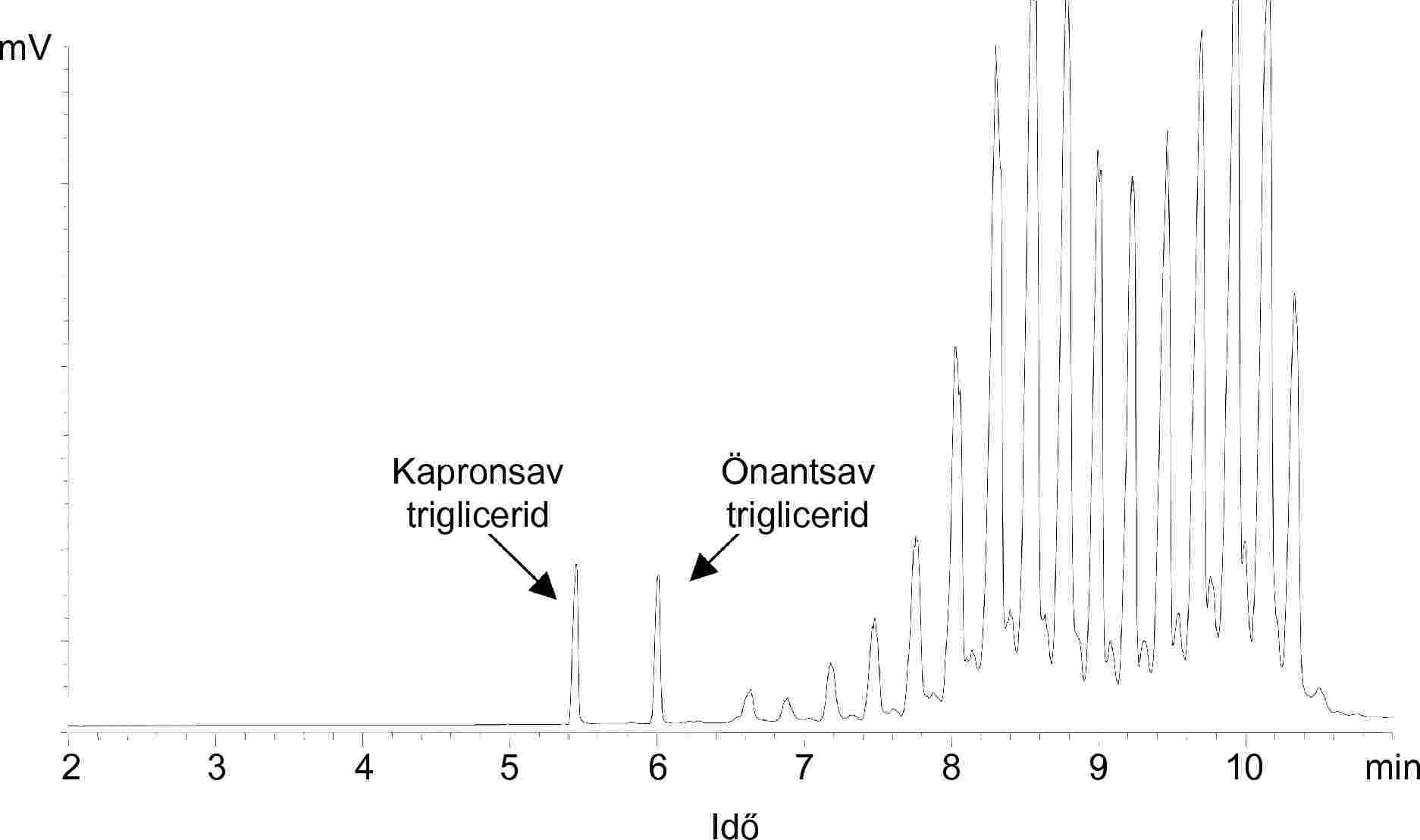
Az ISO 14156 IDF 172:2001 szabvány szerint a különböző termékekből tejzsírt vonunk ki. A kivont tejzsírban az önantsav-triglicerid-tartalom mennyiségi meghatározása kapilláris gázkromatográfiával (GC) történik. A mintára kapott eredmény értékelése a belső standardként használt kapronsav-trigliceriddel való összevetés útján történik.
Megjegyzés: A tributirin is kielégítő belső standardnak bizonyult.
4. VEGYSZEREK
Minden vegyszernek elismert analitikai tisztaságúnak kell lennie.
4.1. n-hexán
4.2. Standard kapronsav-triglicerid, legalább 99 %-os tisztaságú
4.3. Standard önantsav-triglicerid, legalább 99 %-os tisztaságú
4.4. Vízmentes nátrium-szulfát (Na2SO4).
5. ESZKÖZÖK
A szokásos laboratóriumi berendezések és különösen a következők:
5.1. Analitikai mérleg, 1 mg-os pontosságú
5.2. 10 ml és 20 ml űrtartalmú mérőlombikok
5.3. 30 ml űrtartalmú centrifugacsövek
5.4. Rotációs bepárló
5.5. 50 °C ± 5 °C hőmérsékleten tartható kemence
5.6. Szűrőpapír, közepes porozitással, körülbelül 15 cm átmérővel
5.7. A gázkromatográfiás berendezés
5.7.1. Gázkromatográf "split/splitless" vagy "on-column" injektorral és lángionizációs detektorral (FID)
5.7.2. GC oszlop, olyan állófázissal, amelyben sikeresen végrehajtottak triglicerid-elválasztást (100 % dimetil-polisziloxán vagy 5 % fenil-95 % metil-polisziloxán). A laboratóriumi tapasztalat és az alkalmazott injekciós rendszer figyelembevételével kiválasztjuk az állófázist, az oszlophosszat (4 m és 15 m között), a belső átmérőt (0,22 mm és 0,50 mm között) és a filmvastagságot (legalább 0,12 μm). A kiválasztott oszloppal mindenképp teljesen el kell különülnie az alapvonalon az oldószer és a kapronsav-triglicerid csúcsának, valamint a kapronsav-triglicerid- és az önantsav-triglicerid-csúcsának. Az alkalmazandó feltételek példáinak felsorolása alább található.
5.7.2.1. Alkalmazandó feltételek példája "split" injektor használata esetén
- Vivőgáz: hélium
- Oszlopfej nyomása: 100 kPa
- Oszlop: 12 m hosszú, 0,5 mm belső átmérőjű, 0,1 μm filmvastagságú olvasztott szilícium-dioxid oszlop
- Állófázis: 100 % dimetil-polisziloxán vagy 5 % fenil-95 % dimetil-polisziloxán (pl. HT5)
- Oszlophőmérséklet: kezdeti hőmérséklet 130 °C, fenntartva 1 percig, ezután felemelve 20 °C/min sebességgel 260 °C-ra majd felemelve 30 °C/min sebességgel 360 °C-ra; 360 °C fenntartása 10 percig
- Detektor hőmérséklete: 370 °C
- Injektor hőmérséklete: 350 °C
- Megosztási arány 1:30
- Injektált minta mennyisége: 1 μl.
5.7.2.2. Alkalmazandó feltételek példája "on-column" injektor használata esetén:
- Vivőgáz: hidrogén (állandó áramlású rendszer)
- Oszlopfej nyomása: 89 kPa
- Oszlop: 4 m hosszú, 0,32 mm belső átmérőjű, 0,25 μm filmvastagságú olvasztott szilícium-dioxid-oszlop
- Állófázis: 5 % fenil, 95 % dimetil-polisziloxán
- Oszlophőmérséklet: kezdeti hőmérséklet 60 °C, fenntartva 2 percig majd felemelve 35 °C/min sebességgel 340 °C-ra, ezen a hőmérsékleten tartva 5 percig;
- Detektor hőmérséklete: 350 °C
- Injektált minta mennyisége: 1 μl.
5.8. 5 μl űrtartalmú injekciós fecskendő.
6. MINTAVÉTEL
Fontos, hogy a labor valóban reprezentatív mintát kapjon, amely nem sérült vagy nem változott meg a szállítás vagy a tárolás során.
A mintavétel nem képezi az ebben a nemzetközi szabványban leírt módszer részét. Ajánlott mintavételi módszer található az IDF 50C:1995 vagy ISO 707-1997 - Tej és tejtermékek - Mintavételi módszerek szabványban.
7. ELJÁRÁS
7.1. A vizsgálati minta és a vizsgálati adag előkészítése
Eljárás az ISO 14156IDF 172:2001 szerint.
7.1.1. Vajolaj, vaj
7.1.1.1. Olvasszunk fel a vizsgálati mintából 50-100g-ot a kemencében (5.5.)
7.1.1.2. Helyezzünk 0,5-1,0g vízmentes nátrium-szulfátot (5.4.) redőzött papírszűrőre
7.1.1.3. Szűrjük át a zsírt a vízmentes nátrium-szulfátot tartalmazó szűrőpapíron, gyűjtsük a szűrletet kemencében (5.5.) tartott főzőpohárba. Az olvasztott vaj szűrőpapírra való dekantálásakor vigyázzunk, hogy a szérumból semmi ne kerüljön át
7.1.2. Tejszín
7.1.2.1. Melegítsük fel a vizsgálati mintát 20 °C ± 2 °C-ra
7.1.2.2. Alaposan keverjük vagy rázzuk össze a mintát
7.1.2.3. Hígítsunk fel a vizsgálati mintából megfelelő mennyiséget ahhoz, hogy 100 ml vizsgálati adagot kapjunk, amelyben a zsír tömegtörtje megközelítőleg 4 %
7.1.2.4. Folytassuk az eljárást a zsír tejszínből való kivonásához úgy, mint a nyers tej és a homogenizált tej esetében (lásd ISO 14156 IDF 172:2001, §8.3)
7.1.2.5. Mérjünk ki egy 10 ml-es mérőlombikba (5.2.) 1 mg-os pontossággal 1 g-ot a kivont zsírból. Adjunk hozzá a 7.2.2. pont szerinti oldatból 1 ml-t. Töltsük fel 10 ml-re n-hexánnal (4.1.) és homogenizáljuk
7.1.2.6. Tegyünk a 7.1.1.2. pont szerinti oldatból 1 ml-t egy 10 ml-es mérőlombikba (5.2.) és hígítsuk fel 10 ml-re n-hexánnal (4.1.)
7.2. A kalibrációs standardok elkészítése
7.2.1. Oldjunk fel 100 mg önantsav-trigliceridet (4.3.) 10 ml n-hexánban (4.1.)
7.2.2. Oldjunk fel 100 mg kapronsav-trigliceridet (4.2.) 10 ml n-hexánban (4.1.)
7.2.3. Tegyünk a 7.2.2. pont szerinti oldatból 1 ml-t egy 10 ml-es mérőlombikba (5.2.) Töltsük fel 10 ml-re n-hexánnal (4.1.)
7.2.4. Tegyünk a 7.2.1. pont szerinti oldatból 1 ml-t és a 7.2.2. pont szerinti oldatból 1 ml-t egy 10 ml-es mérőlombikba (5.2.). Töltsük fel 10 ml-re n-hexánnal (4.1.)
7.2.5. Tegyünk a 7.2.4. pont szerinti oldatból 1 ml-t egy 10 ml-es mérőlombikba (5.2.) és hígítsuk fel 10 ml-re n-hexánnal (4.1.)
7.3. Kromatográfiás meghatározás
7.3.1. Injektáljunk a 7.2.5. pont szerinti standard oldatból 1 μl-t kétszer
7.3.2. Injektáljunk minden mintaoldatból 1 μl-t
Megjegyzés: Ha az "on-column" injektort használjuk, nagyobb hígítást kell alkalmazni a standard és a mintaoldatok esetében is.
7.3.3. Ismételjük meg a 7.3.1. pont szerinti műveletet minden harmadik mintánál annak érdekében, hogy a mintákat kettős standard injektálások fogják közre. Az eredmények a standard kromatogramból származó válaszjelek számtani közepén alapulnak
8. AZ EREDMÉNYEK KISZÁMÍTÁSA
Minden egyes kromatogram esetében integrálni kell az önantsav és a kapronsav trigliceridjeihez kapcsolódó csúcsterületeket.
Kövessük ezeket az utasításokat minden egyes közrefogott sorozatra vonatkozóan, azaz a közrefogott mintasorozatok esetében a közvetlenül előttük kétszer injektált standard az STD1 és a közvetlenül utánuk kétszer injektált standard az STD2.
8.1. Kalibrálás
8.1.1. Számítsuk ki az Rf1(a) és Rf1(b) válaszjel értékét minden kettős STD1-re
Rf1 (a) vagy (b) = (csúcsterület kapronsav-triglicerid/csúcsterület önantsav-triglicerid) × 100
Számítsuk ki a közepes Rf1 válaszjelet
Rf1 = (Rf1(a) + Rf1(b)) / 2
8.1.2. Hasonlóképp számítsuk ki a közepes STD2, Rf2 válaszjelet
8.1.3. Számítsuk ki a közepes Rf válaszjelet
Rf = (Rf1 + Rf2) /2
8.2. Vizsgálati minták
Az STD1 és STD2 között kapott minden egyes mintakromatogramra számítsuk ki a C (kg/t) önantsavtaralmat:
C = (önantsav-triglicerid csúcsterülete × Rf × 100)/(kapronsav-triglicerid csúcsterülete × Wt × 1 000)
ahol:
- Wt = a vett zsír súlya (g),
- 100 = a minta hígítási térfogata,
- 1 000 = átváltási tényező (μg/g átváltása kg/t-ra)
A vajminták esetében vegyük figyelembe a vaj zsírtartalmát és számítsunk korrigált koncentrációs értéket, Cvaj (kg/t vaj)
Cvaj = Czsír × F
ahol F a vaj zsírtartalma.
9. PONTOSSÁG
A pontossági módszerre vonatkozó ISO 5725-1 és ISO 5725-2 szabványnak megfelelő laboratóriumközi vajvizsgálatok részletei a 12. pontban találhatók.
Az ismételhetőségi és reprodukálhatósági határértékek 95 %-os valószínűségi szinten vannak kifejezve, és előfordulhat, hogy nem alkalmazhatók a megadottakon kívüli koncentrációtartományokra és mátrixokra.
9.1. Ismételhetőség
Ugyanazon módszerrel azonos vizsgálati anyagon, ugyanazon laboratóriumban ugyanazon kezelőszemély által, ugyanazon berendezésen, és a két teszt elvégzése között eltelt rövid idő alatt kapott két független egyedi vizsgálati eredmény közötti abszolút különbség az esetek legfeljebb 5 %-ában lesz nagyobb 0,35 kg/t-nál.
9.2. Reprodukálhatóság
Ugyanazon módszerrel azonos vizsgálati anyagon, különböző laboratóriumban különböző kezelőszemély által, különböző berendezésen kapott két független egyedi vizsgálati eredmény közötti abszolút különbség az esetek legfeljebb 5 %-ában lesz nagyobb 0,66 kg/t-nál.
10. TŰRÉSHATÁROK: ALSÓ HATÁROK (NEM ELÉGSÉGES MENNYISÉGEK ESETE)
10.1. A megjelölt termékből három mintát kell venni annak ellenőrzésére, hogy a termék megjelölése helyesen történt-e
10.2. Vaj és vajkoncentrátum
10.2.1. A hozzákeverési arány 11 kg legalább 95 %-os tisztaságú önantsav-triglicerid, a vaj minden tonnájára, vagyis 10,45 kg/t
10.2.2. A termék elemzéséből kapott három minta eredményét a jelölőanyag-hozzákeverés arányának és homogenitásának ellenőrzésére lehet alkalmazni, és ezek közül az eredmények közül a legalacsonyabbat a következő határértékekkel kell egybevetni:
- 9,51 kg/t (a 95 %-os tisztaságú önantsav-triglicerid legkisebb hozzákeverési arányának 95 %-a, egyedi meghatározás),
- 6,89 kg/t (a 95 %-os tisztaságú önantsav-triglicerid legkisebb hozzákeverési arányának 70 %-a, egyedi meghatározás),
- A legalacsonyabb eredményt adó minta jelölőanyag-koncentrációját használjuk a 9,51 kg/t és a 6,89 kg/t közötti interpolációval kapcsolatosan.
10.3. Tejszín
10.3.1. A hozzákeverési arány 10 kg legalább 95 %-os tisztaságú önantsav-triglicerid, a tejzsír minden tonnájára, vagyis 9,50 kg/t jelölt tejzsír
10.3.2. A termék elemzéséből kapott három minta eredményét a jelölőanyag-hozzákeverés arányának és homogenitásának ellenőrzésére lehet alkalmazni, és ezek közül az eredmények közül a legalacsonyabbat a következő határértékekkel kell egybevetni:
- 8,60 kg/t (a 95 %-os tisztaságú önantsav-triglicerid legkisebb hozzákeverési arányának 95 %-a, egyedi meghatározás),
- 6,23 kg/t (a 95 %-os tisztaságú önantsav-triglicerid legkisebb hozzákeverési arányának 70 %-a, egyedi meghatározás),
- A legalacsonyabb eredményt adó minta jelölőanyag-koncentrációját használjuk a 8,60 kg/t és a 6,23 kg/t közötti interpolációval kapcsolatosan.
11. TŰRÉSHATÁROK: FELSŐ HATÁROK (LEGALÁBB 20 %-OS MENNYISÉGTÚLLÉPÉS ESETE)
11.1. A megjelölt termékből három mintát kell venni annak ellenőrzésére, hogy a termék megjelölése helyesen történt-e
11.2. Vaj és vajkoncentrátum
11.2.1. A termék elemzéséből kapott három minta eredményét a jelölőanyag-hozzákeverés arányának és homogenitásának ellenőrzésére lehet alkalmazni, és az eredmények számtani közepét a következő határértékkel kell egybevetni:
- Felső határ 12,96 kg/t.
11.3. Tejszín
11.3.1. A termék elemzéséből kapott három minta eredményét a jelölőanyag-hozzákeverés arányának és homogenitásának ellenőrzésére lehet alkalmazni, és az eredmények számtani közepét a következő határértékkel kell egybevetni:
- Felső határ 11,82 kg/t.
12. TOVÁBBI TÁJÉKOZTATÁS: A VAJZSÍR TRIÖNANTOÁT-TARTALMÁNAK TRIGLICERID-ANALÍZISSEL TÖRTÉNŐ MEGHATÁROZÁSA SORÁN AZ EREDMÉNYEK STATISZTIKAI VIZSGÁLATA
Négy körvizsgálatra kerül sor a jelölt vaj triönantoát-tartalmának meghatározására.
Kilenc laboratórium vett részt az első körvizsgálatban, amelyben nem adtak meg előírásokat a használandó analitikai módszerekről.
Tíz laboratórium vett részt a második körvizsgálatban, amelyben 4 különböző módszert alkalmaztak:
- Metil-heptanoát mennyiségi meghatározása n-nonán vagy metil-nonanoát belső standard használatával
- Triönantoát mennyiségi meghatározása trikaproát belső standard használatával
- Metil-heptanoát mennyiségi meghatározása kalibrációs minta/keverék használatával
- Metil-heptanoát mennyiségi meghatározása kalibrációs keverék használatával.
Ezenfelül, amikor FAME (zsírsav-metil-észter) elemzésére került sor, két különböző metilációs eljárást használtak (De Francesco és Christopherson & Glass).
A kapott eredmények alapján két módszert választottak ki a harmadik körvizsgálat elvégzéséhez:
- Metil-heptanoát mennyiségi meghatározása n-nonán vagy metil-nonanoát belső standard használatával
- Triönantoát mennyiségi meghatározása trikaproát belső standard használatával.
7 laboratórium eredményeiből kiderült, hogy a FAME-módszer nagyobb változékonyságot eredményezett, következésképp az a döntés született, hogy csak a triönantoát, mint triglicerid meghatározását alkalmazzák, a triönantoát trikaproát belső standard használatával való mennyiségi meghatározására szolgáló eljárás alapján. A triglicerid-analízist ezenkívül kapilláris oszloppal kell végezni.
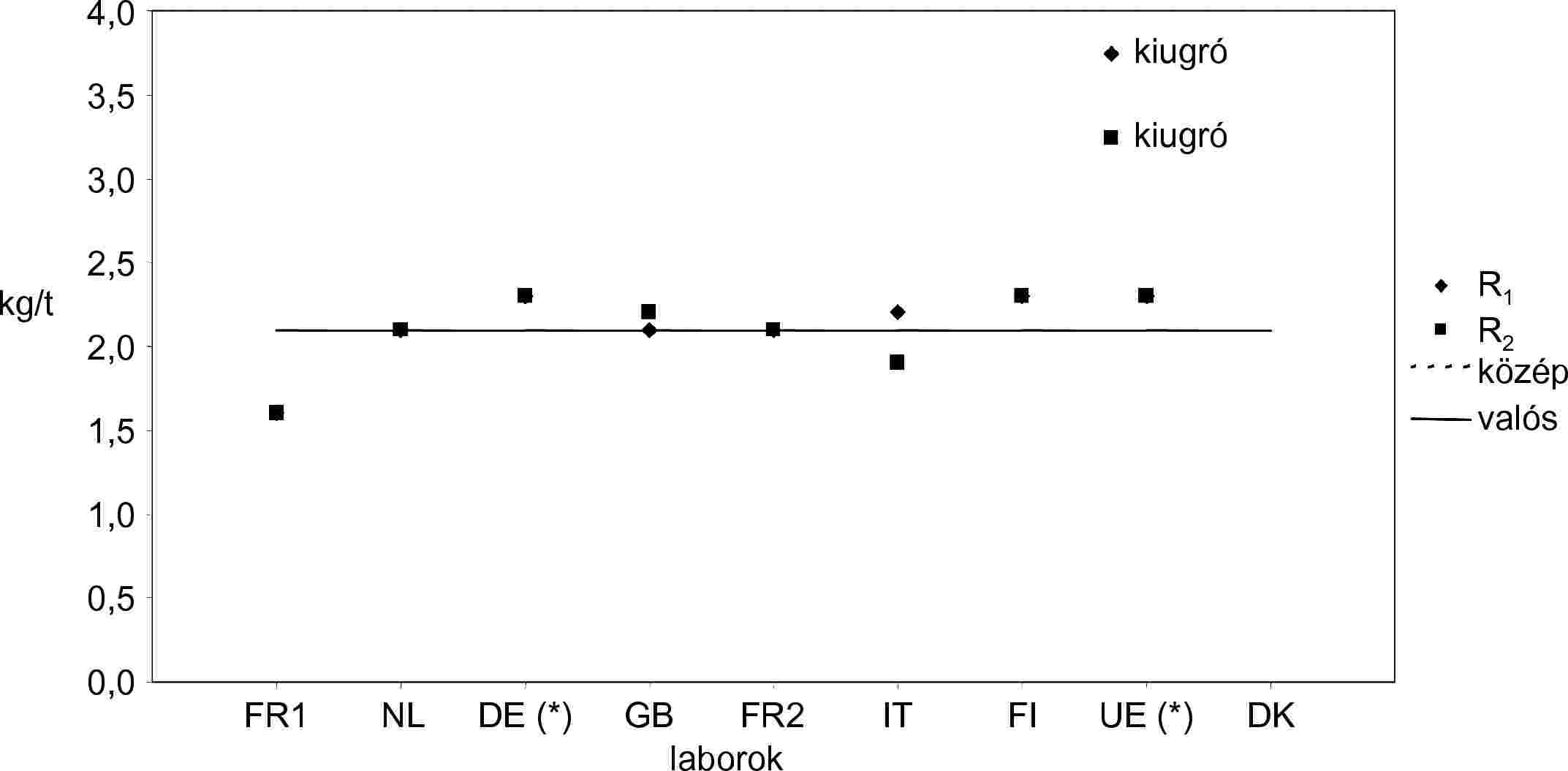
A negyedik körvizsgálatban négy mintát (A., B., C., D.) adtak körbe, és kilenc laboratórium szolgáltatott eredményeket (1-2. táblázat).
Két laboratórium (DE és UE) a FAME-módszer használatával analizálta a mintákat.
A laboratóriumok kis száma miatt a statisztikai számítás elvégzése a FAME-eredményeket is tartalmazó teljes adatkészleten (1-2. ábra) és a TG-analízis során kapott adatokon is megtörtént.
Kiugró értékek kimutatása:
- A. minta: a Dixon-, a Cochran- és a Grubbs-teszt 1 és 5 %-os szinten egy kiugró laboratóriumra mutatott rá.
- B. minta: a Grubbs-teszt 5 %-os szinten egy kiugró laboratóriumra mutatott rá.
- C. minta: a Dixon- és a Grubbs-teszt 1 és 5 %-os szinten egy kiugró laboratóriumra mutatott rá.
- D. minta: a Dixon- és a Grubbs-teszt 1 és 5 %-os szinten egy kiugró laboratóriumra mutatott rá.
A kiugró laboratóriumot kizárták a számításból.
Érdemes megjegyezni, hogy a FAME-módszerrel kapott eredményeket sosem tekintették kiugrónak az alkalmazott vizsgálatoknál.
Pontossági paraméterek
A 1. és 2. táblázat számol be az összes laboratórium eredményeiről és az elfogadható számú (8) laborra kiszámított pontossági paraméterekről, de sajnos az eredmények nem ugyanazon analitikai módszerből származnak.
A 3. és 4. táblázat számol be a csak a TG-módszerből származó eredményekről és a vonatkozó pontossági paraméterekről. E paraméterek elfogadása a laboratóriumok alacsony számának (6) elfogadásától függ.
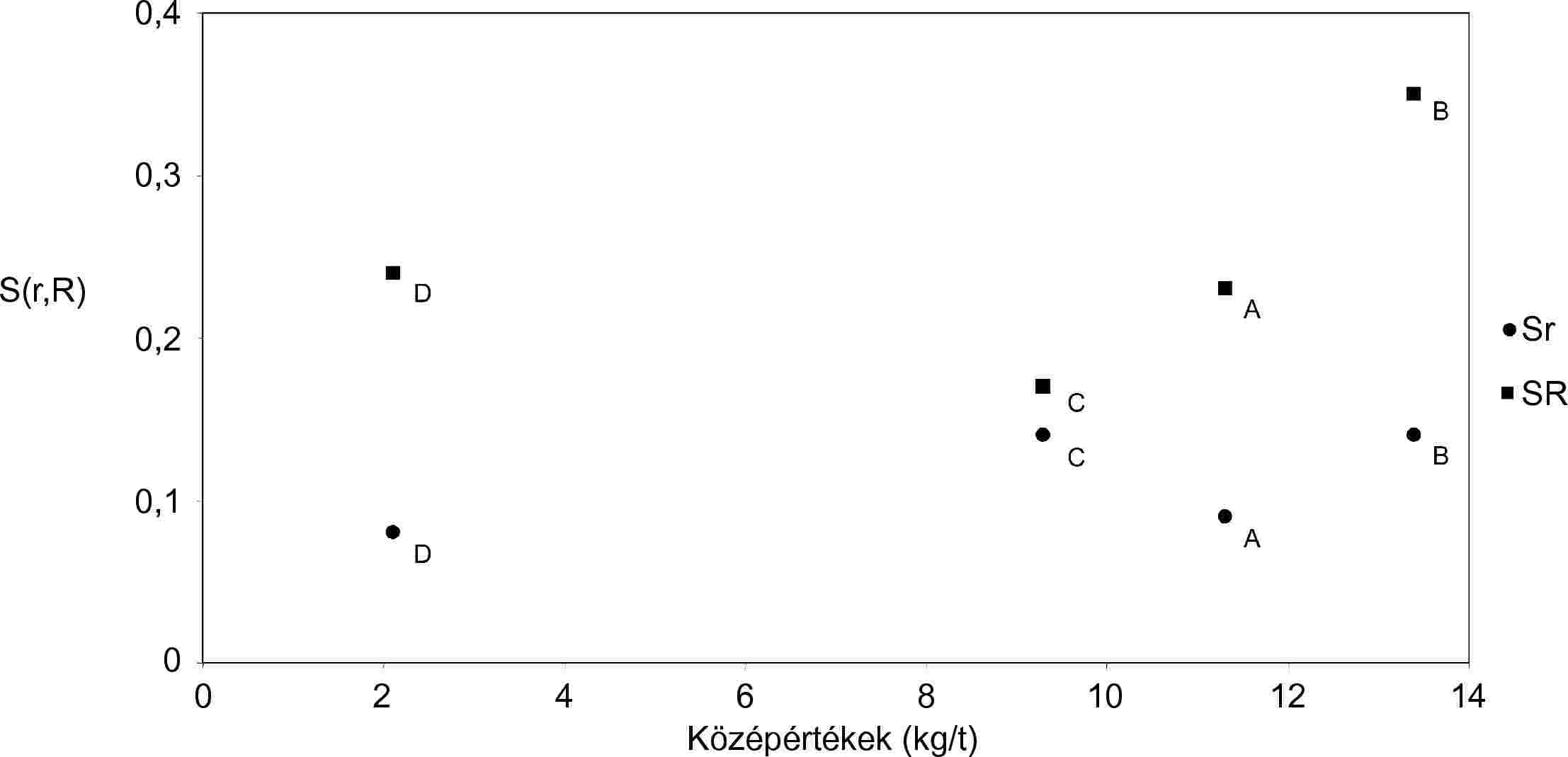
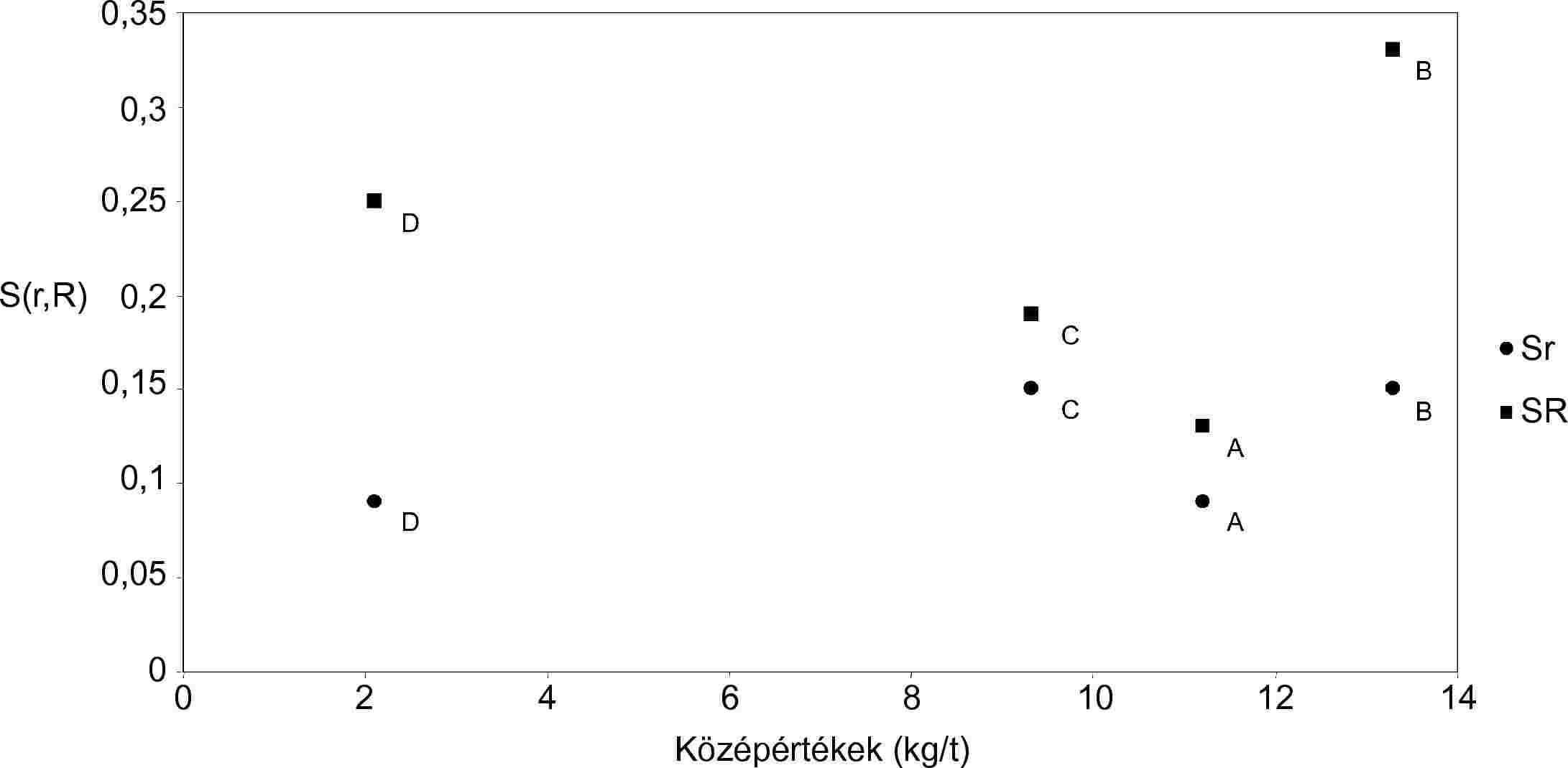
A 2. és 3. ábra a fent leírt két adatsor 4 mintájára kiszámított Sr és SR trendjét mutatja be.
Az 5. táblázat számol be az Sr és SR értékekről a vonatkozó összevont értékkel és az általános r és R paraméterekkel együtt.
Végül sor került a kritikus eltérés kiszámítására 95 %-os valószínűségi szint mellett.
1. táblázat
A TG + FAME* módszerek statisztikai eredményei
| A. minta | R1 | R2 | Közép | Fennmaradó laboratóriumok száma a kiugró adatok eltávolítása után | 8 | |
| RENNES | FR1 | 11,0 | 11,1 | 11,1 | Kiugrók száma | 1 |
| RIKILT | NL | 11,2 | 11,2 | 11,2 | Kiugrók | DК |
| ZPLA | DE* | 11,6 | 11,8 | 11,7 | Középérték | 11,3 |
| ADAS | GB | 11,4 | 11,2 | 11,3 | Valós érték | 11,0 |
| CNEVA | FR2 | 11,4 | 11,4 | 11,4 | Ismételhetőségi szórás (Sr) | 0,09 |
| LODI | IT | 11,1 | 11,3 | 11,2 | Ismételhetőségi relatív szórás (RSDr %) | 0,80 |
| EELA | FI | 11,3 | 11,2 | 11,3 | Ismételhetőség r (95 %) | 0,26 |
| ISPRA | UE* | 11,0 | 11,0 | 11,0 | Relatív ismételhetőség r % | 2,24 |
| D.V.F.A. | DK | 13,3 | 11,8 | 12,6 | Reprodukálhatósági szórás (SR) | 0,23 |
| Reprodukálhatósági relatív szórás (RSDR %) | 2,04 | |||||
| Reprodukálhatóság R (95 %) | 0,84 | |||||
| Relatív reprodukálhatóság R % | 5,71 | |||||
| B. minta | R1 | R2 | Közép | Fennmaradó laboratóriumok száma a kiugró adatok eltávolítása után | 8 | |
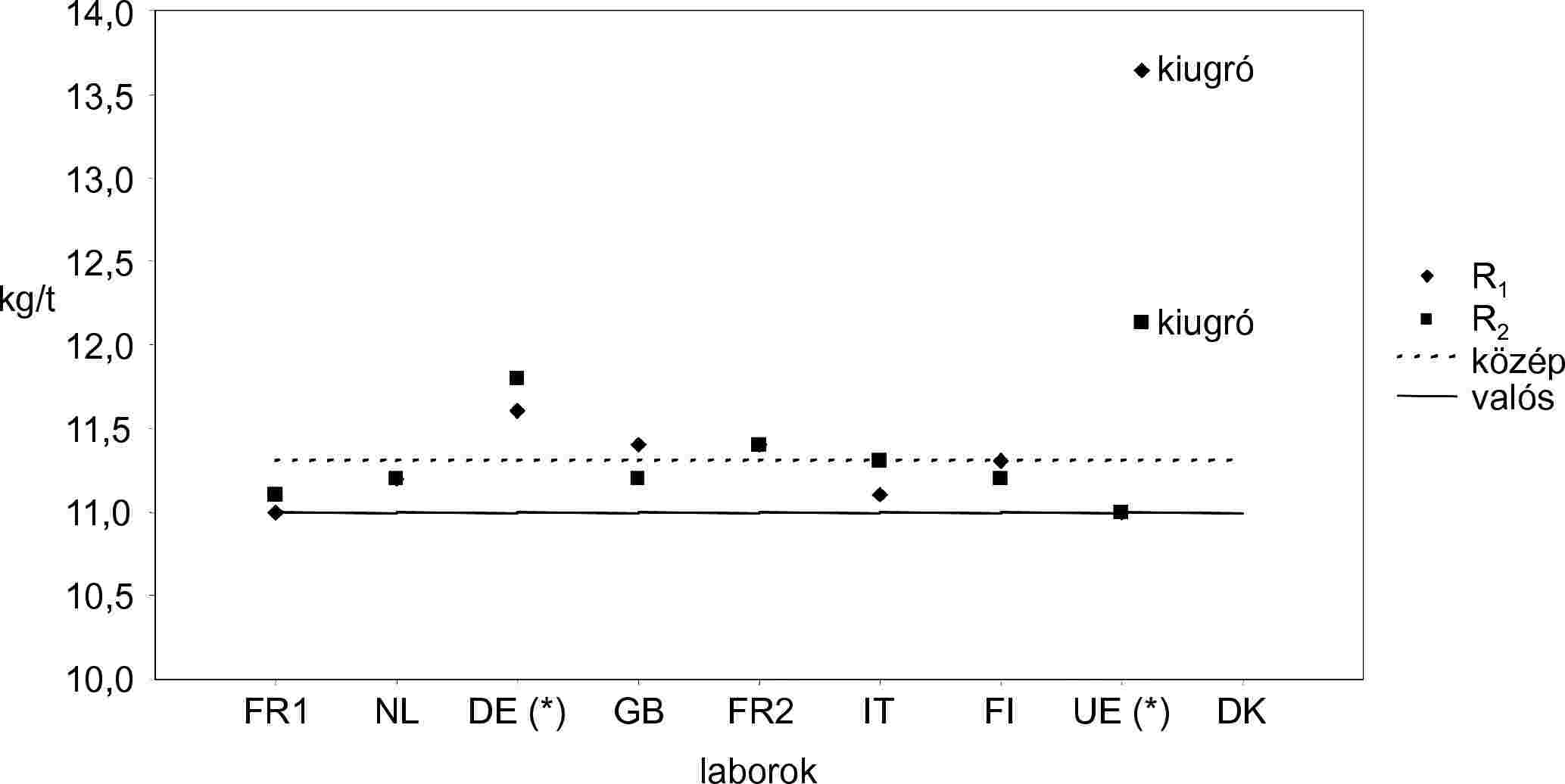
| RENNES | FR1 | 12,7 | 12,8 | 12,8 | Kiugrók száma | 1 |
| RIKILT | NL | 13,5 | 13,3 | 13,4 | Kiugrók | DK |
| ZPLA | DE* | 14,0 | 13,8 | 13,9 | Középérték | 13,4 |
| ADAS | GB | 13,4 | 13,5 | 13,5 | Valós érték | 13,5 |
| CNEVA | FR2 | 13,3 | 13,4 | 13,4 | Ismételhetőségi szórás (Sr) | 0,14 |
| LODI | IT | 13,9 | 13,5 | 13,7 | Ismételhetőségi relatív szórás (RSDr %) | 1,04 |
| EELA | FI | 13,4 | 13,2 | 13,3 | Ismételhetőség r (95 %) | 0,40 |
| ISPRA | UE* | 13,2 | 13,3 | 13,3 | Relatív ismételhetőség r % | 2,91 |
| D.V.F.A. | DK | 14,1 | 14,8 | 14,5 | Reprodukálhatósági szórás (SR) | 0,35 |
| Reprodukálhatósági relatív szórás (RSDR %) | 2,61 | |||||
| Reprodukálhatóság R (95 %) | 0,99 | |||||
| Relatív reprodukálhatóság R % | 7,31 |
2. táblázat
A TG + FAME* módszerek statisztikai eredményei
| C. minta | R1 | R2 | Közép | Fennmaradó laboratóriumok száma a kiugró adatok eltávolítása után | 8 | |
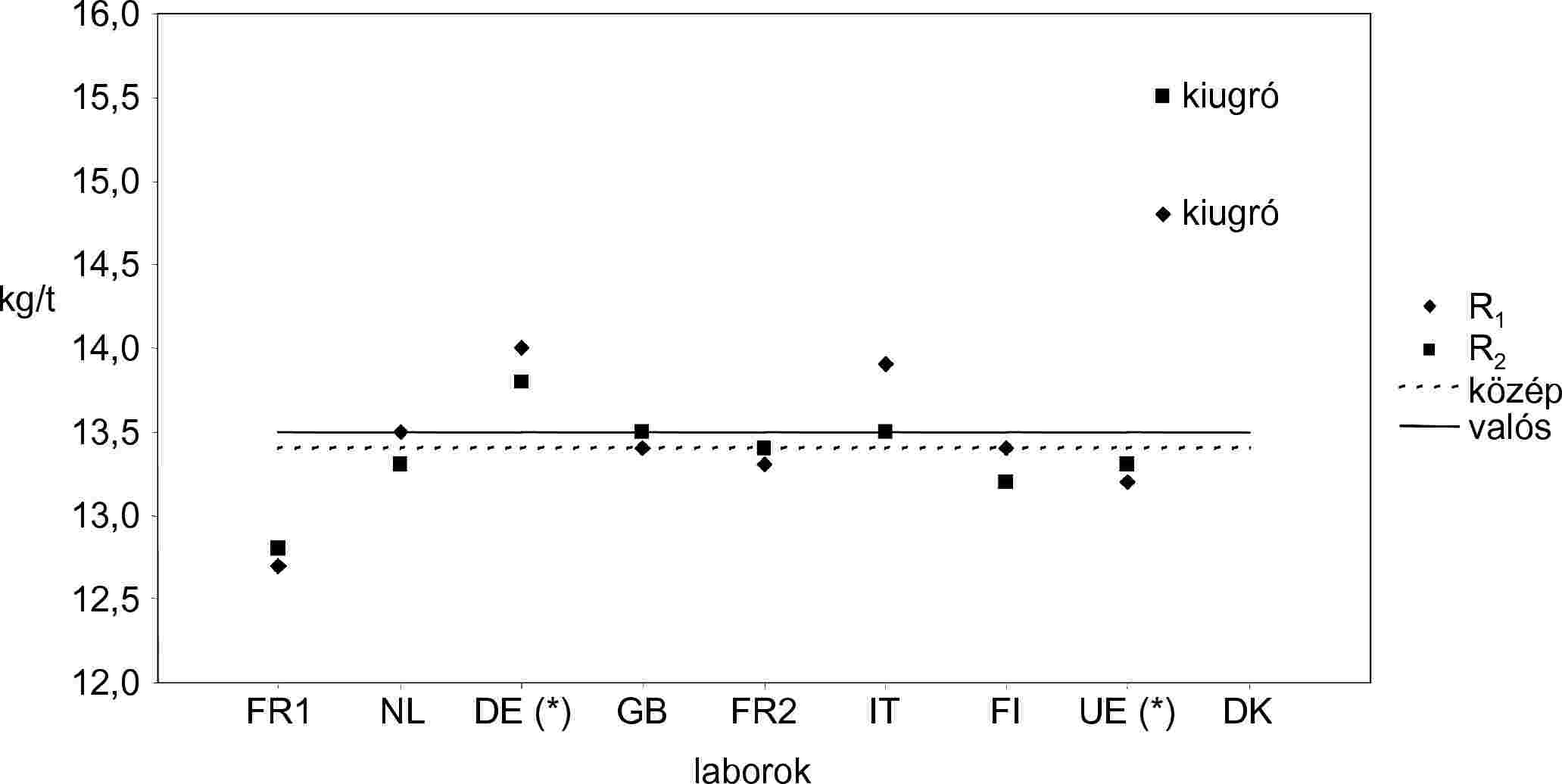
| RENNES | FR1 | 8,9 | 9,2 | 9,1 | Kiugrók száma | 1 |
| RIKILT | NL | 9,2 | 9,3 | 9,3 | Kiugrók | DK |
| ZPLA | DE* | 9,2 | 9,4 | 9,3 | Középérték | 9,3 |
| ADAS | GB | 9,5 | 9,3 | 9,4 | Valós érték | 9,3 |
| CNEVA | FR2 | 9,4 | 9,4 | 9,4 | Ismételhetőségi szórás (Sr) | 0,14 |
| LODI | IT | 9,2 | 9,5 | 9,4 | Ismételhetőségi relatív szórás (RSDr %) | 1,50 |
| EELA | FI | 9,4 | 9,6 | 9,5 | Ismételhetőség r (95 %) | 0,40 |
| ISPRA | UE* | 9,4 | 9,3 | 9,4 | Relatív ismételhetőség r % | 4,20 |
| D.V.F.A. | DK | 10,7 | 10,9 | 10,8 | Reprodukálhatósági szórás (SR) | 0,17 |
| Reprodukálhatósági relatív szórás (RSDR %) | 1,82 | |||||
| Reprodukálhatóság R (95 %) | 0,47 | |||||
| Relatív reprodukálhatóság R % | 5,10 | |||||
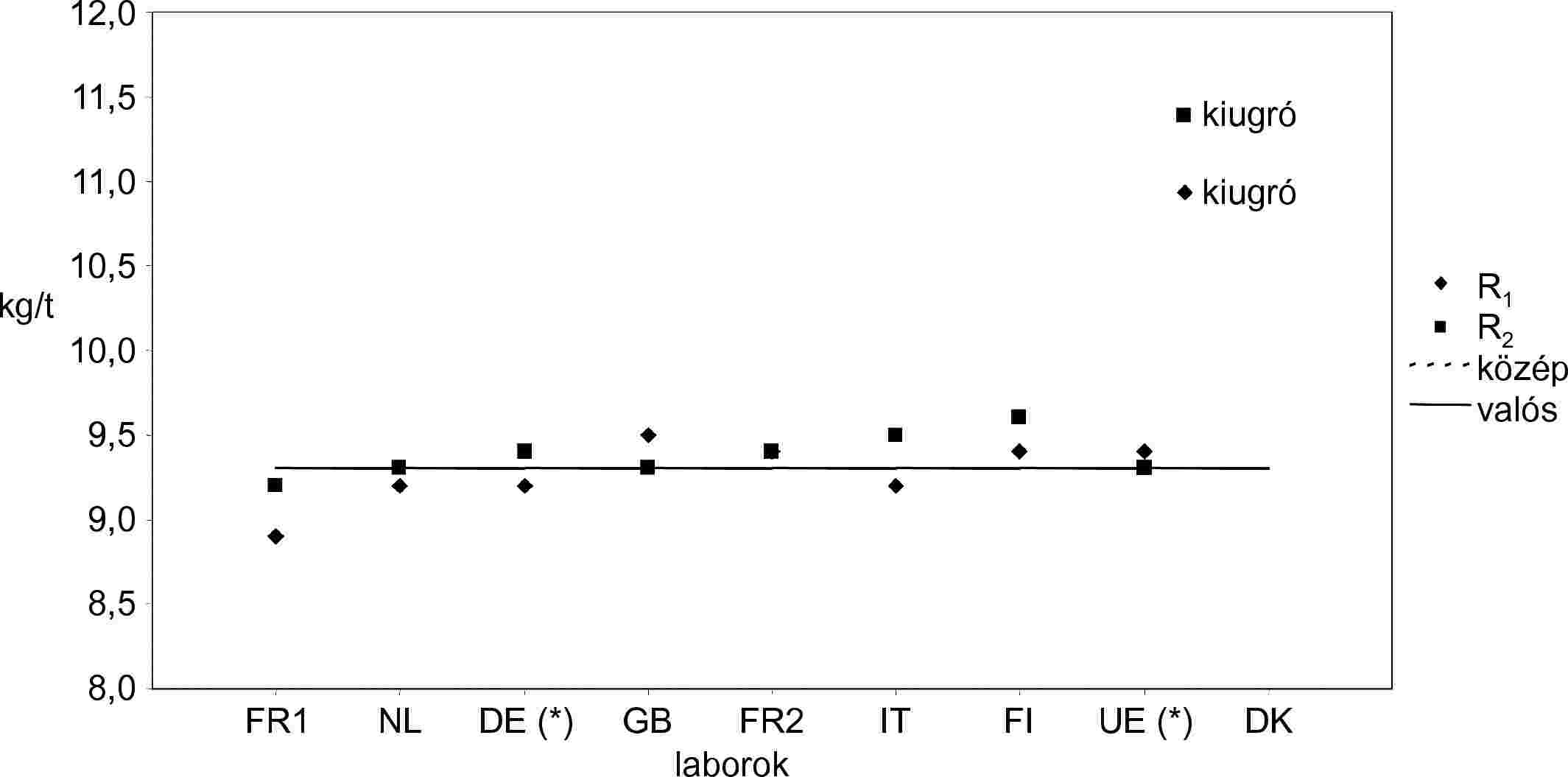
| D. minta | R1 | R2 | Közép | Fennmaradó laboratóriumok száma a kiugró adatok eltávolítása után | 8 | |
| RENNES | R1 | 1,6 | 1,6 | 1,6 | Kiugrók száma | 1 |
| RIKILT | NL | 2,1 | 2,1 | 2,1 | Kiugrók | DK |
| ZPLA | DE* | 2,3 | 2,3 | 2,3 | Középérték | 2,1 |
| ADAS | GB | 2,1 | 2,2 | 2,2 | Valós érték | 2,1 |
| CNEVA | FR2 | 2,1 | 2,1 | 2,1 | Ismételhetőségi szórás (Sr) | 0,08 |
| LODI | IT | 2,2 | 1,9 | 2,1 | Ismételhetőségi relatív szórás (RSDr %) | 3,81 |
| EELA | FI | 2,3 | 2,3 | 2,3 | Ismételhetőség r (95 %) | 0,22 |
| ISPRA | UE* | 2,3 | 2,3 | 2,3 | Relatív ismételhetőség r % | 10,67 |
| D.V.F.A. | DK | 3,4 | 2,9 | 3,2 | Reprodukálhatósági szórás (SR) | 0,24 |
| Reprodukálhatósági relatív szórás (RSDR %) | 11,43 | |||||
| Reprodukálhatóság R (95 %) | 0,67 | |||||
| Relatív reprodukálhatóság R % | 32,00 |
3. táblázat
A TG + FAME* módszerek statisztikai eredményei
| A. minta | R1 | R2 | Közép | Fennmaradó laboratóriumok száma a kiugró adatok eltávolítása után | 6 | |
| RENNES | FR1 | 11,0 | 11,1 | 11,1 | Kiugrók száma | 1 |
| RIKILT | NL | 11,2 | 11,2 | 11,2 | Kiugrók | DК |
| ADAS | GB | 11,4 | 11,2 | 11,3 | Középérték | 11,2 |
| CNEVA | FR2 | 11,4 | 11,4 | 11,4 | Valós érték | 11,0 |
| LODI | IT | 11,1 | 11,3 | 11,2 | Ismételhetőségi szórás (Sr) | 0,09 |
| EELA | FI | 11,3 | 11,2 | 11,3 | Ismételhetőségi relatív szórás (RSDr %) | 0,80 |
| D.V.F.A. | DK | 13,3 | 11,8 | 12,6 | Ismételhetőség r (95 %) | 0,25 |
| Relatív ismételhetőség r % | 2,24 | |||||
| Reprodukálhatósági szórás (SR) | 0,13 | |||||
| Reprodukálhatósági relatív szórás (RSDR %) | 1,16 | |||||
| Reprodukálhatóság R (95 %) | 0,36 | |||||
| Relatív reprodukálhatóság R % | 3,25 | |||||
| B. minta | R1 | R2 | Közép | Fennmaradó laboratóriumok száma a kiugró adatok eltávolítása után | 6 | |
| RENNES | FR1 | 12,7 | 12,8 | 12,8 | Kiugrók száma | 1 |
| RIKILT | NL | 13,5 | 13,3 | 13,4 | Kiugrók | DК |
| ADAS | GB | 13,4 | 13,5 | 13,5 | Középérték | 13,3 |
| CNEVA | FR2 | 13,3 | 13,4 | 13,4 | Valós érték | 13,5 |
| LODI | IT | 13,9 | 13,5 | 13,7 | Ismételhetőségi szórás (Sr) | 0,15 |
| EELA | FI | 13,4 | 13,2 | 13,3 | Ismételhetőségi relatív szórás (RSDr %) | 1,13 |
| D.V.F.A. | DK | 14,1 | 14,8 | 14,5 | Ismételhetőség r (95 %) | 0,42 |
| Relatív ismételhetőség r % | 3,16 | |||||
| Reprodukálhatósági szórás (SR) | 0,33 | |||||
| Reprodukálhatósági relatív szórás (RSDR %) | 2,48 | |||||
| Reprodukálhatóság R (95 %) | 0,93 | |||||
| Relatív reprodukálhatóság R % | 6,94 |
4. táblázat
A TG módszer statisztikai eredményei
| C. minta | R1 | R2 | Közép | Fennmaradó laboratóriumok száma a kiugró adatok eltávolítása után | 6 | |
| RENNES | FR1 | 8,9 | 9,2 | 9,1 | Kiugrók száma | 1 |
| RIKILT | NL | 9,2 | 9,3 | 9,3 | Kiugrók | DК |
| ADAS | GB | 9,5 | 9,3 | 9,4 | Középérték | 9,3 |
| CNEVA | FR2 | 9,4 | 9,4 | 9,4 | Valós érték | 9,3 |
| LODI | IT | 9,2 | 9,5 | 9,4 | Ismételhetőségi szórás (Sr) | 0,15 |
| EELA | FI | 9,4 | 9,6 | 9,5 | Ismételhetőségi relatív szórás (RSDr %) | 1,61 |
| D.V.F.A. | DK | 10,7 | 10,9 | 10,8 | Ismételhetőség r (95 %) | 0,42 |
| Relatív ismételhetőség r % | 4,51 | |||||
| Reprodukálhatósági szórás (SR) | 0,19 | |||||
| Reprodukálhatósági relatív szórás (RSDR %) | 2,04 | |||||
| Reprodukálhatóság R (95 %) | 0,53 | |||||
| Relatív reprodukálhatóság R % | 5,71 | |||||
| D. minta | R1 | R2 | Közép | Fennmaradó laboratóriumok száma a kiugró adatok eltávolítása után | 6 | |
| RENNES | FR1 | 1,6 | 1,6 | 1,6 | Kiugrók száma | 1 |
| RIKILT | NL | 2,1 | 2,1 | 2,1 | Kiugrók | DK |
| Középérték | 2,1 | |||||
| ADAS | GB | 2,1 | 2,2 | 2,2 | Valós érték | 2,1 |
| CNEVA | FR2 | 2,1 | 2,1 | 2,1 | Ismételhetőségi szórás (Sr) | 0,09 |
| LODI | IT | 2,2 | 1,9 | 2,1 | Ismételhetőségi relatív szórás (RSDr %) | 4,29 |
| EELA | FI | 2,3 | 2,3 | 2,3 | Ismételhetőség r (95 %) | 0,26 |
| D.V.F.A. | DK | 3,4 | 2,9 | 3,2 | Relatív ismételhetőség r % | 12,01 |
| Reprodukálhatósági szórás (SR) | 0,25 | |||||
| Reprodukálhatósági relatív szórás (RSDR %) | 11,90 | |||||
| Reprodukálhatóság R (95 %) | 0,69 | |||||
| Relatív reprodukálhatóság R % | 33,32 |
5. táblázat
Ismételhetőség és reprodukálhatóság (FAME-val)
| Laborok száma | Kiugrók | Ismételhetőség Sr (95 %) | Reprodukálhatóság SR (95 %) | |
| A. minta | 8 | 1 | 0,09 | 0,23 |
| Β. minta | 8 | 1 | 0,14 | 0,35 |
| C. minta | 8 | 1 | 0,14 | 0,17 |
| D. minta | 8 | 1 | 0,08 | 0,24 |
| Összevont érték | 0,116 | 0,256 | ||
| R | R | |||
| Összevont érték x 2,8 | 0,324 | 0,716 | ||
| CrD95 = 0,40 A triönantoát kijelentett minimális tisztasága = 95 % A vajzsírban található triönantoát kijelentett minimális határértéke = 11 kg/t Figyelembe véve a kritikus eltérést 95 %-os valószínűségi szint esetén, a két eredmény számtani közepe nem lehet kevesebb mint: 95 %-os tisztaságú triönantoát hozzákeverése esetén 10,05 kg/t | ||||
Ismételhetőség és reprodukálhatóság (FAME nélkül)
| Laborok száma | Kiugrók | Ismételhetőség Sr (95 %) | Reprodukálhatóság SR (95 %) | |
| A. minta | 6 | 1 | 0,09 | 0,13 |
| B. minta | 6 | 1 | 0,15 | 0,33 |
| C. minta | 6 | 1 | 0,15 | 0,19 |
| D. minta | 6 | 1 | 0,09 | 0,25 |
| Összevont érték | 0,124 | 0,237 | ||
| r | R | |||
| Összevont érték x 2,8 | 0,347 | 0,663 | ||
| CrD95 = 0,36 A triönantoát kijelentett minimális tisztasága = 95 % A vajzsírban található triönantoát kijelentett minimális határértéke = 11 kg/t Figyelembe véve a kritikus eltérést 95 %-os valószínűségi szint esetén, a két eredmény számtani közepe nem lehet kevesebb mint: 95 %-os tisztaságú triönantoát hozzákeverése esetén 10,09 kg/t. | ||||
1. ábra ( 8 )
Kísérleti eredmények: A. minta
Kísérleti eredmények: B. minta
Kísérleti eredmények: C. minta
Kísérleti eredmények: D. minta
2. ábra
Ismételhetőségi és reprodukálhatósági szórás különböző szinteken (TG+FAME)
3. ábra
Ismételhetőségi és reprodukálhatósági szórás különböző szinteken (TG)
4. ábra
Példa "on-column" injektor használata esetén
VI. MELLÉKLET
(5. cikk)
VANILLINTARTALOM MEGHATÁROZÁSA NAGYTELJESÍTMÉNYŰ FOLYADÉKKROMATOGRÁFIÁVAL VAJKONCENTRÁTUMBAN, VAJBAN VAGY TEJSZÍNBEN
1. TÁRGY ÉS ALKALMAZÁSI TERÜLET
Ez a módszer a vajkoncentrátum, a vaj és a tejszín vanillintartalmának mennyiségi meghatározására alkalmas eljárást ismerteti.
2. A MÓDSZER ELVE
Ismert mennyiségű minta kivonása izopropanol, etil-alkohol, valamint acetonitril 1:1:2 arányú keverékével. A zsír nagy részének kicsapása -15 °C és -20 °C közötti hőmérsékletre történő hűtéssel és azt követő centrifugálással.
Vízzel való hígítás után a vanillintartalmat nagyteljesítményű folyadékkromatográfiás módszerrel lehet meghatározni (HPLC).
3. ESZKÖZÖK
A szokásos laboratóriumi berendezés, valamint különösen a következők:
3.1. Fagyasztó, amely -15 °C és -20 °C hőmérséklet közötti tartományban üzemel
3.2. Eldobható, 2 ml űrtartalmú fecskendők
3.3. Membrános mikroszűrők 0,45 μm pórusmérettel, amelyek ellenállóak az 5 % extraháló oldatot tartalmazó folyadékkal szemben (4.4.)
3.4. Folyadékkromatográfiás rendszer, amely egy szivattyúból (1,0 ml/min átfolyási teljesítmény), egy injektorból (20 μl automatikus vagy kézi befecskendezés), egy UV-detektorból (306 nm-en üzemelő, 0,01 Å a teljes skálán), egy regisztráló készülékből vagy integrátorból, valamint egy 25 °C-on üzemelő oszloptermosztátból áll
3.5. Analitikai oszlop (250 mm × 4,6 mm ID) LiChrospher RP 18 (Merck, 5 μm) vagy azzal egyenértékű anyaggal töltve
3.6. Védőoszlop (kb. 20 mm × 3 mm ID) LiChrospher RP 18 (5-10 μm) hordozóval vagy azzal egyenértékű anyaggal szárazon töltve
3.7. 2 000-es percenkénti fordulatszámon működő centrifuga.
4. VEGYSZEREK
Az összes felhasznált vegyszernek elismert analitikai minőségűnek kell lennie.
4.1. Izopropanol
4.2. Etil-alkohol 96 % (térfogatszázalék)
4.3. Acetonitril
4.4. Extraháló oldat
Keverjük össze az izopropanolt (4.1.), etil-alkoholt (4.2.) és acetonitrilt (4.3.) 1:1:2 (v/v) térfogat arányban.
4.5. Vanillin (4-hidroxi-3-metoxi-benzaldehid) ≥ 98 %
4.5.1. Vanillin törzsoldat (= 500 μg/ml)
Mérjünk ki egy 100 ml-es mérőlombikba 0,1 mg-os pontossággal 50 mg (CM mg) vanillint (4.5.), adjunk hozzá 25 ml extraháló oldatot (4.4.), és töltsük fel vízzel.
4.5.2. Vanillinstandard-oldat (= 10 μg/ml)
Pipettázzunk 5 ml vanillin törzsoldatot (4.5.1.) egy 250 ml űrtartalmú mérőlombikba, és töltsük fel vízzel.
4.5.3. Metil-alkohol, HPLC-minőség
4.5.4. Vízmentes ecetsav (jégecet)
4.5.5. Víz, HPLC-minőség
4.5.6. HPLC-mozgófázis Keverjünk el egy 1 000 ml-es mérőlombikban 300 ml metil-alkoholt (4.5.3.) és 20 ml ecetsavat (4.5.4.) körülbelül 500 ml vízzel (4.5.5.) és töltsük fel vízzel (4.5.5.). Szűrjük át a 0,45 μm pórusátmérőjű szűrőn (3.3.).
5. ELJÁRÁS
5.1. A minta előkészítése
5.1.1. Vaj
Addig melegítsük a mintát, amíg olvadni kezd. A 30 °C hőmérséklet feletti helyi túlmelegedéseket el kell kerülni. A vaj semmiképp nem válhat szét két fázisra. Amikor a minta már kellően lággyá vált, rázással homogenizáljuk. A vajat 15 másodpercen át keverjük, mielőtt mintát vennénk belőle. Mérjünk le egy 100 ml-es mérőlombikba 1 milligrammos pontossággal körülbelül 5 g (SM g) vajat.
5.1.2. Vajkoncentrátum
Közvetlenül mintavétel előtt helyezzük a vajkoncentrátumot tartalmazó edényt 40 és 50 °C közötti hőmérsékleten egy szárítószekrénybe, amíg teljesen meg nem olvad. A mintát ezután keveréssel, rázogatással keverjük el, kerülve a túl heves rázás okozta légbuborékok képződését. Mérjünk ki egy 100 ml-es mérőlombikba 1 milligrammos pontossággal körülbelül 4 g (SM g) vajkoncentrátomot.
5.1.3. Tejszín
Vízfürdőben vagy inkubátorban melegítsük a mintát 35-40 °C hőmérsékletre. Keveréssel vagy - szükség esetén - rázogatással, egyenletesen oszlassuk el a zsírt. Hűtsük le gyorsan a mintát 20 ± 2 °C hőmérsékletre. A mintának homogénnek kell kinéznie; ellenkező esetben az eljárást ismételjük meg. Mérjünk le egy 100 ml-es mérőlombikba 1 milligrammos pontossággal körülbelül 10 g (SM g) tejszínt.
5.2. A vizsgálati oldat elkészítése.
Adjunk hozzá a vizsgálati mintához (5.1.1., 5.1.2. vagy 5.1.3.), 75 ml extraháló oldatot (4.4.) erősen kavarjuk vagy rázzuk fel körülbelül 15 percig és töltsük fel az extraháló oldattal (4.4.). Ebből az extraktumból vigyünk át körülbelül 10 ml mennyiséget egy dugóval ellátott kémcsőbe. Tegyük a kémcsövet fagyasztóba (3.1.), és hagyjuk ott állni körülbelül 30 percen át. Centrifugáljuk a hideg extraktumot 5 percig megközelítőleg percenkénti 2 000-es fordulatszámon és azonnal dekantáljuk. A dekantált oldatot hagyjuk szobahőmérsékletűvé válni. Pipettázzunk 5 ml dekantált oldatot egy 100 ml űrtartalmú mérőlombikba, és töltsük fel vízzel. Egy aliquot részt szűrjünk le a membrános mikroszűrőn (3.3.) fecskendő (3.2.) használatával. A szűrlet most már készen áll a HPLC-s vizsgálatra.
5.3. Kalibrálás
Pipettázzunk 5 ml vanillinstandard-oldatot (4.5.2.) egy 100 ml űrtartalmú mérőlombikba. Adjunk hozzá 5 ml extraháló oldatot (4.4.), és töltsük fel a jelig vízzel. Ez az oldat 0,5 μg/ml vanillint tartalmaz.
5.4. HPLC-meghatározás (futtatás)
Hagyjuk a kromatográfiás rendszert körülbelül 30 percig stabilizálódni. Injektáljuk a standard oldatot (5.3.). Addig ismételjük ezt a műveletet, míg két egymást követő befecskendezés után a csúcs alatti területek, illetve a csúcsok magassága közötti különbség kisebb nem lesz mint 2 %. A meghatározott feltételek mellett a vanillin retenciós ideje körülbelül 9 perc. A standard oldatot (5.3.) 20 μl befecskendezésével kettős próbában elemezzük. Injektáljunk 20 μl vizsgálati oldatot (5.2.). Határozzuk meg a kapott vanillincsúcs magasságát vagy a görbe alatti területet. A vizsgálati minta (5.2.) tíz befecskendezését követően ismételjük meg a standard oldat kettős befecskendezését (5.3.).
6. AZ EREDMÉNYEK KISZÁMÍTÁSA
Számítsuk ki az egyes vizsgálati oldattételeket közrefogó kettős standardhoz tartozó vanillincsúcsok csúcsterületének átlagos nagyságát (vagy magasságát) (AC) (összesen négy terület, illetve magasság).
Számítsuk ki a területi tényezőt (R):
ahol CM a vanillin tömege milligrammban (4.5.1.).
A vizsgálati mintában található vanillintartalom (mg/kg) (C) a következő összefüggés alapján számítható ki:
ahol:
AS = a vizsgálati minta vanillincsúcsának csúcsterülete vagy magassága
SM = a vizsgálati minta tömege grammban (5.1.1., 5.1.2. vagy 5.1.3.).
Megjegyzés: Ha tejszínt elemzünk vanillintartalom meghatározása céljából, a jelölőanyag koncentrációját az egy kilogramm tejzsírra eső jelölőanyag milligrammjában kell kifejezni. Ezt úgy tehetjük meg, hogy a C-értéket megszorozzuk 100/f-el. Az f a tejszín zsírtartalma százalékban megadva (m/m).
20 = a standard oldat, valamint a vizsgálati minta hígítását figyelembe vevő korrekciós tényező
0,96 = a vizsgálati minta első hígításában található zsírtartalomra meghatározott korrekciós tényező
Megjegyzés: A csúcsterület helyett a csúcsok magasságát is használhatjuk (lásd a 8.3. pontot).
7. A MÓDSZER PONTOSSÁGA
7.1. Ismételhetőség (r)
A lehető legrövidebb időeltéréssel, ugyanazon laboratóriumi személy által, azonos eszközökkel, azonos vizsgálati anyagon elvégzett két meghatározás eredményének különbsége ne haladja meg a 16 mg/kg-ot.
7.2. Reprodukálhatóság (R)
Különböző laboratóriumokban lévő személyzet által, különböző eszközökkel, azonos vizsgálati anyagon elvégzett két meghatározás eredményének különbsége ne haladja meg a 27 mg/kg-ot.
8. TŰRÉSHATÁROK
8.1. A megjelölt termékből a homogenitás ellenőrzése érdekében három mintát kell venni.
8.2. A jelölőanyagot vagy vaníliából vagy szintetikus vanillinből nyerjük
8.2.1. A 4-hidroxi-3-metoxibenzaldehid hozzákeverési aránya 250 g a vajkoncentrátum vagy a vaj tonnájára. Amennyiben a tejszínnél alkalmazzuk a megjelölést, a hozzákeverési arány 250 g a tejzsír tonnájára számolva
8.2.2. A termék elemzéséből kapott három minta eredménye szolgál a jelölőanyag-hozzákeverés arányának és homogenitásának ellenőrzésére, és ezek közül az eredmények közül a legalacsonyabbat a következő határértékekkel kell egybevetni:
- 220,8 mg/kg (a legkisebb hozzákeverési arány 95 %-a),
- 158,3 mg/kg (a legkisebb hozzákeverési arány 70 %-a).
A legalacsonyabb eredményt adó minta jelölőanyag-koncentrációját a 220,8 mg/kg és 158,3 mg/kg közötti interpolációval együtt használjuk.
8.3. Kizárólag vaníliarúdból vagy annak teljes értékű kivonatából előállított jelölőanyag:
8.3.1. A 4-hidroxi-3-metoxibenzaldehid hozzákeverési aránya 100 g a vajkoncentrátum vagy a vaj tonnájára. Amennyiben a tejszínnél alkalmazzuk a megjelölést, a hozzákeverési arány 100 g a tejzsír tonnájára számolva
8.3.2. A termék elemzéséből kapott három minta eredménye szolgál a jelölőanyag-hozzákeverés arányának és homogenitásának ellenőrzésére, és ezek közül az eredmények közül a legalacsonyabbat a következő határértékekkel kell egybevetni:
- 78,3 mg/kg (a legkisebb hozzákeverési arány 95 %-a),
- 53,3 mg/kg (a legkisebb hozzákeverési arány 70 %-a).
A legalacsonyabb eredményt adó minta jelölőanyag-koncentrációját a 78,3 mg/kg és 53,3 mg/kg közötti interpolációval együtt használjuk.
9. MEGJEGYZÉS
9.1. A hozzáadott vanillin visszanyerési aránya 250 mg/kg vajolajnál 97,0 és 103,8 között ingadozik. A kapott átlagos vanillintartalom 99,9 % volt 2,7 % szórás mellett.
9.2. A vizsgálati mintában található 5 %-os extraháló oldat jelenléte által okozott csúcskiszélesedés ellensúlyozása érdekében a standard oldat 5 %-os extraháló oldatot tartalmaz. Ezáltal lehetővé válik a mennyiségi meghatározás a csúcsmagasság alapján is.
9.3. Az elemzés origón áthaladó lineáris kalibrációs görbén alapszik.
9.4. A standard oldat megfelelő hígításainak alkalmazásával (4.5.2.) a linearitást az elemzés első alkalommal történő elvégzésekor ellenőrizni kell, a későbbiekben rendszeres időközönként, valamint a HPLC-eszközben történt változtatás, illetve annak javítása után. A vanillint a pasztörizálatlan tejszínben vagy abból készült termékekben lévő enzimek lebonthatják vanillinsavvá, divanillinná és más vegyületekké.
VII. MELLÉKLET
(5. cikk)
BÉTA-APO-8'-KAROTINSAV ETIL-ÉSZTERÉNEK MEGHATÁROZÁSA VAJKONCENTRÁTUMBAN ÉS VAJBAN SPEKTROMÉTERREL
1. TÁRGY ÉS ALKALMAZÁSI TERÜLET
A módszer a béta-apo-8'-karotinsav etil-észterének (apo-karotén-észter) vajkoncentrátumban és vajban való mennyiségi meghatározására alkalmas eljárást ismerteti. Az apo-karotén-észter mindazon anyagok összessége, amelyek a módszerben meghatározott feltételek mellett készített minta kivonatában jelen vannak, és a fényt 440 nm-en elnyelik.
2. A MÓDSZER ELVE
A vajzsírt petroléterben feloldjuk, és megmérjük a fényelnyelését 440 nm-es hullámhosszon. Az apo-karotén-észter-tartalom meghatározása egy külső standarddal való egybevetés útján történik.
3. ESZKÖZÖK
3.1. Pipetták - beosztással ellátott, 0,25, 0,50, 0,75 és 1,0 ml űrtartalommal
3.2. Spektrofotométer - 440 nm (és 447-449 nm) hullámhosszon való mérésre alkalmas és 1 cm hosszú fényúttal rendelkező küvettákkal felszerelve
3.3. 20 ml és 100 ml űrtartalmú mérőlombikok
3.4. Analitikai mérleg, 0,1 mg-os érzékenységű, 1 mg-os pontosságú mérésre képes, 0,1 mg-os leolvashatósággal
3.5. Kemence, 45 °C ± 1 °C
3.6. Gyorsszűrő hamumentes szűrőpapírok
4. VEGYSZEREK
Az összes vegyszernek elismerten analitikai tisztaságúnak kell lennie.
4.1. Apo-karotén-észter szuszpenzió (megközelítőleg 20 %-os)
4.1.1. A szuszpenzió tartalmát a következőképpen állapítjuk meg:
Melegítsük fel a szuszpenziót 45 °C és 50 °C közé, és homogenizáljuk a felnyitatlan, eredeti tárolóedényben. Mérjünk ki megközelítőleg 400 mg-ot egy mérőlombikba (100 ml), oldjuk fel 20 ml kloroformban (4.4.), és töltsük fel jelig ciklohexánnal (4.5.). Ebből az oldatból 5 ml-t ciklohexánnal hígítsunk fel 100 ml-re (A. oldat). Az A. oldatból 5 ml-t ciklohexánnal hígítsunk fel 100 ml-re. Mérjük meg az elnyelését 447-449 nm hullámhosszon (a maximumot ciklohexánnal mint vakpróbával szemben mérjük ki 1 cm hosszú fényúttal rendelkező üvegküvettákat használva).
Apo-karotén-észter-tartalom P (%) = (Abs max × 40 000) / (Msusp × 2 550) vagy: (Absmax / 2 550) × (100 / 5) × (100 / 5) × (100 / Msusp)
Absmax = a mérőoldat elnyelési maximuma
Msusp = a szuszpenzió tömege (g)
2 550 = elnyelési referenciaérték (1 %, 1 cm)
P = A szuszpenzió tisztasága (tartalma) P (%)
Megjegyzés: Az apo-karotén-észter levegőre, hőre és fényre egyaránt érzékeny. A felnyitatlan, eredeti tároló edényben (amelyet nitrogén atmoszférában zárnak le), hűvös helyen körülbelül 12 hónapig tárolható. A felnyitás után azonban az edény tartalmát rövid időn belül el kell használni.
4.1.2. Apo-karotén-észter-standardoldat, megközelítőleg 0,2 mg/ml töménységben
Mérjünk ki 1 mg pontossággal körülbelül 0,100 grammot az apo-karotén-észter-szuszpenzióból (4.1.1.) (W), oldjuk fel petroléterben (4.2.), majd az egész mennyiséget vigyük át egy 100 ml űrtartalmú mérőlombikba és petroléterrel töltsük fel a jelig.
Ez az oldat (W × P)/10 mg/ml apo-karotén-észtert tartalmaz.
Megjegyzés: Az oldatot sötét és hűvös helyen kell tárolni. A fel nem használt oldatot egy hónap elteltével ki kell önteni.
4.2. Petroléter (40-60 °C).
4.3. Kristályvizet nem tartalmazó szemcsés nátrium-szulfát, amelyet előzetesen két órán keresztül 102 °C-on szárítottak
4.4. Kloroform
4.5. Ciklohexán
5. ELJÁRÁS
5.1. A minta előkészítése
5.1.1. Vajkoncentrátum
A mintát kemencében megközelítőleg 45 °C-os hőmérsékleten olvasszuk meg.
5.1.2. Vaj
A mintát szárítószekrényben megközelítőleg 45 °C-os hőmérsékleten olvasszuk meg, majd egy reprezentatív adagot szűrjünk le belőle egy körülbelül 10 gramm, kristályvízmentes nátrium-szulfátot (4.3.) tartalmazó szűrőn, erős természetes és mesterséges fénytől védett környezetben, és továbbra is megtartva a 45 °C-os hőmérsékletet. Gyűjtsünk össze elegendő mennyiségű vajzsírt.
5.2. Meghatározás
Mérjünk ki 1 mg pontossággal körülbelül 1 gramm vajkoncentrátumot (vagy kivont vajzsírt (5.1.2.), (M). Az egész mennyiséget vigyük át egy 20 ml (V) űrtartalmú mérőlombikba, és petroléterrel (4.2.) töltsük fel a jelig, majd alaposan keverjük el.
Egy aliquot részt tegyünk bele egy 1 cm-es küvettába, és mérjük meg az elnyelését 440 nm hullámhosszon petroléterrel, mint vakpróbával szemben. Az apo-karotén-észter koncentrációját az oldatban a kapott kalibrációs görbével összevetve kapjuk meg (C μ/ml).
5.3. Kalibrálás
Pipettázzunk 0, 0,25, 0,5, 0,75, valamint 1,0 ml apo-karotén-észter-standardoldatot (4.1.2.) öt darab, 100 ml űrtartalmú mérőlombikba. Töltsük fel teljes térfogatra petroléterrel (4.2.), és keverjük el.
Az oldatok közelítőleges koncentrációi 0-tól 2 μg/ml-ig terjednek, és a standard oldat koncentrációjával egybevetve pontosan számoljuk ki (4.1.2.) (W × P)/10 mg/ml. Mérjük meg a fényelnyelést 440 nm-en a petroléterrel, mint vakpróbával szemben (4.2.).
Az elnyelési értékeket ábrázoljuk az y tengelyen, míg az apo-karotén-észter-koncentrációk az x tengelyen legyenek feltüntetve. Számítsuk ki a kalibrációs görbe egyenletét.
6. AZ EREDMÉNYEK KISZÁMÍTÁSA
6.1. Az apo-karotén-észter-tartalom mg/kg termékre megadva a következőképpen számítható ki:
Vajkoncentrátum: (C × V)/M
Vaj: 0,82 (C × V)M
ahol:
C = a kalibrációs görbéről leolvasott apo-karotén-észter-tartalom μg/ml-ben (5.3.)
V = a vizsgálati oldat mennyisége milliliterben (5.2.)
M = a vizsgálati minta tömege grammban (5.2.)
0,82 = korrekciós tényező a vaj vajzsírtartalmára.
7. A MÓDSZER PONTOSSÁGA
7.1. Ismételhetőség
7.1.1. Vajelemzés
A lehető legrövidebb időeltéréssel, ugyanazon laboratóriumi személy által, azonos eszközökkel, azonos vizsgálati anyagon elvégzett két meghatározás eredményének különbsége ne haladja meg az 1,4 mg/kg-ot.
7.1.2. Vajkoncentrátum-elemzés
A lehető legrövidebb időeltéréssel, ugyanazon laboratóriumi személy által, azonos eszközökkel, azonos vizsgálati anyagon elvégzett két meghatározás eredményének különbsége ne haladja meg az 1,6 mg/kg-ot.
7.2. Reprodukálhatóság
7.2.1. Vajelemzés
Különböző laboratóriumokban lévő személyzet által, különböző eszközökkel, azonos vizsgálati anyagon elvégzett két meghatározás eredményének különbsége ne haladja meg a 4,7 mg/kg-ot.
7.3. Vajkoncentrátum-elemzés
Különböző laboratóriumokban lévő személyzet által, különböző eszközökkel, azonos vizsgálati anyagon elvégzett két meghatározás eredményének különbsége ne haladja meg az 5,3 mg/kg-ot.
7.4. A pontossági adatok forrása
A pontossági adatokat egy 1995-ben elvégzett kísérlet szolgáltatta, amelyben 11 laboratórium vett részt, és tizenkét, jelölőanyagot tartalmazó mintát (hat kettős vakpróbát) használtak vaj vonatkozásában, illetve 12 jelölőanyagot tartalmazó mintát (hat kettős vakpróbát) vajkoncentrátum vonatkozásában.
8. TŰRÉSHATÁROK
8.1. A jelölőanyagot tartalmazó termékből három mintát kell venni annak ellenőrzésére, hogy a termék megjelölése helyesen történt-e.
8.2. Vaj
8.2.1. A vaj esetében a hozzákeverési arány, a háttérelnyelést is figyelembe véve, 22 mg/kg
8.2.2. A termék elemzéséből kapott három minta eredménye szolgál a jelölőanyag-hozzákeverés arányának és homogenitásának ellenőrzésére, és ezek közül az eredmények közül a legalacsonyabbat a következő határértékekkel kell egybevetni:
- 17,7 mg/kg (a legkisebb hozzákeverési arány 95 %-a),
- 12,2 mg/kg (a legkisebb hozzákeverési arány 70 %-a).
A legalacsonyabb eredményt adó minta jelölőanyag-koncentrációját a 17,7 mg/kg és 12,2 mg/kg közötti interpolációval együtt használjuk.
8.3. Vajkoncentrátum
8.3.1. A vajkoncentrátum esetében a hozzákeverési arány, a háttérelnyelést is figyelembe véve, 24 mg/kg.
A termék elemzéséből kapott három minta eredménye szolgál a jelölőanyag-hozzákeverés arányának és homogenitásának ellenőrzésére, és ezek közül az eredmények közül a legalacsonyabbat a következő határértékekkel kell egybevetni:
- 19,2 mg/kg (a legkisebb hozzákeverési arány 95 %-a),
- 13,2 mg/kg (a legkisebb hozzákeverési arány 70 %-a).
A legalacsonyabb eredményt adó minta jelölőanyag-koncentrációját a 19,2 mg/kg és 13,2 mg/kg közötti interpolációval együtt használjuk.
VIII. MELLÉKLET
(5. cikk)
SZITOSZTERIN VAGY SZTIGMASZTERIN MEGHATÁROZÁSA VAJBAN VAGY VAJKONCENTRÁTUMBAN KAPILLÁRIS GÁZKROMATOGRÁFIÁVAL
1. TÁRGY ÉS ALKALMAZÁSI TERÜLET
Ez a módszer a szitoszterin vagy a sztigmaszterin vajkoncentrátumból és vajból való mennyiségi kimutatására alkalmas eljárást ismerteti. A szitoszterint a β-szitoszterin és 22 dihidro-β-szitoszterin összegének tekintjük, a többi szitoszterint jelentéktelennek tételezzük fel.
2. A MÓDSZER ELVE
A vajat vagy a vajkoncentrátumot kálium-hidroxiddal etil-alkohol-oldatban elszappanosítjuk, és az el nem szappanosítható részt dietil-éterrel kivonjuk.
A szterinvegyületek trimetil-szilil-éterré alakulnak át, és kapilláris oszloppal, gázkromatográfiás úton elemezhetők belső standarddal/betulinnal összevetve.
3. ESZKÖZÖK
3.1. 150 ml-es szappanosító lombik visszafolyó hűtővel és csiszoltüveg-csatlakozásokkal ellátva
3.2. 500 ml-es leválasztó tölcsérek
3.3. 250 ml-es lombikok
3.4. Nyomáskiegyenlítő tölcsérek 250 ml vagy ahhoz hasonló űrtartalommal a felesleges dietiléter felfogására
3.5. Zsugorítottüveg-dugóval ellátott 350 mm × 20 mm méretű üvegoszlop
3.6. Vízfürdő vagy hőntartó köpeny
3.7. Reakciófiola, 2 ml-es
3.8. Kapillárissal is használható gázkromatográf, a következő elemekből álló leválasztó rendszerrel ellátva:
3.8.1. hőszabályozó kamra az oszlopokhoz, amely a kívánt hőmérsékletet ±1 °C pontossággal tartani képes;
3.8.2. szabályozható hőmérsékletű párologtató egység;
3.8.3. lángionizáló detektor és átalakító-erősítő;
3.8.4. regisztráló integrátor, amely alkalmas az átalakító-erősítővel (3.8.3.) való használatra
3.9. Teljes egészében BP1-el vagy azzal egyenértékű anyaggal borított kvarcüveg kapilláris oszlop (vagy bármilyen legalább ugyanolyan felbontású oszlop), egyformán 0,25 μm vastagon; az oszlop legyen képes a lanoszterin és szitoszterin trimetilszilil-származékainak elválasztására. E célra 12 méter hosszú, 0,2 mm belső átmérőjű oszlop alkalmas
3.10. Edzett hegyű tűvel rendelkező 1 μl-es gázkromatográfiás mikrofecskendő.
4. VEGYSZEREK
Az összes vegyszernek elismerten analitikai tisztaságúnak kell lennie. A felhasznált víznek desztillált víznek vagy azzal legalább egyenértékű tisztaságú víznek kell lennie.
4.1. Legalább 95 %-os tisztaságú etil-alkohol
4.2. Kálium-hidroxid 60 %-os oldata: oldjunk fel 600 g (legalább 85 %-os tisztaságú) kálium-hidroxidot vízben, és vízzel töltsük fel egy literre
4.3. Legalább 99 %-os tisztaságú betulin
4.3.1. Betulin dietil-éterben (4.4.) oldott oldata
4.3.1.1. A szitoszterin meghatározásra használt betulinoldat töménysége 1,0 mg/ml
4.3.1.2. A sztigmaszterin meghatározásra használt betulinoldat töménysége 0,4 mg/ml
4.4. Analitikai tisztaságú dietil-éter (peroxidtól és egyéb maradványoktól mentes)
4.5. Vízmentes szemcsés nátrium-szulfát, amelyet előzetesen két órán keresztül 102 °C-on szárítottak
4.6. Szililező reagens, például TRI-SIL (beszerezhető a Pierce Chemical vállalattól, katalógusszáma 49001) vagy ezzel egyenértékű más vegyszer. (Fontos tudnivaló: a TRI-SIL gyúlékony, toxikus, maró és esetlegesen rákkeltő anyag. A laboratórium személyzetének ismernie kell a TRI-SIL biztonságossági adatait, és meg kell tennie a szükséges óvintézkedéseket.)
4.7. Lanoszterin
4.8. Szitoszterin, ismert tisztaságú, tisztasága legalább 90 % (P)
1. megjegyzés: A kalibráláshoz alkalmazott standardanyagok tisztaságát normalizálási módszerrel kell meghatározni. Feltételezzük, hogy az összes, a mintában jelen lévő szterinvegyület megjelenik a kromatogramon, tehát a csúcsok teljes területe a szterinösszetevőket 100 %-ban képviseli, és a szterinek ugyanolyan detektorválaszt váltanak ki. A rendszer linearitását a releváns koncentrációtartományokban érvényesíteni kell.
4.8.1. Szitoszterinstandard-oldat - készítsünk dietil-éterben (4.4.) 0,001 mg/ml pontossággal, megközelítőleg 0,5 mg/ml (W1) szitoszterint (4.8.) tartalmazó oldatot.
4.9. Sztigmaszterin, ismert tisztaságú, tisztasága legalább 90 % (P)
4.9.1. Sztigmaszterinstandard-oldat - készítsünk dietil-éterben (4.4.) 0,001 mg/ml pontossággal, megközelítőleg 0,2 mg/ml (W1) sztigmaszterint (4.9.) tartalmazó oldatot.
4.10. Felbontóképességet ellenőrző keverék. Készítsünk dietil-éterben (4.4.) 0,05 mg/ml lanoszterint (4.7.) és 0,5 mg/ml szitoszterint (4.8.) tartalmazó oldatot.
5. MÓDSZER
5.1. Standard oldatok készítése kromatográfiás céllal.
A belső standard oldatot (4.3.1.) ugyanakkor kell hozzáadni a megfelelő szterinstandard-oldathoz, mint amikor az elszappanosított mintához hozzáadjuk (lásd az 5.2.2. pontot)
5.1.1. Kromatográfiás szitoszterinstandard-oldat: vigyünk át két reakciófiolába (3.7.) 1-1 ml szitoszterinstandard-oldatot (4.8.1.), és a dietil-étert nitrogénárammal távolítsuk el. Adjunk hozzá 1 ml belső mintát (4.3.1.1.) és a dietil-étert nitrogénárammal távolítsuk el
5.1.2. Kromatográfiás sztigmaszterinstandard-oldat: vigyünk át két reakciófiolába (3.7.) 1-1 ml sztigmaszterinstandard-oldatot (4.9.1.), és a dietil-étert nitrogénárammal távolítsuk el. Adjunk hozzá 1 ml belső standardot (4.3.1.2.), és a dietil-étert nitrogénárammal távolítsuk el
5.2. Nem szappanosítható összetevők előkészítése
5.2.1. A vajmintát 35 °C-ot meg nem haladó hőmérsékleten olvasszuk meg, majd kavarással alaposan keverjük össze
Egy 150 ml űrtartalmú lombikba (3.1.) mérjünk ki 1 mg pontossággal megközelítőleg 1 g vajat (W2) vagy vajkoncentrátumot (W2). Adjunk hozzá 50 ml etil-alkoholt (4.1.) és 10 ml kálium-hidroxid-oldatot (4.2.). Szereljük fel a visszafolyó hűtőt, és melegítsük a lombik tartalmát 30 percig, megközelítőleg 75 °C-os hőmérsékletre. Válasszuk le a hűtőt, és hűtsük le a lombikot szobahőmérsékletre
5.2.2. Adjunk hozzá 1,0 ml belső standard oldatot (4.3.1.1.) a lombik tartalmához, ha szitoszterint akarunk meghatározni, vagy ugyanennyit a 4.3.1.2. pontban leírt belső standard oldatból, ha sztigmaszterin akarunk meghatározni. Keverjük össze alaposan. A lombikok tartalmát teljes mennyiségben vigyük át egy 500 ml-es leválasztó tölcsérre (3.2.), majd öblítsük ki a lombikot 50 ml vízzel, majd 250 ml dietil-éterrel (4.4.). A leválasztó tölcsért két percen át hevesen rázzuk, majd hagyjuk, hogy a fázisok elváljanak egymástól. Engedjük le az alsó vizes fázist, az éteres fázist pedig négyszer egymás után 100 ml vízzel rázva mossuk ki
2. megjegyzés: Az emulzióképződés elkerülése érdekében lényeges, hogy az első két vizes öblítést gyengéden végezzük (10 felfordítás). A harmadikat már végezhetjük 30 másodpercig tartó erős rázással. Ha mégis képződik emulzió, azt 5-10 ml etil-alkohol hozzáadásával le lehet bontani. Viszont ha etil-alkoholt adtuk hozzá, akkor még két erőteljes vizes öblítésre van szükség.
5.2.3. A tiszta, szappanmentes éteres fázist vigyük keresztül egy 30 g, vízmentes nátrium-szulfáttal (4.5.) töltött üvegoszlopon (3.5.). Az étert egy 250 ml űrtartalmú lombikban gyűjtsük (3.3.). Adjunk hozzá egy darabka forrkövet, és vízfürdőben vagy hőntartó köpenyben majdnem teljesen párologtassuk el, gondosan összegyűjtve a maradék oldószert
3. megjegyzés: Ha a kivonatmintákat teljes száradásig túlságosan magas hőmérsékleten tartjuk, előfordulhat, hogy a szterinek egy része elvész.
5.3. A trimetil-szilil-éterek előkészítése
5.3.1. A lombikban maradt éteres oldatot 2 ml dietil-éterrel vigyük át egy 2 ml-es reakciósfiolába (3.7.), majd az étert nitrogénárammal távolítsuk el. A lombikot még kétszer 2 ml dietil-éter-adaggal öblítsük, az öblítőfolyadékot szintén a fiolába öntjük, majd az étert nitrogénnel eltávolítjuk
5.3.2. A mintát 1 ml TRI-SIL (4.6.) hozzáadásával szililezzük. Zárjuk le a fiolát és erőteljesen rázzuk, hogy feloldódjon a reagens. Ha az oldódás nem teljes, 65-70 °C-ra kell felmelegíteni. Hagyjuk legalább öt percig állni, mielőtt befecskendeznénk a gázkromatográfba. A standardokat ugyanúgy szililezzük, mint a mintákat. Szililezzük a felbontóképességet ellenőrző keveréket (4.10.) is, ugyanúgy, ahogy a mintákat
4. megjegyzés: A szililezést vízmentes környezetben kell végezni. A betulin nem teljes szililezését a betulin csúcsához közeli, második csúcs kialakulása jelzi.
Az etil-alkohol jelenléte a szililező szakaszban megzavarja a szililezést. Ez a kivonási szakaszt követő, nem megfelelő öblítés következménye lehet. Amennyiben a probléma nem oldódik meg, a kivonási szakaszba egy ötödik öblítést is be lehet iktatni, amely 30 másodpercig tartó erőteljes rázással történjen.
5.4. Gázkromatográfiás elemzés
5.4.1. Az üzemeltetési feltételek megválasztása
A gázkromatográfot a gyártó utasításai szerint állítsuk össze.
Az üzemeltetési feltételekre vonatkozó irányelvek a következők:
- az oszlop hőmérséklete: 265 °C
- az injektor hőmérséklete: 265 °C
- a detektor hőmérséklete: 300 °C
- a vivőgáz áramlási sebessége: 0,6 ml/min.
- a hidrogén nyomása: 84 kPa
- a levegő nyomása: 155 kPa
- minta megosztása: 10:1 és 50:1 között; a megosztási arányt a gyártó utasításai szerint, valamint a detektorválasz linearitása alapján kell optimalizálni, majd a releváns koncentrációtartományon belül érvényesíteni.
-
5. megjegyzés: Különösen fontos, hogy az injektáló béléscsövet rendszeresen tisztítsuk.
- injektált anyag mennyisége: 1 μl TMSE-oldat.
Mielőtt bármiféle elemzéshez hozzákezdenénk, hagyjuk, hogy a rendszer egyensúlyba kerüljön, és kapjunk kielégítő stabil választ.
Ezeket a feltételeket az oszlop, valamint a gázkromatográf jellemző tulajdonságaira tekintettel úgy kell változatni, hogy olyan kromatogramot kapjunk, amely megfelel a következő követelményeknek:
- a szitoszterincsúcsnak a lanoszterintől kellőképpen el kell válnia. Az 1. ábra azt mutatja, hogy milyennek kell egy felbontóképességet ellenőrző szililezett keverékből (4.10.) nyert tipikus kromatogramnak lennie,
- az alább felsorolt szterinek viszonylagos visszatartási ideje megközelítőleg a következő legyen:
-
koleszterin: 1,0
sztigmaszterin: 1,3
szitoszterin: 1,5
betulin: 2,5
- a betulin retenciós ideje megközelítőleg 24 perc.
5.4.2. Analitikai eljárás
Fecskendezzünk be 1 μl szililezett standard oldatot (sztigmaszterin vagy szitoszterin), majd állítsuk be az integrátor kalibrációs paramétereit.
Fecskendezzünk be további 1 μl szililezett standard oldatot a betulinra vonatkozó válaszjelek meghatározása érdekében.
Fecskendezzünk be 1 μl szililezett mintaoldatot, és mérjük meg a csúcsterületeket. Minden kromatográfiás szakaszt a standardok befecskendezésével kell közrefogni.
Általában egy-egy közrefogott szakaszban hat mintát kell injektálni.
6. megjegyzés: A sztigmaszterincsúcs integrálásakor bele kell számítani a 2b. ábra 1., 2. és 3. pontjában meghatározott egybefolyó értékeket is.
Ha a teljes szitoszterinmennyiséget akarjuk értékelni, akkor a szitoszterincsúcs integrálásakor bele kell számítani a 22 dihidro-β-szitoszterin- (sztigmaszterin-) csúcsot is, amely rögtön a szitoszterin után eluálódik (lásd a 3b. ábrát).
6. AZ EREDMÉNYEK KISZÁMÍTÁSA
6.1. Határozzuk meg a szterincsúcsok és a betulincsúcsok alatti terület nagyságát mindkét - a tételt közrefogó - standardban, majd számítsuk ki az R1-t:
R1 = (a szterincsúcs alatti átlagos terület a standardban)/(a betulincsúcs alatti átlagos terület a standardban)
Határozzuk meg a szterincsúcs alatti területet (sztigmaszterin és szitoszterin), majd a betulincsúcs alatti területet a mintában, és számítsuk ki az R2-t:
R2 = (a szterincsúcs alatti terület a mintában)/(a betulincsúcs alatti terület a mintában)
W1 = a standard szterintartalma (mg) 1 ml standard oldatban (4.8.1. vagy 4.9.1.)
W2 = a minta súlya (g) (5.2.1.)
P = a standard szterin tisztasági foka (4.8. vagy 4.9.)
A minta szterintartalma (mg/kg) = ((R 2) / (R 1)) × ((W 1) / (W 2)) × P × 10.
7. A MÓDSZER PONTOSSÁGA
7.1. Vaj
7.1.1. Ismételhetőség
7.1.1.1. Sztigmaszterin
A lehető legrövidebb időeltéréssel, ugyanazon laboratóriumi személy által, azonos eszközökkel, azonos vizsgálati anyagon elvégzett két meghatározás eredményének különbsége ne haladja meg a 19,3 mg/kg-ot.
7.1.1.2. Szitoszterin
A lehető legrövidebb időeltéréssel, ugyanazon laboratóriumi személy által, azonos eszközökkel, azonos vizsgálati anyagon elvégzett két meghatározás eredményének különbsége ne haladja meg a 23,0 mg/kg-ot.
7.1.2. Reprodukálhatóság
7.1.2.1. Sztigmaszterin
Különböző laboratóriumokban lévő személyzet által, különböző eszközökkel, azonos vizsgálati anyagon elvégzett két meghatározás eredményének különbsége ne haladja meg a 31,9 mg/kg-ot.
7.1.2.2. Szitoszterin
Különböző laboratóriumokban lévő személyzet által, különböző eszközökkel, azonos vizsgálati anyagon elvégzett két meghatározás eredményének különbsége ne haladja meg a meghatározás középértékéhez viszonyított 8,7 %-ot.
7.1.3. A pontossági adatok forrása
A pontossági adatokat egy 1992-ben elvégzett kísérlet szolgáltatta, amelyben nyolc laboratórium vett részt, és a sztigmaszterin vonatkozásában hat mintát (három azonosítatlan), a szitoszterin tekintetében pedig hat mintát (három azonosítatlan minta) használtak.
7.2. Vajkoncentrátum
7.2.1. Ismételhetőség
7.2.1.1. Sztigmaszterin
A lehető legrövidebb időeltéréssel, ugyanazon laboratóriumi személy által, azonos eszközökkel, azonos vizsgálati anyagon elvégzett két meghatározás eredményének különbsége ne haladja meg a 10,2 mg/kg-ot.
7.2.1.2. Szitoszterin
A lehető legrövidebb időeltéréssel, ugyanazon laboratóriumi személy által, azonos eszközökkel, azonos vizsgálati anyagon elvégzett két meghatározás eredményének különbsége ne haladja meg a meghatározások középértékéhez viszonyított 3,6 %-ot.
7.2.2. Reprodukálhatóság
7.2.2.1. Sztigmaszterin
Különböző laboratóriumokban lévő személyzet által, különböző eszközökkel, azonos vizsgálati anyagon elvégzett két meghatározás eredményének különbsége ne haladja meg a 25,3 mg/kg-ot.
7.2.2.2. Szitoszterin
Különböző laboratóriumokban lévő személyzet által, különböző eszközökkel, azonos vizsgálati anyagon elvégzett két meghatározás eredményének különbsége ne haladja meg a meghatározás középértékéhez viszonyított 8,9 %-ot.
7.2.3. A pontossági adatok forrása
A pontossági adatokat egy 1991-ben elvégzett kísérlet szolgáltatta, amelyben kilenc laboratórium vett részt, és a sztigmaszerin vonatkozásában hat mintát (három azonosítatlan minta), a szitoszterin tekintetében hat mintát (három kettős vakpróba) használtak.
8. TŰRÉSHATÁROK
8.1. A megjelölt termékből három mintát kell venni annak ellenőrzésére, hogy a termék megjelölése helyesen történt-e.
8.2. Vaj
8.2.1. Sztigmaszterin
8.2.1.1. A sztigmaszterin esetében a hozzákeverési arány 150 g legalább 95 %-os tisztaságú sztigmaszterin a vaj minden tonnájára, vagyis 142,5 mg/kg, illetve 170 g legalább 85 %-os tisztaságú sztigmaszterin a vaj minden tonnájára, vagyis 144,5 mg/kg
8.2.1.2. A termék elemzéséből kapott három minta eredménye szolgál a jelölőanyag-hozzákeverés arányának és homogenitásának ellenőrzésére, és ezek közül az eredmények közül a legalacsonyabbat a következő határértékekkel kell egybevetni:
- 115,8 mg/kg (a legkisebb hozzákeverési arány 95 %-a a 95 %-os tisztaságú sztigmaszerinre),
- 117,7 mg/kg (a legkisebb hozzákeverési arány 95 %-a a 85 %-os tisztaságú sztigmaszerinre),
- 80,1 mg/kg (a legkisebb hozzákeverési arány 70 %-a a 95 %-os tisztaságú sztigmaszerinre),
- 81,5 mg/kg (a legkisebb hozzákeverési arány 70 %-a a 85 %-os tisztaságú sztigmaszerinre).
A legalacsonyabb eredményt adó minta jelölőanyag-koncentrációját a 115,8 mg/kg és 80,1 mg/kg közötti, illetve a 117,7 mg/kg és 81,5 mg/kg közötti interpolációval együtt használjuk.
8.2.2. Szitoszterin
8.2.2.1. A szitoszterin esetében a hozzákeverési arány 600 g legalább 90 %-os tisztaságú szitoszterin a vaj minden tonnájára, vagyis 540 mg/kg
8.2.2.2. A termék elemzéséből kapott három minta eredménye szolgál a jelölőanyag-hozzákeverés arányának és homogenitásának ellenőrzésére, és ezek közül az eredmények közül a legalacsonyabbat a következő határértékekkel kell egybevetni:
- 482,6 mg/kg (a legkisebb hozzákeverési arány 95 %-a a 90 %-os tisztaságú szitoszerinre),
- 347,6 mg/kg (a legkisebb hozzákeverési arány 70 %-a a 90 %-os tisztaságú szitoszerinre).
A legalacsonyabb eredményt adó minta jelölőanyag-koncentrációját a 482,6 mg/kg és 347,6 mg/kg közötti interpolációval együtt használjuk.
8.3. Vajkoncentrátum
8.3.1. Sztigmaszterin
8.3.1.1. A sztigmaszterin esetében a hozzákeverési arány 150 g legalább 95 %-os tisztaságú sztigmaszterin, a vajkoncentrátum minden tonnájára, vagyis 142,5 mg/kg, illetve 170 g legalább 85 %-os tisztaságú sztigmaszterin a vajkoncentrátum minden tonnájára, vagyis 144,5 mg/kg
8.3.1.2. A termék elemzéséből kapott három minta eredménye szolgál a jelölőanyag-hozzákeverés arányának és homogenitásának ellenőrzésére, és ezek közül az eredmények közül a legalacsonyabbat a következő határértékekkel kell egybevetni:
- 118,5 mg/kg (a legkisebb hozzákeverési arány 95 %-a a 95 %-os tisztaságú sztigmaszerinre),
- 120,4 mg/kg (a legkisebb hozzákeverési arány 95 %-a a 85 %-os tisztaságú sztigmaszerinre),
- 82,9 mg/kg (a legkisebb hozzákeverési arány 70 %-a a 95 %-os tisztaságú sztigmaszerinre),
- 84,3 mg/kg (a legkisebb hozzákeverési arány 70 %-a a 85 %-os tisztaságú sztigmaszerinre).
A legalacsonyabb eredményt adó minta jelölőanyag-koncentrációját a 118,5 mg/kg és 82,9 mg/kg közötti, illetve a 120,4 mg/kg és 84,3 mg/kg közötti interpolációval együtt használjuk.
8.3.2. Szitoszterin
8.3.2.1. A szitoszterin esetében a hozzákeverési arány 600 g legalább 90 %-os tisztaságú szitoszterin a vajkoncentrátum minden tonnájára, vagyis 540 mg/kg
8.3.2.2. A termék elemzéséből kapott három minta eredménye szolgál a jelölőanyag-hozzákeverés arányának és homogenitásának ellenőrzésére, és ezek közül az eredmények közül a legalacsonyabbat a következő határértékekkel kell egybevetni:
- 480,9 mg/kg (a legkisebb hozzákeverési arány 95 %-a a 90 %-os tisztaságú szitoszerinre),
- 345,9 mg/kg (a legkisebb hozzákeverési arány 70 %-a a 90 %-os tisztaságú szitoszerinre).
A legalacsonyabb eredményt adó minta jelölőanyag-koncentrációját a 480,9 mg/kg és 345,9 mg/kg közötti interpolációval együtt használjuk.
A teljes felbontsási kép a kïvánatos, vagyis a lanoszterinnek megfelelő kromatográfiás csúcsnak előbb vissza kell térnie as alapvonalra, mielőtt a szitoszterint jelző másik csúcs elindulna, habár a nem egészen teljes felbontás is elfogadható. [Kép #1]
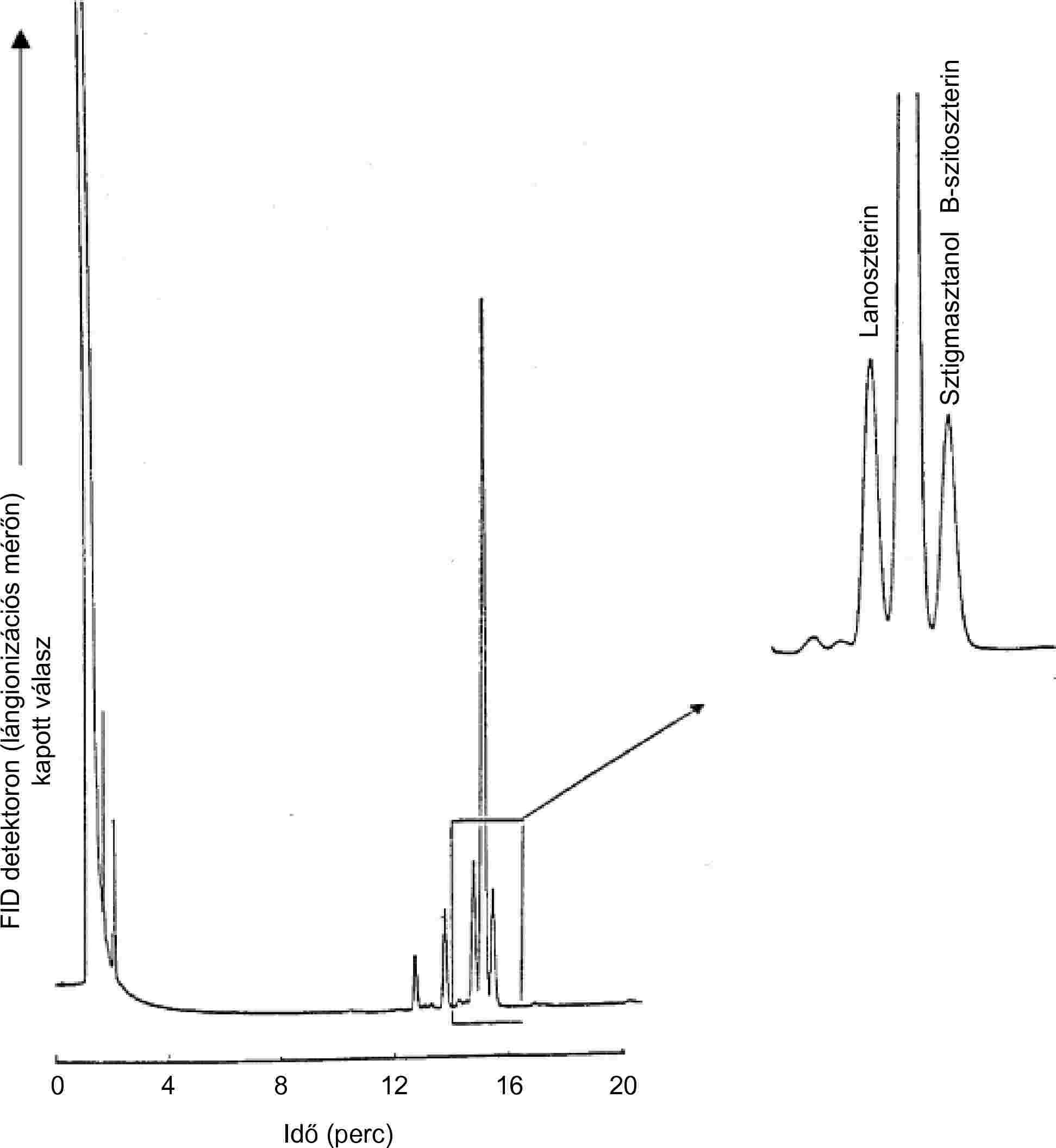
Kép #1
| 2a. ábra | 2b. ábra |
| Sztigmaszterin standard | Sztigmaszterin denaturált vajminta |
| [Kép #2] | [Kép #3] |
| Megjegyzés: A sztigmaszterin csúcs integrálásakor az 1., 2., valmint 3. pontok által meghatározott kilógó (elmosódott) értékeket is bele kell számolni | |
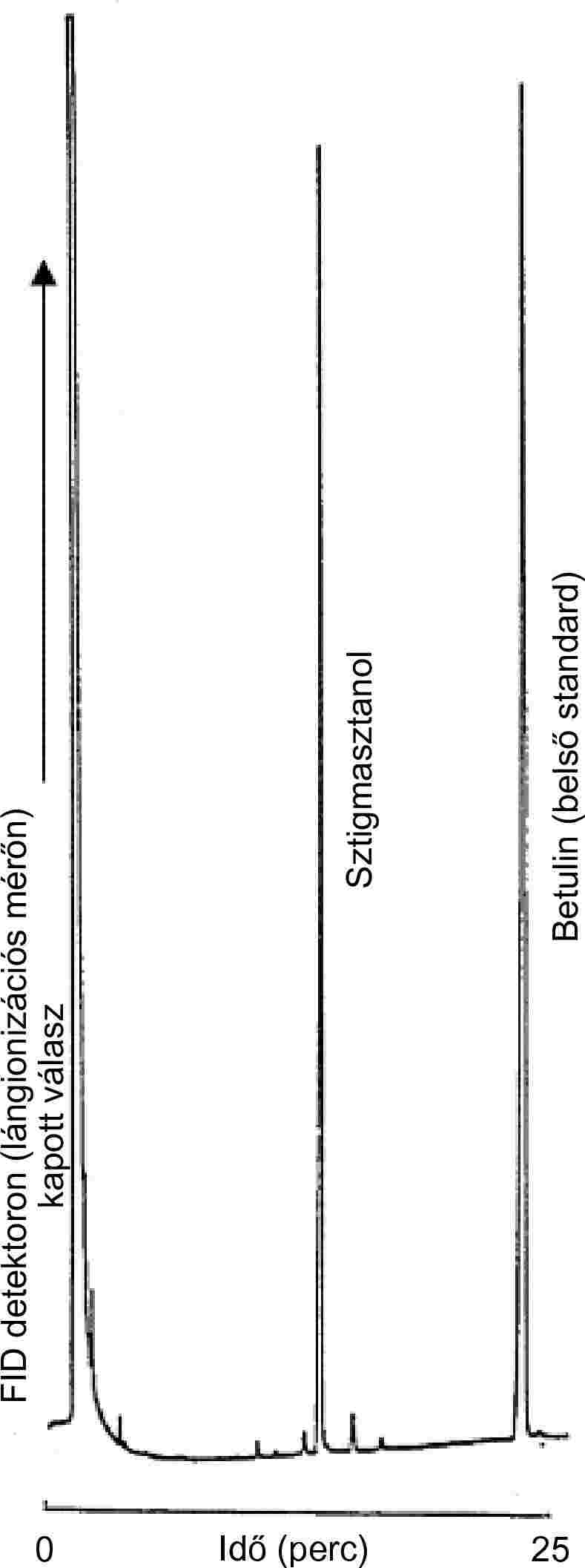
Kép #2
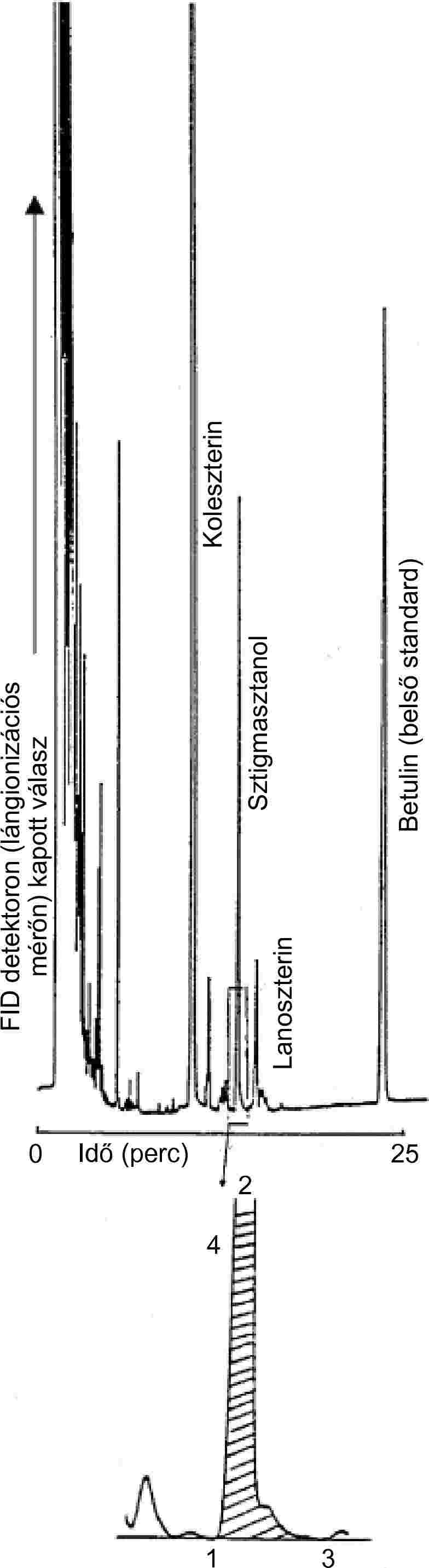
Kép #3
| 3a. ábra | 3b. ábra |
| Sztigmaszterin standard | Sztigmaszterin denaturált vajminta |
| [Kép #4] | [Kép #5] |
| Megjegyzés: A β-szitoszterin gyakran tartalmaz szennyeződéseket (sztigmaszterunként azonosïtható), amelyek azonnal a β-szitoszterin után eluálodnak. Ennélfogva ennek a két csúcsnak a területét össze kell adni, amikor a jelenlévő összes β-szitoszterin mennyiséget értékeljük. | |
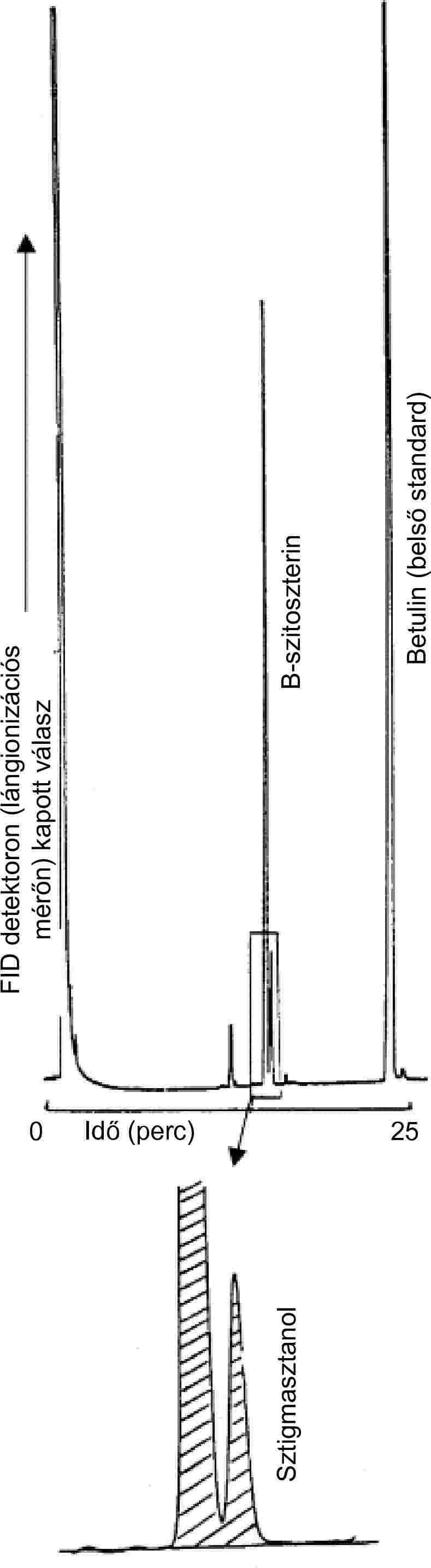
Kép #4

Kép #5
IX. MELLÉKLET
(6. cikk)
JUHTEJBŐL, KECSKETEJBŐL VAGY BIVALYTEJBŐL, ILLETVE JUH-, KECSKE- ÉS BIVALYTEJ ELEGYÉBŐL KÉSZÍTETT SAJTOKBAN TALÁLHATÓ TEHÉNTEJ ÉS KAZEINÁT KIMUTATÁSÁRA ALKALMAS REFERENCIA-MÓDSZER
1. TÁRGY
Juhtejből, kecsketejből vagy bivalytejből, illetve juh-, kecske- és bivalytej elegyéből készített sajtokban található tehéntej és kazeinátok kimutatása a γ-kazeinekre alkalmazott, plazmolízist követő izoelektromos fókuszálással.
2. ALKALMAZÁSI KÖR
Ez a módszer alkalmas a kezeletlen és hőkezelt tehéntej, valamint kazeinát nagy érzékenységű és specifikus kimutatására juhtejből, kecsketejből vagy bivalytejből, illetve juh-, kecske- és bivalytej elegyéből készített friss és érlelt sajtokban. Nem alkalmas azonban a tej és a sajt hőkezelt tehéntejsavófehérje-koncentrátumokkal történt hamisításának kimutatására.
3. A MÓDSZER ELVE
3.1. A kazeinek izolálása a sajtból és a referenciastandardból.
3.2. Az izolált kazeinek feloldása és alávetése a plazminos (EC.3.4.21.7.) bontásnak.
3.3. Izoelektromos fókuszálás alkalmazása a plazminnal kezelt kazeinekre karbamid jelenlétében, és a fehérjék megfestése.
3.4. A megfestett (a tehéntej jelenlétét bizonyító) γ3 és γ2-kazeinmintázatoknak az értékelése a vizsgálati mintából nyert mintázatok és az ugyanazon a gélen, a 0 %, illetve az 1 % tehéntejet tartalmazó referenciastandardokból kapott mintázatok összehasonlításával.
4. VEGYSZEREK
Ellenkező rendelkezés hiányában, analitikai tisztaságú vegyszereket kell alkalmazni. A víznek kétszeresen desztilláltnak vagy azzal egyenértékű tisztaságúnak kell lennie.
Megjegyzés: A következő részletek a laboratóriumban elkészített, karbamidtartalmú poliakrilamid gélekre vonatkoznak, amelyek méretei 265 × 125 × 0,25 mm. Ha más nagyságú vagy más típusú gélt használunk, a leválasztási körülményeket külön be kell állítani.
Izoelektromos fókuszálás
4.1. Vegyszerek a karbamidtartalmú poliakrilamid gél előállításához
4.1.1. Géltörzsoldat
Oldjuk fel a következőket vízben:
4,85 g akrilamid
0,15 g N, N'-metilén-bisz-akrilamid (BIS)
48,05 g karbamid
15,00 g glicerin (87 % m/m),
töltsük fel 100 ml-re, és hűtőszekrényben, barna üvegben tároljuk.
Megjegyzés: A fent említett fix tömegű, neurotoxikus akrilamidok helyett előnyösebb lehet a kereskedelemben kapható, előre bekevert akrilamid/BIS-oldat használata. Ha az ilyen oldat 30 % vegyesszázalék (m/v) akrilamidot és 0,8 % m/v BIS-t tartalmaz, 16,2 ml térfogatot kell belőle felhasználni a fix tömegek helyett. A törzsoldat eltarthatósági ideje legfeljebb 10 nap; ha vezetőképessége több mint 5 μS, 2 g Amberlite MB-3 vegyszerrel kell 30 percen keresztül történő keverés útján ionmentesíteni, majd egy 0,45 μm pórusátmérőjű membránszűrőn átszűrni.
4.1.2. Géloldat
Készítsük el a géloldatot úgy, hogy az adalékanyagokat és az amfolitokat elkeverjük a géltörzsoldattal (lásd a 4.1.l. pontot).
9,0 ml törzsoldat
24 mg β-alanin
500 μl 3,5-9,5 pH közötti amfolit ( 9 )
250 μl 5-7 pH közötti amfolit (9)
250 μl 6-8 pH közötti amfolit (9)
Keverjük össze a géloldatot, és vákuumban vagy ultrahangos fürdőben gázmentesítsük két vagy három percen át.
Megjegyzés: A géloldatot közvetlenül a kiöntés előtt készítsük el (lásd a 6.2. pontot).
4.1.3. Katalizátoroldatok
4.1.3.1. N, N, N' N' - tetrametil-etilén-diamin (Temed).
4.1.3.2. 40 % m/v ammónium-perszulfát (PER):
Oldjunk fel vízben 800 mg PER-t, és töltsük fel vízzel 2 ml-re.
Megjegyzés: Mindig frissen készített PER-oldatot használjunk.
4.2. Kontaktfolyadék
Világítópetróleum vagy folyékony paraffin
4.3. Anódoldat
Oldjunk fel vízben 5,77 g foszforsavat (85 % m/m), és töltsük fel vízzel 100 ml-re.
4.4. Katódoldat
Oldjunk fel vízben 2,00 g nátrium-hidroxidot, és hígítsuk fel vízzel 100 ml-re.
A minta előkészítése
4.5. Vegyszerek a fehérjék izolálására
4.5.1. Hígított ecetsav (25,0 ml jégecet vízzel 100 ml-re hígítva)
4.5.2. Diklórmetán
4.5.3. Aceton
4.6. Proteinoldó puffer
Oldjuk fel vízben a következőket:
5,75 g glicerin (87 % m/m)
24,03 g karbamid
250 mg ditio-treitol,
és töltsük fel 50 ml-re.
Megjegyzés: Hűtőszekrényben tárolva eltarthatósági ideje legfeljebb egy hét.
4.7. A kazeinek plazminos bontására használt vegyszerek
4.7.1. Ammónium-karbonát-puffer
Titráljunk 0,05 mol/l etilén-diamin-tetraecetsavat (EDTA, 1,46 g/100 ml) tartalmazó 0,2 mol/l ammónium-hidrogénkarbonát-oldatot (1,58 g/100 ml víz) 0,05 mol/l EDTA-t tartalmazó 0,2 mol/l ammónium-karbonát-oldattal (1,92 g/100 ml víz) 8 pH-ig.
4.7.2. Szarvasmarhaplazmin (EC.3.4.21.7.), aktivitása legalább 5 U/ml
4.7.3. ε-aminokapronsav-oldat enzimgátlásra Oldjunk fel 2,624 g ε-aminokapronsavat (6-amino-n-hexánsav) 100 ml 40 térfogatszázalékos etil-alkoholban.
4.8. Standardok
4.8.1. Bizonylattal ellátott, 0 %, illetve 1 % tehéntejet tartalmazó, tejoltóenzimmel kezelt fölözött juhtej- és kecsketejkeverék referenciastandardot a Referenciaanyagok és Mérések Bizottsági Intézeténél (B-2440 Geel, Belgium) lehet beszerezni.
4.8.2. Tejoltóenzimmel kezelt bivalytejből előállított, 0 % és 1 % tehéntejet tartalmazó laboratóriumi belső standard készítése
A fölözött tejet nyers, ömlesztett bivalytejből, illetve szarvasmarhatejből 37 °C-on, 2 500 g-vel 20 percen át történő centrifugálással készítjük. A centrifugacsövet a tartalmával együtt hirtelen 6-8 °C-ra lehűtjük, majd a felső, zsíros réteget teljes egészében eltávolítjuk. Az 1 %-os standard elkészítéséhez 1 literes főzőpohárban adjunk 5,00 ml fölözött szarvasmarhatejet 495 ml fölözött bivalytejhez, pH-ját 10 vegyesszázalékos hígított tejsav hozzáadásával állítsuk be 6,4-re. A hőmérsékletet állítsuk be 35 °C-ra és adjunk hozzá 100 μl borjúgyomorból nyert oltóenzimet (aktivitása 1:10 000, c. 3 000 U/ml), egy percen keresztül kavarjuk, majd a főzőpoharat alufóliával letakarva hagyjuk egy órán át 35 °C-on pihenni az alvadék kialakulásának elősegítésére. Miután az alvadék kialakult, a teljes beoltott tejet előzetes homogenizálás és a savó leszívása nélkül fagyasztva szárítjuk. A fagyasztva szárítást követően az anyagot finomra daráljuk, amíg homogén port nem kapunk. A 0 %-os standard elkészítéséhez ugyanezt a műveletsort a tiszta fölözött bivalytejjel kell elvégezni. A standardokat -20 °C-on kell tárolni.
Megjegyzés: Ajánlatos a bivalytej tisztaságát a standardok elkészítése előtt a plazminos emésztéssel előkészített kazeinek izoelektromos fókuszálásával ellenőrizni.
A fehérje festéséhez használt vegyszerek
4.9. Fixálószer
Oldjunk fel vízben 150 g triklórecetsavat, és töltsük fel vízzel 1 000 ml-re.
4.10. Festékeltávolító oldat
Oldjunk fel 500 ml metil-alkoholt és 200 ml jégecetet 2 000 ml desztillált vízben.
Megjegyzés: A festékeltávolító oldatot minden nap frissen készítsük el; elkészíthető 50 % térfogatszázalékos metilalkohol- és 20 térfogatszázalékos jégecettörzsoldat egyenlő mennyiségeinek az összekeverésével.
4.11. Festékoldatok
4.11.1. Festékoldat (1. törzsoldat)
Mágneses keverő alkalmazásával oldjunk fel 3,0 g Coomassie G-250 brillantkéket (C.I. 42655) 1 000 ml 90 térfogatszázalékos metil-alkoholban (megközelítőleg 45 percen át), és két, közepes sebességű, redős szűrőn szűrjük át.
4.11.2. Festékoldat (2. törzsoldat)
Oldjunk fel 5,0 g rézszulfát-pentahidrátot 1 000 ml 20 térfogatszázalékos jégecetben.
4.11.3. Festékoldat (munkaoldat)
Közvetlenül festés előtt öntsünk össze 125 ml-t mindkét törzsoldatból (4.11.1., 4.11.2.).
Megjegyzés: A festékoldatot a felhasználás napján kell elkészíteni.
5. ESZKÖZÖK
5.1. Üveglemezek (265 × 125 × 4 mm); gumihenger (15 cm széles); szintező asztal
5.2. Géltartó lemez (265 × 125 mm)
5.3. Fedőlemez (280 × 125 mm). Mindkét hosszanti végére ragasszunk fel egy-egy csíkban ragasztószalagot (280 × 6 × 0,25 mm) (lásd az 1. ábrát)
5.4. Elektrofókuszáló kamra hűtőlemezzel (pl. 265 × 125 mm) és megfelelő áramellátással (≥ 2,5 kV) vagy automata elektroforézis-készülék
5.5. Cirkulációs kriosztát, termosztátos vezérléssel 12 ±0,5 °C-on
5.6. 3 000 g-ig állítható centrifuga
5.7. Elektródacsíkok (≥ 265 mm hosszban)
5.8. Műanyag csepegtetőpalackok az anód- és a katódoldat számára
5.9. Mintafelvivő eszköz (10 × 5 mm, viszkózus vagy alacsony fehérjeadszorpciós szűrőpapír)
5.10. Rozsdamentes acélolló, szike és műszerészcsipesz
5.11. Rozsdamentes acél vagy üvegfestő- és festékeltávolító-edények (például 280 × 150 mm műszertálca)
5.12. Szabályozható rúdhomogenizátor (10 mm tengelyátmérővel), percenkénti fordulatszám-tartomány 8 000-20 000 között
5.13. Mágneses keverő
5.14. Ultrahangos fürdő
5.15. Filmhegesztő gép
5.16. 25 μl-es mikropipetta
5.17. Vákuumbepárló vagy fagyasztva-szárító
5.18. Termosztatikus vezérlésű, 35 és 40 ±1 °C-ra állítható vízfürdő rázógéppel
5.19. Denzitométer, λ = 634 nm-es leolvasóval felszerelve
6. ELJÁRÁS
6.1. A minta előkészítése
6.1.1. Kazeinek izolálása
Egy 100 milliliteres centrifugacsőbe mérjünk ki 5 g szárazanyag-mennyiségnek megfelelő sajtot vagy a referenciastandardot, adjunk hozzá 60 ml desztillált vizet és homogenizáljuk a rúdhomogenizátorral (8 000-10 000-es percenkénti fordulatszám között). Állítsuk be hígított ecetsavval (4.5.1.) 4,6 pH-ra, és centrifugáljuk (5 perc, 3 000 g). A zsírt és a savót öntsük le, a maradékot pedig 20 000-es percenkénti fordulatszámon homogenizáljuk 40 ml desztillált vízben, amelynek a pH-ját előzetesen hígított ecetsavval (4.5.1.) 4,5 pH-ra állítottuk be, adjunk hozzá 20 ml diklórmetánt (4.5.2.), ismét homogenizáljuk és centrifugáljuk (5 perc, 3 000 g). Egy spatulával távolítsuk el a kazeinréteget, amely a vizes és a szerves fázis között található (lásd a 2. ábrát), és dekantáljuk mindkét fázist. A kazeint ismét homogenizáljuk 40 ml desztillált vízben (a fentiek szerint), és 20 ml diklórmetánnal (4.5.2.) centrifugáljuk. Addig ismételjük ezt a műveletet, amíg mindkét extraháló fázis színtelen nem lesz (két-három alkalommal). A fehérjemaradékot 50 ml acetonban (4.5.3.) homogenizáljuk, és egy közepes átfolyási sebességű, redős papírszűrőn szűrjük át. A szűrőn maradt maradékot két, külön 25 ml-es adagban acetonnal mossuk le, hagyjuk a levegőn megszáradni, vagy nitrogénnel szárítsuk, majd törjük finom porrá egy mozsárban.
Megjegyzés: A száraz kazeinizolátumokat -20 °C hőmérsékleten kell tartani.
6.1.2. A β-kazeinek plazminos bontása a γ-kazeinek kifejezettebbé tétele érdekében
Oszlassunk el 25 mg izolált kazeint (6.1.1.) 0,5 ml ammónium-karbonát-pufferben (4.7.1.) és 20 percig homogenizáljuk, például ultrahangos kezeléssel. Melegítsük fel 40 °C-ra, és adjunk hozzá 10 μl plazmint (4.7.2.), keverjük össze, majd folyamatos rázatás mellett inkubáljuk egy órán át 40 °C-on. Az enzim késleltetése érdekében adjunk hozzá 20 μl ε-aminokapronsav-oldatot (4.7.3.), majd 200 mg szilárd karbamidot és 2 mg ditio-treitolt.
Megjegyzés: A fókuszált kazeincsíkok nagyobb szimmetriájának elérése érdekében ajánlatos az oldatot a ε-aminokapronsav hozzáadását követően liofilizálni, majd a maradékot 0,5 ml proteinoldó-pufferben oldani (4.6.).
6.2. A karbamidtartalmú poliakrilamid gél elkészítése
Néhány csepp víz segítségével terítsük ki a géltartó lemezt (5.2.) egy üveglapra (5.1.) egy papírtörölközővel vagy itatóssal eltávolítva a felesleges vizet. A fedőlemezt (5.3.) egy másik üveglapra terítsük ki a távtartókkal (0,25 mm) együtt azonos módon. Fektessük a lapot vízszintesen egy szintező asztalra.
Adjunk hozzá az előkészített és légmentesített géloldathoz (4.1.2.) 10 μl Temed-oldatot (4.1.3.1.), keverjük össze és adjunk még hozzá 10 μl PER-oldatot (4.1.3.2.), ezt is alaposan keverjük össze, majd azonnal öntsük ki egyenletesen a fedőlemez közepére. A géltartó lemez egyik szélét arccal lefelé fektessük a fedőlemez mellé, és lassan engedjük rá úgy, hogy a két lemez között egy gélfilm képződjön, amely egyenletesen és buborékmentesen kitölti a helyet (3. ábra). Egy vékony spatula segítségével gondosan, egészen engedjük le a gélhordozó lemezt, és helyezzünk súlyként még három üveglemezt a tetejére. Miután a polimerizáció befejeződött (mintegy 60 perc elteltével), a polimerizálódott gélt az üveglemezek felfordításával emeljük át a hordozólemezről a fedőlemezre. A hordozólemez alsó felét gondosan tisztítsuk meg a maradványok és a karbamid eltávolítása érdekében. A gélszendvicset hegesszük bele egy filmcsőbe, és tároljuk hűtőszekrényben (legfeljebb hat hétig).
Megjegyzés: A fedőlemez a távtartókkal újra felhasználható. A poliakrilamid gél kisebb darabokra is felvágható, amely akkor ajánlott, ha kevesebb minta áll rendelkezésünkre, vagy egy automatikus elektroforézis készüléket használunk (két gél, 4,5 × 5 cm méretben).
6.3. Izoelektromos fókuszálás
A hűtőtermosztátot állítsuk be 12 °C-ra. A géltartó lemez hátulját petróleummal töröljük le, majd cseppentsünk néhány csepp petróleumot (4.2.) a hűtőblokk közepére. Ezután a gélszendvicset terítsük rá hordozó felülettel lefelé, óvatosan ügyelve arra, hogy ne képződjenek levegőbuborékok. Törüljünk le minden felesleges petróleumot, és távolítsuk el a fedőlemezt. Az elektródcsíkokat áztassuk bele az elektródos oldatokba (4.3., 4.4.), vágjuk le a gél hosszára, és fektessük le a megadott helyzetbe (az elektródok távolsága 9,5 cm).
Az izoelektromos fókuszálás feltételei:
6.3.1. Gélméret 265 × 125 × 0,25 mm
| Lépés | Idő (perc) | Feszültség (V) | Áramerősség (mA) | Teljesítmény (W) | Volt-óra (Vh) |
| 1. Előfókuszálás | 30 | maximum 2 500 | maximum 15 | konstans 4 | c. 300 |
| 2. A minta fókuszálása (1) | 60 | maximum 2 500 | maximum 15 | konstans 4 | c. 1 000 |
| 3. Zárófókuszálás | 60 | maximum 2 500 | maximum 5 | maximum 20 | c. 3 000 |
| 40 | maximum 2 500 | maximum 6 | maximum 20 | c. 3 000 | |
| 30 | maximum 2 500 | maximum 7 | maximum 25 | c. 3 000 | |
| (1) Mintaalkalmazás: Az előfókuszálást követően (1. lépés) pipettázzunk 18 μl mintát és standard oldatokat a mintafelvivőre (10 × 5 mm), azokat egymástól legalább 1 mm-es távolságra, az anódtól pedig hosszirányban legalább 5 mm-re helyezzük rá a gélre, és enyhén nyomjuk meg. A fenti körülmények mellett végezzük el a fókuszálást, és óvatosan távolítsuk el a mintafelvivő eszközt a minta 60 perces futtatását követően. | |||||
Megjegyzés: Ha a gél vastagságát vagy szélességét megváltoztatjuk, az áramerősség és a teljesítmény értékeit megfelelő módon kell módosítani (például kettőzzük meg az áramerősség és teljesítmény értékeit, ha 265 × 125 × 0,5 mm-es gélt használunk).
6.3.2. A következőkben egy automatikus elektroforéziskészülék feszültségprogramja látható (2 db 5,0 × 4,5 cm gél), az elektródokat csíkok nélkül, azonnal a gélre helyezzük fel
| Lépés | Feszültség | Áramerősség | Teljesítmény | Hőmérséklet | Volt-óra |
| 1. Előfókuszálás | 1 000 V | 10,0 mA | 3,5 W | 8 °C | 85 Vh |
| 2. A minta fókuszálása | 250 V | 5,0 mA | 2,5 W | 8 °C | 30 Vh |
| 3. Fókuszálás | 1 200 V | 10,0 mA | 3,5 W | 8 °C | 80 Vh |
| 4. Fókuszálás | 1 500 V | 5,0 mA | 7,0 W | 8 °C | 570 Vh |
A mintafelvivőt a 2. lépésben 0 Vh-nál helyezzük el.
A mintafelvivőt a 2. lépésben 30 Vh-nál távolítsuk el.
6.4. Fehérjefestés
6.4.1. Fehérjefixálás
Az áram kikapcsolása után azonnal távolítsuk el az elektródcsíkokat, és a gélt rögtön tegyük a 200 ml fixálószerrel (4.9.) töltött festő/festékeltávolító edénybe; folyamatos rázatás mellett hagyjuk benne 15 percig.
6.4.2. A géllemez mosása és festése
Alaposan öntsük le a fixálószert, és a géllemezt kétszer harminc másodpercig mossuk le alkalmanként 100 ml festékeltávolító szerben (4.10.). Öntsük le az eltávolító oldatot, és töltsük meg az edényt 250 ml festőoldattal (4.11.3.); 45 percig fessünk, miközben gyengén rázogatjuk az edényt.
6.4.3. Festék eltávolítása a géllemezről
Öntsük le a festőoldatot, kétszer mossuk le alkalmanként 100 ml festékeltávolító szerben (4.10.), majd rázzuk 15 percen át 200 ml festékeltávolító szerben, és ismételjük meg a festékeltávolítást legalább két-három alkalommal egészen addig, amíg a háttér tiszta és színtelen nem lesz. Ezt követően desztillált vízzel öblítsük le a géllemezt (2 × 2 perc), és levegőn (2-3 óra) vagy hajszárítóval (10-15 perc) szárítsuk meg.
1. megjegyzés: A fixálást, mosást, festést és festékeltávolítást 20 °C-on végezzük. Ne használjunk magas hőmérsékletet.
2. megjegyzés: Ha az érzékenyebb ezüstfestést (például a Pharmacia Biotech ezüstfestő fehérjekészlete, kódszáma 17-1150-01) választjuk, a plazminnal kezelt kazeinmintákat 5 mg/ml-re kell hígítani.
7. ÉRTÉKELÉS
Az értékelést az ismeretlen minta által adott fehérjemintázatnak a referenciastandardból ugyanazon a gélen kapott mintázattal történő összehasonlítása révén végezhetjük el. A juh-, kecske- és bivalytejből, illetve juh-, kecske- és bivalytej keverékéből készített sajtokban található tehéntej kimutatása a γ3- és γ2-kazeinek révén történik, amelyek izoelektromos fókuszálási pontjai a 6,5-ös pH és 7,5-ös pH közötti tartományba esnek (4a., 4b. és 5. ábra). A kimutatási határ kisebb mint 0,5 %.
7.1. Szemrevételezéssel történő értékelés
A szarvasmarha-eredetű tej mennyiségének szemrevételezéssel történő értékeléséhez ajánlatos a minták és a standardok koncentrációit úgy beszabályozni, hogy ugyanolyan intenzitást kapjunk a juh-, kecske- és/vagy bivalyeredetű γ2- és γ3-kazeinekre (lásd a "γ2 E,G,B" és "γ3 E,G,B" csíkokat a 4a., 4b. és 5. ábrán). Ezt követően az ismeretlen minta szarvasmarha-eredetű tejtartalma (1 %-nál kevesebb, egyenlő vagy több) közvetlenül megítélhető, ha a szarvasmarha-eredetű γ3- és γ2-kazeineket (lásd a "γ3 C" és "γ2 C" csíkot a 4a., 4b. és az 5. ábrán) egybevetjük a 0 % és az 1 %-os referenciastandardokkal (juh és kecske esetében), valamint a laboratórium belső standardjával (bivalynál).
7.2. Denzitometriás értékelés
Ha rendelkezésre áll, használjunk denzitométert (5.19.) a szarvasmarha-eredetű és a juh-, kecske-, illetve bivalyeredetű γ3- és γ2-kazeinek adta csúcsok egymáshoz viszonyított arányának mérésére (lásd az 5. ábrát). Hasonlítsuk össze ezt az értéket az ugyanezen a gélen elemzett 1 %-os referenciastandard (juh, kecske), illetve a laboratóriumi belső standard (bivaly) γ3- és γ2-kazeincsúcsa által lefedett terület arányával.
Megjegyzés: A módszer akkor működik megfelelően, ha mindkét szarvasmarha-eredetű kazeinre, a γ3- és γ2-kazeinekre egyértelműen pozitív a jelzés az 1 %-os referenciastandardban, de nincs jel a 0 %-os standardban. Ellenkező esetben, az eljárást optimalizálni kell a módszer részletes leírását követve.
A mintát akkor fogjuk pozitívnak tekinteni, ha benne a szarvasmarha-eredetű γ3- és γ2-kazein, illetve az ezeknek megfelelő csúcsok alatti terület nagysága nagyobb vagy egyenlő az 1 %-os referenciastandardnál kapott szintnél.
8. HIVATKOZÁSOK
1. Addeo F., Moio L., Chianese L., Stingo C., Resmini P., Berner I, Krause I., Di Luccia A., Bocca A.: Use of plasmin to increase the sensitivity of the detection of bovine milk in ovine and/or caprine cheese by gel isoelectric focusing of γ2-caseins. Milchwissenschaft 45, 708-711 (1990).
2. Addeo F., Nicolai M.A., Chianese L., Moio L., Spagna Musso S., Bocca A., Del Giovine L.: A control method to detect bovine milk in ewe and water buffalo cheese using immunoblotting. Milchwissenschaft 50, 83-85 (1995).
3. Krause I., Berner I, Klostermeyer H.: Sensitive detection of cow milk in ewe and goat milk and cheese by carrier ampholyte - and carrier ampholyte/immobilized pH gradient - isoelectric focusing of γ-caseins using plasmin as signal amplifier. in: Electrophoresis-Forum 89 (B. J. Radola, ed.) pp 389-393, Bode-Verlag, München (1989).
4. Krause Ι., Belitz H.-D., Kaiser K.-P.: Nachweis von Kuhmilch in Schaf and Ziegenmilch bzw. -käse durch isoelektrische Fokussierung in harnstoffhaltigen Polyacrylamidgelen. Z. Lebensm. Unters. Forsch. 174, 195-199 (1982).
5. Radola B.J.: Ultrathin-layer isoelectric focusing in 50-100 μm polyacrylamide gels on silanised glass plates or polyester films. Electrophoresis 1, 43-56 (1980).
1. ábra
A fedőlemez sematikus rajza
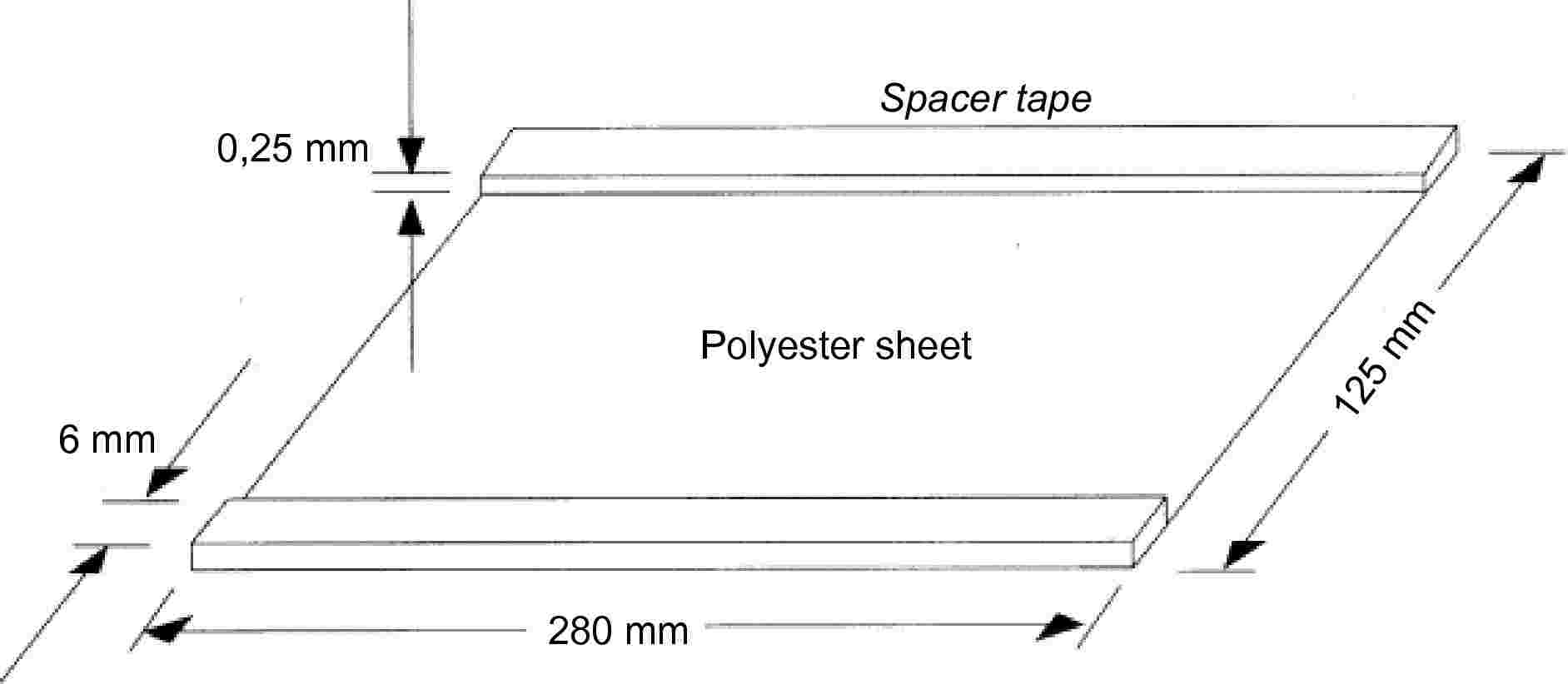
2. ábra
A vizes és a szerves fázis között a centrifugálást követően kialakuló kazeinréteg
3. ábra
Ultravékony poliakrilamid gél öntésére alkalmas terítéses eljárás
a = távtartószalag (0,25 mm); b = fedőlemez (5.3.); c, e = üveglapok (5.1.); d = géloldat (4.1.2.); f = géltartó lemez (5.2.).
4a. ábra
A juhsajtból és a kecskesajtból származó és különböző mennyiségű tehéntejet tartalmazó, plazminnal kezelt kazeinek izoelektromos fókuszálása
% CM = tehéntej százalékos aránya, C = tehén, E = juh, G = kecske
Az IEF-gél felső fele látható.
4b. ábra
A juhtej, kecsketej és bivalytej keverékéből készült sajtokból származó és különböző mennyiségű tehéntejet tartalmazó, plazminnal kezelt kazeinek izoelektromos fókuszálása
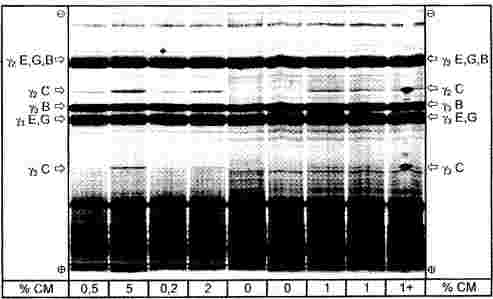
% CM = tehéntej százalékos aránya, 1 + = 1 % tehéntejet tartalmazó és a tiszta szarvasmarhakazein-csúccsal a fókuszált távolság felénél jelentkező minta C = tehén, E = juh, G = kecske, B = Bivaly
Az IEF-gél teljes elválasztási távolsága látható.
5. ábra
A standardok (STD) és a juh-, illetve kecsketej keverékéből készült sajtokból származó minták denzitogrammjai izoelektromos fókuszálást követően
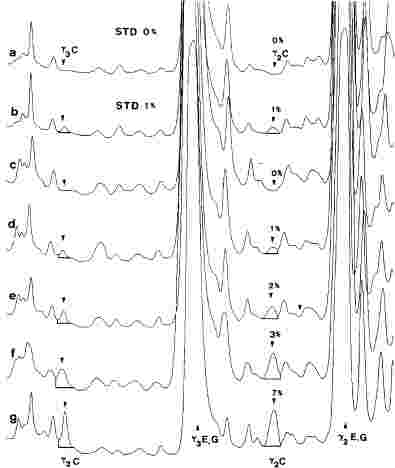
a, b = 0 és 1 % tehéntejet tartalmazó standardok; c-g = 0, 1, 2, 3 és 7 % tehéntejet tartalmazó sajtminták, C = tehén, E = juh, G = kecske.
Az IEF-gél felső felét a λ = 634 nm hullámhosszon mértük le.
X. MELLÉKLET
(7. cikk)
REFERENCIA-MÓDSZER KÓLIBAKTÉRIUMOK KIMUTATÁSÁRA VAJBAN, SOVÁNY TEJPORBAN, KAZEINBEN ÉS KAZEINÁTOKBAN
1. A MINTÁK ELŐKÉSZÍTÉSE
8261 ISO-szabvány.
2. ELJÁRÁS
4831 ISO-szabvány.
A táptalajt 1 g vajnak megfelelő vagy 0,1 g sovány tejpornak vagy kazeinnek/kazeinátoknak megfelelő mintákkal oltjuk be.
Mintánként három kémcsövet oltunk be.
3. EREDMÉNYEK
Ha a 3 kémcső három negatív eredményt hoz, az eredmény "megfelelő"
Ha a 3 kémcső kettő vagy három pozitív eredményt hoz, az eredmény "nem megfelelő"
Ha a 3 kémcső kettő negatív eredményt hoz, az analízist kétszer meg kell ismételni (két kémcsővel)
- Ha a 2 eredmény negatív, az eredmény "megfelelő"
- Ha legalább 1 eremény pozitív, az eredmény "nem megfelelő".
XI. MELLÉKLET
(8. cikk)
LAKTÓZ MEGHATÁROZÁSA ÖSSZETETT TAKARMÁNYOKBAN
1. TÁRGY ÉS ALKALMAZÁSI TERÜLET
Laktóz meghatározása összetett takarmányokban.
2. HIVATKOZÁS
A laktóztartalom a megadott eljárás alapján meghatározott, tömegszázalékban kifejezett mennyiség.
3. DEFINÍCIÓ
A vízmentes laktóztartalmat g/100g-ban fejezzük ki.
4. A MÓDSZER ELVE
Az összetett takarmányt vízzel felöntjük. Adjunk "Biggs"-oldatot egy hígított lemért aliquot-hoz, hogy kicsapódjon az összetett takarmányból a zsír és a fehérjekomponens-frakció. A mintát leszűrjük (vagy centrifugáljuk), és a szűrletet (vagy felülúszót) injektáljuk egy ólomionos kationcserélő HPLC-oszlopra mozgófázisként HPLC-minőségű víz használatával. Az eluált laktózt differenciál refraktométerrel ( 10 ) mutatjuk ki.
5. VEGYSZEREK
5.1. Általános
Ellenkező rendelkezés hiányában csak elismert analitikai tisztaságú vegyszert és gázmentes HPLC-minőségű vizet használjunk.
5.2. Laktóz
A d-laktóz monohidrát ((C12H22)O11H2O) többletnedvességet tud felvenni. Használat előtt mérjük meg a tényleges vízmennyiséget a Karl-Fisher-módszerrel vagy távolítsuk el a többletnedvességet úgy, hogy 8 órára 105 °C-os kemencébe helyezzük a laktózt (a laktóz ennél a kezelésnél nem veszíti el kristályvíz-tartalmát).
5.3. Koncentrált Biggs/Szijarto-oldat ( 11 )
Oldjunk fel 9,10 g cink-acetát-dihidrátot (Zn(CH3COO)2.2H2O) és 5,46 g foszfor-wolframsav-monohidrátot (H3[P(W3O10)4.xH2O]) körülbelül 70 ml HPLC-minőségű vízben egy 100 ml-es mérőlombikban.
Adjunk hozzá 5,81 ml jégecetet (CH3COOH). Hígítsuk fel a 100 ml jelig HPLC-minőségű vízzel (6.8.), és keverjük össze. Ez az oldat szobahőmérsékleten egy évig tárolható.
5.4. Hígított Biggs/Szijarto-oldat
Hígítsunk fel 25 ml koncentrált Biggs/Szijarto-oldatot (5.3.) vízzel 500 ml-re mérőlombik használatával. Ez az oldat szobahőmérsékleten egy hónapig tárolható.
5.5. HPLC-minőségű víz előkészítése
Szűrjük le az ultratiszta vizet (6.8.) vákuumos szűrőrendszer használatával (6.9.). A pumpa teljesítményének javításához és stabil alapvonal eléréséhez a mozgófázist naponta gázmentesítsük valamilyen rendelkezésre álló technikával, mint pl. hélium bevezetése, ultrahangos kezelés, vákuumos vagy in-line gázmentesítés.
Megjegyzés: Az oszlop élettartamának meghosszabítása érdekében fontos, hogy az eluens szén-dioxid-tartalma a lehető legalacsonyabb legyen és hogy az újrafelvétel ne legyen lehetséges.
6. ESZKÖZÖK
A szokásos laboratóriumi berendezés, valamint különösen a következők:
6.1. Ioncserélő gyantát tartalmazó HPLC-oszlop
Oszloptöltet: ólomionos kationcserélő csoportokkal aktivált 8 % keresztkötésű polisztirol-divinil-benzol kopolimer.
Oszlop méretei: 300 mm hosszú, belső átmérő kb. 8mm.
Más átmérőt is lehet használni, ha az áramlási sebességet megfelelően beállítjuk.
6.2. Előtétoszlop
Az előtétoszlop egy külön kationcserélő (H+) és egy anioncserélő (CO3-) kombinációja, mindegyik kb. 30 mm × 4,6 mm-es (hossz × belső átmérő) oszlopokba töltve (pl. mikro-előtétoszlopok mikroelőtétoszlop-tartóban)és sorba kötve vagy "mixed-bed" formában, amelyet egy AG 50W-X4, - 400 mesh (H+) és egy AG3-X4A, 200-400 mesh (OH-) alkot 35:65 (m/m) arányban, manuálisan egy kb. 20 × 9 mm-es (hossz × belső átmérő) oszlopba töltve.
6.3. Termosztatikus oszlopkamra
85 °C ± 1 °C állandó hőmérséklet fenntartására alkalmas kamra.
6.4. HPLC-pumpa
0,2-1,0 m/min állandó áramlási sebesség (< 0,5 % ingadozás) előállítására képes pumpa.
6.5. HPLC-injektáló eszköz
25 μl injektálására képes automata mintaadagoló < 0,5 % ismételhetőséggel.
Kézi eszköz is használható (ugyanazokkal a követelményekkel, mint az automata adagoló).
6.6. HPLC-detektor
Magas érzékenységű refraktív index detektor, melynek zajszintje < 5,10-9 RI egység.
6.7. Integrátor
Szoftver vagy az arra fenntartott integrátor az adatgyűjtéshez és feldolgozáshoz és olyan csúcsterületek és csúcsmagasságok előállításához, amelyet át lehet számítani laktózkoncentrációra.
6.8. Víztisztító egység
> 14 MΩ.cm ellenállású ultratiszta víz (1. típus) előállítására képes rendszer.
6.9. Oldószerszűrő egység
0,45 μm pórusméretű membránszűrő használatával való vízszűrést lehetővé tévő rendszer.
Megjegyzés: Sok víztisztító egység (6.8.) rendelkezik beépített 0,45 vagy 0,2 μm-es szűrővel. A további szűrés elhagyható, ha ezt a vizet közvetlenül felhasználjuk.
6.10. Analitikai mérleg
0,1 mg leolvashatóságú mérleg.
6.11. Vízfürdő
40 °C (±0,5) hőmérséklet tartására képes vízfürdő.
6.12. Centrifuga
Eppendorf-csövek vagy azzal egyenértékű, vagy nagyobb típusú csövek esetén legalább 3 000 g előállítására képes centrifuga.
6.13. 50 ml-es mérőlombik
50 ml űrtartalom, A osztály
Megjegyzés: Más űrtartalmú lombik is használható a térfogattényező figyelembevételével.
6.14. 100 ml-es mérőlombik
100 ml űrtartalom, A osztály
6.15. Osztott pipetta
10 ml-es osztott pipetta.
Megjegyzés: 5 ml-es kézi pipettázó feltét is használható, kétszer 5 ml vegyszer (5.3.) hozzáadásával.
7. MINTAVÉTEL
Fontos, hogy a laboratórium az ISO 707/IDF 50 ( 12 ) szerint vett, valóban reprezentatív mintát kapjon, amely nem sérült a szállítás vagy a tárolás során.
8. A LAKTÓZSTANDARD-OLDAT ELŐKÉSZÍTÉSE
8.1.
1. standard
Oldjunk fel kb. 50 mg mennyiségű, pontosan (0,1 mg leolvashatóság) lemért laktóz monohidrátot (5.2.) egy 100 ml-es mérőlombikban (6.14.) és töltsük fel vízzel a jelzésig.
8.2.
2. standard
Oldjunk fel kb. 100 mg mennyiségű, pontosan (0,1 mg leolvashatóság) lemért laktóz monohidrátot (5.2.) egy 100 ml-es mérőlombikban (6.14.), és töltsük fel vízzel a jelzésig.
Megjegyzés: A standard oldatok legfeljebb egy hétig tárolhatók kb. 5 °C-on.
9. A VIZSGÁLATI MINTA ELŐKÉSZÍTÉSE
9.1. A minta elkészítése
Mérjünk ki kb. 5 g port egy 5 ml-es lombikba (6.13.) és 1 mg-os pontossággal jegyezzük fel a tömegét (W1, (11)). Adjunk hozzá 50 ml vizet, és 0,01 g pontossággal jegyezzük fel a tömeget (W2, (11)). A lezárt lombikot helyezzük vízfürdőbe (6.11.) 30 percre, és ezalatt fordítsuk át néhányszor. Ezt követően hagyjuk szobahőmérsékletre hűlni.
9.2. A minta kezelése
Vegyünk kb. 1 g-ot ebből az oldatból, és helyezzük egy 50 ml-es mérőlobikba (6.13.), jegyezzük fel 1 mg-os pontossággal a tömegét (W3, (11)), adjunk hozzá 20 ml vizet majd 10 ml hígított Biggs/Szijarto oldatot (5.4.), és töltsük fel a jelig vízzel. Az első 30 perc alatt ötször gyengéden fordítsuk át a lombikot.
1 óra elteltével vegyünk egy aliquot részt, és centrifugáljuk (6.12.) 3 000 g-n 10 percig (magasabb g alkalmazható megfelelően rövidebb ideig). Használjunk egy aliquot részt a felülúszóból a HPLC-vizsgálathoz.
10. HPLC-MEGHATÁROZÁS
10.1. A HPLC előzetes előkészítése
10.1.1. Az oszlop és az előtétoszlop felszerelése
Szereljük fel az előtétoszlopot (6.2.) az oszlopkamrán (6.3.) kívül és az oszlopot (6.1.) a kamrában.
Megjegyzés: Ha a kamrában nicsen csövezés az eluens előmelegítéséhez, az eluensnek 15 cm rozsdamentes acél csövezésen keresztül kell a kamrába jutnia, mielőtt az oszlopba kerül (az eluensnek mindenképp fel kell melegednie, mielőtt az oszlopba kerül, különben csúcskiszélesedés fordul elő).
10.1.2. Detektor és kezdeti áramlás
Annak érdekében, hogy stabil legyen az alapvonal, kapcsoljuk be a detektort (6.6.) legalább 24 órával a vizsgálat megkezdése előtt. Állítsuk a detektor belső hőmérsékletét 35 °C-ra. Állítsuk az áramlást 0,2 ml/min sebességre (6.4.) legalább 20 percre, ezalatt az oszlopkamrát (6.3.) szobahőmérsékletre állítjuk.
10.1.3. Oszlopkamra és végleges áramlási sebesség
Állítsuk az oszlopkamrát (6.3.) 85 °C-ra. Ha elérte ezt a hőmérsékletet, 30 perc után fokozatosan emeljük az áramlási sebességet 0,2ml/min-ről 0,6 ml/min-re (6.4.). Hagyjuk, hogy a rendszer egyensúlyba kerüljön ezzel az áramlási sebességgel és 85 °C-on 2 órán át vagy amíg stabil alapvonalat nem kapunk.
10.1.4. Integrálás
Gondosan válasszuk ki az adatgyűjtés és -integráció paramétereit (6.7.), úgy mint adatátvitel, érzékenység, időkonstans, csúcsszélesség és küszöbérték.
A laktóz retenciós ideje kb. 11 perc.
Megjegyzés: Számos adatgyűjtő szoftver (6.7.) lehetővé teszi az elméleti tányérszám könnyű mérését. Rendszeresen mérjük meg 1. standard (8.1.) elméleti tányérszámát, és cseréljük ki az oszlopot (6.1.), ha a tányérszám 25 %-kal alacsonyabb egy új oszlop kezdeti értékénél.
10.1.5. Előtétoszlop-vizsgálat
25 μl 0,05 %-os nátrium-klorid-oldat injektálásával rendszeresen (sorozatonként legalább egyszer) ellenőrizzük, hogy az előtétoszlop (6.2.) képes-e eltávolítani a sókat a mintából. Ha csúcsok jelennek meg, az előtétoszlopot ki kell cserélni.
10.2. Standardok futtatása
Minden egyes vizsgálatsorozat elején injektáljunk 25 μl-t (6.5.) az 1. standardból (8.1.) majd a 2. standardból (8.2.). Ismételjük ezt meg 10-20 mintánként és a sorozat végén is.
10.3. Minták futtatása
Injektáljunk 25 μl-t a minta felülúszójából (9.2.).
11. SZÁMÍTÁS ÉS AZ EREDMÉNYEK KIFEJEZÉSE
11.1. Kalibrálás
Rendes esetben a csúcsok magasságát használjuk az eredmények számításához, de ha a jel túl zajos, a csúcsterületet is használhatjuk (a csúcsmagasság szerinti mennyiségi meghatározást kevésbé befolyásolják az alacsony koncentrációjú összetevők csúcsai, amelyek részben, de nem elegendő mértékben elválasztódnak a laktózcsúcstól).
A szoftvernek (6.7.) egy lineáris kalibrációs görbét kell kiszámítania, amely keresztül vezet az origón. Ellenőrizzük a görbét, hogy esetleg eltér-e a linearitástól (a linearitástól való eltérést legnagyobb valószínűséggel az 1. (8.1.) és 2. (8.2.) standard hibás előkészítése, a rossz integráció és kevésbé valószínű módon az injektor hibás működése okozza).
Kiindulási adatként az 1. (8.1.) és 2. (8.2.) standard mg/ml-ben kiszámított laktózkoncentrációit használjuk vízmentes laktózként.
A kalibrációs vonal meredekségének (RF) meghatározása terület/koncentráció mg/ml-ben.
11.2. Minták
Az analízis eredményét g/100g-ra kapjuk meg, és a szoftver (6.7.) használatával vagy a következő képlet használatával számítjuk:
ahol:
C : laktózkoncentráció g/100 g por
H : a minta laktózcsúcsának magassága
RF : a kalibrációs görbe válaszjele (vagy meredeksége) mV/mg/ml-ben
W1 : a porminta súlya g-ban (9.1.)
W2 : a pormintához adott víz súlya g-ban (9.1.)
W3 : a pormintából elkészített oldat súlya g-ban (9.2.)
50 : a (9.2.) pont szerint használt mérőlombik térfogata
0,1 : az eredmény átváltása g/100 g-ra
12. PONTOSSÁG
Az ebből a laboratóriumközi tesztből származó értékek valószínűleg nem alkalmazhatók a megadottakon kívül más koncentrációtartományokra és mátrixokra. Az ismételhetőség és a reprodukálhatóság értékei egy, az ISO 5725 ( 13 ) szerint elvégzett laboratóriumközi teszt eredményéből származnak.
12.1. Ismételhetőség
Ugyanazon módszerrel azonos vizsgálati anyagon, ugyanazon laboratóriumban ugyanazon kezelőszemély által, ugyanazon berendezésen, és a két teszt elvégzése között eltelt rövid idő alatt kapott két egyedi vizsgálati eredmény közötti abszolút különbség az esetek legfeljebb 5 %-ában lesz nagyobb xxx-nál (körvizsgálattal meghatározandó) ( 14 ).
12.2. Reprodukálhatóság
Ugyanazon módszerrel azonos vizsgálati anyagon, különböző laboratóriumban különböző kezelőszemély által, különböző berendezésen kapott két egyedi teszteredmény közötti abszolút különbség az esetek legfeljebb 5 %-ában lesz nagyobb 0,5 g/100 g-nál (körvizsgálattal meghatározandó).
13. HIVATKOZÁSOK
XII. MELLÉKLET
(9. cikk)
OLTÓS SAVÓ KIMUTATÁSA INTERVENCIÓS RAKTÁROZÁSRA SZÁNT SOVÁNY TEJPORBAN A KAZEINOMAKROPEPTIDEK NAGYTELJESÍTMÉNYŰ FOLYADÉKKROMATOGRÁFIÁS ELJÁRÁSSAL (HPLC) VALÓ MEGHATÁROZÁSA ÚTJÁN
1. TÁRGY ÉS ALKALMAZÁSI TERÜLET
Ez a módszer lehetővé teszi az oltós savó kimutatását intervenciós raktározásra szánt sovány tejporból a kazeinomakropeptidek meghatározása révén.
2. HIVATKOZÁS
ISO 707 nemzetközi szabvány - Tej és tejtermékek - Mintavételi módszerek, az I. melléklet 2. c) pontjának utolsó bekezdésében foglalt irányelvek szerint.
3. DEFINÍCIÓ
Az oltóssavószárazanyag-tartalom a megadott eljárás alapján, kazeinomakropeptid-tartalom kimutatásával meghatározott, tömegszázalékban kifejezett mennyiség.
4. A MÓDSZER ELVE
- A sovány tejpor helyreállítása, zsír és fehérjék eltávolítása triklórecetsavas kicsapással és ezt követő centrifugálással vagy szűréssel.
- A kazeinomakropeptidek (CMP) mennyiségének meghatározása a felülúszóból nagyteljesítményű folyadékkromatográfiás eljárással (HPLC).
- A mintából kapott eredmények értékelése sovány tejporból és akár ismert mennyiségű savópor hozzáadásával, akár anélkül készült standard mintákkal való összehasonlítás révén.
5. VEGYSZEREK
Az összes vegyszernek elismerten analitikai tisztaságúnak kell lennie. A felhasznált víznek desztillált víznek vagy azzal legalább egyenértékű tisztaságú víznek kell lennie.
5.1. Triklór-ecetsav-oldat
Oldjunk fel 240 g trikór-ecetsavat (CCl3COOH) vízben, és töltsük fel vízzel 1 000 ml-re. Az oldatnak tisztának és színtelennek kell lennie.
5.2. Eluáló-oldat, 6,0 pH
Oldjunk fel 1,74 g kálium-hidrogén-foszfátot (K2HPO4), 12,37 g kálium-dihidrogén-foszfátot (KH2PO4), valamint 21,41 g nátrium-szulfátot (Na2SO4) megközelítőleg 700 ml vízben. Szükség esetén foszforsav vagy kálium-hidroxid felhasználásával állítsuk be a pH-ját 6,0-ra.
Töltsük fel vízzel 1 000 ml-re, és homogenizáljuk.
Megjegyzés: Az eluens összetétele megváltoztatható, hogy megfeleljen a standardok tanúsítványának vagy az oszlop-töltőanyag gyártója ajánlásainak.
Felhasználás előtt az eluens-oldatot 0,45 μm pórusátmérőjű membránszűrőn szűrjük le.
5.3. Öblítőoldat
Keverjünk el egy rész acetonitrilt (CH3CN) kilenc rész vízzel. A keveréket szűrjük át egy 0,45 μm pórusátmérőjű membránszűrőn.
Megjegyzés: Bármilyen más, csíraölő hatású öblítőoldat is alkalmazható, amely nem rontja az oszlopok elválasztóképességét.
5.4. Standard minták
5.4.1. Az e rendeletben foglalt követelményeknek megfelelő sovány tejpor (azaz [0]).
5.4.2. Ugyanez a sovány tejpor 5 % (m/m) szabványos összetételű oltós savóporral módosítva (azaz [5]).
6. ESZKÖZÖK
6.1. Analitikai mérleg
6.2. 2 200 g centrifugális erő elérésére képes opcionális centrifuga, körülbelül 50 ml-es, ledugózott vagy kupakkal ellátott centrifugacsövekkel felszerelve.
6.3. Mechanikus rázógép
6.4. Mágneses keverő
6.5. Körülbelül hét centiméter átmérőjű üvegtölcsérek
6.6. Szűrőpapírok, közepes szűrőképességgel, körülbelül 12,5 cm átmérővel
6.7. Üvegszűrő-berendezés 0,45 μm pólusátmérőjű membránszűrővel
6.8. Beosztással ellátott pipetták, amelyek alkalmasak 10 ml adagolására (ISO 648, A osztály vagy ISO/R 835), vagy olyan adagolórendszer, amely két perc alatt képes 10,0 ml adagolására
6.9. 20,0 ml víz kb. 50 °C-on történő adagolására képes adagolórendszer
6.10. Termosztátos szabályozással ellátott vízfürdő 25 ± 0,5 °C-ra állítva
6.11. HPLC-eszköz, részei:
6.11.1. Szivattyú
6.11.2. Injektor, kézi vagy automata, 15-30 μl űrtartalommal.
6.11.3. Két sorosan kötött TSK 2 000-SW kromatográfiás oszlop (30 cm hosszú, belső átmérő 0,75 cm) vagy ezzel egyenértékű oszlopok (pl. egy TSK 2 000-SWxl, egy Agilent Technologies Zorbax GF 250), és egy I 125-el vagy azzal egyenértékű anyaggal töltött előtétoszlop (3 cm×0,3 cm)
6.11.4. Termosztált oszlopkamra, 35 ± 1 °C-ra állítva
6.11.5. Változtatható hullámhosszúságú UV-detektor, amely 205 nm-en 0,008 A érzékenységű méréseket tesz lehetővé
6.11.6. Völgytől-völgyig történő integrálásra alkalmas integrátor
Megjegyzés: Lehetséges szobahőmérsékleten tartott oszlopokkal is dolgozni, de ekkor az elválasztóképességük valamivel kisebb. Ebben az esetben a hőmérséklet az elemzés egyik tartományában sem változhat ± 5 °C-nál többet.
7. MINTAVÉTEL
7.1. A mintákat az ISO 707 nemzetközi szabványban meghatározottak szerint kell venni. A tagállamok azonban alkalmazhatnak ettől eltérő mintavételi módszert is, feltéve hogy az teljesíti a fent említett szabványban foglalt alapelveket.
7.2. Tároljuk a mintát olyan körülmények között, amely kizárja, hogy az összetétele megváltozzon vagy lebomoljon.
8. ELJÁRÁS
8.1. A minta előkészítése
A tejport tegyük a tejpor terjedelménél kétszer nagyobb űrtartalmú edénybe, amely légmentesen zárható. Azonnal zárjuk le az edényt. Az edény ismételt felfordításával alaposan keverjük fel a benne lévő tejport.
8.2. Vizsgálati minta
Egy centrifugacsőbe (6.2.) vagy 50 ml-es ledugaszolt lombikba mérjünk ki a mintából 2 000 ± 0,001 g-ot.
8.3. Zsír és fehérje eltávolítása
8.3.1. Adjunk 20,0 ml meleg vizet (50 °C) a vizsgálati mintához. Oldjuk fel a port mechanikus rázógép segítségével ötperces rázatással (6.3.). Helyezzük a csövet vízfürdőbe (6.10.), és engedjük 25 °C-ra hűlni
8.3.2. Adjunk hozzá két perc alatt 10,0 ml kb. 25 °C-os triklór-ecetsav-oldatot (5.1.), miközben a mágneses keverővel erőteljesen keverjük (6.4.). Helyezzük a csövet vízfürdőbe (6.10.), és hagyjuk ott 60 percig
8.3.3. Centrifugáljuk (6.2.) 10 percig 2 200 g-n, vagy szűrjük át a szűrőpapíron (6.6.), kidobva az első 5 ml szűrletet
8.4. Kromatográfiás meghatározás
8.4.1. Fecskendezzünk 15-30 μl pontosan kimért felülúszót vagy szűrletet (8.3.3.) a HPLC-készülékbe (6.11.), amelynek percenként 1,0 ml eluáló oldat (5.2.) áramlási sebességgel kell működnie
1. megjegyzés: A használt oszlopok belső átmérőjétől vagy az oszlop gyártójának utasításaitól függően más áramlási sebesség is használható.
2. megjegyzés: Az eluáló oldatot (5.2.) a kromatográfiás elemzés egész időtartama alatt tartsuk 85 °C-on, hogy gázmentes maradjon, és hogy megakadályozzuk a mikroorganizmusok szaporodását. Más, hasonló eredményre vezető, elővigyázatossági intézkedés is alkalmazható.
3. megjegyzés: Minden megszakítás alkalmával öblítsük át vízzel az oszlopokat. Soha ne hagyjunk bennük az eluálószert (5.2.).
Mielőtt 24 óránál hosszabb időre megszakítanánk a vizsgálatot, előbb öblítsük át az oszlopokat vízzel, majd mossuk legalább három órán át öblítőoldattal (5.3.) percenként 0,2 ml-es folyási sebesség mellett.
8.4.2. Az [E] vizsgálati minta kromatográfiás elemzésének eredményét egy kromatogram formájában kapjuk meg, ahol minden csúcsot a rá jellemző retenciós idő (RT) alapján tudunk azonosítani a következők szerint:
| II. csúcs: | a kromatogram második csúcsa, kb. 12,5 perces retenciós idővel |
| III. csúcs: | a kromatogram harmadik csúcsa, a CMP-nek megfelelően, kb. 15,5 perces retenciós idővel |
Az oszlopválasztás jelentősen befolyásolhatja az egyes csúcsok retenciós idejét.
Az integrátor (6.11.6.) minden csúcsra automatikusan kiszámolja az A területet:
| AII: | II. csúcs területe |
| AIII: | III. csúcs területe |
Lényeges, hogy még a mennyiségi értelmezés előtt minden kromatogram külalakját megvizsgáljuk, hogy felfedezzünk minden olyan rendellenességet, amely vagy a készülék vagy az oszlopok hibás működéséből ered, vagy a minta eredetére és jellegére vezethető vissza.
Kétség esetén az elemzést meg kell ismételni.
8.5. Kalibrálás
8.5.1. A standard mintára (5.4.) is alkalmazzuk pontosan ugyanazt az eljárást, mint amely a 8.2. és a 8.4.2. pont közötti leírásban szerepel.
Használjunk frissen készített oldatokat, mert a CMP 8 %-os triklórecetsavas-környezetben hamar bomlik. A veszteség becsült értéke 30 °C-on óránként 0,2 %.
8.5.2. A minták kromatográfiás vizsgálatát megelőzően az oszlopokat az oldatban (8.5.1.) lévő standard minta (5.4.2.) ismételt befecskendezésével addig kondicionáljuk, amíg a CMP-nek megfelelő csúcs retenciós ideje konstanssá nem válik.
8.5.3. Ugyanannyi mennyiségű szűrletnek (8.5.1.) az injektálásával, mint amennyit a mintáknál használtunk, határozzuk meg az R-válaszjeleket.
9. AZ EREDMÉNYEK KIFEJEZÉSE
9.1. A számítás módszere és a képletek
9.1.1. Az R-válaszjelek kiszámítása:
| II. csúcs: | RII = 100/(AII[0]) |
ahol:
RII = a II. csúcs válaszjelei,
AII [0] = a standard mintából [0] a 8.5.3. pont szerint nyert II. csúcs területe
| III. csúcs: | RIII = W/(AIII[5] – AIII[0]) |
ahol:
RIII = a III. csúcs válaszjele,
AIII [0] és AIII [5] = a [0], illetve [5] standard minták esetén a 8.5.3. pontban nyert III. csúcs alatti területek
W = a savó mennyisége a [5] standard mintában, azaz 5.
9.1.2. A csúcsok relatív területe a mintában [E]
SII[E] = RII × AII[E]
SIII[E] = RIII × AIII[E]
SIV[E] = RIV × AIV[E]
ahol:
SII [E], SIII [E], SIV [E] = a II., III. és IV. csúcs alatti relatív területek a mintában [E],
AII [E], AIII [E] = a 8.4.2. pontban nyert II. és III. csúcs alatti területek a mintában [E],
RII, RIII = a 9.1.1. pontban számított választjelek.
9.1.3. A mintából [E] kapott III. csúcs relatív retenciós idejének kiszámítása: RRTIII[E] = (RTIII[E])/(RTIII[5])
ahol:
RRTIII [E] = a minta [E] III. csúcsának relatív retenciós ideje,
RTIII [E] = a mintából [E] a 8.4.2. pont szerint nyert III. csúcs retenciós ideje,
RTIII [5] = a kontrollmintából [5] a 8.5.3. pont szerint nyert III. csúcs retenciós ideje.
9.1.4. A kísérletek azt mutatták ki, hogy a III. csúcs relatív retenciós ideje, vagyis RRTIII [E] és a hozzáadott savópor mennyisége között 10 %-ig lineáris összefüggés van.
- Az RRTIII [E] < 1,000, ha a savótartalom > 5 %;
- az RRTIII [E] ≥ 1,000, ha a savótartalom ≤ 5 %.
Az RRTIII értékeire megengedhető bizonytalansági tényező ±0,002.
Rendes esetben az RRTIII [0] értéke 1,034-től csak kis mértékben tér el. Az oszlopok állapotától függően az érték közeledhet az 1,000-hez, de annál mindig nagyobbnak kell lennie.
9.2. A mintában található oltós savópor százalékos mennyiségének kiszámítása:
W = SIII[E] - [1, 3 + (SIII[0] - 0, 9)]
ahol:
W = az oltós savó tömegszázaléka a mintában [E];
SIII [E] = az [E] vizsgálati minta 9.1.2. pont szerint nyert III. csúcsának relatív területe;
1,3 = a III. csúcs relatív átlagos területe a különféle származású nem módosított sovány tejporban meghatározott oltós savó gramm/100 g-ban kifejezve. Ez egy kísérletben nyert számadat;
SIII [0] = a III. csúcs relatív területe, amely egyenlő RIII × AIII [0]. Ezek a 9.1.1. pontban és a 8.5.3. pontban kapott értékek;
(SIII [0] - 0,9) = az 1,3 relatív átlagos területre elvégzendő korrekció, ha SIII [0] nem egyenlő 0,9. Kísérleti úton a kontrollminta [0] III. csúcsának relatív átlagos területe 0,9.
9.3. Az eljárás pontossága
9.3.1. Ismételhetőség
Az egyidőben vagy rövid időeltéréssel egymás után, ugyanazon laboratóriumi személyzet által, ugyanazzal az eszközzel, azonos vizsgálati anyagon elvégzett két meghatározás különbsége nem haladhatja meg a 0,2 % m/m értéket.
9.3.2. Reprodukálhatóság
A két különböző laboratóriumban, azonos vizsgálati anyagon kapott két egyedi, egymástól független eredmény különbsége ne haladja meg a 0,4 % m/m értéket.
9.4. Értelmezés
9.4.1. Feltételezzük a savó hiányát a mintában, ha a SIII [E] III. csúcsának a termék 100 grammjára jutó oltós savó grammban kifejezett relatív területe ≤ 2,0 + (SIII [0] - 0,9) ahol
2,0 = a III. csúcs relatív területére megengedett legnagyobb érték, figyelembe véve a III. csúcs relatív területét, azaz 1,3-at, a sovány tejpor eltérő összetétele miatti bizonytalanságot és a módszer reprodukálhatóságát (9.3.2.),
(SIII [0] - 0,9) = elvégzendő korrekció, ha az SIII [0] terület különbözik 0,9-től (lásd a 9.2. pontot).
9.4.2. Ha a III. csúcs, SIII [E] relatív területe > 2,0 + (SIII [0] - 0,9) és a II. csúcs, SII [E] relatív területe ≤ 160, az oltóssavó-tartalmat a 9.2. pontban jelzett módon határozzuk meg.
9.4.3. Ha a III. csúcs, SIII [E] relatív területe > 2,0 + (SIII [0] - 0,9) és a II. csúcs, SII [E] relatív területe ≤ 160, határozzuk meg az összes fehérjetartalmat (P %); majd vizsgáljuk meg az 1. és 2. grafikont.
9.4.3.1. A magas fehérjetartalommal rendelkező, módosítás nélküli sovány tejpor mintáinak elemzésével nyert adatok összeállítását az 1. és 2. grafikon mutatja.
A folyamatos vonal lineáris regressziót jelent, amelynek együtthatóit a legkisebb négyzetek módszerével számították ki.
A szaggatott egyenes vonal a III. csúcs relatív területének felső határát az esetek 90 %-ában meg nem haladott valószínűséggel rögzíti.
Az 1. és 2. grafikon szaggatott egyenes vonalainak egyenletei a következők:
| SIII = 0,376 P % – 10,7 | (1. grafikon), |
| SIII = 0,0123 SII [E] + 0,93 | (2. grafikon), |
ahol:
SIII = a III. csúcs relatív területe, az összes fehérjetartalom vagy az SII [E] csúcs relatív területe szerint számítva,
P % = az összes fehérjetartalom tömegszázalékban kifejezve,
SII [E] = a minta 9.1.2. pontban kiszámított relatív területe
Ezek az egyenletek a 9.2. pontban említett 1,3-as számmal egyenértékűek.
A kapott SIII [E] relatív területe és az SIII relatív területe közötti különbség (T1 és T2) a következők szerint adódik: T1 = SIII[E] - [(0,376 P % - 10,7) + (SIII[0] - 0,9)]T2 = SIII[E] - [(0,0123 SII[E] + 0,93) + (SIII[0] - 0,9)]
9.4.3.2. Ha T1 és/vagy T2 nulla vagy kevesebb, nem határozható meg oltós savó jelenléte.
Ha T1 és T2 nullánál több, oltós savó van jelen.
Az oltós savó mennyiségét a következő képlet alapján számíthatjuk ki: W = T2+0,91
ahol:
0,91 a szaggatott és folyamatos vonal között a függőleges tengelyen mért távolság.
XIII. MELLÉKLET
(9. cikk)
OLTÓSSAVÓ-SZÁRAZANYAG MEGHATÁROZÁSA SOVÁNY TEJPORBAN ÉS A 2799/1999/EK RENDELETBEN EMLÍTETT KEVERÉKEKBEN
1. CÉL: OLTÓSSAVÓ-SZÁRAZANYAG HOZZÁADÁSÁNAK KIMUTATÁSA:
a) a 2799/1999/EK rendelet 2. cikkében meghatározott sovány tejporból; és
b) a 2799/1999/EK rendelet 4. cikkében meghatározott keverékekből.
2. HIVATKOZÁSOK: ISO 707 NEMZETKÖZI SZABVÁNY
3. FOGALOMMEGHATÁROZÁS
Az oltóssavószárazanyag-tartalom a megadott eljárás alapján, kazeinomakropeptid-tartalom kimutatásával meghatározott, tömegszázalékban kifejezett mennyiség.
4. A MÓDSZER ELVE
A kazeinomakropeptid-tartalmat a XII. melléklet szerint határozzuk meg. A pozitív eredményt mutató mintákat fordított fázisú nagy teljesítményű folyadékkromatográfiás eljárással (HPLC) vizsgáljuk meg kazeinomakropeptid-A jelenlétére. A minták közvetlenül is vizsgálhatók a fordított fázisú HPLC-eljárással. A mintából kapott eredmények értékelése standard mintákkal való összehasonlítás révén történik, amelyek sovány tejporból, ismert mennyiségű savópor hozzáadásával, vagy anélkül készülnek. Az 1 %-nál (m/m) magasabb eredmények azt mutatják, hogy oltóssavó-szárazanyag van jelen.
5. VEGYSZEREK
Az összes vegyszernek analitikai tisztaságúnak kell lennie. A felhasznált víznek desztillált víznek vagy azzal legalább egyenértékű tisztaságú víznek kell lennie. Az acetonitrilnek spektroszkópiai minőségűnek vagy HPLC-minőségűnek kell lennie.
Az eljáráshoz szükséges vegyszerek leírását ennek a rendeletnek a XII. melléklete tartalmazza.
Vegyszerek fordított fázisú HPLC-hez.
5.1. Triklór-ecetsav oldat
Oldjunk fel 240 g trikór-ecetsavat (CCl3COOH) vízben, és egészítsük ki vízzel 1 000 ml-re. Az oldatnak tisztának és színtelennek kell lennie.
5.2. A. és B. eluens
A. eluens: Helyezzünk 150 ml acetonitrilt (CH3CN), 20 ml izopropanolt (CH3CHOHCH3), és 1,00 ml trifluor-ecetsavat (TFA, CF3COOH) egy 1 000 ml-es mérőlombikba. Egészítsük ki vízzel 1 000 ml-re.
B. eluens: Helyezzünk 550 ml acetonitrilt, 20 ml izopropanolt és 1,00 ml TFA-t egy 1 000 ml-es mérőlombikba. Egészítsük ki vízzel 1 000 ml-re. Felhasználás előtt az eluenst 0,45 μm pórusátmérőjű membránszűrőn szűrjük le.
5.3. Az oszlop konzerválása
Használatot követően az oszlopot a B-eluenssel (egy gradiens útján) át kell mosni és ezt követően acetonitrillel elöblíteni (egy gradiens útján, 30 percig). Az oszlopot acetonitrilben kell tárolni.
5.4. Standard minták
5.4.1. Az intervenciós raktározás követelményeinek megfelelő sovány tejpor (azaz [0]).
5.4.2. Ugyanez a sovány tejpor 5 % (m/m) szabványos összetételű oltós savóporral módosítva (azaz [5]).
5.4.3. Ugyanez a sovány tejpor 50 % (m/m) szabványos összetételű oltós savóporral módosítva (azaz [50]) ( 15 ).
6. ESZKÖZÖK
Az ehhez az eljáráshoz használatos eszközöket ennek a rendeletnek a XII. melléklete ismerteti.
6.1. Analitikai mérleg
6.2. 2 200 g centrifugális erő elérésére képes opcionális centrifuga, körülbelül 50 ml-es, ledugózott vagy kupakkal ellátott centrifugacsövekkel felszerelve
6.3. Mechanikus rázógép
6.4. Mágneses keverő
6.5. Körülbelül hét centiméter átmérőjű üvegtölcsérek
6.6. Szűrőpapírok, közepes szűrőképességgel, körülbelül 12,5 cm átmérővel
6.7. Üvegszűrő-berendezés 0,45 μm pórusátmérőjű membránszűrővel
6.8. Beosztással ellátott pipetták, amelyek alkalmasak 10 ml adagolására (ISO 648, A osztály vagy ISO/R 835), vagy olyan adagolórendszer, amely két perc alatt képes 10,0 ml adagolására
6.9. 20,0 ml víz kb. 50 °C-on történő adagolására képes adagolórendszer.
6.10. Termosztáttal ellátott vízfürdő 25 ±0,5 °C-ra állítva
6.11. HPLC-berendezés, részei:
6.11.1. Bináris gradiensű szivattyúrendszer
6.11.2. Injektor, kézi vagy automata, 100 μl kapacitással
6.11.3. Agilent Technologies Zorbax 300 SB-C3 kromatográfiás oszlop (25 cm hosszú, belső átmérő 0,46 cm) vagy ezzel egyenértékű, nagy belső átmérőjű, szilika alapú fordított fázisú oszlop
6.11.4. Termosztatikus oszlopkamra, 35 ± 1 °C-ra állítva
6.11.5. Változtatható hullámhosszúságú UV-detektor, amely 210 nm-en (szükség esetén magasabb hullámhosszon is, 220 nm-ig) 0,02 Å érzékenységű méréseket tesz lehetővé
6.11.6. A közös alapvonalra vagy völgytől-völgyig történő integrálásra alkalmas integrátor
Megjegyzés: Lehetséges szobahőmérsékleten tartott oszlopokkal is dolgozni, de ekkor a hőmérséklet az elemzés során sem változhat ± 1 °C-nál többet, ellenkező esetben túl nagy lesz a CMPA retenciós idejében fellépő variancia.
7. MINTAVÉTEL
7.1. A mintákat az ISO 707 nemzetközi szabványban meghatározottak szerint kell venni. A tagállamok azonban alkalmazhatnak ettől eltérő mintavételi módszert is, feltéve hogy az teljesíti a fent említett szabványban foglalt alapelveket.
7.2. Tároljuk a mintát olyan körülmények között, amely kizárja, hogy az összetétele megváltozzon vagy lebomoljon.
8. ELJÁRÁS
8.1. A vizsgálati minta előkészítése
A tejport tegyük a tejpor terjedelménél kétszer nagyobb űrtartalmú edénybe, amely légmentesen zárható. Azonnal zárjuk le az edényt. Az edény ismételt felfordításával alaposan keverjük fel a benne lévő tejport.
8.2. Vizsgálati adag
Egy centrifugacsőbe (6.2.) vagy egy alkalmas 50 ml-es ledugaszolt lombikba mérjünk ki a mintából 2,00 ±0,001 g-ot.
Megjegyzés: Keverékek esetében a vizsgálati mintából olyan mennyiséget mérjünk ki, hogy a zsírtalanított mintaadag 2,00 g-nak feleljen meg.
8.3. Zsír és fehérje eltávolítása
8.3.1. Adjunk 20,0 ml meleg vizet (50 °C) a vizsgálandó mennyiséghez. Oldjuk fel a port ötperces rázatással, vagy savanyú író esetében harmincperces rázatással mechanikus rázógépen (6.3.). Helyezzük a centrifugacsövet vízfürdőbe (6.10.), és engedjük 25 °C-ra hűlni.
8.3.2. Adjunk hozzá 10,0 ml 25 °C hőmérsékletű triklórecetsav-oldatot (5.1.) folyamatosan két percen át, miközben erőteljesen keverjük a mágneses keverő segítségével (6.4.). Helyezzük a csövet vízfürdőbe (6.10.), és hagyjuk ott 60 percig.
8.3.3. Centrifugáljuk (6.2.) 10 percig 2 200 g-n, vagy szűrjük át szűrőpapíron (6.6.). Öntsük ki az első 5 ml szűrletet.
8.4. Kromatográfiás meghatározás
8.4.1. A HPLC-elemzést a XII. mellékletben leírtak szerint végezzük el. Ha negatív eredményt kapunk, az elemzett minta nem tartalmaz kimutatható mennyiségű oltóssavó-szárazanyagot. Ha az eredmény pozitív, a következőkben meghatározott fordított fázisú HPLC-eljárást kell alkalmazni. A fordított fázisú HPLC-módszer közvetlenül is alkalmazható. Savanyú írópor jelenléte hamis pozitív eredményt adhat a XII. mellékletben leírt módszer használatával. A fordított fázisú HPLC-eljárás kizárja ezt a lehetőséget.
8.4.2. Mielőtt végrehajtanánk a fordított fázisú HPLC-elemzést, a gradienskörülményeket optimalizálni kell. A körülbelül 6 ml holttérfogattal (a térfogat az oldatok összefutási pontjától az injektorhurok térfogatáig, bezárólag) rendelkező gradiensrendszerekhez 26 ± 2 perces CMPA-retenciós idő az ideális. Az ennél kisebb holttérfogattal rendelkező gradiensrendszerekhez (például 2 ml) optimális retenciós időnek 22 percet használjunk.
Vegyük az 50 % oltós savót tartalmazó, illetve nem tartalmazó standard mintákat (5.4.).
Injektáljunk 100 μl felülúszót vagy szűrletet (8.3.3.) a HPLC-készülékbe, amely az 1. táblázat szerint megadott felderítő gradienskörülmények között működik.
1. táblázat
Felderítő gradienskörülmények a kromatográfiás vizsgálatok optimalizálásához
| Idő (perc) | Áramlási sebesség (ml/min) | % A | % B | Görbe |
| Kiindulás | 1,0 | 90 | 10 | * |
| 27 | 1,0 | 60 | 40 | lineáris |
| 32 | 1,0 | 10 | 90 | lineáris |
| 37 | 1,0 | 10 | 90 | lineáris |
| 42 | 1,0 | 90 | 10 | lineáris |
A két kromatogram összehasonlítása megmutatja a CMPΑ-csúcs helyét.
A következőkben meghatározott képlet alkalmazásával kiszámítható, hogy milyen kiindulási oldószer-összetételt kell alkalmazni a normál gradienshez (lásd 8.4.3.). % B = 10 - 2,5 + (13,5 + (RTcmpA - 26) / 6) × 30 / 27 % B = 7,5 + (13,5 + (RTcmpA - 26) / 6) × 1,11
ahol:
RTcmpA : CMPΑ retenciós ideje a felderítő gradiensben
10 : a felderítő gradiens kiindulási % B-je
2,5 : a % B a középpontnál mínusz a % B a kiinduláskor a normál gradiensben
13,5 : a felderítő gradiens fél futamideje
26 : a CMPΑ szükséges retenciós ideje
6 : a felderítő és normál gradiens meredekségeinek aránya
30 : A % B kiinduláskor mínusz a % B 27 percnél a felderítő gradiensben
27 : a felderítő gradiens futamideje.
8.4.3. Vegyünk oldatot a vizsgálati mintákból
Fecskendezzünk 100 μl pontosan kimért felülúszót vagy szűrletet (8.3.3.) a percenként 1,0 ml eluens (5.2.) áramlási sebességgel üzemelő HPLC-készülékbe.
A vizsgálat kezdetén az eluens összetételét a 8.4.2. pont adja meg. Ez rendszerint közelíti az A:B = 76:24 arányt (5.2.). Közvetlenül az injektálás után egy lineáris gradiens indul, amely 27 perc elteltével a B-oldat 5 %-kal magasabb százalékos arányát eredményezi. Ezután elindul egy lineáris gradiens, amely az eluens összetételét öt perc múlva 90 % B arányra alakítja. Ez az összetétel öt percen keresztül fennmarad, ezt követően egy lineáris gradiens mentén öt perc alatt a kiindulási összetételre változik. A szivattyúrendszer belső térfogatától függően a következő injektálást az eredeti állapot visszaállása után 15 perccel lehet elvégezni.
1. megjegyzés: A CMPA retenciós idejének 26 ± 2 percnek kell lennie. Ezt az első gradiens kiindulási és végponti feltételeinek változtatásával lehet elérni. Mindazonáltal a % B különbsége a kiindulási és végponti körülmények között az első gradiensben 5 % B maradjon.
2. megjegyzés: Az eluenseknek kellőképpen gázmenteseknek kell lenniük és így is kell maradniuk. Ez létfontosságú a gradiens-szivattyúrendszer megfelelő működéséhez. A CMPA-csúcs retenciós idejének szórása kisebb kell legyen 0,1 percnél (n = 10).
3. megjegyzés: Minden öt minta után ismét a referenciamintát (5) kell beinjektálni, és segítségével új R-válaszjelet kell meghatározni (9.1.1.).
8.4.4. A vizsgálati minta (E) kromatográfiás elemzésének eredményét egy kromatogram formájában kapjuk meg, ahol a CMPA-csúcsot a körülbelül 26 perces retenciós idő alapján lehet beazonosítani.
Az integrátor (6.11.6.) automatikusan kiszámolja a CMPA-csúcs H-csúcsmagasságát. Az alapvonal helyzetét minden kromatogramon ellenőrizni kell. Ha az alapvonal illesztése nem volt pontos, az elemzést vagy az integrálást ismételni kell.
Megjegyzés: ha a CMPA-csúcs kellőképp elkülönül a többi csúcstól, völgytől-völgyig történő alapvonal-hozzárendelést kell alkalmazni, egyéb esetben merőlegeseket kell egy közös alapvonalra állítani, a kiindulási pont legyen közel a CMPA-csúcshoz (azaz nem t = 0 percnél!). Használjuk ugyanazt az integrálási módszert a standardnál és a mintáknál is, és közös alapvonal esetén ellenőrizzük a konzisztenciáját a minták és a standard esetében is.
Lényeges, hogy még a mennyiségi értelmezés előtt valamennyi kromatogram külalakját megvizsgáljuk, hogy felfedezzünk minden olyan rendellenességet, amely vagy a készülék vagy az oszlopok hibás működéséből ered, vagy a vizsgált minta eredetére és jellegére vezethető vissza. Kétség esetén az elemzést meg kell ismételni.
8.5. Kalibrálás
8.5.1. A standard mintákra (5.4.1. és 5.4.2.) is alkalmazzuk pontosan ugyanazt az eljárást, mint amely a 8.2.-8.4.4. pontig tartó leírásban szerepel. Használjunk frissen készített oldatokat, mert a CMP 8 %-os triklór-ecetsavas környezetben, szobahőmérsékleten hamar bomlik. Az oldat 4 °C-on 24 órán át stabil marad. Hosszú elemzési sorok esetében ajánlatos az automatikus injektorban hűtött mintatálcát használni.
Megjegyzés: A 8.4.2. pont kihagyható, ha a kiindulási feltételre korábbi elemzésekből ismert a % B értéke.
A referenciaminta [5] kromatogramjának az 1. ábrával analógnak kell lennie. Ezen a görbén a CMPA-csúcsot két kisebb csúcs előzni meg. Lényeges ugyanilyen elválasztást elérni.
8.5.2. A minták kromatográfiás meghatározása előtt injektáljunk 100 μl oltós savó nélküli standard mintát [0] (5.4.1.).
Ekkor a kromatogramon a CMPA-csúcs retenciós idejénél csúcs ne jelenjen meg.
8.5.3. Az R-válaszjelet úgy határozzuk meg, hogy a mintáknál használttal azonos mennyiségű szűrletet injektálunk (8.5.1.).
9. AZ EREDMÉNYEK KIFEJEZÉSE
9.1. A számítás módszere és a képletek
9.1.1. Az R-válaszjel kiszámítása:
CMPA-csúcs: R = W/H
ahol:
R = a CMPA-csúcs válaszjele
H = a CMPA-csúcs magassága
W = a savó mennyisége a standard mintában [5].
9.2. A mintában található oltós savópor százalékos mennyiségének kiszámítása
W(E) = R × H(E)
ahol:
W(E) = az oltós savó százalékos (m/m) mennyisége a mintában (E)
R = a CMPA-csúcs válaszjele (9.1.1.)
H(E) = a CMPA-csúcs magassága a mintában (E)
Amennyiben W(E) nagyobb mint 1 %, és a retenciós ideje, valamint a standard minta [5] retenciós ideje közötti különbség kisebb mint 0,2 perc, akkor oltóssavó-szárazanyag van jelen.
9.3. Az eljárás pontossága
9.3.1. Ismételhetőség
Az egy időben vagy rövid időeltéréssel egymás után, ugyanazon laboratóriumi személyzet által, ugyanazzal az eszközzel, azonos vizsgálati anyagon elvégzett két meghatározás különbsége nem haladhatja meg a 0,2 % m/m értéket.
9.3.2. Reprodukálhatóság
Nincs meghatározva.
9.3.3. Linearitás
A 0-16 % oltóssavó-tartalom esetén lineáris összefüggést kell kapni, amelynek korrelációs együtthatója > 0,99.
9.4. Értelmezés
Az 1 %-os határ összhangban van a 214/2001/EK rendelet XIX. mellékletének 9.2. és 9.4.1. pontjában foglalt rendelkezésekkel, és tartalmazza a reprodukálhatóság miatti bizonytalanságot.
Table 1
Ni -4,6 standard
XIV. MELLÉKLET
(10. cikk)
SOVÁNY TEJPOR: FOSZFATIDILSZERIN ÉS FOSZFATIDIL-ETANOLAMIN MENNYISÉGI MEGHATÁROZÁSA
Módszer: fordított fázisú HPLC.
1. CÉL ÉS ALKALMAZÁSI TERÜLET
A módszer a foszfatidilszerin (PS) és a foszfatidil-etanolamin (PE) mennyiségi meghatározására szolgál sovány tejporból (SMP), és alkalmas a sovány tejporban található írószárazanyag kimutatására.
2. FOGALOMMEGHATÁROZÁS
PS + PE tartalom: az itt meghatározott eljárás alapján meghatározott anyag tömegtörtje. Az eredményt 100 g porra megadott foszfatidil-etanolamin-dipalmitoil (PEDP) milligrammjában fejezzük ki.
3. A MÓDSZER ELVE
Aminofoszfolipidek kivonása tejporból készült tejből metil-alkohollal. A PS és a PE meghatározása fordított fázisú (RP) HPLC-vel és fluoreszcenciaészleléssel o-ftáldialdehid-(OPA-) származék formájában történik. A vizsgálati minta PS- és PE-tartalmát egy ismert mennyiségű PEDP-t tartalmazó standarddal való összehasonlítás alapján kapjuk meg.
4. VEGYSZEREK
Az összes vegyszernek analitikai tisztaságúnak kell lennie. A felhasznált víznek egyéb előírás hiányában desztillált víznek, vagy azzal egyenértékű tisztaságú víznek kell lennie.
4.1. Standardanyag: legalább 99 %-os tisztaságú PEDP
Megjegyzés: A standardanyagot - 18 °C-on kell tárolni.
4.2. A standard mintához és a vizsgálati minta előkészítéséhez használt vegyszerek
4.2.1. HPLC-minőségű metil-alkohol
4.2.2. HPLC-minőségű kloroform
4.2.3. Triptamin-monohidroklorid
4.3. Az o-ftáldialdehid-derivátum készítéséhez használt vegyszerek
4.3.1. NaOH, 12 M vizes oldat
4.3.2. Bórsav, 0,4 M vizes oldat NaOH-val 10,0 pH-ra állítva (4.3.1.)
4.3.3. 2-merkapto-etanol
4.3.4. o-ftáldialdehid (OPA)
4.4. HPLC-eluensek
4.4.1. Az eluenseket HPLC-minőségű vegyszerek felhasználásával kell készíteni
4.4.2. HPLC-minőségű víz
4.4.3. Fluorimetrikus tisztaságú metil-alkohol
4.4.4. Tetrahidrofurán
4.4.5. Nátrium-dihidrogén-foszfát
4.4.6. Nátrium-acetát
4.4.7. Ecetsav
5. ESZKÖZÖK
5.1. Analitikai mérleg, 1 mg-os pontosságú mérésre képes, 0,1 mg-os leolvashatósággal
5.2. Főzőpoharak, 25 és 100 ml űrtartalommal
5.3. 1 és 10 ml adagolására alkalmas pipetták
5.4. Mágneses keverő
5.5. 0,2, 0,5 és 5 ml-es adagolásra képes beosztásos pipetták
5.6. 10, 50 és 100 ml űrtartalmú mérőlombikok
5.7. 20 és 100 μl-es fecskendők
5.8. Ultrahangos fürdő
5.9. 27 000 × g-vel működő centrifuga
5.10. Mintegy 5 ml űrtartalmú üvegfiolák
5.11. 25 ml űrtartalmú beosztásos mérőhenger
5.12. pH-mérő, 0,1 pH egységig pontos
5.13. HPLC-berendezés
5.13.1. Gradiens szivattyúrendszer, amely 200 baron 1,0 ml/min sebességgel tud működni
5.13.2. Automata mintavevő derivátumképző lehetőséggel
5.13.3. Oszlopfűtő egység, képes az oszlopot 30 °C ± 1 °C-on tartani
5.13.4. Fluoreszcencia-detektor, képes 330 nm gerjesztési hullámhosszal és 440 nm kibocsátási hullámhosszal működni
5.13.5. A görbe csúcsai alatti terület mérésére alkalmas integrátor vagy adatfeldolgozó szoftver
5.13.6. Egy Lichrosphere - 100 oszlop (250 × 4,6 mm) vagy azzal egyenértékű oszlop, 5 μm-os részecskenagyságú oktadecilszilánnal (C18) töltve
6. MINTAVÉTEL
A mintavételt a 707 ISO-szabvány szerint kell elvégezni.
7. ELJÁRÁS
7.1. Belső standard oldat készítése
7.1.1. Mérjünk ki egy 100 ml-es mérőlombikba (5.6.) 30,0 ±0,1 mg triptamin-monokloridot (4.2.3.), és töltsük fel jelig metil-alkohollal (4.2.1.)
7.1.2. Pipettázzunk 1 ml-t (5.3.) ebből az oldatból egy 10 ml-es mérőlombikba (5.6.), és töltsük fel jelig metil-alkohollal (4.2.1.), hogy elérjük a 0,15 mM-triptaminkoncentrációt
7.2. A vizsgálati minta oldatának elkészítése
7.2.1. Mérjünk ki egy 25 ml-es főzőpohárba (5.2.) 1,000 ± 0,001 g soványtejpor-mintát. Adjunk hozzá pipettával (5.3.) 10 ml 40 °C ± 1 °C-os desztillált vizet, és a mágneses keverőn (5.4.) keverjük 30 percig, hogy minden csomó feloldódjon
7.2.2. Pipettázzunk 0,2 ml (5.5.) tejporból készült tejet egy 10 ml-es mérőlombikba (5.6.), majd a fecskendő használatával (5.7.) adjunk hozzá 100 μl 0,15 mM-os triptaminoldatot (7.1.), és töltsük fel metil-alkohollal a jelig (4.2.1.). Gondosan keverjük el felfordítással, és 15 percen át kezeljük ultrahanggal (5.8.)
7.2.3. Centrifugáljuk (5.9.) 27 000 × g erővel 10 percig, és a felülúszót egy üvegfiolába gyűjtsük össze (5.10.).
Megjegyzés: A vizsgálati mintából készült oldatot a HPLC-vizsgálat elvégzéséig 4 °C-os hőmérsékleten kell tárolni.
7.3. Külső standard oldat készítése
7.3.1. Mérjünk ki 55,4 mg PEDP-t (4.1.) egy 50 ml-es mérőlombikba (5.6.), és adjunk hozzá a beosztásos mérőhenger (5.11.) használatával 25 ml kloroformot (4.2.2.). Melegítsük a ledugaszolt lombikot 50 °C± 1 °C-ra, és gondosan addig keverjük, míg a PEDP fel nem oldódik. Hűtsük vissza a lombikot 20 °C-ra, metil-alkohollal (4.2.1.) töltsük fel jelig, és felfordítással keverjük el
7.3.2. Pipettázzunk (5.3.) ebből az oldatból 1 ml-t egy 100 ml-es mérőlombikba (5.6.), majd jelig töltsük fel metil-alkohollal (4.2.1.). Pipettázzunk (5.3.) ebből az oldatból 1 ml-t egy 10 ml-es mérőlombikba (5.6.), adjunk hozzá 100 μl (5.7.) 0,15 mM-os triptaminoldatot (7.1.), majd jelig töltsük fel metil-alkohollal (4.2.1.). Keverjük el felfordítással
Megjegyzés: A referenciaoldatot a HPLC-elemzés elvégzéséig 4 °C-os hőmérsékleten kell tárolni.
7.4. A deriváló vegyszer elkészítése
Mérjünk ki 25,0 ±0,1 mg OPA-t (4.3.4.) egy 10 ml-es mérőlombikba (5.6.), adjunk hozzá 0,5 ml (5.5.) metil-alkoholt (4.2.1.), és gondosan keverjük el, hogy az OPA feloldódjon. Jelig töltsük fel bórsavoldattal (4.3.2.), és fecskendővel (5.7.) adjunk hozzá 20 μl 2-merkaptoetanolt (4.3.3.).
Megjegyzés: A deriváló reagenst barna üvegfiolában 4 °C-on kell tárolni, körülbelül egy hétig stabil.
7.5. HPLC-meghatározás
7.5.1. Eluáló-oldószerek (4.4.)
A. oldószer: 0,3 mM nátrium-dihidrogén-foszfát és 3 mM nátrium-acetát-oldat (6,5 ± 0,1-es pH-ra ecetsavval beállítva):metil-alkohol:tetrahidrofurán = 558:440:2 (v/v/v) arányban
B. oldószer: metil-alkohol.
7.5.2. Javasolt eluáló gradiens:
| Idő (perc) | A. oldószer (%) | B. oldószer (%) | Áramlási sebesség (ml/min) |
| Kiindulás | 40 | 60 | 0 |
| 0,1 | 40 | 60 | 0,1 |
| 5,0 | 40 | 60 | 0,1 |
| 6,0 | 40 | 60 | 1,0 |
| 6,5 | 40 | 60 | 1,0 |
| 9,0 | 36 | 64 | 1,0 |
| 10,0 | 20 | 80 | 1,0 |
| 11,5 | 16 | 84 | 1,0 |
| 12,0 | 16 | 84 | 1,0 |
| 16,0 | 10 | 90 | 1,0 |
| 19,0 | 0 | 100 | 1,0 |
| 20,0 | 0 | 100 | 1,0 |
| 21,0 | 40 | 60 | 1,0 |
| 29,0 | 40 | 60 | 1,0 |
| 30,0 | 40 | 60 | 0 |
Megjegyzés: Annak érdekében, hogy az 1. ábrán bemutatott elválasztási képességet elérjük, az eluáló gradiens kismértékű megváltoztatására lehet szükség.
Oszlophőmérséklet: 30 °C.
7.5.3. Befecskendezett mennyiség: 50 μl deriválószer és 50 μl mintaoldat
7.5.4. Oszlop kalibrálása
A rendszer napi indításakor az oszlopot 100 %-os B.oldószerrel mossuk át egy negyed órán át, majd váltsunk át A:B = 40:60 arányú keverékre, és 1 ml/min áramlási sebességgel további negyed órán keresztül kalibráljuk. Végezzünk vak tesztet metil-alkohol injektálásával (4.2.1.).
Megjegyzés: Hosszabb idejű tárolás előtt az oszlopot metil-alkohol:kloroform = 80:20 (v/v) arányú keverékével mossuk 30 percen át.
7.5.5. Határozzuk meg a PS + PE tartalmat a vizsgálati mintában
7.5.6. Végezzük el a kromatográfiás elemzés lépéseit a futtatástól-futtatásig terjedő időt állandó értéken tartva, hogy konstans retenciós időket kapjunk. A válaszjel számításához a külső standard oldatot (7.3.) minden 5-10 vizsgálati minta után fecskendezzük be. Megjegyzés: Az oszlopot a 100 %-os B.oldószerrel (7.5.1.) minden 20-25 futtatás után legalább harminc percen át kell mosnunk.
7.6. Integrálás módja
7.6.1. PEDP-csúcs
A PEDP egyetlen csúcsban eluálódik. A csúcs alatti területet völgytől-völgyig integrálással határozzuk meg.
7.6.2. Triptamincsúcs
A triptamin egyetlen csúcsban eluálódik (1. ábra). A csúcs alatti területet völgytől-völgyig integrálással határozzuk meg.
7.6.3. PS- és PE-csúcscsoportok
A leírt feltételek mellett (1. ábra), a PS két fő, részben egymástól el nem váló csúcsban eluálódik ki, amelyet egy kisebb csúcs előz meg. A PE három fő, részben egymástól el nem váló csúcsban eluálódik ki. Határozzuk meg minden csúcscsoport egész területét az alapvonalnak az 1. ábrán bemutatott illesztésével.
8. SZÁMÍTÁS ÉS AZ EREDMÉNYEK KIFEJEZÉSE
A vizsgálati minta PS- és PE-tartalmát a következők szerint lehet kiszámítani: C = 55,36 × ((A2)/(A1)) × ((T1)/(T2))
ahol:
C = a PS- vagy PE-tartalom a vizsgálati mintában (mg/100 g por)
A1 = a standardminta-oldat PEDP-csúcsa alatti terület (7.3.)
A2 = a PS- vagy PE-csúcs alatti terület a vizsgálati mintából készült oldatban (7.2.)
T1 = a standardminta-oldat triptamincsúcsa alatti terület (7.3.)
T2 = a triptamincsúcs alatti terület a vizsgálati mintából készült oldatban (7.2.).
9. A MÓDSZER PONTOSSÁGA
Megjegyzés: Az ismételhetőségre vonatkozó értékek kiszámítása az IDF nemzetközi szabványnak ( 16 ) megfelelően történt. Az ideiglenes reprodukálhatósági határérték kiszámítása az e rendelet III. mellékletének b) pontja szerint történt.
9.1. Ismételhetőség
Az ismételhetőség relatív szórása, amely az ugyanazon személy által, ugyanannak a készüléknek a használatával, ugyanolyan körülmények között, ugyanazon vizsgálati mintából, rövid időeltéréssel kapott független elemzési eredmények variabilitását fejezi ki, nem haladhatja meg a 2 %-os relatív értéket. Ha ilyen körülmények között két meghatározást végeznek el, akkor a két eredmény viszonylagos eltérése nem lehet nagyobb, mint az eredmények számtani középértékének 6 %-a.
9.2. Reprodukálhatóság
Ha két laboratóriumban más készülékeken, más személyek, más feltételek mellett végeznek el két meghatározást ugyanannak a vizsgálati mintának az elemzése céljából, akkor a két eredmény viszonylagos eltérése nem lehet nagyobb, mint az eredmények számtani középértékének 11 %-a.
10. HIVATKOZÁSOK
10.1. Resmini P., Pellegrino L., Hogenboom J.A., Sadini V., Rampilli M., "Detection of buttermilk solids in skimmilk powder by HPLC quantification of aminophospholipids" Sci. Tecn. Latt.-Cas., 39,395 (1988).
XV. MELLÉKLET
(11. cikk)
A SOVÁNY TEJPORBAN TALÁLHATÓ ANTIMIKROBIÁLIS MARADVÁNYANYAGOK KIMUTATÁSA
A baktérium gátlási szűrővizsgálat céljára a Geobacillus stearothermophilus var. calidolactis ATCC 10149 törzsét (azonos a C953 törzzsel) használjuk teszt-mikroorganizmusnak, amely eléggé érzékeny ahhoz, hogy kimutasson tejkilogrammonként 4 μg benzilpenicillint és 100 μg szulfadimidint. A kereskedelemben kapható vizsgálati készletek is használhatók, ha rendelkeznek a kívánt benzilpencillin- és szulfadimidin-érzékenységgel.
A vizsgálatok céljára tejporból készített tejet használjunk (1 g por +9 ml desztillált víz). A vizsgálatot az ISO/TS 26844:2006 Milk and milk products - Determination of antimicrobial residues - Tube diffusion test, 258/1991. számú IDF-Bulletin, 2. fejezet, 1. szakaszában leírtak szerint, vagy a készlet gyártójának utasításait követve végezzük el ( 17 ).
A pozitív eredményeket a következő módon kell értelmezni:
1. A β-laktámok jelenléte megerősíthető a vizsgálat megismétlésével, penicillinázt adva a kísérleti rendszerhez ( 18 ).
Negatív eredmény: A gátlóanyag β-laktám típusú antibiotikum.
Pozitív eredmény marad: Ezzel az eljárással gátlóanyag nem mutatható ki, folytassuk a 2. ponttal.
2. A szulfonamidok jelenléte megerősíthető a vizsgálat megismétlésével, p-amino-benzoesavat adva a kísérleti rendszerhez:
Negatív eredmény: A gátlóanyag szulfonamid.
Pozitív eredmény marad: Ezzel az eljárással gátlóanyag nem mutatható ki, folytassuk a 3. ponttal.
3. A β-laktámok és szulfonamidok kombinációjának jelenléte megerősíthető a vizsgálat megismétlésével, penicillinázt + p-amino-benzoesavat adva a kísérleti rendszerhez.
Negatív eredmény: A gátlóanyag β-laktám típusú antibiotikum és szulfonamid.
Pozitív eredmény: Ezzel az eljárással gátlóanyag nem mutatható ki.
XVI. MELLÉKLET
(12. cikk)
A SOVÁNY TEJPOR MENNYISÉGÉNEK MEGHATÁROZÁSA ÖSSZETETT TAKARMÁNYOKBAN A PARA-KAZEIN ENZIMES KICSAPÁSA ÚTJÁN
1. CÉL
A sovány tejpor mennyiségének meghatározása összetett takarmányokban a para-kazein enzimes kicsapása útján.
2. TÁRGY
Ez a módszer legalább 10 % sovány tejport tartalmazó összetett takarmányokra vonatkozik; nagy mennyiségű író és/vagy bizonyos nem tej eredetű fehérjék jelenléte interferenciához vezethet.
3. A MÓDSZER ELVE
3.1. Az összetett takarmányokban található kazein kioldása nátrium-citrát-oldatos kivonással
3.2. A kalciumion-koncentráció beállítása a kívánt szintre a para-kazein kicsapása érdekében tejoltó hozzáadásával
3.3. A para-kazeines csapadék nitrogéntartalmát az ISO 8968-2:2001IDF 20-2:2001 szabványban meghatározott Kjeldahl-módszerrel állapítjuk meg; a sovány tejpor mennyiségét a minimális 27,5 %-os kazeintartalom alapján lehet kiszámítani (lásd 8.1.).
4. VEGYSZEREK
Minden vegyszernek analitikai minőségűnek kell lennie. A felhasznált víznek desztillált víznek vagy azzal legalább egyenértékű tisztaságú víznek kell lennie. A tejoltó kivételével (4.5.) az összes vegyszernek és oldatnak nitrogénmentesnek kell lennie.
4.1. Trinátrium-citrát, dihidrát (1 % w/v oldat)
4.2. Kalcium-klorid (kb. 5 M-os oldat)
Oldjunk fel 75g CaCl2·2 H2O-t 100 ml desztillált vízben rázással (figyeljünk az exoterm reakcióra). Hagyjuk állni egy éjszakára, majd szűrjük le az oldatot. Az oldatot hűtőszekrényben tároljuk.
4.3. 0,1 N nátrium-hidroxid
4.4. 0,1 N sósav
4.5. Borjúeredetű folyékony tejoltó (oltóerőssége kb. 100 IMCU/ml az ISO 11815IDF 157 szabvány szerint). Hűtőszekrényben 4-6 °C között tartsuk
4.6. A nitrogén mennyiségi meghatározásához használatos vegyszerek az ISO 8968-2:2001IDF 20-2:2001 szabványban leírt Kjeldahl-módszer szerint
5. ESZKÖZÖK
Szokásos laboratóriumi eszközök, beleértve a következőket:
5.1. Dörzsmozsár vagy homogenizátor
5.2. Analitikai mérleg, 1 mg-os pontosságú mérésre képes, 0,1 mg-os leolvashatósággal
5.3. Asztali centrifuga (500 g vagy 2 000 és 3 000 közötti percenkénti fordulatszámmal), 50 ml-es csövekkel és 2 000 g-vel
5.4. Mágneses keverő (10-15 mm) keverőbotokkal
5.5. 150 és 200 ml-es főzőpoharak
5.6. 250 és 500 ml-es lombikok
5.7. 60 és 80 mm átmérő közötti üvegtölcsérek
5.8. Gyorsszűrő hamumentes szűrők 150 mm átmérővel (Whatman N° 41 vagy egyenértékű)
5.9. Különböző névleges térfogatú pipetták
5.10. Termosztatikusan szabályozott vízfürdő 37 °C ± 1° C-on
5.11. pH-mérő, 0,1 pH egységig pontos
5.12. Hőmérők, 1 °C-os pontossággal
6. ELJÁRÁS
6.1. A minta előkészítése
A minta 10-20 grammját őröljük a mozsárban vagy homogenizáljuk a darálóban, amíg homogén keveréket nem kapunk.
6.2. A tejpor feloldása és a feloldhatatlan maradványok eltávolítása
6.2.1. Mérjünk ki 1,000 ± 0,002 g alaposan homogenizált összetett takarmányt (6.1.) közvetlenül egy 50 ml-es centrifugacsőbe. Adjunk hozzá előzetesen 45 °C ± 2 °C-ra melegített 30 ml trinátrium-citrát oldatot (4.1.). A mágneses keverő segítségével keverjük legalább öt percen át vagy rázzuk kézzel erőteljesen.
6.2.2. Centrifugáljuk 500 g-vel (2 000 és 3 000 közötti percenkénti fordulatszámon) 10 percen át, majd öntsük le a tiszta, vizes felülúszót egy 150-200 ml-es főzőpohárba, miközben vigyázunk, hogy a fenéken található laza anyagból semmi ne kerüljön át.
6.2.3. Végezzünk el ugyanezzel az eljárással még két extrakciót a maradékon, és a kivont anyagot öntsük hozzá az elsőhöz.
6.2.4. Ha olajos réteg képződik a felszínen, addig hűtsük a hűtőszekrényben, amíg a zsír meg nem dermed, és egy spatulával vegyük le a szilárd réteget.
6.3. A kazein kicsapása az oltóenzimekkel
6.3.1. Folyamatos kavargatás mellett adjunk hozzá cseppenként 2 ml kalcium-kloridot (4.2.) az összes vizes kivonathoz (mintegy 100 ml). A pH-értéket NaOH- (4.3.) vagy HCl-oldat (4.4.) felhasználásával állítsuk be 6,4-6,5-re. 15-20 percre állítsuk be a termosztatikusan szabályozott, 37 °C ± 1 °C-ra állított vízfürdőbe a sóegyensúly kialakulása érdekében. A sóegyensúly kialakulása enyhe zavarosodás fellépésével válik egyértelművé.
6.3.2. A folyadékot tegyük át egy centrifugacsőbe, majd 10 percen át centrifugáljuk 2 000 g erővel a kicsapódott anyag eltávolítása érdekében. A felülúszót az üledék kimosása nélkül tegyük át egy másik centrifugacsőbe.
6.3.3. A felülúszó hőmérsékletét állítsuk be ismét 37 °C ± 1 °C-ra. A kivonat keverése mellett cseppenként tegyünk bele 0,5 ml folyékony tejoltót (4.5.). A kicsapás két percen belül megtörténik.
6.3.4. A mintát tegyük vissza a vízfürdőbe, és 15 percig hagyjuk ott 37 °C ± 1 °C-os hőmérsékleten. Azután vegyük ki a vízfürdőből, és kavargatással törjük meg az alvadékot. Centrifugáljuk 10 percig 2 000 g-vel. Alkalmas szűrőpapíron (5.8.) szűrjük át a felülúszót, és tartsuk meg a szűrőpapírt. 50 ml, megközelítőleg 35 °C-os vízzel mossuk ki a csapadékot a centrifugacsőben miközben kavargatjuk.
Ismét centrifugáljuk 10 percen át 2 000 g-vel. Szűrjük meg a korábban megőrzött szűrőpapíron a felülúszót.
6.4. A kazein nitrogéntartalmának meghatározása
6.4.1. Mosást követően a csapadékot desztillált víz felhasználásával teljes mennyiségben vigyük át a 6.3.4. pont végrehajtása során megőrzött szűrőpapírra. A szárított szűrőpapírt tegyük bele a Kjeldahl-lombikba. A nitrogéntartalmat a Kjeldahl-módszerrel, az ISO 8968-2:2001IDF 20-2:2001 szabványban leírtak szerint határozzuk meg.
7. VAKPRÓBA
7.1. Rendszeresen vakpróbát kell végezni az ISO 8968-2:2001IDF 20-2:2001 szabványban meghatározott Kjeldahl-módszer szerinti mineralizálással. 90 ml (4.1.) nátrium-citrát-oldat, 2 ml kalcium-klorid-oldat (4.2.), valamint 0,5 ml folyékony tejoltó (4.5.) keverékével megnedvesítünk egy hamumentes szűrőpapírt (5.8.), amelyet ezután 3 × 15 ml desztillált vízzel kimosunk
7.2. A vakpróbához elhasznált sav mennyiségét le kell vonni a minta titrálására használt sav (4.4.) mennyiségéből
8. AZ EREDMÉNYEK KIFEJEZÉSE
8.1. Az összetett takarmányokban jelen lévő sovány tejpor százalékos mennyiségét a következő képlettel számíthatjuk ki:
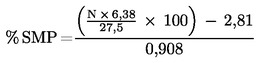
ahol:
N a para-kazein százalékos nitrogéntartalma;
27,5 a meghatározott kazeinnek a sovány tejpor százalékára való átszámítási tényezője;
2,81 és 0,908 a regresszióanalízis során nyert korrekciós tényezők.
9. A MÓDSZER PONTOSSÁGA
9.1. Ismételhetőség
A tanulmányozott esetek legalább 95 %-ában az ugyanazon a mintán, ugyanazon laborszemélyzet által, ugyanabban a laboratóriumban elvégzett kettős elemzések eredményei között megfigyelhető eltérés 100 gramm összetett takarmányra számítva nem lehet nagyobb 2,3 g sovány tejpornál.
9.2. Reprodukálhatóság
A tanulmányozott esetek legalább 95 %-ában az ugyanazon a mintán, különböző laboratóriumban elvégzett elemzések eredményei között megfigyelhető eltérés 100 gramm összetett takarmányra számítva nem lehet nagyobb 6,5 g sovány tejpornál.
10. ÉSZREVÉTELEK
10.1. Bizonyos nem tej eredetű fehérjék és különösen szójafehérjék nagy százalékban való hozzáadása, ha azokat a sovány tejporral együtt hevítik, a tej para-kazeinjával való együttes kicsapódás miatt túlságosan magas eredményeket okozhat.
10.2. Író adagolása némileg alacsonyabb számokat eredményezhet, annak következtében, hogy csak a zsírmentes frakciót határozzuk meg. Bizonyos mennyiségű savanyú író hozzáadása meglehetősen alacsony számértékeket eredményezhet, mert az író a citrátos oldatban nem oldódik fel teljesen.
10.3. A 0,5 % vagy annál nagyobb mennyiségben hozzáadott lecitin szintén alacsonyabb eredményhez vezethet.
10.4. A magas hőmérsékleten kezelt sovány tejpor bekeverése túlságosan magas értékekhez vezethet bizonyos savófehérjék és a tej para-kazeinje közös kicsapódása következtében.
XVII. MELLÉKLET
(13. cikk)
KEMÉNYÍTŐ KIMUTATÁSA SOVÁNY TEJPORBAN, DENATURÁLT TEJPORBAN ÉS ÖSSZETETT TAKARMÁNYOKBAN
1. TÁRGY
Ez a módszer a denaturált tejporokhoz jelölőanyagként adagolt keményítő kimutatására alkalmas.
A módszer kimutatási határa megközelítőleg 0,05 g keményítő a minta 100 grammjában.
2. A MÓDSZER ELVE
A reakció a jodometriában alkalmazott alapelven nyugszik:
- a szabad jód megkötése vizes oldatban kolloidok által,
- abszorbció keményítőszemcsék és elszíneződés által.
3. VEGYSZEREK
3.1. Jódoldat
- jód: 1,0 g,
- kálium-jodid: 2,0 g,
- desztillált víz: 100 ml,
- Oldjunk fel 1,0 g jódot és 2,0 g kálium-jodidot vízben egy 100 ml-es egyjelzésű mérőlombikban. Hígítsuk fel a 100 ml jelig vízzel és keverjük össze.
4. ESZKÖZÖK
4.1. Analitikai mérleg
4.2. Forró vízfürdő
4.3. Kémcsövek, 25 mm × 200 mm
5. ELJÁRÁS
Mérjünk ki 1,0 g mintát 0,1 g-os pontossággal, és tegyük bele a kémcsőbe (4.3.).
Adjunk hozzá 20 ml desztillált vizet, és rázogatással oszlassuk el benne a mintát.
Helyezzük forrásban lévő vízfürdőbe, és hagyjuk ott öt percig (4.2.).
Vegyük ki a vízfürdőből, és hűtsük szobahőmérsékletűre.
Adjunk hozzá 0,5 ml jódoldatot (3.1.), rázzuk fel, és figyeljük meg az eredményül kapott színt.
6. AZ EREDMÉNYEK KIFEJEZÉSE
A kék elszíneződés azt jelzi, hogy a mintában nyers keményítő van jelen.
Ha a minta módosított keményítőt tartalmaz, a szín nem biztos, hogy kék lesz.
7. MEGJEGYZÉS
A keményítő színe, a szín intenzitása és a keményítő mikroszkópos megjelenése a nyers keményítő eredete (például kukorica vagy burgonya), illetve a mintában jelen lévő módosított keményítő típusa szerint változhat.
A módosított keményítők jelenléte a kapott színt ibolyára, vörösre vagy barnára változtathatja, attól függően, hogy a nyers keményítő kristályszerkezete milyen mértékben módosult.
XVIII. MELLÉKLET
(14. cikk)
A SZÁRÍTOTT TEJSZÍN NEDVESSÉGTARTALMÁNAK MEGHATÁROZÁSA
1. TÁRGY
Ez a melléklet a szárított tejszín nedvességtartalmának megállapítására alkalmas módszert határozza meg.
2. KIFEJEZÉSEK ÉS FOGALOMMEGHATÁROZÁSOK
E melléklet alkalmazásában:
Nedvességtartalom: az ebben a nemzetközi szabványban előírt eljárás által meghatározott tömegveszteség.
Tömegszázalékban fejezzük ki.
3. A MÓDSZER ELVE
A vizsgálati adagot 102 ± 2 °C-on tömegállandóságig szárítjuk, és megmérjük a tömegveszteség meghatározásához.
4. ESZKÖZÖK
A szokásos laboratóriumi berendezés, valamint különösen a következők:
4.1. Analitikai mérleg, 1 mg-os pontosságú mérésre képes, 0,1 mg-os leolvashatósággal.
4.2. Jól szellőző szárítószekrény, termosztáttal 102 ± 2 °C tartására képes a teljes munkatérben.
4.3. Exszikkátor, frissen szárított szilikagéllel, higrometrikus indikátorral vagy egyéb hatékony szárítószerrel.
4.4. Lapos aljú edények, kb. 25 mm mélységgel, kb. 50 mm átmérővel és a megfelelő anyagból készítve (például üvegből, rozsdamentes acélból, nikkelből vagy alumíniumból) jól záródó, könnyen levehető fedővel.
4.5. Palackok, szorosan záródó dugóval, a laboratóriumi minták keveréséhez.
5. MINTAVÉTEL
Fontos, hogy a labor valóban reprezentatív vizsgálati mintát kapjon, amely nem sérült vagy nem változott meg a szállítás vagy a tárolás során.
A mintavétel nem képezi az ebben a nemzetközi szabványban leírt módszer részét. Egy ajánlott mintavételi módszer az ISO 707IDF 50 szabványban szerepel.
A mintát úgy tároljuk, hogy elkerüljük az összetétel romlását és változását.
6. A VIZSGÁLATI MINTA ELŐKÉSZÍTÉSE
Alaposan keverjük össze a mintát a tartóedény ismételt rázogatásával és felfordításával (szükség esetén előzőleg tegyük az összes mintát egy elegendő kapacitású légmentesen záró tartóedénybe, hogy el tudjuk végezni ezt a műveletet).
Ha ezzel az eljárással nem érünk el teljes homogenitást, vegyük a vizsgálati adagokat (két egyedi meghatározáshoz) az előkészített vizsgálati minta két lehető legtávolabbi pontjából.
7. ELJÁRÁS
7.1. Az edény előkészítése
7.1.1. Melegítsünk fel egy lefedetlen edényt és a fedelét (4.4.) a 102 ± 2 °C-ra állított szárítószekrényben (4.2.) legalább egy órán át.
7.1.2. A fedőt helyezzük rá az edényre, és a lefedett edényt tegyük bele az exszikkátorba (4.3.), majd hagyjuk a mérőszoba hőmérsékletére hűlni, mérjük le 1 mg pontossággal, és jegyezzük fel a tömeget 0,1 mg pontossággal.
7.2. Vizsgálati adag
Tegyünk az előkészített vizsgálati mintából (6) kb. 1-3 g-ot az edénybe, tegyük rá a fedőt, és mérjük le 1 mg pontossággal, és 0,1 mg-os pontossággal jegyezzük fel a tömeget.
7.3. Meghatározás
7.3.1. Nyissuk ki az edényt, és a fedővel együtt tegyük a 102 ± 2 °C-ra állított szárítószekrénybe (4.2.) legalább 2 órára.
7.3.2. A fedőt helyezzük rá az edényre, és a lefedett edényt tegyük bele az exszikkátorba, majd hagyjuk a mérőszoba hőmérsékletére hűlni, mérjük le 1 mg pontossággal, és jegyezzük fel a tömeget 0,1 mg pontossággal.
7.3.3. Nyissuk fel az edényt, és melegítsük újra a fedelével együtt a szárítószekrényben egy órán át. Ezután ismételjük meg a 7.3.2. műveletet.
7.3.4. A melegítési és mérési eljárást addig ismételjük, amíg a tömeg két egymást követő mérés között 1 mg-gal vagy kevesebbel csökken vagy nő.
A számításhoz a legalacsonyabb feljegyzett tömeget használjuk.
8. SZÁMÍTÁS ÉS AZ EREDMÉNYEK KIFEJEZÉSE
8.1. Kiszámítás
A g/100g-ban kifejezett nedvességtartalom egyenlő:
ahol:
m0 az edény és a fedő tömege grammban (7.1.2);
m1 az edény, a fedő és a vizsgálati adag tömege szárítás előtt grammban (7.2.);
m2 az edény, a fedő és a vizsgálati adag tömege szárítás után grammban (7.3.4.).
Az eredményt két tizedesjeggyel kell megadni.
9. PONTOSSÁG
Megjegyzés: Az ismételhetőségi és reprodukálhatósági értékek egy, a 135B:1991 Tej és tejtermékek - Analitikai módszerek pontossági jellemzői - Vázlat a kollaboratív vizsgálati eljáráshoz IDF-szabvánnyal összhangban elvégzett laboratóriumközi vizsgálat eredményeiből származnak (lásd Steiger, G. Bulletin of IDF No 285/1993, 21-28. o.).
9.1. Ismételhetőség
Ugyanazon módszerrel azonos vizsgálati anyagon, ugyanazon laboratóriumban ugyanazon kezelőszemély által, ugyanazon berendezésen, és a két teszt elvégzése között eltelt rövid idő alatt kapott két független egyedi teszteredmény közötti abszolút különbség az esetek legfeljebb 5 %-ában lesz nagyobb a 0,20 g nedvességnél 100 g termékben.
9.2. Reprodukálhatóság
Ugyanazon módszerrel azonos vizsgálati anyagon, különböző laboratóriumokban különböző kezelőszemélyek által, különböző berendezéseken kapott két független egyedi teszteredmény közötti abszolút különbség az esetek legfeljebb 5 %-ában lesz nagyobb 0,40 g nedvességnél 100 g termékben.
10. VIZSGÁLATI JELENTÉS
A vizsgálati jelentésnek tartalmaznia kell az alábbiakat:
- a minta teljes azonosításához szükséges minden információt,
- a használt mintavételi módszert, ha ismert,
- a használt vizsgálati módszert hivatkozással erre a nemzetközi szabványra,
- minden olyan alkalmazott részletet, amely e nemzetközi szabvány leírásában nem szerepel vagy amely nem kötelező, olyan egyéb körülmények részletezésével, amelyek a vizsgálati eredmény(eke)t befolyásolhatták,
a kapott eredmény(eke)t, és ha ellenőriztük az ismételhetőséget, a kapott végleges eredményt.
XIX. MELLÉKLET
(15. cikk)
SAVANYÚ ÍRÓPOR NEDVESSÉGTARTALMÁNAK MEGHATÁROZÁSA
1. TÁRGY
Eredetileg állati takarmányozásra szánt savanyú írópor nedvességtartalmának meghatározása.
2. A MÓDSZER ELVE
A mintát vákuumban szárítjuk. A tömegveszteséget méréssel állapítjuk meg.
3. ESZKÖZÖK
3.1. Analitikai mérleg, 1 mg-os pontosságú mérésre képes, 0,1 mg-os leolvashatósággal
3.2. Edények rozsdamentes fémből vagy üvegből, légmentes zárást biztosító fedőkkel; a munkafelületnek lehetővé kell tennie a minta 0,3 g/cm2 vastagságban történő eloszlatását
3.3. Állítható, elektromos fűtésű vákuumos szárítószekrény olajszivattyúval felszerelve, és egy mechanizmus, amellyel például kalcium-oxidot vagy kalcium-szulfátot tartalmazó tornyon keresztül szárított forró levegő vezethető be (nedvességjelzővel ellátva)
3.4. Hatékony szárítószerrel ellátott exszikkátor
3.5. Termosztátos szabályozással ellátott, 102 ± 2 °C-ra állított, levegőztetett szárítószekrény
4. ELJÁRÁS
Az egyik edényt (3.2.) fedőjével együtt legalább egy órán keresztül melegítsük a szárítószekrényben (3.5.). A fedőt helyezzük rá az edényre, és azonnal tegyük bele az exszikkátorba (3.4.), majd hagyjuk szobahőmérsékletűre hűlni, mérjük le 1 mg pontossággal és jegyezzük fel a tömeget 0,1 mg pontossággal.
vegyük le az edény fedelét, helyezzünk az edénybe kb. 5 g mintát, mérjük le 1 mg pontossággal, és jegyezzük fel a tömeget 0,1 mg pontossággal. Az edényt fedővel együtt helyezzük a 83 °C-ra előfűtött vákuumos szárítószekrénybe (3.3.). A vákuumos szárítószekrény belső hőmérsékletének nem kívánatos csökkenését elkerülendő, az edényt a lehető leggyorsabban helyezzük bele.
A nyomást állítsuk be 100 Torr-ra (13,3 kPa), hagyjuk állandó tömegig száradni (kb. 4 óra) ezen a nyomáson forró, száraz légáram alatt.
A szárítási időt attól a pillanattól kell számítani, amikor a szárítószekrény visszaáll 83 °C-ra. Óvatosan állítsuk vissza a szárítószekrényben a légköri nyomást. Nyissuk ki a szárítószekrényt, azonnal helyezzük a fedőt az edényre, az edényt távolítsuk el a szárítószekrényből, hagyjuk 30-45 percig hűlni az exszikkátorban (3.4.), és mérjük meg 1 mg-os pontossággal, majd jegyezzük fel a tömeget 0,1 mg-os pontossággal. Szárítsuk még egyszer 30 percig a vákuumos szárítószekrényben (3.3.) 83 °C-on, majd ismét mérjük meg. A melegítési és mérési eljárást addig ismételjük, amíg az edény tömege fedővel együtt két egymást követő mérés között 1 mg-gal vagy kevesebbel csökken vagy nő. A számításhoz a legalacsonyabb feljegyzett tömeget használjuk.
5. KISZÁMÍTÁS
% nedvesség = (m1 - m2) / (m1 - m0) x 100 %
ahol
m0 az edény és a fedő tömege;
m1 az edény, a fedő és a vizsgálati adag tömege szárítás előtt;
m2 az edény, a fedő és a vizsgálati adag tömege szárítás után;
Jegyezzük fel az eredményeket 0,1 g / 100 g-os pontossággal.
6. PONTOSSÁG
6.1. Ismételhetőségi határérték
Ugyanazon módszerrel azonos vizsgálati anyagon, ugyanazon laboratóriumban ugyanazon kezelőszemély által, ugyanazon berendezésen, és a két teszt elvégzése között eltelt rövid idő alatt kapott két független egyedi teszteredmény közötti abszolút különbség az esetek legfeljebb 5 %-ában lesz nagyobb 0,4 g víznél 100 g íróporban.
6.2. Reprodukálhatósági határérték
Ugyanazon módszerrel azonos vizsgálati anyagon, különböző laboratóriumokban különböző kezelőszemélyek által, különböző berendezéseken kapott két független egyedi teszteredmény közötti abszolút különbség az esetek legfeljebb 5 %-ában lesz nagyobb 0,6 g víznél 100 g íróporban.
6.3. A pontossági adatok forrása
A pontossági adatokat egy 1995-ben elvégzett kísérlet szolgáltatta, amelyben 8 laboratórium és 12 minta (hat azonosítatlan minta) vett részt.
XX. MELLÉKLET
(16. cikk)
REFERENCIA-MÓDSZER A TEJZSÍRTISZTASÁG MEGHATÁROZÁSÁRA TRIGLICERIDEK GÁZKROMATOGRÁFIÁS ELEMZÉSÉVEL - 2. ÁTDOLGOZOTT VÁLTOZAT
1. TÁRGY ÉS ALKALMAZÁSI TERÜLET
Ez a szabvány a tejzsírtisztaság trigliceridek gázkromatográfiás elemzésével történő meghatározására szolgáló referencia-módszert határoz meg. Mind növényi, mind állati zsírok, mint a marhafaggyú vagy a sertészsír kimutathatók.
Előre meghatározott trigliceridegyenletek alkalmazásával meghatározható a tejzsír integritása. A módszer alapvetően ömlesztett szarvasmarhatejre, vagy abból készült termékekre alkalmazandó, függetlenül a takarmányozástól, fajtától vagy a laktációs körülményektől. Csak a kivételesen magas arányban tiszta növényi olajjal, mint például repceolajjal történő takarmányozás okozhat hamis pozitív eredményt. Az egyes tehenektől származó tejtermékek is okozhatnak hamis pozitív eredményt.
A módszer különösen alkalmazható a változatlan összetételű tiszta tejzsírt tartalmazó tejtermékekből - úgy mint vaj, tejszín, tej és tejpor - kivont zsírra. A tejzsír technológiai kezelése, például a koleszterin eltávolítása vagy a frakcionálás, hamis pozitív eredményhez vezethet. Ez igaz a sovány tejből vagy íróból nyert tejzsírra is. A módszer nem mindig alkalmazható sajtból kivont zsírra, mert az érési folyamat olyan erősen befolyásolhatja a zsír összetételét, hogy hamis pozitív eredményt kaphatunk.
1. megjegyzés: A vajsav (n-vajsav, C4) kizárólag tejzsírban fordul elő, és lehetővé teszi a növényi és állati zsírokban megtalálható kis és közepes mennyiségű tejzsír mennyiségi becslését. Mivel a C4 tömeghányada igen tág határok között, megközelítőleg 3,1-3,8 % százaléknyi mennyiség között ingadozik, a tiszta tejzsírhoz hozzáadott idegen zsírról - 20 tömegszázaléknyi mennyiségig - nehezen szerezhető minőségi és mennyiségi információ[1].
2. megjegyzés: A növényi zsírok szterintartalmából a gyakorlatban nem lehet mennyiségi eredményeket nyerni, mert ezek a termelési és feldolgozási körülményektől függenek. Ezenkívül a növényi zsírok szterinek használatával történő minőségi meghatározása sem egyértelmű.
2. FOGALOMMEGHATÁROZÁS
Tejzsírtisztaság: a növényi és állati zsírok ebben a szabványban leírt eljárással meghatározott hiánya.
Megjegyzés: A tisztaság meghatározása S-értékek használatával történik, melyeket a triglicerid-összetételből számítunk ki. A triglicerid tömeghányadát százalékban fejezzük ki.
3. A MÓDSZER ELVE
A tejből vagy tejtermékekből nyert zsírt gázkromatográfiával analizáljuk a trigliceridek (TG) meghatározásához töltetes vagy rövid kapilláris oszlopot használva, elkülönítésükhöz a szénatomszámot véve alapul. A különböző méretű zsírmolekulák (C24-C54, csak páros C-számokat használva) százalékban kifejezett tömegtörtjének a megfelelő TG-egyenletekbe való behelyettesítésével S-értékeket számítunk ki. Ha az S-értékek túllépik a tiszta tejzsírral megállapított határértékeket, idegen zsír jelenlétét mutatjuk ki.
1. megjegyzés: Mind a töltetes, mind a kapilláris oszlopok alkalmassága már korábban bebizonyosodott [2-4].
2. megjegyzés: Az S-érték a meghatározott tényezőkkel megszorzott TG-tömegtörtek összege.
4. VEGYSZEREK
Az összes vegyszernek analitikai tisztaságúnak kell lennie.
4.1. Vivőgáz: nitrogén, vagy hélium vagy hidrogén, mind legalább 99,995 %-os tisztaságú
4.2. Zsírstandardok, a 7.3.3. pont szerinti tejzsírstandard beállításához
4.2.1. Triglicerid standardok, telített, megfelelő termékek kereskedelmi forgalomban kaphatók
4.2.2. Koleszterinstandard
4.3. Vízmentes metil-alkohol (CH3OH)
4.4. n-hexán (CH3(CH2)4CH3)
4.5. n-heptán (CH3(CH2)5CH3)
4.6. Egyéb gázok: hidrogén, legalább 99,995 %-os tisztaságú, szerves szennyeződésektől mentes (CnHm < 1 μl/l); szintetikus levegő, szerves szennyeződésektől mentes (CnHm < 1 μl/l)
4.7. Vízmentes nátrium-szulfát (Na2SO4)
5. ESZKÖZÖK
A szokásos laboratóriumi berendezés, valamint különösen a következők:
5.1. Magas hőmérsékleten működő gázkromatográf
A magas hőmérsékleten működő gázkromatográf legyen alkalmas legalább 400 °C-os hőmérséklethez és legyen lángionizációs detektorral (FID) felszerelve. Az injektorban használt szeptumoknak nagy hőmérsékletnek is ellenállónak kell lenniük, és csupán igen kis mértékű "elszivárgást" szabad mutatniuk. A kapilláris gázkromatográfhoz használjunk "on-column" injektort. Mindig használjunk grafittömítéseket az oszlopok csatlakoztatásához, valamint az injektor- és/vagy detektorbetétekhez (adott esetben).
5.2. A kromatográfiás oszlop
5.2.1. Töltetes oszlop
Használjunk 2 mm belső átmérőjű és 500 mm hosszú üvegoszlopot, 3 % OV-1, 125μm-150 μm (100-120 mesh) Gas ChromQ ( 19 ) állófázissal töltve. A töltetes oszlop előkészítése, szilanizálása, töltése és kondicionálása az A. mellékletben szerepel.
Kapilláris oszlop is használható (5.2.2.).
5.2.2. Kapilláris oszlop
Használjunk pl. 5 m hosszúságú rövid kapilláris oszlopot, nem poláris állófázissal, amely ellenálló 400 °C vagy magasabb hőmérséklettel szemben ( 20 ). Kondicionáljuk az oszlopot 20 tejzsíroldat-analízis (7.2.) elvégzésével 2-3 napon belül a 7.3.4.2. pont szerinti beállítások használatával. Ezután a válaszjeleknek 1-hez kell közelíteniük és 1,20-nál kevesebbnek kell lenniük.
Megjegyzés: Eltérő méretekkel és eltérő, apoláris, magas hőmérsékletnek ellenálló fázissal is használhatók oszlopok, ha teljesítményük összhangban van ezzel a szabvánnyal. Lásd még a 7.3.4.2. pontot is.
5.3. Szilikagéllel töltött, 1-3 ml-es Extrelut-oszlop, csak a 7.1.3. szerinti tejzsírkivonáshoz szükséges
5.4. Grafittömítések, legalább 400 °C-os hőmérsékletnek ellenállóak; a GC-oszlop csatlakoztatásához valamint az injektor- és/vagy detektorbetétekhez használandó
5.5. 50 °C ± 2 °C-os hőmérséklet tartására képes vízfürdő
5.6. 50 °C ± 2 °C-on és 100 °C ± 2 °C-on működőképes szárítószekrény
5.7. Mikroliteres pipetta
5.8. 5 ml-es beosztásos pipetta
5.9. 50 ml-es gömblombik
5.10. Erlenmeyer-lombik 250 ml névleges űrtartalommal
5.11. Tölcsér
5.12. Finom-pórusos szűrőpapír
5.13. Forgó bepárló
5.14. 1 ml nominális űrtartalmú ampullák, politetrafluoretilén bevonatú alumínium peremes kupakkal vagy csavaros kupakkal
5.15. Injekciós fecskendő, a fecskendő adagolódugattyúja nem érhet bele a tű csúcsába (töltetes oszlopos gázkromatográf)
Megjegyzés: Ezekkel a fecskendőkkel az eredmények jobb ismételhetősége érhető el.
5.16. Analitikai mérleg, 1 mg-os pontosságú mérésre képes, 0,1 mg-os leolvashatósággal
6. MINTAVÉTEL
A laboratórium számára reprezentatív mintát kell küldeni, amely a szállítás vagy tárolás során nem sérülhet vagy változhat.
A mintavétel nem képezi az ebben a nemzetközi szabványban leírt módszer részét. Egy ajánlott mintavételi módszer az ISO 707IDF 50[5] szabványban szerepel.
7. ELJÁRÁS
7.1. A vizsgálati minták előkészítése
A vizsgálati minta előkészítéséhez az alábbi három tejzsírkivonási módszer egyikét használjuk.
7.1.1. Izoláció vajból vagy vajolajból
Vízfürdő (5.5.) vagy szárítószekrény (5.6.) használatával olvasszunk fel 50-100 g vizsgálati mintát 50 °C-on. Helyezzünk 0,5-1,0g nátrium-szulfátot (4.7.) redőzött papírszűrőre (5.12.). Melegítsünk elő egy 250 ml-es Erlenmeyer-lombikot (5.10.) és egy tölcsért (5.11.) belehelyezett szűrőpapírral az 50 °C-ra állított szárítószekrényben (5.6.). Szűrjük le a megolvadt mintáról a zsírréteget, miközben az előmelegített lombikot, tölcsért és a behelyezett szűrőpapírt a szárítószekrényben tartjuk. Figyeljünk, hogy a szérumból semmi ne kerüljön át.
Kisebb vizsgálati minta csak ebben az esetben használható - az eljárás megfelelő kiigazításával -, ha korlátozott mennyiségű vizsgálati minta áll rendelkezésre. Mindazonáltal a kisebb vizsgálati adagok kezelésekor a nem reprezentatív minta nyerésének kockázata magasabb.
1. megjegyzés: A tejszínből vajat kaphatunk köpüléssel és az ebből eredő vajszemcsék alapos mosásával.
2. megjegyzés: A 7.1.1. szerinti eljárással kapott tejzsír majdnem teljesen mentes a foszfolipidektől.
7.1.2. Kivonás a Röse-Gottlieb gravimetriás módszer szerint
Az ISO 1211IDF 001D, ISO 2450IDF 016C vagy ISO 7328IDF 116A szabványok egyikében leírt gravimetriás módszerrel kivonjuk a zsírfrakciót a vizsgálati mintából.
Megjegyzés: Ha foszfolipidek vannak jelen a nyert tejzsírban, megközelítőleg 0,1 %-kal megnövelt koleszterincsúcsot kapunk. A koleszterinnel 100 %-ra beállított TG-összetételre gyakorolt hatás ezáltal elhanyagolható mértékű.
7.1.3. Kivonás tejből szilikagél oszlopokkal
Egy mikroliteres pipettával (5.7.) adjunk 0,7 ml 20 °C-ra temperált vizsgálati mintát egy 1-3 ml-es Extrelut-oszlopra (5.3.). Hagyjuk, hogy megközelítőleg öt perc alatt egyenletesen eloszoljon a szilikagélen.
A protein-lipid-komplexek denaturálásához adjunk beosztott pipetta (5.8.) használatával 1,5 ml metil-alkoholt (4.3.) az Extrelut-oszlophoz. Ezt követően a vizsgálati mintából 20 ml n-hexánnal (4.4.) kivonjuk a zsírfrakciót. Az n-hexánt lassan, kis mennyiségekben adjuk hozzá. A lecsöpögő oldószert egy 50 ml-es gömblombikban (5.9.) fogjuk fel, amelyet előzetesen ismert, állandó tömegig szárítottunk, amelyet 1 mg-os pontossággal megmértünk, a tömeget 0,1 mg pontossággal feljegyeztük.
A kivonás után hagyjuk az oszlopot üresre csöpögni. Az eluátumból rotációs bepárlóban (5.13.), 40-50 °C hőmérsékletű vízfürdőben kidesztilláljuk az oldószereket. Miután az oldószereket kidesztilláltuk, szárítsuk meg, majd mérjük le a gömblombikot és tartalmát 1 mg pontossággal, feljegyezve a tömeget 0,1 mg pontossággal. Határozzuk meg a zsírtömeget úgy, hogy a kapott tömegből levonjuk a száraz üres gömblombik tömegét.
Megjegyzés: A Gerber-, Weibull-Berntrop- vagy Schmid-Bondzynski-Ratzlaff-módszer szerinti zsírkivonás vagy a tejzsír izolálása detergensekkel (BDI-módszer) nem megfelelő TG-analízisre, mivel jelentős mennyiségű részleges gliceridek vagy foszfolipidek mehetnek át a zsírfázisba. Következésképpen ennek a nemzetközi szabványnak az alkalmazása korlátozott egyes termékek, különösen a sajt vonatkozásában.
7.2. A mintaoldat elkészítése
A töltetes oszlopos gázkromatográfiához készítsük el a (a 7.1. szerint nyert) zsír 5 %-os (térfogatszázalék) oldatát n-hexánban (4.4.) vagy n-heptánban (4.5.). Az oszlop méreteitől függően kapilláris oszlopos "on-column" injektáláshoz legfeljebb 1 %-os koncentrációt (0,53 mm, wide-bore belső átmérő) használjunk.
A használt oszlop és a 7.1.3. szerint kapott zsír tömege alapján határozzuk meg az oldószermennyiséget (4.4. vagy 4.5.), amelyet a vizsgálati mintaanyaghoz kell adnunk a lombikban; ehhez mérjük meg 1 mg pontossággal és jegyezzük fel a tömeget 0,1 mg pontossággal. A maradékot teljesen oldjuk fel.
Töltsünk át körülbelül 1 ml mintaoldatot egy ampullába (5.14.).
7.3. A trigliceridek gázkromatográfiás meghatározása
7.3.1. Az alapvonal eltolódása
Az alapvonal emelkedésének minimalizálása érdekében az oszlopot az 5.2.2. pont szerint (kapilláris oszlop) vagy az A. melléklet A.4. pontja szerint (töltetes oszlop) kell kondicionálni.
Megjegyzés: A magas oszlophőmérséklet miatt a TG-analízis során különösen könnyen fordul elő alapvonal emelkedés a magas szénszámok esetében.
7.3.2. Injektálási technika
7.3.2.1. Töltetes oszlop
A különbséget okozó hatások elkerülésére, a magas forráspontú triglicerid-komponenseknél a mennyiségi meghatározás javítása érdekében a "forró injektálás" módszerét alkalmazzuk. Töltsük meg a tűt levegővel a zsíroldat fecskendőbe való felszívása révén. Helyezzük a tűt az injektorba. Megközelítőleg három másodpercig forrósítjuk a tűt a befecskendezést megelőzően. Ezt követően a fecskendő tartalmát gyorsan beinjektáljuk.
7.3.2.2. Kapilláris oszlop
Ha hideg on-column injektálást (7.3.4.2.) alkalmazunk, helyezzük be a fecskendő tűjét, és azonnal injektáljunk. A tű tartózkodási ideje az injektáló nyílásban annyi legyen, hogy elkerüljük az oldószercsúcsok széles elhúzódását (tailing).
Megjegyzés: Az optimális tartózkodási idő jellemzően kb. 3 másodperc.
7.3.3. Kalibrálás
7.3.3.1. Általános
A vizsgálati minták kalibrálásához a standardizált tejzsírt minden nap kezdetén két-háromszor analizáljuk. A standardizált tejzsír legutolsó vizsgálatát használjuk a TG és a koleszterin RFsi (tömeghányad/területhányad) válaszjeleinek meghatározására, és ezeket alkalmazzuk a következő vizsgálati mintákra (lásd 9.1.):
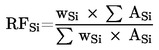
(1) ahol
w si a standardizált tejzsírban lévő egyes trigliceridek vagy a koleszterin tömeghányada százalékban kifejezve;
A si a standardizált tejzsírban lévő egyes trigliceridek vagy a koleszterin csúcsterületének numerikus értéke.
Ismert TG-összetételű standardizált tejzsír nyeréséhez alkalmazzuk a 7.3.3.2. vagy 7.3.3.3. pontokat.
7.3.3.2. Kereskedelmi tejzsírstandard
A vizsgálati minta egyes alkotóelemei válaszjelének legjobb meghatározási módja, ha tanúsított TG-összetételű standardizált tejzsírt használunk.
Megjegyzés: Megfelelő standard a CRM 519 (vízmentes tejzsír), amely a Referenciaanyagok és Mérések Intézetéből (IRMM), a belgiumi Geelből szerezhető be ( 21 ).
7.3.3.3. Laboratóriumi tejzsírstandard
Készítsünk kb. 1 g keveréket a zsírstandardokból (lásd 4.2., tartalmazza legalább a telített C24, C30, C36, C42, C48 és C54 TG-ket, valamint koleszterint, ezen felül lehetőleg C50-t és C52-t is) 1 mg pontosságú méréssel, jegyezzük fel a tömeget 0,1 mg pontossággal, hogy a tejzsírhoz hasonló relatív TG-összetételt kapjunk.
Ismételten elemezzük a zsírstandard-keverék oldatát n-hexánban (4.4.) vagy n-heptánban (4.5.) a 7.3.4. pont szerint. Ugyanebben a sorozatban ismételten elemezzünk átlagos összetételű tejzsírt.
Határozzuk meg a zsírstandardkeverék TG-választjeleit. A keverékben nem jelen lévő közbeeső TG-válaszjeleket matematikai interpolációval számíthatjuk ki. Alkalmazzuk a kapott válaszjeleket a tejzsírra, hogy standardizált összetételt kapjunk. Az így kapott standardizált tejzsír több évig eltartható, ha legfeljebb - 18 °C-on nitrogénben tároljuk.
7.3.4. Kromatografálási feltételek
Megjegyzés: A töltetes vagy kapilláris oszlopok használata általában az 1. ábrához hasonló elválasztást eredményez. A páros számú TG-k felosztása normál esetben nem történik meg, és kerülendő.
7.3.4.1. Töltetes oszlop
a) Hőmérsékletprogram: Állítsuk a szárítószekrény kezdeti hőmérsékletét 210 °C-ra. Tartsuk fenn ezt a hőmérsékletet 1 percig. Ezután 6 °C/min sebességgel emeljük a hőmérsékletet 350 °C-ra. Tartsuk fenn ezt a (végső) hőmérsékletet 5 percig.
b) Detektor- és injektor-hőmérséklet: Állítsuk mindkettőt 370 °C-ra.
c) Vivőgáz: Használjunk nitrogént 40 ml/min állandó áramlási sebességgel. Igazítsuk ki a pontos vivőgázáramlást úgy, hogy a C54 341 °C-on eluálódjon.
d) Az analízis időtartama: 29,3 perc.
e) Injektált térfogat: Injektáljunk 0,5 μl 5 %-os (térfogatfrakció) mintaoldatot.
Ha nem végzünk TG-analízist, tartsuk állandó szinten az a) pontban megadott kezdeti szárítószekrény-hőmérsékletet, a b) pontban megadott detektor- és injektor-hőmérsékletet és a c) pontban megadott vivőgáz áramlási sebességet, éjszaka, hétvégén és munkaszüneti napokon is. Ez biztosítja az oszlop legjobb teljesítményét.
7.3.4.2. Kapilláris oszlop
a) Hőmérsékletprogram: Állítsuk a szárítószekrény kezdeti hőmérsékletét 80 °C-ra. Tartsuk fenn ezt a hőmérsékletet fél percig. Ezután 50 °C/min sebességgel emeljük a hőmérsékletet 190 °C-ra, ezt követően 6 °C/min sebességgel 350 °C-ra. Tartsuk fenn ezt a (végső) hőmérsékletet 5 percig.
b) Detektor hőmérséklete: Állítsuk 370 °C-ra.
c) Vivőgáz: Használjunk nitrogént 3 ml/min állandó áramlási sebességgel.
d) Az analízis időtartama: 34,4 perc.
e) Injektált térfogat: Injektáljunk 0,5 μl 1 %-os (térfogatfrakció) mintaoldatot.
Tartsuk fenn ezeket a beállításokat készenlét alatt is a legjobb teljesítmény biztosítása érdekében (lásd 7.3.4.1.).
A 7.3.4.2. pontban megadott analitikai beállítások alkalmasak az 5.2.2. pontban megadott nagy belső átmérőjű (wide-bore, 0,53 mm) oszlopokhoz is. Eltérő feltételek alkalmazhatók más oszlopméret vagy más fázis használata esetén.
8. INTEGRÁLÁS, ÉRTÉKELÉS ÉS AZ ANALITIKAI TELJESÍTMÉNY ELLENŐRZÉSE
Értékeljük a kromatogram csúcsait alapvonal meghúzására és újraintegrálásra képes integrálórendszerrel. Az 1. ábrán egy helyesen integrált kromatogram látható, míg a 2. ábra sporadikus hibát mutat be az alapvonal végződésében a C54 után, amely befolyásolja az összes TG százalékos arányát. A C54 után következő csúcsokat mindazonáltal kizárjuk az értékelésből.
Páratlan acilált szénatomszámmal rendelkező triglicerideket (2n + 1) az előttük álló páros szénatomszámú trigliceriddel kombináljuk (2n). Ne vegyük figyelembe az alacsony C56-tartalmat. Szorozzuk meg a fennmaradó trigliceridek és a koleszterin területeinek százalékarányait a standardizált tejzsír megfelelő válaszjeleivel (utolsó kalibráció) és normalizáljuk együtt 100 %-ra a 9.1. szerint.
1. ábra
Tejzsír triglicerid-kromatogramja helyesen meghatározott alapvonallal
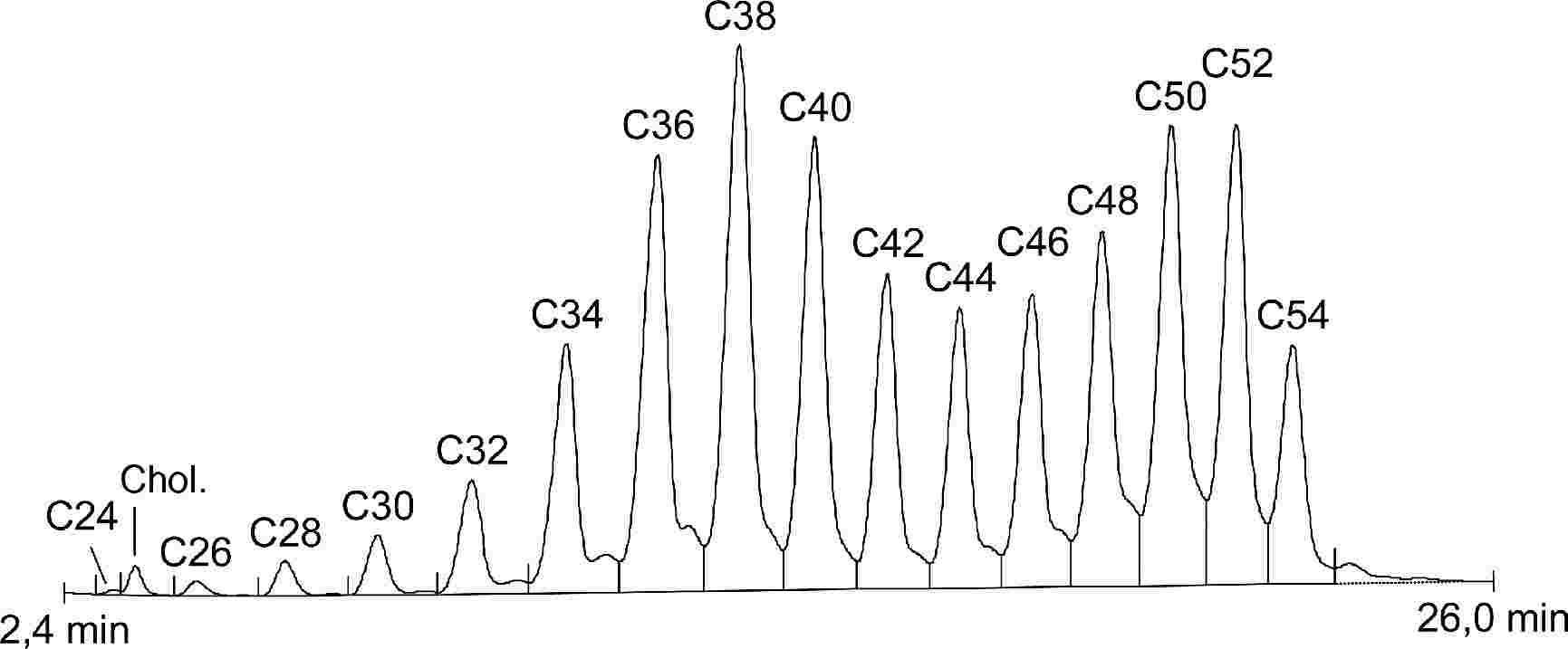
2. ábra
Tejzsír triglicerid-kromatogramja helytelenül meghatározott alapvonallal
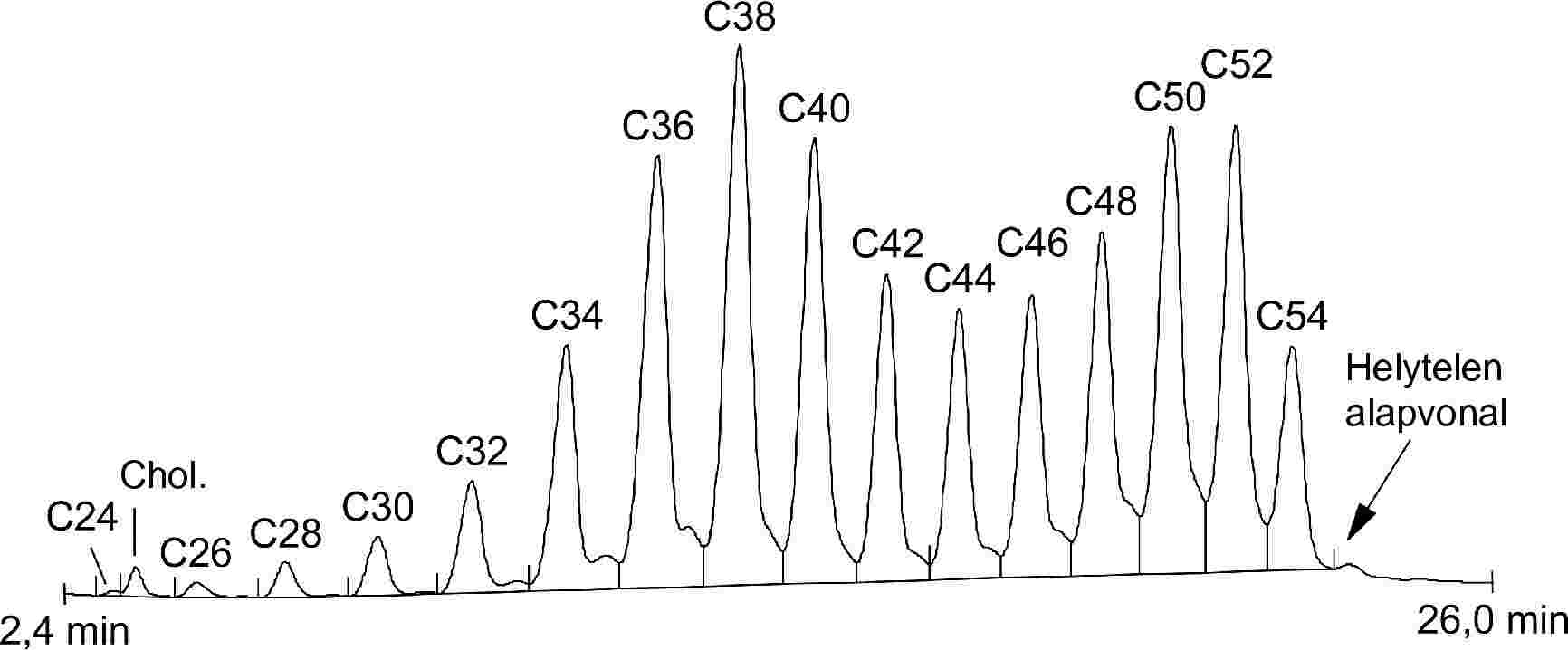
A mérési körülmények ellenőrzése érdekében hasonlítsuk össze az 1. táblázatban a különféle trigliceridekre megadott százalékban kifejezett variációs együtthatókkal (CV), amelyek ugyanannak a tejzsírmintának egymás után elvégzett 19 elemzésén alapulnak.
Ha a variációs együtthatók jóval magasabbak az 1. táblázatban megadott értékeknél, a kromatográfiás feltételek nem megfelelőek.
Megjegyzés: Az 1. táblázatban megadott értékek nem kötelezőek, de minőség-ellenőrzési szempontból jelzésértékűek.
Mindazonáltal, ha magasabb CV-értékeket fogadunk is el, be kell tartani a 10. pontban megadott ismételhetőségi és reprodukálhatósági határértékeket.
1. táblázat
Trigliceridtartalmak variációs együtthatói (19 egymást követő analízis)
| Triglicerid | CV % |
| C24 | 10,00 |
| C26 | 2,69 |
| C28 | 3,03 |
| C30 | 1,76 |
| C32 | 1,03 |
| C34 | 0,79 |
| C36 | 0,25 |
| C38 | 0,42 |
| C40 | 0,20 |
| C42 | 0,26 |
| C44 | 0,34 |
| C46 | 0,37 |
| C48 | 0,53 |
| C50 | 0,38 |
| C52 | 0,54 |
| C54 | 0,60 |
9. SZÁMÍTÁS ÉS AZ EREDMÉNYEK KIFEJEZÉSE
9.1. Triglicerid-összetétel
9.1.1. Kiszámítás
Számítsuk ki az egyes trigliceridek plusz a koleszterin wi tömeghányadát (i = C24, C26, C28, C30, C32, C34, C36, C38, C40, C42, C44, C46, C48, C50, C52, és C54), a vizsgálati minta teljes TG-tartalmának százalékaként kifejezve, a következő egyenlet használatával:
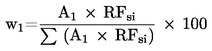
(2) ahol
Ai a vizsgálati mintában lévő egyes trigliceridek csúcsterületének numerikus értéke;
RF si az egyes TG-k kalibrálással (7.3.3.) meghatározott válaszjele.
9.1.2. A vizsgálati eredmények kifejezése
Az eredményeket két tizedesjeggyel kell megadni.
9.2. S-értékek
9.2.1. Kiszámítás
9.2.1.1. A százalékban kifejezett S-értékeket úgy számítjuk ki, hogy a megfelelő TG-százalékarányok kapott wi (9.1.1.) értékeit behelyettesítjük a (3)-(7) egyenletbe. Minden egyenlet használható függetlenül a feltételezett idegen zsír fajtájától.
9.2.1.2. Szójabab-, napraforgó-, olíva-, repcemag-, lenmag-, búzacsíra-, kukoricacsíra-, gyapotmag- és halolaj.
S = 2,098 3 · w C30 + 0,728 8 · w C34 + 0,692 7 · w C36 + 0,635 3 · w C38 + 3,745 2 · w C40 - 1,292 9 · w C42 + 1,354 4 · w C44 + 1,701 3 · w C46 + 2,528 3 · w C50 (3)
9.2.1.3. Kókuszdió és pálmamagzsír.
S = 3,745 3 · w C32 + 1,113 4 · w C36 + 1,364 8 · w C38 + 2,154 4 · w C42 + 0,427 3 · w C44 + 0,580 9 · w C46 + 1,292 6 · w C48 + 1,030 6 · w C50 + 0,995 3 · w C52 + 1,239 6 · w C54 (4)
9.2.1.4. Pálmaolaj és marhafaggyú.
S = 3,664 4 · w C28 + 5,229 7 · w C30 - 12,507 3 · w C32 + 4,428 5 · w C34 - 0,201 0 · w C36 + 1,279 1 · w C38 + 6,743 3 · w C40 - 4,271 4 · w C42 + 6,373 9 · w C46 (5)
9.2.1.5. Sertészsír.
S = 6,512 5 · w C26 + 1,205 2 · w C32 + 1,733 6 · w C34 + 1,755 7 · w C36 + 2,232 5 · w C42 + 2,800 6 · w C46 + 2,543 2 · w C52 + 0,989 2 · w C54 (6)
9.2.1.6. Összesítve.
S = - 2,757 5 · w C26 + 6,407 7 · w C28 + 5,543 7 · w C30 - 15,324 7 · w C32 + 6,260 0 · w C34 + 8,010 8 · w C40 - 5,033 6 · w C42 + 0,635 6 · w C44 + 6,017 1 · w C46 (7)
9.2.2. A vizsgálati eredmények kifejezése
Az eredményeket két tizedessel kell megadni.
9.3. Idegen zsírok kimutatása
Hasonlítsuk össze a 9.2.1. pontban kapott öt S-értéket a 2. táblázatban megadott megfelelő S-határértékekkel.
Akkor tekintjük a vizsgálati mintát tiszta tejzsírnak, ha mind az öt S-érték a 2. táblázatban szereplő határértékeken belül van. Ha azonban bármely S-érték a megfelelő határértéken kívül esik, a mintát idegen zsírt tartalmazónak kell tekinteni.
Bár a (3)-(6) egyedi egyenletek érzékenyebbek bizonyos idegen zsírokra mint a (7) összesített egyenlet (lásd a B.1. táblázatot), a csak a (3)-(6) egyenletek közül egyikkel kapott pozitív eredményből nem vonhatók le következtetések az idegen zsír típusára vonatkozóan.
A B. melléklet leír egy eljárást a növényi vagy állati zsírtartalom kiszámítására a módosított tejzsírban. Ez a módszer nem validált ezért csak tájékoztató jellegű.
2. táblázat
Tiszta tejzsír S-határértékei
| Idegen zsír | Egyenlet | S-határértékek () |
| Szójabab-, napraforgó-, olíva-, repcemag-, lenmag-, búzacsíra-, kukoricacsíra- gyapotmag- és halolaj | (3) | 98,05–101,95 |
| Kókuszdió és pálmamagzsír | (4) | 99,42–100,58 |
| Pálmaolaj és marhafaggyú | (5) | 95,90–104,10 |
| Sertészsír | (6) | 97,96–102,04 |
| Összesítve | (7) | 95,68–104,32 |
| (1) 99 %-os megbízhatósági szinten számítva, így az idegen zsír hozzáadása csak akkor feltételezhető, ha az adott egyenlet kimutatási határértékei túllépésére került sor (lásd a B.1. táblázatot). | ||
10. PONTOSSÁG
10.1. Laboratóriumok közötti vizsgálatok
Az ismételhetőségi és reprodukálhatósági értékek meghatározása a (3)-(7) egyenletek alapján tiszta tejzsír felhasználásával történt, és nem alkalmazható a megadottaktól eltérő mátrixokra.
10.2. Ismételhetőség
Ugyanazon módszerrel azonos vizsgálati anyagon, ugyanazon laboratóriumban ugyanazon kezelőszemély által, ugyanazon berendezésen, és a két teszt elvégzése között eltelt rövid idő alatt kapott két egyedi vizsgálati eredmény közötti abszolút különbség az esetek legfeljebb 5 %-ában lépi túl a 3. táblázatban felsorolt határértékeket.
3. táblázat
Ismételhetőségi határértékek, r, a (3)-(7) egyenlethez
| Idegen zsír | Egyenlet | r % |
| Szójabab-, napraforgó-, olíva-, repcemag-, lenmag-, búzacsíra-, kukoricacsíra-, gyapotmag- és halolaj | (3) | 0,67 |
| Kókuszdió és pálmamagzsír | (4) | 0,12 |
| Pálmaolaj és marhafaggyú | (5) | 1,20 |
| Sertészsír | (6) | 0,58 |
| Összesítve | (7) | 1,49 |
10.3. Reprodukálhatóság
Ugyanazon módszerrel azonos vizsgálati anyagon, különböző laboratóriumban különböző kezelőszemély által, különböző berendezésen kapott két egyedi vizsgálati eredmény közötti abszolút különbség az esetek legfeljebb 5 %-ában lépi túl a 4. táblázatban felsorolt határértékeket.
4. táblázat
Reprodukálhatósági határértékek, R, a (3)-(7) egyenlethez
| Idegen zsír | Egyenlet | R % |
| Szójabab-, napraforgó-, olíva-, repcemag-, lenmag-, búzacsíra-, kukoricacsíra-, gyapotmag- és halolaj | (3) | 1,08 |
| Kókuszdió és pálmamagzsír | (4) | 0,40 |
| Pálmaolaj és marhafaggyú | (5) | 1,81 |
| Sertészsír | (6) | 0,60 |
| Összesítve | (7) | 2,07 |
11. A MÉRÉS BIZONYTALANSÁGA
Az r ismételhetőséggel és az R reprodukálhatósággal kiszámítható egy S-érték kiterjesztett bizonytalansága.
Ha belevesszük a kiterjesztett bizonytalanságot (kettős analízisekre alapozva) a 2. táblázat S-határértékeibe, akkor kiterjesztett S-határértékeket kapunk, amelyek az 5. táblázatban szerepelnek.
5. táblázat
Tiszta tejzsír kiterjesztett bizonytalanságot tartalmazó kiterjesztett S-határértékei
| Idegen zsír | Egyenlet | Kiterjesztett S-határértékek |
| Szójabab, napraforgó, olíva, repcemag, lenmag, búzacsíra, kukoricacsíra, gyapotmag és halolaj | (3) | 97,36–102,64 |
| Kókuszdió és pálmamagzsír | (4) | 99,14–100,86 |
| Pálmaolaj és marhafaggyú | (5) | 94,77–105,23 |
| Sertészsír | (6) | 97,65–102,35 |
| Összesítve | (7) | 94,42–105,58 |
12. VIZSGÁLATI JELENTÉS
A vizsgálati jelentésnek tartalmaznia kell az alábbiakat:
- a minta teljes azonosításához szükséges minden információt,
- a használt mintavételi módszert, ha ismert,
- a használt vizsgálati módszert hivatkozással erre a nemzetközi szabványra,
- minden olyan alkalmazott részletet, amely e nemzetközi szabvány leírásában nem szerepel vagy amely nem kötelező, olyan egyéb körülmény részletezésével, amely a vizsgálati eredmény(eke)t befolyásolhatta,
- a kapott vizsgálati eredmény(eke)t, és ha ellenőriztük az ismételhetőséget, a kapott végleges eredményt.
A. MELLÉKLET
(normatív)
A TÖLTETES OSZLOP ELŐKÉSZÍTÉSE
A.1. VEGYSZEREK ÉS ESZKÖZÖK
A.1.1. Toluol (C6H5CH3)
A.1.2. Dimetil-diklórszilán [Si(CH3)2Cl2] oldat
Oldjunk fel 50 ml dimetil-diklórszilánt 283 ml toluolban (A1.1.)
A.1.3. Kakaóvaj-oldat, tömeghányada 5 % kakaóvaj n-hexánban (4.4) vagy n-heptánban (4.5)
A.1.4. Állófázis, 3 % OV-1, 125-150 μm (100-120 mesh) Gas ChromQ ( 22 )
Megjegyzés: A szemcseméret megjelölése a BS 410 szerint mikrométerre átváltva történt (minden rész)[6]
A.1.5. Üvegoszlop, 2 mm belső átmérővel és 500 mm hosszal, U-alakú
A.1.6. Eszköz a töltetes oszlop megtöltéséhez
A.1.6.1. Töltőoszlop a végére csavarható lezáró kupakkal, a betöltendő állófázis-mennyiséget jelző jellel ellátva
A.1.6.2. Finomszűrő, mesh mérete megközelítőleg 100 μm, csavaros kupakkal, amely alkalmas az üvegoszlop A.3. ábra szerinti lezárására
A.1.6.3. Szilanizált üveggyapot, deaktivált
A.1.6.4. Vibrátor, az állófázis egyenletes eloszlatására a töltés folyamán
A.1.6.5. Szilanizáló eszközök, az oszlop üvegfelületének szilanizálásához
A.1.6.6. Woulff-lombik
A.1.6.7. Vízsugárszivattyú
A.2. SZILANIZÁLÁS (AZ ÜVEGFELÜLET DEAKTIVÁLÁSA)
Miután a Woulff-lombikot (A.1.6.6.) csatlakoztattuk a vízsugárszivattyúhoz (A.1.6.7.), a 2. csövet (lásd az A.1. ábrát) belemerítjük a dimetil-diklórszilán-oldatba (A.1.2.). A zárócsap lezárásával feltöltjük az oszlopot (A.1.5.) az oldattal. Újra kinyitjuk a zárócsapot, majd eltávolítjuk a két csövet. Az oszlopot egy állványon rögzítjük. Egy pipetta segítségével teljesen megtöltjük dimetil-diklórszilán-oldattal (A.1.2.)
A.1. ábra
A szilanizálás eszközei

Jelmagyarázat
1. cső
2. cső
3. vízsugárszivattyú
4. zárócsap
5. üvegoszlop
6. dimetil-diklórszilán és toluol
Hagyjuk az oszlopot állni 20-30 percig. Ezután a Woulff-lombikot egy szűrőlombikkal helyettesítjük. Az oszlopot a vízsugárszivattyúra (A.1.6.7.) csatlakozatva kiürítjük (lásd az A.2. ábrát). Az üres oszlopot egymás után 75 ml toluol (A.1.1.) és 50 ml metil-alkohol (4.3.) felhasználásával kiöblítjük, a 2. cső oldószerbe merítésével. Szárítsuk meg a kiöblített oszlopot a 100 °C-ra állított kemencében (5.6.) megközelítőleg 30 percig.
A.2. ábra
Az öblítés eszközei
Jelmagyarázat
1. cső
2. cső
3. vízsugárszivattyú
4. szűrőlombik
5. üvegoszlop
6. öblítőszer
A.3. TÖLTÉS
Az A.3. ábrán bemutatott eszközök használatával töltsük meg az oszlopot. Töltsük az állófázist (A.1.4.) a töltőoszlopba (A.1.6.1.) a jelzésig. A megtöltendő üvegoszlop alsó végét lezárjuk egy megközelítőleg 1 cm hosszú szilanizált, tömörített üveggyapot dugóval (A.1.6.3.). Zárjuk le az oszlop végét a finomszűrővel (A.1.6.2.).
A.3. ábra
Az üvegoszlop megtöltése
Jelmagyarázat
1. nitrogénbemenet
2. töltőoszlop, a jelzésig feltöltendő OV-1-gyel
3. megtöltendő üvegoszlop
4. csavaros kupak szűrővel, amelyre nyomást fejt ki az üvegszál és az állófázis
Az oszlopot nyomás alatt (300 kPa-s nitrogénáram) megtöltjük az állófázissal. Az egységes, folyamatos és erős kitöltés elérése érdekében a töltés ideje alatt egy vibrátort mozgatunk fel-alá az üvegoszlopon. A töltés befejeztével egy szilárd szilanizált üveggyapot dugót (A.1.6.3.) nyomunk a megtöltött oszlop másik végébe. Levágjuk a kitüremkedő végeket. A dugót egy spatulával néhány milliméterre belenyomjuk az oszlopba.
A.4. KONDICIONÁLÁS
Az a)-c) lépések során a szennyeződések elkerülése végett az oszlop tetejét nem csatlakoztatjuk a detektorhoz. A következő módon kondicionáljuk a töltött oszlopot (A.3.):
a) öblítsük át az oszlopot nitrogénnel 15 percig, 40 ml/min-re állított áramlási sebességgel és 50 °C-ra állított GC-kemencével;
b) melegítsük az oszlopot 1 °C/min sebességgel 355 °C-ra, 10 ml/min nitrogénáramlási sebesség mellett;
c) tartsuk az oszlopot 355 °C-on 12-15 órán át;
d) fecskendezzünk kétszer 1 μl kakaóvaj-oldatot (A.1.3.) a töltetes oszlophoz a 7.3.4.1. pontban megadott hőmérséklet-program használatával;
Megjegyzés: A kakaóvaj majdnem kizárólag magas forráspontú (C50-C54) trigliceridekből áll, így a vonatkozó válaszjelek tekintetében csökkenti az oszlopkondicionálásra fordított erőfeszítést.
e) fecskendezzünk 20-szor 0,5 μl, a 7.2. szerinti tejzsíroldatot 2-3 napon belül a 7.3.4.1. pontban a töltetes oszlopra megadott beállítások alkalmazásával.
- Csak az 1-hez közelítő válaszjellel rendelkező oszlopokat használjuk a vizsgálati minták analíziséhez. A válaszjelek ne legyenek nagyobbak 1,20-nál.
B. MELLÉKLET
(informatív)
IDEGENZSÍR-TARTALOM MENNYISÉGI MEGHATÁROZÁSA
B.1. ÁLTALÁNOS
A B.1. táblázatban a különböző idegen zsírok kimutathatósági határértéke 99 %-os megbízhatósági szinten számítva szerepel. A középső oszlop mutatja a (3)-(6)-ig a legjobb egyedi egyenlet kimutathatósági határértékeit.
A (7) összesített egyenlet jobb szélső oszlopban szereplő kimutathatósági határértékei valamivel magasabbak. Elviekben a (7) egyenlet csak az idegen zsírok mennyiségi kimutatásához szükséges.
Valamennyi egyenlettel ki lehet mutatni a különböző idegen zsírok kombinációit is. Az azonos típusú idegen zsírok egyedi mintái közötti triglicerid-összetétel eltérése nincs jelentős hatással a kimutathatósági határértékekre.
Ha az egyedi egyenleteket és az összesített egyenletet is használjuk, az egyedi egyenletek kimutathatósági határértékei érvényesek. Bizonyos esetekben azonban az összesített egyenlet S-értékére is szükség van a mennyiségi meghatározáshoz (B.2.).
B.1. táblázat
99 %-os kimutathatósági határértékek a tejzsírhoz adott idegen zsír százalékában
| Idegen zsír | Egyedi egyenlet % | Összesített egyenlet % |
| Szójababolaj | 2,1 | 4,4 |
| Napraforgóolaj | 2,3 | 4,8 |
| Olívaolaj | 2,4 | 4,7 |
| Kókuszdióolaj | 3,5 | 4,3 |
| Pálmaolaj | 4,4 | 4,7 |
| Pálmamagzsír | 4,6 | 5,9 |
| Repceolaj | 2,0 | 4,4 |
| Lenmagolaj | 2,0 | 4,0 |
| Búzacsíraolaj | 2,7 | 6,4 |
| Kukoricacsíra-olaj | 2,2 | 4,5 |
| Gyapotmagolaj | 3,3 | 4,4 |
| Sertészsír | 2,7 | 4,7 |
| Marhafaggyú | 5,2 | 5,4 |
| Hidrogénezett halolaj | 5,4 | 6,1 |
B.2. SZÁMÍTÁS
Csak legalább egy S-határérték (2. táblázat vagy 5. táblázat) túllépésekor végezzünk mennyiségi idegen zsír meghatározást. Mennyiségi információ nyeréséhez a vizsgálati mintában az idegen zsír vagy idegenzsír-keverék w f, százalékban kifejezett tömeghányadának kiszámításához a következő egyenletet használjuk fel:
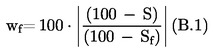
ahol
S úgy kapott eredmény, hogy a (3)-(7) egyenletek egyikébe behelyettesítjük az olyan tejzsír triglicerid adatait, amelyhez idegen zsírt vagy idegenzsír-keveréket adnak hozzá,
S f a hozzáadott idegen zsír típusától függő konstans.
Ha nem ismert a tejzsírhoz adott idegen zsír típusa, használjuk az általános 7,46 S f-értéket (B.2. táblázat). Mindig a (7) egyenletből kapott S-értéket használjuk, még ha az S-határértékei túllépése nem is történt meg, de más egyenleté igen.
Ismert idegen zsírok esetén helyettesítsük be az egyéni S f-értékeiket (B.2. táblázat) a (B.1.) egyenletbe. Válasszuk ki a vonatkozó idegenzsír-egyenletet a (3)-(6) egyenletek közül az S kiszámításához.
B.2. táblázat
Sf-értékek a különféle idegen zsíroknál
| Idegen zsír | Sf |
| Ismeretlen | 7,46 |
| Szójababolaj | 8,18 |
| Napraforgóolaj | 9,43 |
| Olívaolaj | 12,75 |
| Kókuszdióolaj | 118,13 |
| Pálmaolaj | 7,55 |
| Pálmamagolaj | 112,32 |
| Repceolaj | 3,30 |
| Lenmagolaj | 4,44 |
| Búzacsíraolaj | 27,45 |
| Kukoricacsíra-olaj | 9,29 |
| Gyapotmagolaj | 41,18 |
| Sertészsír | 177,55 |
| Marhafaggyú | 17,56 |
| Halolaj | 64,12 |
B.3. A VIZSGÁLATI EREDMÉNYEK KIFEJEZÉSE
A vizsgálati eredményeket két tizedesjeggyel kell megadni.
Bibliográfia
(1) Molkentin, J., Precht, D. Representative determination of the butyric acid content in European milk fats. Milchwissenschaft, 52, 1987, pp. 82-85
(2) Precht, D., Molkentin, J. Quantitative triglyceride analysis using short capillary columns. Chrompack News, 4, 1993, pp. 16-17
(3) Molkentin, J., Precht, D. Comparison of packed and capillary columns for quantitative gas chromatography of triglycerides in milk fat. Chromatographia, 39, 1994, pp. 265-270
(4) Molkentin, J., Precht, D. Equivalence of packed and capillary GC columns with respect to suitability for foreign fat detection in butter using the triglyceride formula method. Chromatographia, 52, 2000, pp. 791-797
(5) ISO 707IDF 50, Milk and milk products - Guidance on sampling
(6) BS 410:1988, Test sieves - Technical requirements and testing
(7) Precht, D. Control of milk fat purity by gas chromatographic triglyceride analysis. Kieler Milchwirtsch. Forschungsber., 43, 1991, pp. 219-242
(8) Precht, D. Detection of adulterated milk fat by fatty acid and triglyceride analyses. Fat Sci. Technol., 93, 1991, pp. 538-544
(9) DIN 10336:1994, Nachweis und Bestimmung von Fremdfetten in Milchfett anhand einer gaschromatographischen Triglyceridanalyse [Detection and determination of foreign fats in milk fat using a gas chromatographic triglyceride analysis]
(10) Commission of the European Communities: Consideration of results from the first, second, third, fourth, fifth and sixth EEC collaborative trial: Determination of triglycerides in milk fat; Doc. No VI/2644/91, VI/8.11.91, VI/1919/92, VI 3842/92. VI/5317/92, VI/4604/93
(11) Molkentin, J. Detection of foreign fat in milk fat from different continents by triacylglycerol analysis. Eur. J. Lipid Sci. Technol., 109, 2007, pp. 505-510
XXI. MELLÉKLET
(18. cikk)
VALAMELY ELEMZÉS EREDMÉNYEINEK VITATOTTSÁGA ESETÉN ALKALMAZANDÓ ELJÁRÁS (VEGYI ELEMZÉS)
1. A gyártó kérésére még egy elemzést el lehet végezni a vonatkozó módszer használatával, az illetékes hatóság által jóváhagyott másik laboratóriumban, feltéve hogy a termékből leplombált ellenminták állnak rendelkezésre, és az illetékes hatóság azokat az előírásoknak megfelelően tárolta. A kérést az első elemzés eredményeiről történt értesítést követő hét munkanapon belül meg kell tenni. A vizsgálatot a kérés beérkezését követően 21 munkanapon belül el kell végezni. Az illetékes hatóság ezeket a mintákat a gyártó kérésére és költségén egy másik laboratóriumba küldi. Ennek a laboratóriumnak hivatalos elemzések végzésére vonatkozó engedéllyel, és a kérdéses elemzéssel kapcsolatosan bizonyított szakértelemmel kell rendelkeznie.
2. Az 1. laboratóriumban az
megismételt mérés
átlagának és a 2. laboratóriumban az
megismételt mérés
átlagának (
k
= 2) kiterjesztett bizonytalansága
KÉP HIÁNYZIK
és
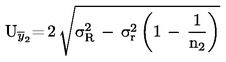
ahol
az adott módszer ismételhetőségi szórása és
a reprodukálhatósági szórása. Ha az
y
laboratóriumi mérési végeredményt az
,
,
vagy
formájú képletek alkalmazásával számítjuk ki, a szórások kombinálásának szokásos eljárását kell ilyen esetekben követni, hogy megkapjuk a bizonytalanságot.
4. Annak vizsgálata érdekében, hogy a két laboratórium eredményei megfelelnek-e a módszer
reprodukálhatósági szórásának, ki kell számítani a
különbség kiterjesztett bizonytalanságát:
KÉP HIÁNYZIK
Ha a laboratóriumi átlagok különbségének abszolút értéke,
nem nagyobb a
bizonytalanságánál,
a két laboratórium eredményei megfelelnek a
reprodukálhatósági szórásnak, és a két laboratóriumi átlag számtani közepe:
tekinthető végeredménynek. Kiterjesztett bizonytalansága:
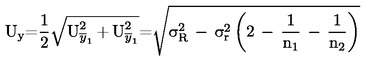
A szállítmány akkor utasítható vissza az UL előírt felső határértéknek nem megfelelőként, ha
egyéb esetben az UL-nek megfelelőként el kell fogadni.
A szállítmány akkor utasítható vissza az LL előírt alsó határértéknek nem megfelelőként, ha
egyéb esetben az LL-nek megfelelőként el kell fogadni.
Ha a laboratóriumi átlagok különbségének abszolút értéke,
nagyobb a
bizonytalanságánál,
a két laboratórium eredményei nem felelnek meg a reprodukálhatósági szórásnak.
Ebben az esetben a szállítmányt nem megfelelőként vissza kell utasítani, ha a második analízis megerősíti az elsőt. Egyéb esetben a szállítmányt megfelelőként el kell fogadni.
A végeredményről az illetékes hatóság köteles haladéktalanul értesíteni a gyártót. Amennyiben a szállítmányt elutasítják, a második elemzés költségeit a gyártó viseli.
XXII. MELLÉKLET
MEGFELELÉSI TÁBLÁZAT
| 213/2001/EK rendelet | Ez a rendelet |
| 1. cikk | 1. cikk |
| 2. cikk | 1. cikk |
| 3. cikk | 2. cikk |
| — | 3. cikk |
| 4. cikk | — |
| 5. cikk | — |
| 6. cikk | 4. cikk |
| 7. cikk | 18. cikk |
| 8. cikk | — |
| 9. cikk | 5. cikk |
| 10. cikk | 6. cikk |
| 11. cikk | 7. cikk |
| 12. cikk | 8. cikk |
| 13. cikk | 9. cikk |
| 14. cikk | 10. cikk |
| 15. cikk | 11. cikk |
| 16. cikk | 12. cikk |
| 17. cikk | 13. cikk |
| — | 14. cikk |
| 18. cikk | 15. cikk |
| 19. cikk | 16. cikk |
| 17. cikk | |
| 19. cikk | |
| 20. cikk | — |
| 21. cikk | — |
| 22. cikk | 20. cikk |
| 23. cikk | 21. cikk |
( 1 ) HL L 160., 1999.6.26., 48. o. A legutóbb az 1152/2007/EK rendelettel (HL L 258., 2007.10.4., 3. o.) módosított rendelet. 2008. július 1-jétől a 1255/1999/EK rendelet helyébe az 1234/2007/EK rendelet (HL L 299., 2007.11.16., 1. o.) lép.
( 2 ) HL L 37., 2001.2.7., 1. o.
( 3 ) HL L 308., 2005.11.25., 1. o. A legutóbb az 1546/2007/EK rendelettel (HL L 337., 2007.12.21., 68. o.) módosított rendelet.
( 4 ) HL L 279., 1990.10.11., 22. o. A legutóbb az 1487/2006/EK rendelettel (HL L 278., 2006.10.10., 8. o.) módosított rendelet.
( 5 ) HL L 256., 1987.9.7., 1. o. A legutóbb az 1352/2007/EK bizottsági rendelettel (HL L 303., 2007.11.21., 3. o.) módosított rendelet.
( 6 ) HL L 165., 2004.4.30., 1. o.
( 7 ) HL L 228., 2009.9.1., 3. o.
( 8 ) = FAME-módszer.
( 9 ) Az Ampholine® pH 3,5-9,5-es (Pharmacia) és a Resolyte® pH 5-7, valamint pH 6-8-as (BDH, Merck) termékek különösen alkalmasnak bizonyultak a γ -kazeinek kívánt szeparációjának elérésére.
( 10 ) J. Koops en C. Olieman, Netherlands Milk and Dairy Journal, 39 (1985) 89-106.
( 11 ) D.A. Biggs en L. Szijarto, Journal of Dairy Science, 46 (1963) 1196.
( 12 ) ISO 707 (IDF 50), Tej és tejtermékek - Mintavételi módszerek.
( 13 ) ISO 5725-1, Mérési módszerek és eredmények pontossága (valódiság és precizitás). 1. rész: Általános elvek és meghatározások.
( 14 ) ISO 5725-2, Mérési módszerek és eredmények pontossága (valódiság és precizitás). 2. rész: Alapmódszer egy mértékadó mérési módszer ismételhetőségének és reprodukálhatóságának meghatározására.
( 15 ) Szabványos összetételű oltóssavópor és a módosított sovány tejpor is beszerezhető a NIZO-tól, Kernhemseweg 2, PO Box 20, NL-6710 BA Ede. Azonban a NIZO-porokkal egyenértékű eredményeket nyújtó porok is felhasználhatók.
( 16 ) IDF 135B/1991 nemzetközi szabvány. Tej és tejtermékek. Analitikai módszerek pontossági jellemzői. Vázlat a kollaboratív vizsgálati eljáráshoz.
( 17 ) Fontos figyelmeztetés: hamis pozitív eredményt kaphatunk, ha sovány tejpor elemzését végezzük. Ezért igen fontos meggyőződnünk arról, hogy a használt vizsgálati rendszer nem ad hamis pozitív eredményt.
( 18 ) Néhány β-laktám kevésbé érzékeny a β-laktamázokra. Ilyen esetekben a minta további előmelegítése ajánlott (1 ml vizsgálati minta 0,3 ml penázkoncentrátummal 37 °C-on két órán át).
( 19 ) Példa a kereskedelemben kapható megfelelő termékre. Ez a tájékoztatás csupán a nemzetközi szabvány felhasználóinak kényelmét szolgálja, és semmiképpen nem jelenti e termék ajánlását.
( 20 ) A CP-Ultimetal SimDist (5 m x 0,53 mm x 0,17 μm) példa egy kereskedelemben kapható alkalmas termékre. Ez a tájékoztatás csupán a nemzetközi szabvány felhasználóinak kényelmét szolgálja, és semmiképpen nem jelenti e termék ajánlását.
( 21 ) Példa a kereskedelemben kapható megfelelő termékre. Ez a tájékoztatás csupán a nemzetközi szabvány felhasználóinak kényelmét szolgálja, és semmiképpen nem jelenti e termék ajánlását.
( 22 ) Példa a kereskedelemben kapható megfelelő termékre. Ez a tájékoztatás csupán a nemzetközi szabvány felhasználóinak kényelmét szolgálja, és semmiképpen nem jelenti e termék ISO vagy IDF általi ajánlását.
Lábjegyzetek:
[1] A dokumentum eredetije megtekinthető CELEX: 32008R0273 - https://eur-lex.europa.eu/legal-content/HU/ALL/?uri=CELEX:32008R0273&locale=hu Utolsó elérhető, magyar nyelvű konszolidált változat CELEX: 02008R0273-20130701 - https://eur-lex.europa.eu/legal-content/HU/ALL/?uri=CELEX:02008R0273-20130701&locale=hu