
8/2001. (I. 26.) FVM rendelet
a termésnövelő anyagok engedélyezéséről, tárolásáról, forgalmazásáról és felhasználásáról
A növényvédelemről szóló 2000. évi XXXV. törvény (a továbbiakban: Tv.) 65. §-a (3) bekezdésének b) pontjában foglalt felhatalmazás alapján - az egészségügyi miniszterrel és a környezetvédelmi miniszterrel egyetértésben - a következőket rendelem el:
Értelmező rendelkezések
1. §
A rendelet alkalmazásában:
1. Műtrágya: a növények tápanyagellátását szolgáló, iparilag, kémiai úton előállított termésnövelő anyag.
2.[1] Engedélyköteles szerves trágya: a növények tápanyagellátását, illetve a talaj szerkezetének javítását szolgáló, növényi vagy állati eredetű szerves anyagból iparilag feldolgozott, meghatározott beltartalmú termésnövelő anyag.
3. Ásványi trágya: a növények tápanyagellátását, illetve a talaj szerkezetének javítását szolgáló, ásványi eredetű, iparilag előállított termésnövelő anyag.
4. Komposzt: a növények tápanyagellátásának, illetve a talaj tápanyagszolgáltató képességének javítására szolgáló, szerves, szervetlen és ásványi eredetű anyagokból komposztálás útján előállított termésnövelő anyag.
5. Gilisztahumusz: a növények tápanyagellátását javító, illetve a talaj termőképességének növelését befolyásoló, gilisztaürülék komposztálása során keletkező termésnövelő anyag.
6. Talajjavító anyag: a talaj kedvezőtlen tulajdonságainak megváltoztatására, illetve a kedvező tulajdonságok fenntartására szolgáló, iparilag előállított termésnövelő anyag.
7. Talajkondicionáló készítmény: a talaj fizikai, kémiai, illetve biológiai tulajdonságaira kedvezően ható, iparilag előállított termésnövelő anyag.
8.[2] Mikrobiológiai készítmény: a talaj termékenységét javító mikroszervezeteket (baktériumokat, gombákat, algákat) tartalmazó termésnövelő anyag, amely mentes az emberre fertőzőképes és a talaj természetes mikroflóráját kedvezőtlenül befolyásoló szervezetektől.
9. Növényvédő szernek nem minősülő növekedés-szabályozó készítmény: növényi eredetű vagy szintetikusan előállított, növényi hormont tartalmazó termésnövelő anyag.
10. Termékcsalád: termésnövelő anyagok, termesztő közegek és növénykondicionáló szerek azonos gyártó által, azonos halmazállapotban, azonos felhasználási céllal gyártott kombinációi.
11.[3] EC jelölésű műtrágya: az Európai Unió tagállamai között, külön engedélyezési eljárás nélkül, szabadon forgalmazható műtrágya.
Engedélyezési eljárás
2. §
(1) A termésnövelő anyagok, termesztő közegek és növénykondicionáló szerek (a továbbiakban együtt: termésnövelő anyag) forgalomba hozatalát és felhasználását a Földművelésügyi és Vidékfejlesztési Minisztérium (a továbbiakban: minisztérium) engedélyezi.
(2) A termésnövelő anyagok termékcsaládként is engedélyezhetőek. ,
(3) A növényvédő szert tartalmazó termésnövelő anyagokra a növényvédő szerek engedélyezésére vonatkozó külön jogszabályban foglaltakat is alkalmazni kell.
3. §
(1) A termésnövelő anyag forgalomba hozatali és felhasználási engedélye (a továbbiakban: engedély) iránti kérelmet az 1. számú melléklet szerint, 6 példányban kell a minisztériumhoz benyújtani.
(2) A kérelmezőnek az engedély iránti kérelemmel együtt a 2. számú mellékletben meghatározott vizsgálati eredményeket és adatokat kell benyújtania.
(3)[4] A 2. számú mellékletben előírt vizsgálatokat az ezen vizsgálatokra akkreditált magyarországi laboratórium végezhet. A mikroelemek és toxikus elemek vizsgálatában alkalmazott módszer adott elemre vonatkozó kimutatási határa legfeljebb a bejelentett érték vagy a határérték 10%-a lehet.
(4)[5] A 3. számú melléklet 1. számú függeléke szerinti műtrágyák vizsgálatait a 7. számú melléklet szerinti módszerekkel, erre akkreditált magyarországi laboratóriumnak kell végeznie.
(5)[6] A magas nitrogéntartalmú ammónium-nitrát műtrágyáknak a 8. számú mellékletben meghatározott vizsgálati módszerek szerinti vizsgálatát az erre akkreditált laboratórium végezheti.
(6)[7] A külön jogszabály szerint veszélyes hulladéknak minősülő összetevőt tartalmazó termésnövelő anyagnál az engedély iránti kérelemmel együtt be kell nyújtani a veszélytelenség megállapításáról szóló határozatot is.
(7)[8] Termékcsalád engedélyezése esetén a 2. számú mellékletben előírt fizikai, kémiai és mikrobiológiai minősítő vizsgálatokat, valamint a toxikus elemek és a csírázásgátló hatás vizsgálatát valamennyi termék esetében el kell végeztetni. A 2. számú mellékletben felsorolt további vizsgálatokat a termékek típusától függően, a közegészségügyi és környezeti kockázatot jelentő összetevőket a legnagyobb arányban tartalmazó termék(ek)nél kell elvégeztetni.
(8)[9] Azokból a változó összetételű műtrágya termékcsaládokból, amelyek csak makro- vagy mezoelemeket tartalmaznak, legalább három, amelyek mikroelemeket is tartalmaznak, legalább öt, a minisztérium által kijelölt jellemző összetételű termék-2. számú melléklet szerinti-fizikai, kémiai és toxikus elem vizsgálatát kell elvégeztetni.
(9)[10] A minisztérium által már engedélyezett termésnöve-10 anyagokból keveréssel előállított termesztőközegek és műtrágyák esetében, elegendő egy termék-2. számú melléklet szerinti - vizsgálata.
(10)[11] Az engedélyezési eljárás során az engedélyező hatóság kiegészítő adatszolgáltatást és további vizsgálatok elvégzését rendelheti el.
4. §
(1)[12] A minisztérium az engedélyezési eljárás során beszerzi a Környezetvédelmi és Vízügyi Minisztérium (a továbbiakban: KvVM) és a Fodor József Országos Közegészségügyi Központ (a továbbiakban: OKK) szakhatósági állásfoglalását.
(2)[13] A szakhatósági állásfoglalás a következőket tartalmazza:
a) a KvVM szakhatósági állásfoglalása kiterjed a termésnövelő anyag felhasználásának, tárolásának, csomagolásának környezet- és természetvédelmi (a továbbiakban együtt: környezetvédelem) szempontból biztonságos megvalósítására, továbbá a hulladékká vált, illetve lejárt szavatosságú készítmény, a maradékanyag, valamint a termésnövelő anyaggal szennyezett csomagolóeszköz kezelésére vonatkozó környezetvédelmi feltételekre, termékcsaládok esetében a legnagyobb környezeti kockázatot jelentő összetevő megnevezésére,
b) az OKK szakhatósági állásfoglalása tartalmazza a készítmény tárolásának, felhasználásának környezet-egészségügyi feltételrendszerét, valamint a kémiai biztonságról szóló törvényben foglaltak szerint a készítmény veszélyességi besorolását, a veszély jeleket, a kockázatra, valamint a biztonságra utaló mondatokat, a készítmény környezet-egészségügyi megítélését, az előkészítéshez és a télhasználáshoz előírt védőeszközöket és az elsősegélynyújtási eljárást; termékcsaládok esetében a legnagyobb közegészségügyi kockázatot jelentő összetevő megnevezését.
(3) A Növény- és Talajvédelmi Központi Szolgálat (a továbbiakban: Központi Szolgálat) a kérelem elbírálása során szakértőként jár el.
Az engedély formája és tartalma
5. §
(1) Az engedély akkor adható meg, ha a termésnövelő anyag megfelel
a) a kérelemben megjelölt felhasználási célnak,
b) a Tv. 37. §-ának a) pontjában foglaltaknak,
c) a 3. számú mellékletben terméktípusonként meghatározott minőségi előírásoknak.
(2) Termésnövelő anyagként nem engedélyezhető a mezőgazdasági művelés alól kivont terület talaja, illetve az ezt tartalmazó termésnövelő anyag.
(3) Az engedélyt a minisztérium adja ki, amelyről engedélyokiratot állít ki.
(4)[14] Az engedélyokirat
a) eredeti példányát az engedélyesnek,
b) egy-egy másolati példányt az engedélyezésben részt vevő szakhatóságoknak, a Központi Szolgálatnak, a megyei (fővárosi) növény- és talajvédelmi szolgálatoknak (a továbbiakban: Szolgálat), továbbá
c) az engedélyezett import műtrágyák adatait informatikai úton a Vám- és Pénzügyőrség Országos Parancsnokságának
kell megküldeni.
(5) Az engedélynek a következő adatokat kell tartalmaznia:
a) a termésnövelő anyag kereskedelmi nevét,
b) a termésnövelő anyag típusát,
c) a gyártó nevét, címét és statisztikai számjelét,
d) az engedélyes nevét, címét és statisztikai számjelét,
e) a készítmény teljes összetételét, a 3. számú mellékletben előírt minőségi és mennyiségi jellemzőket,
f) a felhasználási előírást,
g) a munka-egészségügyi előírásokat, a veszélyességi besorolást, a veszély jeleket, a kockázatra (R) és biztonságra (S) utaló mondatokat, a környezet-egészségügyi előírásokat, az alkalmazás során előírt védőeszközöket, az elsősegélynyújtási eljárást,
h) a környezetvédelmi előírásokat,
i) a termésnövelő anyag tűzveszélyességi osztályba sorolását,
j) a tárolás, a csomagolás, az eltarthatóság jelölését,
k) az engedély érvényességi idejét (év, hó, nap),
l) a csomagolóeszközön, illetve a kísérőokmányon a 4. számú melléklet szerint kötelezően feltüntetendő adatokat,
m)[15] a termésnövelő anyag - kérelmező által megadott - vámtarifaszámát hat számjegyig,
n) a lejárt szavatossági idejű termésnövelő anyag kezelésére vonatkozó előírásokat,
o) a minisztérium által előírt egyéb előírásokat.
(6) Az engedélyes engedélyokiratának megsemmisülése esetén a minisztériumtól kérheti az engedély másodlatának kiadását.
Az engedély ismételt megadása
6. §
A termésnövelő anyag forgalomba hozatalára és felhasználására vonatkozó engedély ismételt megadását a 3. §-ban foglaltak szerint kell kérni. Az engedély ismételten megadható, ha a készítmény megfelel az 5. § (1) bekezdésében előírt feltételeknek.
Az engedély módosítása, szüneteltetése
7. §
(1) Az engedély módosítását az engedélyes kérheti. A módosítási kérelemhez csatolni kell azokat az adatokat, mellékleteket és mintákat, amelyek megalapozzák az engedély módosítását. A kérelmet a minisztérium a szakhatóságok bevonásával bírálja el.
(2) A módosítást a minisztérium a szakhatósági állásfoglalások beszerzése nélkül kiadja,
a) ha a módosítás iránti kérelem az 5. § (5) bekezdésének a), c) és d) pontjára vonatkozik,
b) termékcsalád bővítése esetén, ha az új összetételű termék legnagyobb közegészségügyi és környezeti kockázatot jelentő összetevőjének koncentrációja kisebb a termékcsaládban engedélyezett legmagasabb értéknél, és nem tartalmaz új alapanyagot.
(3) A minisztérium és az engedélyezési eljárásban közreműködő szakhatóságok hivatalból kezdeményezhetik az engedély módosítását a tudományos eredmények vagy az igazolt gyakorlati tapasztalatok alapján.
(4) Az engedélyt az engedélyes kérelmére vagy hivatalból lehet szüneteltetni. A minisztérium hivatalból rendeli el az engedély szüneteltetését
a) az engedélyesnek a termésnövelő anyag forgalomba hozatalához való jogosultságával kapcsolatos per esetén, annak jogerős befejezéséig, illetve megszűnéséig,
b)[16] a termékállandóság vizsgálatakor és a forgalmazás ellenőrzésekor megállapított hiányosságok, minőségi kifogások esetén azok megszüntetéséig.
(5) Az engedély módosítása, illetve szüneteltetése az engedély érvényességi idejét nem hosszabbítja meg.
Az engedélyezéshez benyújtott iratok kezelése
8. §
Az engedély iránti kérelemben foglalt adatok, a kérelemmel együtt benyújtott vizsgálati eredmények és adatok kezelésére a személyes adatok védelméről és a közérdekű adatok nyilvánosságáról szóló külön jogszabály, valamint a Tv. 22. §-ának rendelkezései irányadóak.
A termésnövelő anyagok tárolása
9. §
(1) A tárolás során meg kell akadályozni a termésnövelő anyag hatóanyag tartalmának csökkenését, fizikai és kémiai tulajdonságainak romlását, és meg. kell tartani a környezetvédelmi előírásokat.
(2) Termésnövelő anyag raktározására szolgáló tároló kizárólag olyan helyen létesíthető, ahol a talajvíz az év egyetlen szakában sem emelkedik a tárlószint fölé, és a csapadékvíz elvezetése biztosított oly módon, hogy a tárolt készítménnyel ne érintkezzen.
(3) Szilárd műtrágya kizárólag olyan fedett, szilárd aljzatburkolatú helyen tárolható, ami védett az ár- és belvíz veszélyeztetettségétől, ahol biztosított, hogy a tárolt anyag nem okoz sem közvetlen, sem közvetett bevezetést a felszín alatti vízbe, valamint a kiszóródott anyagok összegyűjtése megoldható.
(4)[17] Különböző hatóanyag tartalmú ömlesztett műtrágyákat egy tároló térben elkülönítetten kell elhelyezni, beazonosítható módon, jelzőtáblán, nem vízoldható feliraton feltüntetve az adott helyen tárolt ömlesztett műtrágya nevét, az engedélyokiratban szereplő hatóanyag megnevezését és tartalmát, gyártóját és az elhelyezés dátumát.
(5)[18] Folyékony műtrágya kizárólag olyan csurgás- és csepegésmentes zárt tartályban tárolható, amelyben a tartály anyaga és a műtrágya között nincs kémiai kölcsönhatás. A tartálynak a túltöltés megakadályozását biztosító műszaki felszereltséggel kell rendelkeznie. Hőtágulás lehetősége miatt a tartályok teletöltése tilos. A tartályon nem vízoldható feliraton fel kell tüntetni a tartályban tárolt folyékony műtrágya nevét, engedélyokiratban szereplő hatóanyag megnevezését, tartalmát, összetételét, gyártóját és a betöltés dátumát.
(6) A felszín alatti vizek minőségét érintő tevékenységekkel összefüggő egyes feladatokról szóló jogszabályban meghatározott. kockázatos anyagot tartalmazó termésnövelő anyag tárolása során be kell tartani a jogszabályban előírt környezetvédelmi követelményeket.
(7) A termésnövelő anyagok tárolására szolgáló létesítmények kialakítására és engedélyezésére a külön jogszabályokban foglalt rendelkezéseket kell alkalmazni.
A termésnövelő anyagok csomagolása és forgalmazása
10. §
(1) A termésnövelő anyag csomagolóeszközének meg kell akadályozni a tartalom kiszóródását, kiszivárgását, illetve kiömlését. A csomagolóeszköznek és a záróelemeknek sérülés nélkül meg kell felelnie a megfelelő anyagmozgatás és bármilyen más, a felhasználás során történő rendeltetésszerű igénybevétel követelményeinek.
(2) A termésnövelő anyag csomagolóeszköze külső megjelenésének olyannak kell lennie, hogy más anyaggal (gyógyszer, élelmiszer, háztartási vegyi anyag) a termésnövelő anyagot ne lehessen összetéveszteni.
(3)[19] A csomagolóeszköz megsérülése miatti átcsomagolás során az eredeti csomagolásra vonatkozó előírásokat kell alkalmazni. Az átcsomagolt, valamint a kis egységbe kiszerelt termésnövelő anyag felhasználhatósági ideje az átcsomagolás, illetve a kiszerelés következtében nem térhet el a termék eredeti felhasználhatósági idejétől.
(4)[20] Az engedélyezett termésnövelő anyag csomagolóeszközén, valamint a 100 kg nettó tömeget meghaladó áru és az ömlesztett áru kísérőokmányán a 4. számú mellékletben felsorolt adatokat magyar nyelven is fel kell tüntetni. A kötelezően feltüntetendő valamennyi adatot egyértelműen el kell különíteni a csomagolóeszközön, címkén és a kísérő okmányon feltüntetett minden egyéb információtól.
11. §
(1) A termésnövelő anyagot az engedélyben megjelölt kereskedelmi névvel kell forgalomba hozni. Az engedélyben foglalt kereskedelmi nevet védjegy vagy fantázianév nem helyettesítheti.
(2) A magas nitrogén tartalmú- 28 tömeg%-nál magasabb nitrogén tartalmú - ammónium nitrát műtrágya kizárólag csomagoltan hozható kiskereskedelmi forgalomba.
(3) A Tv. 38. §-ának (2) bekezdésében előírt bejelentési kötelezettségnek az 5. számú melléklet szerinti nyomtatványon kell eleget tenni.
A termésnövelő anyaggal folytatott tevékenység ellenőrzése[21]
12. §
(1)[22] A termésnövelő anyaggal folytatott bármely tevékenységet az illetékes Szolgálat ellenőrzi, ennek során vizsgálja, különösen
a) a forgalmazott készítmény engedélyének meglétét,
b) a 4. számú mellékletben előírtak feltüntetését a címkén, illetve a kísérő okmányokon,
c) a helyszínen megállapítható minőségi kifogásokat (küllem, csomagolástechnika, a csomagolóeszköz szennyezettsége, tárolási körülmények),
d) a gyártási és eltarthatósági időt.
(2)[23] Az ellenőrzés során a növényvédelmi felügyelő, illetve a talajvédelmi felügyelő a készítmény adott tételére forgalomba hozatali, felhasználási korlátozást, illetve tilalmat rendelhet el, ha a termésnövelő anyag
a) engedéllyel nem rendelkezik,
b) jelölése hiányos vagy a vonatkozó előírásoknak nem felel meg,
c) küllemi, csomagolástechnikai, tárolási szempontból minőségi kifogás alá esik,
d) lejárt szavatossági idejű.
(3) A termékállandóság vizsgálatához az illetékes Szolgálat a gyártónál, a kiszerelőnél, a kereskedőnél és a felhasználónál térítésmentesen mintát vehet. A mintavétel helyén ellenmintát kell hagyni.
(4) A Szolgálat végzi a minták vizsgálatát, amely az engedélyben előírt értékekre terjed ki. A minisztérium egyéb adatok ellenőrző vizsgálatát is elrendelheti.
(5)[24] A Szolgálat igazgatója a vizsgált tételt zárolja, ha a toxikus elem és a szerves szennyezőanyag-tartalom akár egy paraméter vonatkozásában is meghaladja a 3. számú mellékletben meghatározott határértéket. A zárolt tétel tulajdonosa, illetve forgalmazója - a zárlati határozatban előírt határidőre - köteles visszaszállítani a tételt a forgalmazói láncban előtte álló forgalmazóhoz, illetve a gyártóhoz, aki köteles azt átvenni.
(6) Az engedélyben előírt hatóanyag tartalom csökkenése esetén a Szolgálat igazgatója a forgalomba hozott termésnövelő anyag használati érték csökkenésének jelölésére kötelezi a forgalmazót.
13. §
(1) Az ellenőrzés és a vizsgálat kérelemre is lefolytatható, amelyért a külön jogszabályban meghatározott vizsgálati díjat kell fizetni.
(2) A vizsgálati eredmény kizárólag az átadott mintára és nem az egész tételre vonatkozik, ha a mintát nem az ellenőrzésre kijelölt személy vette. Kereskedelmi forgalomban lévő termék esetében a Szolgálat hivatalból további eljárást folytat le, ha a termék eltér az engedélyben megadott jellemzőktől.
14. §
(1) Lejárt szavatosságú készítmény a minőség ellenőrzése nélkül nem hozható forgalomba és nem használható fel. A vizsgálatot a forgalmazónak, illetve a felhasználónak kell elvégeztetnie. Amennyiben a minőség-ellenőrzési vizsgálatok eredményei alapján a készítmény eredeti rendeltetésének megfelelően nem használható fel, a késítményt hulladéknak kell tekinteni.
(2) Az azonosíthatatlanná vált termék kezelésére a veszélyes hulladékra vonatkozó jogszabály rendelkezéseit kell alkalmazni.
A termésnövelő anyagok felhasználása
15. §
(1) Termésnövelő anyag kizárólag az engedélyben foglalt feltételek szerint használható fel.
(2) Termésnövelő anyag ivóvízbázis védőterületén való felhasználásakor a vízbázisok, a távlati vízbázisok, valamint az ivóvízellátást szolgáló vízilétesítmények védelméről szóló külön jogszabályban előírt korlátozásokat is figyelembe kell venni.
(3) A termésnövelő anyagok légi úton történő kijuttatására a növényvédelmi tevékenységről szóló jogszabály rendelkezéseit kell alkalmazni.
(4) Nitrogén tartalmú termésnövelő anyag felhasználása során be kell tartani a külön jogszabályban előírt követelményeket.
(5) A termésnövelő anyagokat csak olyan módon és mennyiségben lehet felhasználni, hogy a talajok külön jogszabály szerinti kockázatos anyagtartalma ne haladja meg tartós használat esetén sem a felszín alatti víz és a földtani közeg minőségi védelméhez szükséges határértékekről szóló 10/2000. (VI. 2.) KöM-EüM-FVM-KHVM együttes rendeletben meghatározott "B" szennyezettségi határértéket, valamint a felszín alatti vizek állapota a termésnövelő anyagok felhasználása következtében ne romoljon.
Kísérleti felhasználás
16. §
(1) Termésnövelő anyag kísérleti felhasználása iránti kérelmet a 6. számú melléklet szerint a minisztériumhoz kell benyújtani.
(2) Engedélyezési célból termésnövelő anyaggal kísérletet kizárólag szakirányú felsőfokú végzettséggel rendelkező személy végezhet.
(3) A kísérleti terület méretét a kérelem elbírálása során a minisztérium határozza meg.
(4) A kísérletet a Szolgálat ellenőrzi.
Záró rendelkezések
17. §[25]
(1) Ez a rendelet 2001. február 1-jén lép hatályba.
(2)[26] A 2003. május 15. előtt kiadott forgalomba hozatali és felhasználási engedéllyel rendelkező termésnövelő anyagokat 2003. július 1-jétől a forgalomba hozatali és felhasználási engedélyokirat (3) bekezdés szerinti felülvizsgálatáig e rendeletben meghatározott toxikus elem határértékeknek és hatóanyag tolerancia értékeknek megfelelően kell gyártani. A 2003. június 30-ig az engedélyokirat szerinti toxikus elem határértékeknek és hatóanyag tolerancia értékeknek megfelelően legyártott termékek 2004. június 30-ig hozhatók forgalomba.
(3)[27] A 2003. május 15. előtt kiadott forgalomba hozatali és felhasználási engedélyokiratokat a toxikus elem határértékek és hatóanyag tolerancia értékek vonatkozásában 2004. június 30-ig az engedélyező hatóságnak felül kell vizsgálnia.
(4)[28] Az EC jelölésű műtrágyák esetében a 2. számú melléklet 1.2. pontját és a 3. számú melléklet 1.2. pontját a Magyar Köztársaságnak az Európai Unióhoz történő csatlakozásáról szóló nemzetközi szerződést kihirdető törvény hatálybalépésétől nem kell alkalmazni.
18. §[29]
Ez a rendelet a Magyar Köztársaság és az Európai Közösségek és azok tagállamai között társulás létesítéséről szóló, Brüsszelben, 1991. december 16-án aláírt Európai Megállapodás tárgykörében, a Megállapodást kihirdető 1994. évi I. törvény 3. §-ával összhangban összeegyeztethető szabályozást tartalmaz a következő közösségi jogszabályokkal:
a) a Tanács 76/116/EGK irányelve a műtrágyákra vonatkozó tagállami jogszabályok közelítéséről,
b) a Bizottság 79/138/EGK, 87/566/EGK, 89/519/EGK, 93/1/EGK és 95/8/EK irányelvével módosított 77/535/EGK irányelve a műtrágyák mintavételi és vizsgálati módszereire vonatkozó tagállami jogszabályok közelítéséről,
c) a Tanács 80/876/EGK irányelve a magas nitrogéntartalmú, tiszta ammónium-nitrát műtrágyákra vonatkozótagállami jogszabályok közelítéséről,
d) a Tanács 87/94/EGK irányelve a magas nitrogéntartalmú, tiszta ammónium-nitrát műtrágyák jellemzőinek, határértékeinek és robbanás ellenálló képességének ellenőrzésére irányuló eljárásokra vonatkozó tagállami jogszabályok közelítéséről.
Dr. Torgyán József s. k.,
földművelésügyi és vidékfejlesztési miniszter
1. számú melléklet a 8/2001. (I. 26.) FVM rendelethez
Forgalomba hozatali és felhasználási engedély iránti kérelem
A 2000. évi XXXV. törvény 38. §-a, valamint a 8/2001. (l. 26.) FVM rendelet 3. §-a alapján kérelmet nyújtok be a ................................ kereskedelmi nevű termék forgalomba hozatali és felhasználási engedélyének megadásához.
1. A termésnövelő anyag kereskedelmi neve.
2. A termésnövelő anyag típusa, az 1. számú melléklet függeléke szerint.
3. A gyártó neve, címe és statisztikai számjele.
4. A kérelmező neve, címe és statisztikai számjele.
5. A készítmény teljes összetétele, hatóanyagok, alapanyagok, szennyező anyagok megnevezése és koncentrációja.
6. A gyártó által szavatolt minőségi jellemzők.
7. A gyártó által megadott felhasználási lehetőségek és alkalmazási dózisok.
8. Tűzveszélyességi osztályba sorolás.
9. A gyártó által megadott tárolási és eltarthatósági feltételek.
10. A gyártó által forgalmazni kívánt kiszerelési egység, a csomagolóeszköz anyaga, címketerv.
11. A termésnövelő anyag vámtarifaszáma hat számjegyig.
12. Ha a termésnövelő anyag a veszélyes áruk szállításáról szóló (ADR/RID) szabályzatok hatálya alá esik, a termék UN száma és ADR/RID osztálya.
13. Bányászott termékek esetén a bányanyitási engedély.
14. A termésnövelő anyag gyártástechnológiájának leírása.
A kérelemhez mellékelt dokumentumok:
1. A készítmény vizsgálati eredményei.
2. A készítménybiztonsági adatlapja, a kémiai biztonságról szóló törvény előírásai szerint.
3. Az engedélyezési eljárási díj befizetésének igazolása.
4. A veszélyes hulladéknak minősülő összetevőt, illetve szennyvíziszapot tartalmazó termésnövelő anyagnál a veszélytelenség megállapításáról szóló határozat.
5. A külön jogszabály szerint kockázatos anyagot tartalmazó termésnövelő anyagok esetén annak bemutatása, hogyan viselkednek a kockázatos anyagok a talajban, milyen hatással vannak a felszín alatti vizekre.
1. számú melléklet függeléke
A termésnövelő anyagok típusai
1. MŰTRÁGYÁK
A. EGYSZERŰ MŰTRÁGYÁK
A/1. Nitrogén műtrágyák
- Kalcium-nitrát
- Kalcium-magnézium-nitrát
- Magnézium-nitrát
- Nátrium-nitrát
- Chilei salétrom
- Kalcium-ciánamid
- Nitrát tartalmú kalcium-ciánamid
- Ammónium-szulfát
- Ammónium-nitrát vagy kalcium-ammónium-nitrát
- Ammónium szulfát-nitrát
- Magnézium-szulfo-nitrát
- Magnézium-ammónium-nitrát
- Karbamid
- Krotonilidén-dikarbamid
- Izobutilidén-dikarbamid
- Karbamid-formaldehid
- Krotonilidén-dikarbamid tartalmú nitrogén műtrágya
- Izobutilidén-dikarbamid tartalmú nitrogén műtrágya
- Karbamid-formaldehid tartalmú nitrogén műtrágya
- Ammónium-szulfát nitrifikáció gátlóval (dicián-
diamid)
- Ammónium-szulfo-nitrát nitrifikáció gátlóval (di-
ciándiamid)
- Karbamid-ammónium-szulfát
A/2. Foszfor műtrágyák
- -Bázikus salak
- Thomas foszfátok
= Thomas salak
= Szuperfoszfát
- Koncentrált szuperfoszfát
- Tripleszuperfoszfát
- Részlegesen feltárt foszfátérc
- Dikalcium-foszfát
- Kalcinált foszfát
- Alumínium-kalcium-foszfát
- Őrölt lágy foszfátérc
A/3. Kálium műtrágyák
- Kainit
- Dúsított kainit só
- Kálium-klorid
- Magnéziumsó tartalmú kálium-klorid
- Kálium-szulfát
- Magnéziumsó tartalmú kálium-szulfát
- Kieserit kálium szulfáttal
B. ÖSSZETETT MŰTRÁGYÁK
- NPK műtrágyák
- NP műtrágyák
- NK műtrágyák
- PK műtrágyák
- NPK műtrágyák krotonilidén-dikarbamid, izobutili-[30]
dén-dikarbamid vagy karbamid-formaldehid tartalommal
- NP műtrágyák krotonilidén-dikarbamid, izobutilidén-dikarbamid vagy karbamid-formaldehid tartalommal[31]
- NK műtrágyák krotonilidén-dikarbamid, izobutilidén-dikarbamid vagy karbamid-formaldehid tartalommal[32]
C. FOLYÉKONY MŰTRÁGYÁK
C/1. Egyszerű folyékony műtrágyák
- Nitrogén műtrágya oldat
- Ammónium-nitrát-karbamid műtrágya oldat
- Kalcium-nitrát oldat
- Magnézium-nitrát oldat
- Kalcium-nitrát szuszpenzió
- Karbamid-formaldehid tartalmú nitrogén műtrágya
oldat
- Karbamid-formaldehid tartalmú nitrogén műtrágya
szuszpenzió
C/2. Összetett folyékony műtrágyák
- NPK műtrágya oldat
- NPK műtrágya szuszpenzió
- NP műtrágya oldat
- NP műtrágya szuszpenzió.
- NK műtrágya oldat
- NK műtrágya szuszpenzió
- PK műtrágya oldat
- PK műtrágya szuszpenzió
D. MEZOELEM TARTALMÚ MŰTRÁGYÁK
- Kalcium-szulfát
- Kalcium-klorid oldat
- Elemi kén
- Kieserit
- Magnézium-szulfát
- Magnézium-szulfát oldat
- Magnézium-hidroxid
- Magnézium-hidroxid szuszpenzió
- Magnézium-klorid oldat
- Kalcium-nitrát oldat
E. MIKROELEM TARTALMÚ MŰTRÁGYÁK
E/1. Egyetlen mikroelemet tartalmazó műtrágyák
- Bór
= Bórsav
= Nátrium-borát
= Kalcium-borát
= Bór-etanol-amin
= Bór alapú műtrágya oldat
= Bór alapú műtrágya szuszpenzió
- Kobalt.
= Kobalt só
= Kobalt kelát
= Kobalt műtrágya oldat
- Réz
= Réz só
= Réz-oxid
= Réz-hidroxid
= Réz-kelát
= Réz alapú műtrágya
= Réz oldat műtrágya
= Réz-oxiklorid
= Réz-oxiklorid szuszpenzió
- Vas
= Vas só
= Vas kelát
= Vas műtrágya oldat
- Mangán
= Mangán só
= Mangán kelát
= Mangán-oxid
= Mangán alapú műtrágya
= Mangán alapú műtrágya oldat
- Molibdén
= Nátrium-molibdenát
= Ammónium-molibdenát
= Molibdén alapú műtrágya
= Molibdén alapú műtrágya oldat
- Cink
= Cink só
= Cink kelát
= Cink-oxid
= Cink alapú műtrágya
= Cink alapú műtrágya oldat
E/2. Összetett mikroelem tartalmú műtrágyák
- Szilárd és folyékony összetett mikroelem tartalmú műtrágyák
- Talajtrágyázásra szolgáló fő és/vagy másodlagos tápelemeket is tartalmazó összetett mikroelem tartalmú műtrágyák
- Levéltrágyázásra szolgáló fő és/vagy másodlagos tápelemeket is tartalmazó összetett mikroelem tartalmú műtrágyák
2. SZERVES TRÁGYÁK
3. ÁSVÁNYI TRÁGYÁK
4. KOMPOSZTOK
5. GILISZTAHUMUSZOK
6. TALAJJAVÍTÓ ANYAGOK
6.1. Lúgos hatású talajjavító anyagok
- Puhamészkő őrlemény
- Keménymészkő őrlemény
- Lápi mész
- Tavi mész
- Meszes lápföld
- Cukorgyári mésziszap
- Alginit
- Dolomit
- Önporló dolomit
6.2. Savas hatású talajjavító anyagok
- Gipszanhidrid (őrölt)
- Lignites gipsz (80% gipsz + 20% lignitpor)
- Lignitpor
6.3. Szerves talajjavító anyago[33]
- Tőzeg
- Lápföld
- Lignitpor
- Alginit
7. TALAJKONDICIONÁLÓ KÉSZÍTMÉNYEK
8. MIKROBIOLÓGIAI KÉSZÍTMÉNYEK
9. TERMESZTŐKÖZEGEK
10. NÖVÉNYKONDICIONÁLÓ KÉSZÍTMÉNYEK
11. NÖVÉNYVÉDŐ SZERNEK NEM MINŐSÜLŐ NÖVEKEDÉSSZABÁLYOZÓ KÉSZÍTMÉNYEK
12. EGYÉB KÉSZÍTMÉNYEK
Az egyes terméktípusok egymással kombinálhatók, jelölésük ebben az esetben a következő:
- előre kerül a készítmény megnevezésében a legnagyobb arányban szereplő alkotórész (alaptípus),[34]
- utána a részarány csökkenő sorrendjében a további típusok a "hozzáadásával" kitétel feltüntetésével (például műtrágya szerves és ásványi trágya hozzáadásával).
2. számú melléklet a 8/2001. (I. 26.) FVM rendelethez
Az engedély iránti kérelemmel együtt benyújtandó vizsgálati eredmények és adatok
1. MŰTRÁGYÁK
1.1. Fizikai, kémiai vizsgálat, adatok
1.1.1. Fizikai, kémiai vizsgálat 3 × 1 kg vagy liter mintából
- küllem: szín, szag, halmazállapot,
- pH (szilárd műtrágyáknál 10%-os vizes szuszpenzióban, folyékony műtrágyáknál eredeti oldatban),
- sűrűség/térfogattömeg,
- nedvesség tartalom/szárazanyag tartalom,
- szemcseméret eloszlás,
- gyártó által deklarált hatóanyagok (N, P205, K2O, CaO, MgO, B, Co, Cu, Fe, Mn, Mo, Zn stb.),
- a 3. számú melléklet 1. számú függelékében meghatározott esetekben oldhatóság,
- nitrogén tartalmú műtrágyák esetén a nitrát, ammónium és amid nitrogén formák,
- kálium tartalmú műtrágyák esetén klorid, illetve szulfát tartalom,
- gyártó által deklarált egyéb tulajdonságok.
1.1.2. Fizikai, kémiai jellemzőkre vonatkozó adatok:
- tapadási hajlam (szilárd műtrágyák),
- higroszkóposság (szilárd műtrágyák),
- dinamikai viszkozitás (oldat műtrágyák; szuszpenziós műtrágyák),
- kristályosodási pont (oldat műtrágyák),
- szuszpenzió stabilitás (szuszpenziós műtrágyák),
- relatív kifolyási sebesség (szuszpenziós műtrágyák),
- hidegtűrés (szuszpenziós műtrágyák),
- korrozív hatás,
- kelátképző anyag.
1.2. Toxikus elemek vizsgálata 3 × 1 kg vagy liter mintából:*
- P, K, NPK műtrágyák: As, Cd, Cr, Hg, Ni, Pb,
- mikroelem - és mikroelemet is tartalmazó műtrágyák: As, Cd, Cr, Hg, Ni, Pb, Se, Co.
1.3. Biztonságtechnikai okból elvégzendő vizsgálatok 3 × 1 kg vagy liter mintából
Ammónium nitrát, illetve 80%-nál több ammónium nitrátot tartalmazó műtrágyáknál:
- porozitás (olaj visszatartás),
- éghető anyag tartalom,
- pH,
- szemcseméret eloszlás,
- klorid tartalom,
- nehézfém (Cu) tartalom vizsgálata.
1.4. Szerves szennyezők vizsgálata
Amennyiben a műtrágya előállításához hulladékot (is) használtak fel, a hulladék minőségétől függően az alábbi összetevők vizsgálata:
- összes PAH tartalom (16 vegyület),
- benz(a)pirén tartalom,
- ásványi olaj tartalom (TPH C5-C40),
- összes PCB tartalom (7 PCB-28, 52, 101, 118, 138, 153, 180),
- összes PCDD/F.
1.5. Biológiai hatás vizsgálata
1.5.1. Adott célra még nem engedélyezett termékeknél 2 szántóföldi, illetve 2 kertészeti tesztnövénnyel, növényenként legalább egy szabadföldi és egy tenyészedényes vizsgálat végzése.[35]
1.5.2. Táprudak esetében külön jogszabályban meghatározott kultúracsoportonként legalább 2 eltérő növényben fitotoxikus hatás vizsgálata.
1.6. Munka- és környezet-egészségügyi adatok: a készítmény veszélyességi besorolása, veszély jelek, a kockázatokra (R) és biztonságra (S) utaló mondatok, tapasztalatok a gyártónál és a felhasználónál, helyi irritációs hatások (bőr, szem, légutak), szenzibilizáció, egyéb tünetek.
1.7 Amennyiben a műtrágya előállításához hulladékot (is) használtak fel, a hulladék minőségétől függően további ökotoxikológiai (Daphnia-teszt, halteszt, algateszt) és egyéb vizsgálatok is elrendelhetőek.[36]
2. SZERVES TRÁGYÁK
2.1. Fizikai, kémiai vizsgálat 3 × 1 kg mintából:
- küllem: szín, szag, halmazállapot,
- pH (10%-os vizes szuszpenzióban),
- térfogattömeg,
- szárazanyag tartalom,
- szerves anyag tartalom,
- vízben oldható összes sótartalom,
Amennyiben a műtrágya előállításához hulladékot (is) használtak fel, a hulladék minőségétől függően további toxikus elem vizsgálat is elrendelhető.
- szemcseméret eloszlás,
- gyártó által deklarált hatóanyagok (N, P2O5, K2O, Ca, Mg stb.).
2.2. Toxikus elemek vizsgálata 3 x 1 kg mintából:[37]
- As, Cd, Cr, Co, Cu, Hg, Ni, Pb, Se*
* Hulladékot is tartalmazó készítmények esetében.
2.3. Szerves szennyezők vizsgálata
Amennyiben a-szerves trágya előállításához hulladékot (is) használtak fel, a hulladék minőségétől függően az alábbi összetevők vizsgálata:
- összes PAH tartalom (16 vegyület),
- benz(a)pirén tartalom,
- ásványi olaj tartalom (TPH C5-C40),
- összes PCB tartalom (7 PCB-28, 52, 101, 118, 138, 153, 180),
- összes PCDD/F.
2.4. Csírázásgátló és gyomosító hatás, valamint növénypatogén kórokozóktól és kártevőktől való mentesség vizsgálata 4 × 3 kg mintából.
2.5. Biológiai hatás vizsgálata
2.5.1. Adott célra még nem engedélyezett termékeknél 2 szántóföldi, illetve 2 kertészeti tesztnövénnyel, növényenként legalább egy szabadföldi és egy tenyészedényes vizsgálat végzése.[38]
2.5.2. A csírázásgátló vizsgálatban nem megfelelőnek bizonyult termékek külön jogszabályban meghatározott kultúracsoportonként, legalább 2 eltérő agroökológiai térségben (szabadföldön vagy zárt termesztő berendezésben) végzett biológiai vizsgálata.
2.6. Munka- és környezet-egészségügyi adatok: a készítmény veszélyességi besorolása, veszély jelek, a kockázatokra (R) és biztonságra (S) utaló mondatok, tapasztalatok a gyártónál és a felhasználónál, helyi irritációs hatások (bőr, szem, légutak), szenzibilizáció, egyéb tünetek.
2.6.1. Higiénés mikrobiológiai vizsgálat 3 × 0,5 kg mintából:
- Fekál coliform szám meghatározása,
- Fekál streptococcus szám meghatározása,
- Salmonella sp. kimutatása,
- Humán parazita bélféreg pete szám meghatározása.
2.6.2. Sugár-biológiai vizsgálat 3 × 0,5 kg mintából.
2.7 Amennyiben a szerves trágya előállításához hulladékot (is) használtak fel, a hulladék minőségétől függően további ökotoxikológiai (Daphnia-teszt, halteszt, algateszt) és egyéb vizsgálatok is elrendelhetőek.[39]
3. ÁSVÁNYI TRÁGYÁK
3.1. Fizikai, kémiai vizsgálat 3 × 1 kg mintából:
- küllem: szín, szag, halmazállapot,
- pH (szilárd terméknél 10%-os vizes szuszpenzióban, folyékony terméknél eredeti oldatban),
* Amennyiben a műtrágya előállításához hulladékot (is) használtak fel, a hulladék minőségétől függően további toxikus elem vizsgálat is elrendelhető.
- sűrűség/térfogattömeg,
- nedvesség tartalom,
- szerves anyag tartalom,
- szemcseméret eloszlás,
- gyártó által deklarált hatóanyagok (N, P2O5, K2O, CaO, MgO, B, Co, Cu, Fe, Mn, Mo, Zn stb.),
- gyártó által deklarált egyéb tulajdonságok (vízoldhatóság stb.).
3.2. Toxikus elemek vizsgálata 3 × 1 kg vagy liter mintából:
- As, Cd, Co, Cr, Cu, Hg, Ni, Pb, Se.
3.3. Biológiai hatás vizsgálata: 2 szántóföldi, illetve 2 kertészeti tesztnövénnyel, növényenként legalább egy szabadföldi és egy tenyészedényes vizsgálat végzése.[40]
3.4. Munka-és környezet-egészségügyi adatok: a készítmény veszélyességi besorolása, veszély jelek, a kockázatokra (R) és biztonságra (S) utaló mondatok, tapasztalatok a gyártónál és a felhasználónál, helyi irritációs hatások (bőr, szem, légutak), szenzibilizáció, egyéb tünetek.
4. KOMPOSZTOK
4.1. Fizikai, kémiai vizsgálat 3 × 1 kg mintából:
- küllem: szín, szag, halmazállapot,
- pH (10%-os vizes szuszpenzióban),
- térfogattömeg,
- szárazanyag tartalom,
- szerves anyag tartalom,
- vízben oldható összes sótartalom,
- szemcseméret eloszlás,
- gyártó által deklarált hatóanyagok (N, P2O5, K2O, Ca, Mg stb.).
4.2. Toxikus elemek vizsgálata 3 x 1 kg mintából:[41]
- As, Cd, Cr, Co, Cu, Hg, Ni, Pb, Se.
4.3. Szerves szennyezők vizsgálata
A komposzt előállításához felhasznált hulladék minőségétől függően az alábbi összetevők vizsgálata:
- összes PAH tartalom (16 vegyület),
- benz(a)pirén tartalom,
- ásványi olaj tartalom (TPH C5-C40),
- összes PCB tartalom (7 PCB-28, 52, 101, 118, 138, 153, 180),
- összes PCDD/F.
4.4. Csírázásgátló és gyomosító hatás, valamint növénypatogén kórokozóktól és kártevőktől való mentesség vizsgálata 4 × 3 kg mintából.
4.5. Biológiai hatás vizsgálata
4.5.1. Adott célra még nem engedélyezett termékeknél külön jogszabályban meghatározott kultúracsoportonként, legalább 2 eltérő agroökológiai térségben (szabadföldön vagy zárt termesztő berendezésben) végzett vizsgálat.[42]
4.5.2. A csírázásgátló vizsgálatban nem megfelelőnek bizonyult termékek külön jogszabályban meghatározott kultúracsoportonként, legalább 2 eltérő agroökológiai térségben (szabadföldön vagy zárt termesztő berendezésben) végzett biológiai vizsgálata.
4.5.3. Gomba komposztoknál és gomba takaróföldeknél gomba tesztnövénnyel.
4.6. Munka-és környezet-egészségügyi adatok: a készítmény veszélyességi besorolása, veszély jelek, a kockázatokra (R) és biztonságra (S) utaló mondatok, tapasztalatok a gyártónál és a felhasználónál, helyi irritációs hatások (bőr, szem, légutak), szenzibilizáció, egyéb tünetek.
4.6.1. Higiénés mikrobiológiai vizsgálat 3 × 0,5 kg mintából:
- Fekál coliform szám meghatározása,
- Fekál streptococcus szám meghatározása,
- Salmonella sp. kimutatása,
- Humán parazita bélféreg pete szám meghatározása.
4.6.2. Sugár-biológiai vizsgálat 3 × 0,5 kg mintából.
4.7 A komposzt előállításhoz felhasznált hulladék minőségétől függően további ökotoxikológiai (Daphnia-teszt, halteszt, algateszt) és egyéb vizsgálatok is elrendelhetőek.[43]
5. GILISZTAHUMUSZOK
5.1. Fizikai, kémiai vizsgálat 3 × 1 kg mintából:
- küllem: szín, szag, halmazállapot,
- pH (10%-os vizes szuszpenzióban),
- térfogattömeg,
- szárazanyag tartalom,
- szerves anyag tartalom,
- vízben oldható összes sótartalom,
- szemcseméret eloszlás,
- gyártó által deklarált hatóanyagok (N, P2O5, K2O, Ca, Mg stb.).
5.2. Toxikus elemek vizsgálata 3 x 1 kg mintából:[44]
- As, Cd, Cr, Co, Cu, Hg, Ni, Pb, Se*
* Hulladékot is tartalmazó készítmények esetében.
5.3. Szerves szennyezők vizsgálata
Amennyiben a gilisztahumusz előállításához hulladékot (is) használtak fel, a hulladék minőségétől függően az alábbi összetevők vizsgálata:
- összes PAH tartalom (16 vegyület),
- benz(a)pirén tartalom,
- ásványi olaj tartalom (TPH C5-C40),
- összes PCB tartalom (7 PCB-28, 52, 101, 118, 138, 153, 180),
- összes PCDD/F.
5.4. Csírázásgátló és gyomosító hatás, valamint növénypatogén kórokozóktól és kártevőktől való mentesség vizsgálata 4 × 3 kg mintából.
* Amennyiben a gilisztahumusz előállításához hulladékot (is) használtak fel, a hulladék minőségétől függően további toxikus elem vizsgálat is elrendelhető.
5.5. Biológiai hatás vizsgálat
5.5.1. Adott célra még nem engedélyezett termékeknél külön jogszabályban meghatározott kultúracsoportonként, legalább 2 eltérő agroökológiai térségben (szabadföldön vagy zárt termesztő berendezésben) végzett vizsgálat.[45]
5.5.2. A csírázásgátló vizsgálatban nem megfelelőnek bizonyult termékek külön jogszabályban meghatározott kultúracsoportonként, legalább 2 eltérő agroökológiai térségben (szabadföldön vagy zárt termesztő berendezésben) végzett biológiai vizsgálata.
5.6. Munka- és környezet-egészségügyi adatok: a készítmény veszélyességi besorolása, veszély jelek, a kockázatokra (R) és biztonságra (S) utaló mondatok, tapasztalatok a gyártónál és a felhasználónál, helyi irritációs hatások (bőr, szem, légutak), szenzibilizáció, egyéb tünetek.
5.6.1. Higiénés mikrobiológiai vizsgálat 3 × 0,5 kg mintából:
- Fekál coliform szám meghatározása,
- Fekál streptococcus szám meghatározása,
- Salmonella sp. kimutatása,
- Humán parazita bélféreg pete szám meghatározása.
5.6.2. Sugár-biológiai vizsgálat 3 × 0,5 kg mintából.
5.7 Amennyiben a gilisztahumusz előállításához hulladékot (is) használtak fel, a hulladék minőségétől függően további ökotoxikológiai (Daphnia-teszt, halteszt, algateszt) és egyéb vizsgálatok is elrendelhetőek.[46]
6. TALAJJAVÍTÓ ANYAGOK
6.1. Fizikai, kémiai vizsgálat 3 × 1 kg mintából:
- küllem: szín, szag, halmazállapot.
6.1.1. Lúgos hatású talajjavító anyagok
6.1.1.1. Puhamészkő őrlemény
- összes karbonát tartalom CaCO3-ban kifejezve,
- MgCO3 tartalom,
- nedvesség tartalom,
- szemcseméret eloszlás,
- 0,8 mm-es szitán áteső rész,
- 2,0 mm-es szitán áteső rész,
- 5,0 mm-es szitán áteső rész.
6.1.1.2. Keménymészkő őrlemény
- összes karbonát tartalom CaCO3-ban kifejezve;
- nedvesség tartalom,
- szemcseméret eloszlás,
- 11,25 mm-es szitán áteső rész,
- 1,0 mm-es szitán áteső rész.
6.1.1.3. Lápi mész, tavi mész, meszes lápföld, cukorgyári mésziszap, alginit
- összes karbonát tartalom CaCO3-ban kifejezve,
- nedvesség tartalom.
6.1.1.4. Dolomit
- összes karbonát tartalom CaCO3-ban kifejezve,
- Ca tartalom,
- Mg tartalom,
- nedvesség tartalom,
- szemcseméret eloszlás,
- 0,1 mm-es szitán áteső rész,
- 0,25 mm-es szitán áteső rész,
- 0,8 mm-es szitán áteső rész.
6.1.1.5. Önporló dolomit
- összes karbonát tartalom CaCO3-ban kifejezve,
- Ca tartalom,
- Mg tartalom,
- nedvesség tartalom,
- szemcseméret eloszlás,
- 0,1 mm-es szitán áteső rész,
- 0,25 mm-es szitán áteső rész,
- 1,0 mm-es szitán áteső rész,
- 2,0 mm-es szitán áteső rész.
6.1.2. Savas hatású talajjavító anyagok
6.1.2.1. Gipszanhidrid (őrölt)
- CaSO4 tartalom CaSO4 × H2O-ban kifejezve,
- szemcseméret eloszlás,
- 0,8 mm-es szitán áteső rész,
- 1,0 mm-es szitán áteső rész.
6.1.2.2. Lignites gipsz (80% gipsz + 20% lignitpor)
- CaSO4 tartalom CaSO4 × H2O-ban kifejezve,
- szemcseméret eloszlás,
- 1,0 mm-es szitán áteső rész,
- 2,5 mm-es szitán áteső rész.
6.1.2.3. Lignitpor
- S tartalom,
- nedvesség tartalom,
- szemcseméret eloszlás,
- 1,0 mm-es szitán áteső rész.
6.1.3. Szerves talajjavító anyagok[47]
6.1.3.1. Tőzeg
- nedvesség tartalom,
- szerves anyag tartalom.
6.1.3.2. Lápföld
- nedvesség tartalom,
- szerves anyag tartalom.
6.1.3.3. Lignitpor
- szerves anyag tartalom.
6.1.3.4. Alginit
- nedvesség tartalom,
- szerves anyag tartalom.
6.2. Toxikus elemek vizsgálata 3 × 1 kg mintából:*
- mészkő, dolomit
- vulkanikus kőzet eredetű talajjavító anyagok (például bazalt, riolit) szervesanyag-tartalmú talajjavító anyagok (tőzeg, lápföld, lignit, lápi mész, meszes lápföld, alginit): As, Cd, Co, Cr, Cu, Hg, Ni, Pb, Se.[48]
6.3. Szerves szennyezők vizsgálata
Amennyiben a talajjavító anyag előállításához hulladékot (is) használtak fel, a hulladék minőségétől függően az alábbi összetevők vizsgálata:
* Amennyiben a talajjavító anyagok előállításához hulladékot (is) használtak fel, a hulladék minőségétől függően további toxikus elem vizsgálat is elrendelhető.
- összes PAH tartalom (16 vegyület),
- benz(a)pirén tartalom,
- ásványi olaj tartalom (TPH C5-C10),
- összes PCB tartalom (7 PCB-28, 52, 101, 118, 138, 153, 180),
- összes PCDD/F.
6.4. Csírázásgátló és gyomosító hatás, valamint növénypatogén kórokozóktól és kártevőktől való mentesség vizsgálata 4 x 3 kg mintából: szervesanyag-tartalmú talajjavító anyagoknál.[49]
6.5. Hatékonyságot igazoló vizsgálatok: 6.1. pontban nem szereplő, talajjavító hatású készítményeknél, talajtípusonként legalább 2, szabadföldön vagy tenyészedényben végzett vizsgálat szükséges.
6.6. Munka- és környezet-egészségügyi adatok: a készítmény veszélyességi besorolása, veszély jelek, a kockázatokra (R) és biztonságra (S) utaló mondatok, tapasztalatok a gyártónál és a felhasználónál, helyi irritációs hatások (bőr, szem, légutak), szenzibilizáció, egyéb tünetek.
6.6.1. Higiénés mikrobiológiai vizsgálat szerves eredetű talajjavító anyagoknál 3 × 0,5 kg mintából:
- Fekál coliform szám meghatározása,
- Fekál streptococcus szám meghatározása,
- Salmonella sp. kimutatása,
- Humán parazita bélféreg pete szám meghatározása.
6.6.2. Sugár-biológiai vizsgálat: 3 x 0,5 kg mintából. (Csak az importból származó tőzeg esetében.)[50]
7. TALAJKONDICIONÁLÓ KÉSZÍTMÉNYEK
7.1. Fizikai, kémiai vizsgálat 3 × 1 kg vagy liter mintából:
- küllem: szín, szag, halmazállapot,
- pH (szilárd talajkondicionáló készítményeknél 10%-os vizes szuszpenzióban, folyékony talajkondicionáló készítményeknél eredeti oldatban),
- sűrűség/térfogattömeg,
- nedvesség tartalom/szárazanyag tartalom,
- szemcseméret eloszlás,
- gyártó által deklarált hatóanyagok (N, P2O5, K2O, CaO, MgO, B, Co, Cu, Fe, Mn, Mo, Zn stb.),
- gyártó által deklarált egyéb tulajdonságok (vízoldhatóság, vízfelvevő képesség stb.).
7.2. Toxikus elemek vizsgálata 3 × 1 kg vagy liter mintából:*
- As, Cd, Co, Cr, Cu, Hg, Ni, Pb, Se.
* Amennyiben a talajkondicionáló készítmény előállításához hulladékot (is) használtak fel, a hulladék minőségétől függően további toxikus elem vizsgálat is elrendelhető.
7.3. Szerves szennyezők vizsgálata
Amennyiben a talajkondicionáló készítmény előállításához hulladékot (is) használtak fel, a hulladék minőségétől függően az alábbi összetevők vizsgálata:
- összes PAH tartalom (16 vegyület),
- benz(a)pirén tartalom,
- ásványi olaj tartalom (TPH C5-C40),
- összes PCB tartalom (7 PCB-28, 52, 101, 118, 138, 153, 180),
- összes PCDD/F.
7.4: Hatékonyságot igazoló vizsgálatok: talajtípusonként legalább 2, szabadföldön vagy tenyészedényben végzett vizsgálat szükséges.
7.5. Munka-és környezet-egészségügyi adatok: a készítmény veszélyességi besorolása, veszély jelek, a kockázatokra (R) és biztonságra (S) utaló mondatok, tapasztalatok a gyártónál és a felhasználónál, helyi irritációs hatások (bőr, szem, légutak), szenzibilizáció, egyéb tünetek.
7.6 Amennyiben a talajkondicionáló készítmény előállításához hulladékot (is) használtak fel, a hulladék minőségétől függően további ökotoxikológiai (Daphnia-teszt, halteszt, algateszt) és egyéb vizsgálatok is elrendelhetőek.[51]
8. MIKROBIOLÓGIAI KÉSZÍTMÉNYEK
8.1. Fizikai, kémiai vizsgálat 3 × 1 kg vagy liter mintából:
- küllem: szín, szag, halmazállapot,
- pH (szilárd készítményben 10%-os vizes szuszpenzióban, folyékony készítményben eredeti oldatban),
- térfogattömeg/sűrűség,
- nedvesség tartalom/szárazanyag tartalom,
- szerves anyag tartalom.
8.2. Toxikus elemek vizsgálata 3 x 1 kg vagy liter mintából:[52]
- As, Cd, Cr, Co, Cu, Hg, Ni, Pb, Se*
* Hulladékot is tartalmazó készítmények esetében.
8.3. Mikrobiológiai minősítés 3 × 1 kg vagy liter mintából.
8.4. Csírázásgátló és gyomosító hatás vizsgálata 4 × 1 kg vagy liter mintából.
8.5. Biológiai hatás vizsgálata[53]
8.5.1. Szimbionta mikroorganizmusok: magcsávázásos felhordás esetén 1 üvegházi vizsgálat (Roux-csöves vizsgálat).
8.5.2. Cellulózbontó mikroorganizmusok.
8.5.2.1. komposztálási célra: 1 üvegházi vizsgálat kétféle talajon (cellulózbontási teszt).
8.5.2.2. tarlókezelési célra: 2 szabadföldi vizsgálat eltérő fizikai féleségű talajokon, növény nélkül (laboratóriumi respiráció mérés).
8.5.3. Elsődlegesen talajon keresztül ható, termésnövelő hatású mikroorganizmusok: 1 tenyészedényés bioteszt 2 eltérő fizikai féleségű talajon, valamint 2 eltérő fizikai féleségű talajon végzett 2-2 szabadföldi vagy üvegházi vizsgálat.
8.5.4. Növényre ható mikroorganizmusok: kultúránként (szakmailag indokolt esetben külön jogszabályban meghatározott kultúracsoportonként) 2, eltérő agroökológiai körülmények között beállított szabadföldi vagy üvegházi vizsgálat.
8.6. Munka- és környezet-egészségügyi adatok: a készítmény veszélyességi besorolása, veszély jelek, a kockázatokra (R) és biztonságra (S) utaló mondatok, tapasztalatok a gyártónál és a felhasználónál, helyi irritációs hatások (bőr, szem, légutak), szenzibilizáció, egyéb tünetek.
8.6.1. Higiénés mikrobiológiai vizsgálat 3 × 0,5 kg mintából:
- Fekál coliform szám meghatározása,
- Fekál streptococcus szám meghatározása,
- Salmonella sp. kimutatása,
- Humán parazita bélféreg pete szám meghatározása.
8.7. GMO mentességről igazolás szükséges.
8.8. A készítményt a Szent István Egyetem Kertészeti Kar törzsgyűjteményében kell elhelyezni.
8.9. A mikroorganizmusokat legalább faj szerint kell megnevezni.
9. TERMESZTŐKÖZEGEK
9.1. Fizikai, kémiai vizsgálat 3 × 1 kg mintából:
- küllem: szín, szag, halmazállapot,
- pH (10%-os vizes szuszpenzióban),
- sűrűség,
- szárazanyag tartalom,
- szerves anyag tartalom,
vízben oldható összes sótartalom,
- szemcseméret eloszlás,
N, P2O5, K2O.
9.2. Toxikus elemek vizsgálata 3 x 1 kg mintából:[54]
- As, Cd, Cr, Co, Cu, Hg, Ni, Pb, Se*
* Hulladékot is tartalmazó készítmények esetében.
9.3. Szerves szennyezők vizsgálata
Amennyiben a termesztőközeg előállításához hulladékot (is) használtak fel, a hulladék minőségétől függően az alábbi összetevők vizsgálata:
- összes PAH tartalom (16 vegyület),
- benz(a)pirén tartalom,
- ásványi olaj tartalom (TPH G5-C40),
- összes PCB tartalom (7 PCB-28, 52, 101, 118, 138, 153, 180),
- összes PCDD/F.
9.4. Csírázásgátló és gyomosító hatás, valamint növénypatogén kórokozóktól és kártevőktől való mentesség vizsgálata 4 × 3 kg mintából.
9.5. Biológiai hatás vizsgálata: a csírázásgátló vizsgálatban nem megfelelőnek bizonyult termékeknél kultúránként legalább 2 biológiai vizsgálat.
9.6. Munka- és környezet-egészségügyi adatok: a készítmény veszélyességi besorolása, veszély jelek, a kockázatokra (R) és biztonságra (S) utaló mondatok, tapasztalatok a gyártónál és a felhasználónál, helyi irritációs hatások (bőr, szem, légutak), szenzibilizáció, egyéb tünetek.
* Amennyiben a termesztőközeg előállításához hulladékot (is) használtak fel, a hulladék minőségétől függően további toxikus elem vizsgálat is elrendelhető.
9.6.1. Higiénés mikrobiológiai vizsgálat 3 × 0,5 kg mintából:
- Fekál coliform szám meghatározása,
- Fekál streptococcus szám meghatározása,
- Salmonella sp. kimutatása;
- Humán parazita bélféreg pete szám meghatározása.
9.6.2. Sugár-biológiai vizsgálat: 3 x 0,5 kg mintából. (Csak az import tőzeget tartalmazó termesztőközegek esetében.)[55]
9.7 Amennyiben a termesztőközeg előállításához hulladékot (is) használtak fel, a hulladék minőségétől függően további ökotoxikológiai (Daphnia-teszt, halteszt, algateszt) és egyéb vizsgálatok is elrendelhetőek.[56]
10. NÖVÉNYKONDICIONÁLÓ KÉSZÍTMÉNYEK
10.1. Fizikai, kémiai vizsgálat 3 × 1 kg vagy liter mintából:
- küllem: szín, szag, halmazállapot,
- pH (szilárd növénykondicionáló készítményeknél 10%-os vizes szuszpenzióban, folyékony növénykondicionáló készítményeknél eredeti oldatban),
- sűrűség/térfogattömeg,
- nedvesség tartalom/szárazanyag tartalom,
- szemcseméret eloszlás,
- gyártó által deklarált hatóanyagok (N, P2O5, K2O, CaO, MgO, B, Co, Cu, Fe, Mn, Mo, Zn stb.),
- gyártó által deklarált egyéb tulajdonságok (vízoldhatóság stb.).
10.2. Toxikus elemek vizsgálata 3 × 1 kg vagy liter mintából:*
As, Cd, Co, Cr, Cu, Hg, Ni, Pb, Se.
10.3. Szerves szennyezők vizsgálata
Amennyiben a növénykondicionáló készítmény előállításához hulladékot (is) használtak fel, a hulladék minőségétől függően az alábbi összetevők vizsgálata:
- összes PAH tartalom (16 vegyület),
- benz(a)pirén tartalom,
- ásványi olaj tartalom (TPH C5-C40),
- összes PCB tartalom (7 PCB-28, 52, 101, 118, 138, 153, 180),
- összes PCDD/F.
10.4. Biológiai hatékonyságot igazoló vizsgálatok: külön jogszabályban meghatározott kultúracsoportonként legalább 2 eltérő agroökológiai térségben (szabadföldi vagy zárt termesztő berendezésben) végzett vizsgálat szükséges.[57]
Csak egy kultúrára történő engedélyezés esetén is legalább kettő, különböző helyszínen végzett vizsgálat szükséges.
10.5. Munka- és környezet-egészségügyi adatok: a készítmény veszélyességi besorolása, veszély jelek, a kockázatokra (R) és biztonságra (S) utaló mondatok, tapasztalatok a gyártónál és a felhasználónál, helyi irritációs hatások (bőr, szem, légutak), szenzibilizáció, egyéb tünetek.
* Amennyiben a növénykondicionáló készítmény előállításához hulladékot (is) használtak fel, a hulladék minőségétől függően további toxikus elem vizsgálat is elrendelhető.
10.5.1. Higiénés mikrobiológiai vizsgálat 3 × 0,5 kg mintából:
- Fekál coliform szám meghatározása,
- Fekál streptococcus szám meghatározása,
- Salmonella sp. kimutatása,
- Humán parazita bélféreg pete szám meghatározása. Amennyiben a növénykondicionáló készítmény előállításához hulladékot (is) használtak fel, a hulladék minőségétől függően további ökotoxikológiai (Daphnia-teszt, halteszt, algateszt) és egyéb vizsgálatok is elrendelhetőek.
11. NÖVÉNYVÉDŐ SZERNEK NEM MINŐSÜLŐ NÖVÉNYI NÖVEKEDÉSSZABÁLYOZÓ KÉSZÍTMÉNYEK
11. l. Fizikai, kémiai vizsgálat 3 × 1 kg vagy liter mintából:
- küllem: szín, szag, halmazállapot,
- pH (10%-os vizes szuszpenzióban),
- sűrűség, térfogattömeg,
- hatóanyag tartalom,
- szemcseméret eloszlás,
- robbanékonyság és oxidációs tulajdonságok,
- lobbanáspont és a gyúlékonyság, illetve öngyulladási hajlam,
- viszkozitás, felületi feszültség,
- tárolási stabilitás, stabilitás és eltarthatóság (fény, hőmérséklet és páratartalom hatása),
- a készítmény technikai paraméterei,
- nedvesíthetőség,
- perzisztens habképződés,
- szuszpendálhatóság és szuszpenzió stabilitás,
- nedves szita és száraz szita vizsgálat,
- emulgeálhatóság, újra-emulgeálhatóság, emulzió stabilitás,
- folyóképesség, kiönthetőség és porzási hajlam,
- fizikai és kémiai kompatibilitás azon termékekkel, melyekkel együtt felhasználása engedélyezett.
11.2. Biológiai hatás vizsgálata
11.2.1. Teljesen új hatóanyagú és új hatásjellegű készítményeknél a vizsgálati idő legalább 3 év, 3 eltérő agroökológiai térségben, kultúránként.
11.2.2. Új hatóanyagú és ismert hatásjellegű készítményeknél a vizsgálati idő legalább 2 év, 2 eltérő agroökológiai térségben, kultúránként.
11.2.3. Ismert hatóanyagú, de új célra felhasználni kívánt készítményeknél a vizsgálati idő legalább 2 év, 2 eltérő agroökológiai térségben, kultúránként.
11.2.4. Hazánkban az adott kultúrában már engedélyezett hatóanyagú, de új készítményeknél a vizsgálati idő legalább 1 év, 2 eltérő agroökológiai térségben, külön jogszabályban meghatározott kultúracsoportonként.[58]
11.2.5. Technológia változtatás már engedélyezett készítményeknél a vizsgálati idő legalább 1 év, 2 eltérő agroökológiai térségben, kultúránként.
11.2.6. Formuláció változtatás már engedélyezett készítményeknél a vizsgálati idő legalább 1 év, 1 agroökológiai térségben, külön jogszabályban meghatározott kultúracsoportonként.[59]
11.2.7. Több hatóanyagot tartalmazó készítmény (kombináció) elbírálása a legszigorúbb szabályozású hatóanyag szerint történik.
11.3. Munka- és környezet-egészségügyi adatok: a készítmény veszélyességi besorolása, veszély jelek, a kockázatokra (R) és biztonságra (S) utaló mondatok, tapasztalatok a gyártónál és a felhasználónál, helyi irritációs hatások (bőr, szem, légutak), szenzibilizáció, egyéb tünetek.
12. EGYÉB ANYAGOK
Az anyag minőségétől függően a minisztérium esetenként adja meg a szükséges vizsgálatok körét.
3. számú melléklet a 8/2001. (I. 26.) FVM rendelethez
Minőségi előírások a termésnövelő anyagokra
1. MŰTRÁGYÁK
1.1. A hatóanyagokra vonatkozó előírások
A műtrágyák hatóanyagaira és minőségi követelményeire vonatkozó előírásokat a 3. számú melléklet 1. számú függeléke tartalmazza.
A 3. számú melléklet 1. számú függelékében nem szereplő műtrágyák hatóanyag tartalmának a 3. számú melléklet 2. számú függelékében megadott tolerancia határon belül meg kell felelnie a gyártó által deklarált értékeknek.
1.2 A toxikus elemkre vonatkozó előírások[60]
| As | Cd | Co | Cr | Hg | Ni | Pb | Se | |
| tartalom legfeljebb mg/kg szárazanyag | ||||||||
| Foszfor műtrágyák | 10 | * | - | 100 | 1 | 50 | 100 | - |
| Kálium műtrágyák | 10 | 2 | - | 100 | 1 | 50 | 100 | - |
| NPK műtrágyák | 10 | * | - | 100 | 1 | 50 | 100 | - |
| Mikroelem műtrágyák | 10 | 2 | 50 | 100 | 1 | 50 | 100 | 5 |
| NPK + mikroelem műtrágyák | 10 | * | 50 | 100 | 1 | 50 | 100 | 5 |
| Hulladékot is tartalmazó műtrágyák | 10 | 2 | 50 | 100 | 1 | 50 | 100 | 5 |
| * A Cd tartalom legfeljebb 20 mg/P2O5 kg lehet (azaz 0,2 mg Cd/1% P2O5 hatóanyag) |
* 20% szárazanyag tartalom alatt a határértékek mg/l dimenzióban értendők.
1.3. Biztonságtechnikai okból megengedhető értékek
Ammónium nitrát, illetve 80%-nál több ammónium nitrátot tartalmazó műtrágyáknál
- porozitás: a műtrágya olaj visszatartása az első két 25-50 °C közötti termikus ciklus után < 4%,
- éghető anyag tartalom szénben kifejezve: a 31,5%-nál nagyobb nitrogén tartalom esetén < 0,2%, 28,0-31,5% közötti nitrogén tartalom esetén < 0,4%,
- pH: 10%-os vizes oldatban > 4,5 ,
- szemcseméret eloszlás: 0,5 mm alatt legfeljebb 3%, 1,0 mm alatt legfeljebb 5%,
- klorid tartalom: < 0,02%,
- Cu tartalom < 10 mg/kg sz.a.
- robbanóképesség: minden egyes robbantás esetén valamennyi ólomhenger összenyomódása meghaladja az 5%-os értéket.[61]
1.4. Szerves szennyezőkre vonatkozó előírások
| – összes PAH tartalom (16 vegyület) – benz(a)pirén tartalom – ásványi olaj tartalom (TPH C5-C40) – összes PCB tartalom (7 PCB–28, 52, 101, 118, 138,153, 180) – összes PCDD/F | < 1,0 mg/kg sz.a. < 0,1 mg/kg sz.a. < 100,0 mg/kg sz.a. < 0,1 mg/kg sz.a. < 5,0 ng/kg sz.a. T.E.Q |
1.5. A biológiai hatékonyság feleljen meg a gyártó által garantált hatásnak.
2. SZERVES TRÁGYÁK
2.1. Hatóanyagokra vonatkozó előírások
| pH (10%-os vizes szuszpenzióban) térfogattömeg (kg/dm3) szárazanyag tartalom (m/m%) szerves anyag tartalom (m/m%) sz:a. vízben oldható összes sótartalom (m/m%) sz.a. szemcseméret eloszlás 25,0 mm alatt . N tartalom (m/m%) sz.a. P2O5 tartalom (m/m%) sz.a. K2O tartalom (m/m%) sz.a. Ca tartalom (m/m%) sz.a. Mg tartalom (m/m%) sz.a. | legfeljebb legalább legalább legfeljebb legalább legalább legalább legalább legalább legalább | 6,5–8,5 0,7 40,0 50,0 4,0 100,0 1,0 0,5 0,5 1,0 0,5 |
2.2. A toxikus elemekre vonatkozó előírások
| As | Cd | Co | Cr | Cu | Hg | Ni | Pb | Se | ||||
| tartalom legfeljebb mg/kg szárazanyag | ||||||||||||
| Szerves trágyák | 10 | 2 | 50 | 100 | 100 | 1 | 50 | 100 | ||||
| Hulladékot is tartalmazó szerves trágyák | 10 | 2 | 50 | 100 | 100 | 1 | 50 | 100 | 5 | |||
| 2.3. Szerves szennyezőkre vonatkozó előírások – összes PAH tartalom (16 vegyület) – benz(a)pirén tartalom – ásványi olaj tartalom (TPH C5–C40) – összes PCB tartalom (7 PCB–28, 52, 101, 118, 138, 153, 180) – összes PCDD/F | < 1,0 mg/kg sz.a. < 0,1 mg/kg sz.a. < 100,0 mg/kg sz.a. < 0,1 mg/kg sz.a. < 5,0 ng/kg sz.a. T.E.Q |
2.4. Nem tartalmazhat a biológiai körforgásba nem vihető idegen anyagot, csírázást, növekedést gátló anyagokat, zárlati nyomok magvait, illetve ezek vegetatív részeit, humán-, állat- és növényegészségügyi szempontból kárós, fertőző makro- és mikroszervezeteket, mérgező, szennyező és radioaktív anyagokat.
2.5. A biológiai hatékonyság feleljen meg a gyártó által garantált hatásnak.
| 2.6. Talajhigiénés mikrobiológiai előírások – Fekál coliform szám – Fekál streptococcus szám – Salmonella sp. – Humán paraz.ita bélféreg pete szám | < 10 db/g vagy 10 db/ml < 10 db/g vagy 10 db/ml 2 × 10 g vagy ml negatív 100 g vagy 100 ml negatív |
3. ÁSVÁNYI TRÁGYÁK
3.1. A hatóanyagokra vonatkozó előírások
A hatóanyag tartalom a tolerancia határon belül feleljen meg a gyártó által megadott értékeknek.
3.2. A toxikus elemekre vonatkozó előírások[63]
| As | Cd | Co | Cr | Cu | Hg | Ni | Pb | Se | |
| tartalom legfeljebb mg/kg szárazanyag | |||||||||
| Ásványi trágyák | 10 | 2 | 50 | 100 | 100 | 1 | 50 | 100 | - |
| Hulladékot is tartalmazó ásványi trágyák | 10 | 2 | 50 | 100 | 100 | 1 | 50 | 100 | 5 |
* 20% szárazanyag tartalom alatt a határértékek mg/l dimenzióban értendők.
3.3. A biológiai hatékonyság feleljen meg a gyártó által garantált hatásnak.
3.4. Nem tartalmazhat a biológiai körforgásba nem vihető idegen anyagot, csírázást, növekedést gátló anyagokat, zárlati gyomok magvait, illetve ezek vegetatív részeit, humán-, állat- és növényegészségügyi szempontból káros, fertőző makro- és mikroszervezeteket, mérgező, szennyező és radioaktív anyagokat.
4. KOMPOSZTOK
| 4.1. Hatóanyagokra vonatkozó előírások pH (10%-os vizes szuszpenzióban) térfogattömeg (kg/dm3) | legfeljebb | 6,5–8,5 0,9 | |
| szárazanyag tartalom (m/m%) | legalább | 50,0 | |
| szerves anyag tartalom (m/m%) sz.a. | legalább | 25,0 | |
| vízben oldható összes sótartalom (m/m%) sz.a. | legfeljebb | 4,0 | |
| szemcseméret eloszlás 25,0 mm alatt | legalább | 100,0 | |
| N tartalom (m/m%) sz.a. | legalább | 1,0 | |
| P2O5 tartalom (m/m%) sz.a. | legalább | 0,5 | |
| K2O tartalom (m/m%) sz.a. | legalább | 0,5 | |
| Ca tartalom (m/m%) sz.a. | legalább | 1,2 | |
| Mg tartalom (m/m%) sz.a. | legalább | 0,5 | |
4.2. A toxikus elemekre vonatkozó előírások[64]
| As | Cd | Co | Cr | Cu | Hg | Ni | Pb | Se | |
| tartalom legfeljebb mg/kg szárazanyag | |||||||||
| Komposztok | 10 | 2 | 50 | 100 | 100 | 1 | 50 | 100 | 5 |
| 4.3. Szerves szennyezőkre vonatkozó előírások – összes PAH tartalom (16 vegyület) – benz(a)pirén tartalom – ásványi olaj tartalom (TPH C5–C40) – összes PCB tartalom (7 PCB–28, 52, 101, 118, 138, 153, 180) – összes PCDD/F | < 1,0 mg/kg sz.a. < 0,1 mg/kg sz.a. < 100,0 mg/kg sz.a. < 0,1 mg/kg sz.a. < 5,0 ng/kg sz.a. T.E.Q |
4.4. Nem tartalmazhat a biológiai körforgásba nem vihető idegen anyagot, csírázást, növekedést gátló anyagokat, zárlati gyomok magvait, illetve ezek vegetatív részeit, humán-, állat- és növényegészségügyi szempontból káros, fertőző makro- és mikroszervezeteket, mérgező, szennyező és radioaktív anyagokat.
4.5. A biológiai hatékonyság feleljen meg a gyártó által garantált hatásnak.
4.6. Talajhigiénés mikrobiológiai előírások
| – Fekál coliform szám – Fekál streptococcus szám – Salmonella sp. – Humán parazita bélféreg pete szám | < 10 db/g vagy 10 db/ml < 10 db/g vagy 10 db/ml 2 × 10 g vagy ml negatív 100 g vagy 100 ml negatív |
5. GILISZTAHUMUSZOK
5.1. Hatóanyagokra vonatkozó előírások
| pH (10%-os vizes szuszpenzióban) térfogattömeg (kg/dm3) szárazanyag tartalom (m/m%) szerves anyag tartalom (m/m%) sz.a. vízben oldható összes sótartalom (m/m%) sz.a. szemcseméret eloszlás 25,0 mm alatt N tartalom (m/m%) sz.a. P2,O5 tartalom (m/m%) sz.a. K2O tartalom (m/m%) sz.a. Ca tartalom (m/m%) sz.a. Mg tartalom (m/m°7°) sz.a. | legfeljebb legalább legalább legfeljebb legalább legalább legalább legalább legalább legalább | 6,5–8,5 1,0 60,0 20,0 4,0 100,0 1,0 1,0 0,8 3,0 1,0 |
5.2. A toxikus elemekre vonatkozó előírások
| As | Cd | Co | Cr | Cu | Hg | Ni | Pb | Se | |||
| tartalom legfeljebb mg/kg szárazanyag | |||||||||||
| Gilisztahumuszok | 10 | 2 | 50 | 100 | 100 | 1 | 50 | 100 | |||
| Hulladékot is tartalmazó gilisztahumuszok | 10 | 2 | 50 | 100 | 100 | 1 | 50 | 100 | 5 | ||
| 5.3. Szerves szennyezőkre vonatkozó előírások | |
| – összes PAH tartalom (16 vegyület) | < 1,0 mg/kg sz.a. |
| – benz(a)pirén tartalom | < 0,1 mg/kg sz.a. |
| – ásványi olaj tartalom (TPH C5-C40) | < 100,0 mg/kg sz.a. |
| – összes PCB tartalom (7 PCB–28, 52,101,118,138,153,180) | < 0,1 mg/kg sz.a. |
| – ősszes PCDD/F | < 5,0 ng/kg sz.a. T.E.Q |
5.4. Nem tartalmazhat a biológiai körforgásba nem vihető idegen anyagot, csírázást, növekedést gátló anyagokat, zárlati gyomok magvait, illetve ezek vegetatív részeit, humán-, állat- és növényegészségügyi szempontból káros, fertőző makro- és mikroszervezeteket, mérgező, szennyező és radioaktív anyagokat.
5.5. A biológiai hatékonyság feleljen meg a gyártó által garantált hatásnak.
| 5.6. Talajhigiénés mikrobiológiai előírások | |||
| – Fekál coliform szám | < 10 db/g vagy 10 db/ml | ||
| – Fekál streptococcus szám | < 10 db/g vagy 10 db/ml | ||
| – Salmonella sp. | 2 × 10 g vagy ml negatív | ||
| – Humán parazita bélféreg pete szám | 100 g vagy 100 ml negatív | ||
| 6. TALAJJAVÍTÓ ANYAGOK | |||
| 6.1. Hatóanyagokra vonatkozó előírások | |||
| Lúgos kémhatású talajjavító anyagok | |||
| Puhamészkő őrlemény | |||
| – összes karbonát tartalom CaCO3-ban kifejezve (m/m%) sz.a. | legalább | 80,0 | |
| – MgCO3 tartalom (m/m%) sz.a. | legfeljebb | 20,0 | |
| – nedvesség tartalom (m/m%) | legfeljebb | 5,0 | |
| – szemcseméret eloszlás | |||
| = 0,8 mm-es szitán áteső rész | legalább | 45,0 | |
| = 2,0 mm-es szitán áteső rész | legalább | 95,0 | |
| = 5,0 mm-es szitán áteső rész | legalább | 100,0 | |
| Keménymészkő őrlemény | |||
| – összes karbonát tartalom CaCO3-ban kifejezve (m/m%) sz.a. | legalább | 90,0 | |
| – nedvesség tartalom (m/m%) | legfeljebb | 5,0 | |
| – szemcseméret eloszlás | |||
| = 0,25 mm-es szitán áteső rész | legalább | 80,0 | |
| = 1,0 mm-es szitán áteső rész | legalább | 95,0 | |
| Lápi mész, tavi mész | |||
| – összes karbonát tartalom CaCO3-ban kifejezve (m/m%) sz.a. | legalább | 50,0 | |
| – nedvesség tartalom (m/m%) | legfeljebb | 40,0 | |
| Meszes lápföld | |||
| – összes karbonát tartalom CaCO3-ban kifejezve (m/m%) sz.a. | legalább | 20,0 | |
| – nedvesség tartalom (m/m%) | legfeljebb | 40,0 | |
| Szikkasztott cukorgyári mésziszap | |||
| – összes karbonát tartalom CaCO3-ban kifejezve (m/m%) sz.a. | legalább | 40,0 | |
| – nedvesség tartalom (m/m%) | legfeljebb | 35,0 , | |
Víztelenített cukorgyári mésziszap
| – összes karbonát tartalom CaCO3-ban kifejezve (m/m%) sz.a. | legalább | 50,0 | |
| – nedvesség tartalom (m/m%) | legfeljebb | 25,0 | |
| Alginit | |||
| – összes karbonát tartalom CaCO3-ban kifejezve (m/m%) sz.a. | legalább | 30,0 | |
| – nedvesség tartalom (m/m%) | legfeljebb | 40,0 | |
| Dolomit | |||
| – összes karbonát tartalom CaCO3-ban kifejezve (m/m%) sz.a. | legalább | 80,0 | |
| – Ca tartalom (m/m%) sz.a. | legalább | 16,0 | |
| – Mg tartalom | legalább | 10,0 | |
| – nedvesség tartalom (m/m%) | legfeljebb | 15,0 | |
| – szemcseméret eloszlás | |||
| = 0,1 mm-es szitán áteső rész | legalább | 20,0 | |
| = 0,25 mm-es szitán áteső rész | legalább | 80,0 | |
| = 0,8 mm-es szitán átesőrész | legalább | 100,0 | |
| Önporló dolomit | |||
| – összes karbonát tartalom CaCO3-ban kifejezve (m/m%) sz.a. | legalább | 80,0 | |
| – Ca tartalom (m/m%) sz.a. | legalább | 16,0 | |
| – Mg tartalom | legalább | 10,0 | |
| – nedvesség tartalom (m/m%) | legfeljebb | 15,0 | |
| – szemcseméret eloszlás | |||
| = 0,1 mm-es szitán áteső rész | legalább | 20,0 | |
| = 0,25 mm-es szitán áteső rész | legalább | 80,0 | |
| = 1,0 mm-es szitán áteső rész | legalább | 95,0 | |
| = 2,0 mm-es szitán áteső rész | legalább | 100,0 | |
| Savas hatású talajjavító anyagok | |||
| Gipszanhidrid (őrölt) | |||
| – CaSO4 tartalom CaSO5 × H2O-ban kifejezve (m/m%) sz.a. | legalább | 50,0 | |
| – szemcseméret eloszlás | |||
| = 0,8 mm-es szitán áteső rész | legalább | 80,0 | |
| = 1,0 mm-es szitán áteső rész | legalább | 100,0 | |
| Lignites gipsz (80% gipsz + 20% lignitpor) | |||
| – CaSO5 tartalom CaSO5 × H2O-ban kifejezve (m/m%) sz.a. | legalább | 40,0 | |
| – szemcseméret eloszlás | |||
| = 1,0 mm-es szitán áteső rész | legalább | 85,0 | |
| = 2,5 mm-es szitán áteső rész | legalább | 100,0 | |
| Lignitpor | |||
| – S tartalom (m/m%) sz.a. | legalább | 7,5 | |
| – szemcseméret eloszlás | |||
| = 2,5 mm-es szitán áteső rész | legalább | 100,0 | |
| Szerves anyag tartalmú talajjavító anyagok | |||
| Tőzeg | |||
| tartalom (m/m%) – nedvesség | legfeljebb | 55,0 | |
| – szerves anyag tartalom (m/m%) sz.a. | legalább | 40,0 | |
| Lápföld | |||
| – nedvesség tartalom (m/m%) | legfeljebb | 55,0 | |
| – szerves anyag tartalom (m/m%) sz.a. | legalább , | 30,0 | |
| Lignitpor – szerves anyag tartalom (m/m%) sz.a. | legalább | 50,0 | |
| Alginit – nedvesség tartalom (m/m%) | legfeljebb | 40,0 | |
| – szerves anyag tartalom (m/m%) sz.a. | legalább | 20,0 | |
6.2. A toxikus elemekre vonatkozó előírások
| As | Cd | Co | Cr | Cu | Hg | Ni | Pb | Se | ||
| tartalom legfeljebb mg/kg szárazanyag | ||||||||||
| Talajjavító anyagok mészkő, dolomit, tőzeg kivételével | 10 | 2 | 50 | 100 | 100 | 1 | 50 | 100 | 5 | |
| Tőzeg | 25 | 2 | 50 | 100 | 100 | 1 | 50 | 100 | 5 | |
| Hulladékot tartalmazó talajjavító anyagok | 10 | 2 | 50 | 100 | 100 | 1 | 50 | 100 | 5 | |
| 6.3. Szerves szennyezőkre vonatkozó előírások | |
| – összes PAH tartalom (16 vegyület) | < 1,0 mg/kg sz.a. |
| – benz(a)pirén tartalom | < 0,1 mg/kg sz.a. |
| – ásványi olaj tartalom (TPH C5-C40) | < 100,0 mg/kg sz.a. |
| – összes PCB tartalom (7 PCB–28, 52, 101, 118, 138, 153, 180) | < 0,1 mg/kg sz.a. |
| – összes PCDD/F | < 5,0 ng/kg sz.a. T.E.Q |
| 6.4. Szerves talajjavító anyagok talajhigiénés mikrobiológiai előírásai | |
| – Fekál coliform szám | < 10 db/g vagy 10 db/ml |
| – Fekál streptococcus szám | < 10 db/g vagy 10 db/ml |
| - Salmonella sp. | 2 x 10 g vagy ml negatív |
| – Humán parazita bélféreg pete szám | 100 g vagy 100 ml negatív |
7. TALAJKONDICIONÁLÓ KÉSZÍTMÉNYEK
7.1. A hatóanyagokra vonatkozó előírások
A hatóanyag tartalom a tolerancia határon belül feleljen meg a gyártó által megadott értékeknek.
7.2. A toxikus elemekre vonatkozó előírások
| As | Cd | Co | Cr | Cu | Hg | Ni | Pb | Se | |
| tartalom legfeljebb mg/kg szárazanyag* | |||||||||
| Talajkondicionáló készítmények | 10 | 2 | 50 | 100 | 100 | 1 | 50 | 100 | - |
| Hulladékot is tartalmazó talajkondicionáló készít- mények | 10 | 2 | 50 | 100 | 100 | 1 | 50 | 100 | 5 |
* 20% szárazanyag tartalom alatt a határértékek mg/l dimenzióban értendők.
| 7.3. Szerves szennyezőkre vonatkozó előírások | |
| – összes PAH tartalom (16 vegyület) | < 1,0 mg/kg sz.a. |
| – benz(a)pirén tartalom | < 0,1 mg/kg sz.a. |
| – ásványi olaj tartalom (TPH Cs-Cao) | < 100,0 mg/kg sz.a: |
| – összes PCB tartalom (7 PCB-28, 52, 101, 118, 138, 153, 180) | < 0,1 mg/kg sza. |
| – összes PCDD/F | < 5,0 ng/kg sz.a. T.E.Q |
7.4. A hatékonyság feleljen meg a gyártó által feltüntetett hatásnak
8. MIKROBIOLÓGIAI KÉSZÍTMÉNYEK
8.1. A hatóanyagokra vonatkozó előírások
A hatóanyag tartalom a tolerancia határon belül feleljen meg a gyártó által megadott értékeknek.
8.2. A toxikus elemekre vonatkozó előírások
| As | Cd | Co | Cr | Cu | Hg | Ni | Pb | Se | |
| tartalom legfeljebb mg/kg szárazanyag* | |||||||||
| Mikrobiológiai készítmények | 10 | 2 | 50 | 100 | 100 | 1 | 50 | 100 | - |
| Hulladékot is tartalmazó mikrobiológiai készítmények | 10 | 2 | 50 | 100 | 100 | 1 | 50 | 100 | 5 |
* 20% szárazanyag tartalom alatt a határértékek mg/l dimenzióban értendők.
8.3. A mikrobiológiai minősítés feleljen meg a gyártó által garantált értékeknek. Nem okozhat toxikus hatást a talajban.
8.4. Nem tartalmazhat a biológiai körforgásba nem vihető idegen anyagot, csírázást, növekedést gátló anyagokat, zárlati gyomok magvait, illetve ezek vegetatív részeit, humán-, állat- és növényegészségügyi szempontból káros, fertőző makro- és mikroszervezeteket, mérgező, szennyező és radioaktív anyagokat.
8.5. A biológiai hatékonyság feleljen meg a gyártó által garantált hatásnak.
8.6. Talajhigiénés mikrobiológiai előírások
| – Fekál coliform szám | < 10 db/g vagy 10 db/ml |
| – Fekál streptococcus szám | < 10 dbg vagy 10 db/ml |
| – Salmonella sp. | 2 × 10 g vagy ml negatív |
| – Humán parazita bélféreg pete szám | 100 g vagy 100 ml negatív |
9. TERMESZTŐKÖZEGEK
9.1. Hatóanyagokra vonatkozó előírások
| pH (10%-os vizes szuszpenzióban) | 5,0–7,5 | |
| térfogattömeg (kg/dm3) | legfeljebb | 0,8 |
| szárazanyag tartalom (m/m%) | legalább | 45,0 |
| szerves anyag tartalom (m/m%) sz.a. | legalább | 12,0 |
| vízben oldható összes sótartalom (m/m%) sz.a. | legfeljebb | 2,0 |
| szemcseméret eloszlás 20,0 mm alatt | legalább | 100,0 |
| N tartalom (m/m%) sz.a. | legalább | 0,3 |
| PzOs tartalom (m/m%) sz.a. | legalább | 0,1 |
| Kz0 tartalom (m/m%) sz.a. | legalább | 0,3 |
9.2. A toxikus elemekre vonatkozó előírások
| As | Cd | Co | Cr | Cu | Hg | Ni | Pb | Se | |
| tartalom legfeljebb mg/kg szárazanyag | |||||||||
| Tőzeget tartalmazó termesztőközeg | 15 | 2 | 50 | 100 | 100 | 1 | 50 | 100 | |
| Tőzeget nem tartalmazó termesztőközeg | 10 | 2 | 50 | 100 | 100 | 1 | 50 | 100 | |
| Hulladékot tartalmazó termesztőközegek | 10 | 2 | 50 | 100 | 100 | 1 | 50 | 100 | 5 |
| 9.3. Szerves szennyezőkre vonatkozó előírások | |
| – összes PAH tartalom (16 vegyület) | < 1,0 mg/kg sz.a. |
| – benz(a)pirén tartalom . | < 0,1 mg/kg sz.a. |
| – ásványi olaj tartalom (TPH C5-C40) | < 100,0 mg/kg sz.a. |
| – összes PCB tartalom (7 PCB-28, 52, 101, 118,.138, 153, 180) | < 0,1 mg/kg sz.a. |
| – összes PCDD/F | < 5,0 ng/kg sz.a. T.E.Q |
9.4. Nem tartalmazhat a biológiai körforgásba nem vihető idegen anyagot, csírázást, növekedést gátló anyagokat, zárlati gyomok magvait, illetve ezek vegetatív részeit, humán-, állat- és növényegészségügyi szempontból káros, fertőző makro- és mikroszervezeteket, mérgező, szennyező és radioaktív anyagokat.
| 9.5. A biológiai hatékonyság feleljen meg a gyártó által garantált hatásnak. | |
| 9.6. Talajhigiénés mikrobiológiai előírások | |
| – Fekál coliform szám | < 10 db/g vagy 10 db/ml |
| – Fekál streptococcus szám | < 10 db/g vagy 10 db/ml |
| – Salmonella sp. | 2 × 10 g vagy ml negatív |
| – Humán parazita bélféreg pete szám | 100 g vagy 100 ml negatív |
10. NÖVÉNYKONDICIONÁLÓ KÉSZÍTMÉNYEK
10.1. A hatóanyagokra vonatkozó előírások
A hatóanyag tartalom a tolerancia határon belül feleljen meg a gyártó által megadott értékeknek.
10.2. A toxikus elemekre vonatkozó előírások
| As | Cd | Co | Cr | Cu | Hg | Ni | Pb | Se | |
| tartalom legfeljebb mg/kg szárazanyag* | |||||||||
| Növénykondicionáló készítmények | 10 | 2 | 50 | 100 | 100 | 1 | 50 | 100 | - |
| Hulladékot is tartalmazó növénykondicionáló készít- mények | 10 | 2 | 50 | 100 | 100 | 1 | 50 | 100 | 5 |
* 20% szárazanyag tartalom alatt a határértékek mg/l dimenzióban értendők.
10.3. Szerves szennyezőkre vonatkozó előírások
| – összes PAH tartalom (16 vegyület) | < 1,0 mg/kg sz.a. |
| – benz(a)pirén tartalom | < 0,1 mg/kg sz.a. |
| – ásványi olaj tartalom (TPH C5–C40) | < 100,0 mg/kg sz.a. |
| – összes PCB tartalom (7 PCB-28, 52,101, 118,138, 153,180) | < 0,1 mg/kg sz.a. |
| – összes PCDD/F | < 5,0 ng/kg sz.a. T.E.Q |
| 10.4. A biológiai hatékonyság feleljen meg a gyártó által garantált hatásnak. | |
| 10.5. Talajhigiénés mikrobiológiai előírások | |
| – Fekál coliform szám | < 10 db/g vagy 10 db/ml |
| – Fekál streptococcus szám | < 10 db/g vagy 10 db/ml |
| – Salmonella sp. | 2 × 10 g vagy ml negatív |
| – Humán parazita bélféreg pete szám | 100 g vagy 100 ml negatív |
11. NÖVÉNYVÉDŐ SZERNEK NEM MINŐSÜLŐ NÖVÉNYI NÖVEKEDÉSSZABÁLYOZÓ KÉSZÍTMÉNYEK
11.1. A hatóanyagokra vonatkozó előírások
A hatóanyag tartalom a tolerancia határon belül feleljen meg a gyártó által megadott értékeknek.
11.2. A biológiai hatékonyság feleljen meg a gyártó által garantált hatásnak.
3. számú melléklet 1. számú függeléke[69]
Műtrágyák besorolása és minőségi követelményei
A. EGYSZERŰ MŰTRÁGYÁK
A/1. NITROGÉN MŰTRÁGYÁK
| A/1. NITROGÉN MŰTRÁGYÁK |
| Szám | Típus | Az előállításra és az alapvető összetevőkre vonatkozó adatok | Előírt minimális tápanyag tartalom (tömegszázalék); a tápanyagok kifejezésére vonatkozó adatok; egyéb követelmények | A típus megjelölésére vonatkozó egyéb adatok | A deklarált tápanyag tartalom; a tápanyagok formái és oldhatósága; egyéb követelmények |
| 1. | 2 | 3. | 4. | 5. | 6. |
| 1. a) | Kalcium-nitrát | Kémiai úton nyert termék, alapvető hatóanyaga kalcium-nitrát és esetleg ammó-nium-nitrátot tartalmazhat. | 15% N Összes nitrogén vagy nitrát és ammónia nitrogén formában kifejezve Ammónia nitrogén tartalom: legfeljebb 1,5%. | Összes nitrogén További megadható adatok: -Nitrát nitrogén Ammónia nitrogén | |
| 1. b) | Kalcium-magnézium-nitrát | Kémiai úton nyert termék, alapvető hatóanyaga kalcium-nitrát és magnézium-nitrát. | 13% N Nitrát nitrogén formában kifejezve 5% MgO Vízben oldódó magnéziumsók, magnézium-oxidban kifejezve. | Nitrát nitrogén Vízben oldódó magnézium-oxid | |
| 1. c) | Magnézium-nitrát | Kémiai úton nyert termék, alapvető hatóanyaga magnézium-nitrát-hexahidrát. | 10% N Nitrát nitrogén formában kifejezve 14% MgO Vízben oldódó magnéziumsók, magnézium-oxidban kifejezve. | Amennyiben kristályos formában forgalmazzák, a „kristályos formában" megjelölést fel lehet tüntetni | Nitrát nitrogén Vízben oldódó magnézium-oxid |
| 2. a) | Nátrium-nitrát | Kémiai úton nyert termék, alapvető hatóanyaga nátrium-nitrát. | 15% N Nitrát nitrogén formában kifejezve. | Nitrát nitrogén | |
| 2. b) | Chilei salétrom | Chilei salétromból készített termék, alapvető hatóanyaga nátrium-nitrát. | 15% N Nitrát nitrogén formában kifejezve. | Nitrát nitrogén | |
| 3. aj | Kalcium-ciánamid | Kémiai úton nyert termék, alapvető hatóanyaga kalcium-ciánamid, kalcium-oxidot és esetleg kis mennyiségben ammónium sókat és karbamidot tartalmaz | 18% N Összes nitrogénben kifejezve A deklarált nitrogénnek legalább 75%-a ciánamid formában kötött. | Összes nitrogén | |
| 3. b) | Nitrát tartalmú kalcium-ciánamid | Kémiai úton nyert termék, alapvető hatóanyaga kalcium-ciánamid, kalcium-oxidot és esetleg kis mennyiségben ammónium sókat és karbamidot, valamint hozzáadott nitrátot tartalmaz | 18% N Összes nitrogénben kifejezve A deklarált nem nitrát nitrogénnek legalább 75%-a ciánamid formában kötött A nitrát nitrogén tartalom — legalább 1 % N, — legfeljebb 3 % N. | Összes nitrogén Nitrát nitrogén | |
| 4. | Ammónium-szulfát | Kémiai úton nyert termék, alapvető hatóanyaga ammónium-szulfát. | 20% N Ammónia nitrogén formában kifejezve. | Ammónia nitrogén | |
| 5. | Ammónium-nitrát | Kémiai úton nyert tennék, alapvető ható- | 20% N | A „kalcium- ammó- | Összes nitrogén |
| vagy kalcium-am- | anyaga ammónium-nitrát, mely olyan ada- | Ammónia és nitrát nitrogén formában kife- | nium-nitrát' ' elne- | Nitrát nitrogén | |
| mónium-nitrát | lékanyagokat tartalmaz, mint az őrölt | jezve | vezés kizárólag | Ammónia nitrogén | |
| mészkő, kalcium-szulfát, őrölt dolomit, | A két nitrogénforma az összes nitrogén | olyan műtrágyák | |||
| magnézium-szulfát, kieserit. | tartalomnak körülbelül a fele-felét teszi ki. | számára van fenn- | |||
| tartva, melyek az | |||||
| ammónium-nitráton | |||||
| kívül csak kalcium- | |||||
| karbonátot (mész- | |||||
| kő) és/vagy magné- | |||||
| zium-karbonátot és | |||||
| kalcium-karbonátot | |||||
| (dolomit) tartalmaz- | |||||
| nak. Ezen karboná- | |||||
| tok mennyisége leg- | |||||
| alább 20%, tisztasá- | |||||
| gi fokuk pedig leg- | |||||
| alább 90%. | |||||
| 6. | Ammónium- | Kémiai úton nyert termék, alapvető ható- | 25% N | Összes nitrogén | |
| szulfát-nitrát - | anyaga ammónium-nitrát és ammónium- | Ammónia és nitrát nitrogén formában kife- | Nitrát nitrogén | ||
| szulfát. | jezve | Ammónia nitrogén | |||
| A nitrát nitrogén tartalom: legalább 5% | |||||
| 7. | Magnézium- | Kémiai úton nyert termék, alapvető ható- | 19% N | Összes nitrogén | |
| szulfo-nitrát | anyaga ammónium-nitrát, ammónium- | Ammónia és nitrát nitrogén formában kife- | Nitrát nitrogén | ||
| szulfát és magnézium-szulfát. | jezve | Ammónia nitrogén | |||
| A nitrát nitrogén tartalom: legalább 6% | Vízben oldódó magnézium-oxid | ||||
| 5% MgO | |||||
| Vízben oldódó magnéziumsók, magné- | |||||
| zium-oxidban kifejezve. | |||||
| 8. | Magnézium- | Kémiai úton nyert termék, alapvető ható- | 19% N | Összes nitrogén | |
| ammónium-nitrát | anyaga ammónium-nitrát, és magnézium | Ammónia és nitrát nitrogén formában kife- | Nitrát nitrogén | ||
| sók (dolomit, magnézium, karbonát | jezve | Ammónia nitrogén | |||
| és/vagy magnéziumszulfát). | A nitrát nitrogén tartalom: legalább 6% | Összes magnézium-oxid és lehetőleg a víz- | |||
| 5% MgO | ben oldódó magnézium-oxid | ||||
| Az összes magnézium magnézium-oxid- | |||||
| ban kifejezve. | |||||
| 9. | Karbamid | Kémiai úton nyert termék, alapvető ható- | 44% N | Összes nitrogén karbamid nitrogénként ki- | |
| anyaga karbonildiamid (karbamid). | Összes karbamid nitrogén (biuretet is bele- | fejezve | |||
| értve) formában kifejezve | |||||
| Biuret tartalom: legfeljebb 1-2% | |||||
| 10. | Krotonilidén-dikarbamid | Karbamid és krotonaldehid kémiai reakciójával nyert termék Monomer vegyület | 28% N Összes nitrogénben kifejezve Legalább 25% nitrogén krotonilidén-di-karbamidban kötött Karbamid nitrogén tartalom: legfeljebb 3%. | Összes nitrogén Karbamid nitrogén, ahol legalább 1 tömeg% Krotonilidén-dikarbamidban kötött nitrogén | |
| 11. | Izobutilidén-dikarbamid | Karbamid és izobutilaldehid kémiai reakciójával nyert termék Monomer vegyület | 28% N Összes nitrogénben kifejezve Legalább 25% nitrogén izobutilidén-dikarbamidban kötött Karbamid nitrogén tartalom: legfeljebb 3%. | Összes nitrogén Karbamid nitrogén, ahol legalább 1 tömeg% Izobutilidén-dikarbamidban kötött nitrogén. | |
| 12. | Karbamid-formaldehid | Karbamid és formaldehid kémiai reakciójával nyert termék Polimer vegyület | 36% N Összes nitrogénben kifejezve A deklarált érték legalább 60%-a oldódjon forró vízben Legalább 31 % nitrogén karbamid-formaldehidben kötött Karbamid nitrogén tartalom: legfeljebb 5%. | Összes nitrogén Karbamid nitrogén, ahol legalább 1 tömeg% Hideg vízben oldódó karbamid formaldehid nitrogén Csak forró vízben oldódó karbamid formaldehid nitrogén | |
| 13. | Krotonilidén-dikarbamid tartalmú nitrogén műtrágya | Kémiai úton nyert termék, amely krotoni-lidén-dikarbamidot és egy egyszerű nitrogén műtrágyát tartalmaz, az A/1. táblázat a 3. a), 3. b) és 5. termékek kivételével. | 18% N Összes nitrogénben kifejezve Legalább 3% nitrogén ammónia és/vagy nitrát és/vagy karbamid nitrogén formában A deklarált összes nitrogén tartalom legalább 1/3-a krotonilidén-dikarbamid formában. Biuret tartalom legfeljebb: (karbamid nitrogén + krotonilidén-dikarbamid N) x 0,026 | Összes nitrogén Az a N forma, amelyik legalább 1 tömeg%: Nitrát nitrogén Ammónia nitrogén Karbamid nitrogén A krotonilidén-dikarbamidban kötött nitrogén. | |
| 14. | Izobutilidén-dikarbamid tartalmú nitrogén műtrágya | Kémiai úton nyert tennék, amely izobutili-dén-dikarbamidot és egy egyszerű nitrogén műtrágyát tartalmaz, az A/1. táblázat a 3. a), 3. b) és 5. termékek kivételével. | 18% N Összes nitrogénben kifejezve Legalább 3% nitrogén ammónia és/vagy nitrát és/vagy karbamid nitrogén formában A deklarált összes nitrogén tartalom legalább 1/3-a izobutilidén-dikarbamid formában Biuret tartalom legfeljebb: (karbamid nitrogén + izobutilidén-dikarbamid nitrogén) x 0,026 | Összes nitrogén Az a N forma, amelyik legalább 1 tömeg%: Nitrát nitrogén Ammónia nitrogén Karbamid nitrogén Az izobutilidén-dikarbamidban kötött nitrogén | |
| 15. | Karbamid-formaldehid tartalmú nitrogén műtrágya | Kémiai úton nyert termék, amely karbamid-formaldehidet és egy egyszerű nitrogén műtrágyát tartalmaz, az A/1. táblázat a 3. a), 3. b) és 5. termékek kivételével. | 18% N Összes nitrogénben kifejezve Legalább 3% nitrogén ammónia és/vagy nitrát és/vagy karbamid nitrogén formában A deklarált összes nitrogén tartalom legalább 1/3-a karbamid-formaldehid formában A deklarált érték legalább 60%-a oldódjon forró vízben Biuret tartalom legfeljebb: (karbamid nitrogén + karbamid-formaldehid nitrogén) x 0,026 | Összes nitrogén Az a N forma, amelyik legalább 1 tömeg%: Nitrát nitrogén Ammónia nitrogén Karbamid nitrogén A karbamid-formaldehidben kötött nitrogén A hideg vízben oldódó karbamid-formaldehidben kötött nitrogén A forró vízben oldódó karbamid-formaldehidben kötött nitrogén | |
| 16. | Ammónium-szulfát nitrifikáció gátlóval (diciándiamid) | Kémiai úton nyert termék, amely ammó-nium-szulfátot és diciándiamidot tartalmaz. | 20% N Összes nitrogénben kifejezve Ammónium nitrogén tartalom legalább 18% Diciándiamid nitrogén tartalom legalább 1,5% | Összes nitrogén Ammónia nitrogén Diciándiamid nitrogén Használati utasítás* | |
| 17. | Ammónium-szulfo-nitrát nitrifikáció gátlóval (diciándiamid) | Kémiai úton nyert termék, amely ammó-nium-szulfo-nitrátot és diciándiamidot tartalmaz. | 24% N Összes nitrogénben kifejezve Nitrát nitrogén tartalom legalább 3% Diciándiamid nitrogén tartalom legalább 1,5% | Összes nitrogén Ammónia nitrogén Nitrát nitrogén Diciándiamid nitrogén Használati utasítás* | |
| 18. | Karbamid- ammóni um-szulfát | Kémiai úton, karbamidból és ammónium-szulfátból nyert termék. | 30% N Ammónia és karbamid nitrogén formában kifejezve Ammónia nitrogén: legalább 4% Kén tartalom kén-trioxidban kifejezve: legalább 12% Biuret tartalom legfeljebb 0,9% | Összes nitrogén Ammónia nitrogén Karbamid nitrogén Vízben oldódó kén-trioxid | |
| * A lehető legteljesebb használati utasítást kell adni minden egyes csomagolási egységhez vagy ömlesztett szállítmányhoz, amelyért a forgalmazó felel. A használati utasításnak olyannak kell lennie, hogy a felhasználó képes legyen meghatározni a kijuttatási dózist, az alkalmazás idejét a termesztett növénytől függően. | |||||
A/2. FOSZFOR MŰTRÁGYÁK
Amennyiben a szemcseméret követelmény előírt a granulátum formájában forgalmazott műtrágyák esetében (1., 3., 4., 5., 6., és 7. műtrágyák), azt megfelelő analitikai módszerrel meg kell határozni
| Szám | Típus | Az előállításra és az alapvető összetevőkre vonatkozó adatok | Előírt minimális tápanyag tartalom (tömegszázalék); a tápanyagok kifejezésére vonatkozó adatok; egyéb követelmények | A típus megjelölésére vonatkozó egyéb adatok | A deklarált tápanyag tartalom; . a tápanyagok tonnái és oldhatósága; egyéb követelmények |
| 1. | 2. | 3. | 4. | 5. | 6. |
| 1. | Bázikus salak — Thomas foszfátok — Thomas salak | A vasolvasztásnál a foszfor tartalmú olvadékok kezelésével nyert termék, amely alapvető hatóanyagként kalcium-sziliko-foszfátokat tartalmaz. | 12% P2O5 Ásványi savakban oldódó, foszfor-pen-toxidban kifejezett foszfor 10% P2O5 2%-os citrom savban oldódó foszfor-pentoxidban kifejezett foszfor Szemcseméret: — legalább 75%-a átesik egy 0,160 mm-es szitán, — legalább 96%-a átesik egy 0,630 mm-es szitán | Összes (ásványi savakban oldódó) foszfor-pentoxid 2%-os citromsavban oldódó foszfor-pentoxid | |
| 2. a) | Szuperfoszfát | Őrölt ásványi foszfát kén savval való rea-gáltatással nyert termék, amely alapvető hatóanyagként monokalcium-foszfátot, valamint kalcium-szulfátot tartalmaz. | 16% P2O5 Semleges ammónium-citrátban oldódó, foszfor-pentoxidban kifejezett foszfor A megadott foszfor-pentoxid tartalom legalább 93%-a oldódik vízben (vizsgálati minta: 1 g) | Semleges ammónium-citrátban oldódó foszfor-pentoxid Vízben oldódó foszfor-pentoxid | |
| 2. b) | Koncentrált szuperfoszfát | Őrölt ásványi foszfát kénsavval és foszforsavval való reagáltatással nyert termék, amely alapvető hatóanyagként monokalcium-foszfátot, valamint kalcium-szulfátot tartalmaz. | 25% P2O5 Semleges ammónium-citrátban oldódó, foszfor-pentoxidban kifejezett foszfor A megadott foszfor-pentoxid tartalom legalább 93%-a oldódik vízben (vizsgálati minta: 1 g) | Semleges ammónium-citrátban oldódó foszfor-pentoxid Vízben oldódó foszfor-pentoxid | |
| 2. c) | Tripleszuperfoszfát | Őrölt ásványi foszfát foszforsavval való reagáltatással nyert termék, amely alapvető hatóanyagként monokalcium-foszfátot tartalmaz. | 38% P2O5 Semleges ammónium-citrátban oldódó, foszfor-pentoxidban kifejezett foszfor A megadott foszfor-pentoxid tartalom legalább 93%-a oldódik vízben (vizsgálati minta: 3 g) | Semleges ammónium-citrátban oldódó foszfor-pentoxid Vízben oldódó foszfor-pentoxid | |
| 3. | Részlegesen feltárt foszfátérc | Őrölt foszfátércnek kénsavval vagy foszforsavval történő részleges feltárásával nyert termék, amely alapvető hatóanyagként monokalcium-foszfátot, trikalcium-foszfátot és kalcium-szulfátot tartalmaz. | 20% P2O5 Ásványi savakban oldódó, foszfor-pentoxidban kifejezett foszfor A megadott foszfor-pentoxid tartalom legalább 40%-a oldódik vízben Szemcseméret: — legalább 90%-a átesik egy 0,160 mm-es szitán, — legalább 98%-a átesik egy 0,630 mm-es szitán | Összes (ásványi savakban oldódó) foszfor-pentoxid Vízben oldódó foszfor-pentoxid | |
| 4. | Dikalcium-foszfát | Ásványi foszfátokból vagy csontból oldott foszforsavval történő kicsapással nyert termék, amely alapvető hatóanyagként dikal-cium-foszfát- dihidrátot tartalmaz. | 38% P2O5 Lúgos ammónium-citrátban oldódó, fosz-for-pentoxidban kifejezett foszfor (Peter-mann) Szemcseméret: — legalább 90%-a átesik egy 0,160 mm-es szitán, — legalább 98%-a átesik egy 0,630 mm-es szitán | Lúgos ammónium-citrátban oldódó foszfor-pentoxid | |
| 5. | Kalcinált foszfát | Őrölt foszfátércnek lúgos vegyületekkel és szilíciumsavval történő hőkezelése útján nyert termék, amely alapvető hatóanyagként lúgos kalcium-foszfátot és kalciumszilikátot tartalmaz. | 25% P2O5 Lúgos ammónium-citrátban oldódó, fosz-for-pentoxidban kifejezett foszfor (Peter-mann) Szemcseméret: — legalább 75%-a átesik egy 0,160 mm-es szitán, — legalább 96%-a átesik egy 0,630 mm-es szitán | Lúgos ammónium-citrátban oldódó foszfor-pentoxid | |
| 6. | Alumíniumkalcium-foszfát | Amorf alakú, hőkezeléssel és őrléssel nyert termék, amely alapvető hatóanyagként alumínium- és kalcium-foszfátot tartalmaz. | 30% P2O5 Ásványi savakban oldódó, foszfor-pent-oxidban kifejezett foszfor A megadott foszfor-pentoxid tartalom legalább 75%-a oldódik lúgos ammónium-citrátban (Joulie) Szemcseméret: — legalább 90%-a átesik egy 0,160 mm-es szitán — legalább 98%-a átesik egy 0,630 mm-es szitán | Összes (ásványi savakban oldódó) foszfor-pentoxid Lúgos ammónium-citrátban oldódó foszfor-pentoxid | |
| 7. | Őrölt lágy foszfátérc | Lágy ásványi foszfátok őrlésével nyert termék, amely alapvető hatóanyagként trikal-cium-foszfátot és kalcium-karbonátot tartalmaz. | 25% P2O5 Ásványi savakban oldódó, foszfor-pent-oxidban kifejezett foszfor A megadott foszfor-pentoxid tartalom legalább 55%-a oldódik 2%-os hangyasavban Szemcseméret: — legalább 90%-a átesik egy 0,063 mm-es szitán, — legalább 98%-a átesik egy 0,125 mm-es szitán | Összes (ásványi savakban oldódó) foszfor-pentoxid 2%-os hangyasavban oldódó foszfor-pentoxid Az anyag tömegszázalékban megadott hányada, amely átesik egy 0,063 mm-es szitán |
A/3. KÁLIUM MŰTRÁGYÁK
| Szám | Típus | Az előállításra és az alapvető összetevőkre vonatkozó adatok | Előírt minimális tápanyag tartalom (tömegszázalék); a tápanyagok kifejezésére vonatkozó adatok; egyéb követelmények | A típus megjelölésére vonatkozó egyéb adatok | A deklarált tápanyag tartalom; a tápanyagok formái és oldhatósága; egyéb követelmények |
| 1. | 2. | 3. | 4. | 5. | 6. |
| 1. | Kainit | Nyers kálisókból nyert termék | 10% K2O Vízben oldódó kálium-oxidban kifejezve 5% MgO Vízben oldódó magnéziumsók, magnézium-oxidban kifejezve | A szokásos kereskedelmi nevekkel kiegészíthető | Vízben oldódó kálium-oxid Vízben oldódó magnézium-oxid |
| 2. | Dúsított kainit só | Kálium-kloriddal való keverés útján dúsított nyers káliumsókból nyert termék | 1.8% K2O Vízben oldódó kálium-oxidban kifejezve | A szokásos kereskedelmi nevekkel kiegészíthető | Vízben oldódó kálium-oxid Megadható a vízben oldódó magnézium oxid tartalom, ahol a MgO több mint 5% |
| 3. | Kálium-klorid | Nyers kálisóból nyert termék, amely alapvető hatóanyagként kálium-kloridot tartalmaz. | 37% K2O Vízben oldódó kálium-oxidban kifejezve | A szokásos kereskedelmi nevekkel kiegészíthető | Vízben oldódó kálium-oxid |
| 4. | Magnéziumsó tartalmú kálium-klorid | Nyers kálisóból magnézium sók hozzáadásával nyert termék, amely alapvető hatóanyagként kálium-kloridot és magnézium sókat tartalmaz. | 37% K2O Vízben oldódó kálium-oxidban kifejezve 5% MgO Vízben oldódó magnézium sók, magnézium-oxidban kifejezve | Vízben oldódó kálium-oxid Vízben oldódó magnézium-oxid | |
| 5. | Kálium-szulfát | Kálisókból kémiai úton nyert termék, amely alapvető hatóanyagként káliumszulfátot tartalmaz. | 47% K2O Vízben oldódó kálium-oxidban kifejezve Klór tartalom: legfeljebb 3% Cl | Vízben oldódó kálium-oxid Megadható a klór tartalom, ahol kisebb, mint 3% Cl | |
| 6. | Magnéziumsó tartalmú kálium-szulfát | Kálisókból kémiai úton, esetleg magnézium sók hozzáadásával nyert termék, amely alapvető hatóanyagként káliumszulfátot és magnézium sókat tartalmaz. | 22% K2O Vízben oldódó kálium-oxidban kifejezve 8% MgO Vízben oldódó magnézium sók, magnézium-oxidban kifejezve Klór tartalom: legfeljebb 3% Cl | A szokásos kereskedelmi nevekkel kiegészíthető | Vízben oldódó kálium-oxid Vízben oldódó magnézium-oxid Megadható a klór tartalom, ahol kisebb, mint 3% Cl |
| 7. | Kieserit káliumszulfáttal | Kálium-szulfáttal dúsított kieserit | 8% MgO Vízben oldódó magnézium-oxidban kifejezve 6% K2O Vízben oldódó kálium-oxidban kifejezve Az összes MgO + K2O: 20% Klór tartalom: legfeljebb 3%. | A szokásos kereskedelmi nevekkel kiegészíthető | Vízben oldódó magnézium-oxid Vízben oldódó kálium-oxid Megadható a klór tartalom, ahol kisebb mint 3% Cl |
B. ÖSSZETETT MŰTRÁGYÁK
B/1. NPK MŰTRÁGYÁK
| Típus | Az előállításra vonatkozó adatok | Minimális tápanyag tartalom (tömegszázalékban) | A műtrágyák 8., 9. és 10. oszlopban meghatározott formái, oldhatósága és a megadott tápanyag tartalom; szemcseméret eloszlás | A műtrágyák azonosítási adatai; egyéb követelmények | |||||||||
| Összes | Az egyes tápanyagok | N | P2O5 | K2O | N | P2O5 | K2O | ||||||
| 1. | 2. | 3. | 4. | 5. | 6. | 7. | 8. | 9. | 10. | ||||
| NPK | Kémiai úton | 20% | 3% N | 1. Összes nitrogén | 1. Vízben oldódó P2O5 | Vízben | 1. Összes nitrogén | 1. Az olyan NPK műtrágyá- | 1. Vízben ol- | ||||
| műtrágyák | vagy keverés- | (N + P2O5 | 5% P2O5 | oldódó | nál, amely nem tartalmaz Tho- | dódó K2O | |||||||
| sel előállított | + K2O) | 5% K2O | 2. Nitrát nitrogén | 2. Semleges ammó- | K2O | 2. Habármelyiknit- | mas salakot, kalcinált foszfát- | ||||||
| termék, állati | nium-citrátban oldódó | rogén forma (2 — 5) | ércet, alumínium-kalcium- | 2. Az „ala- | |||||||||
| vagy növényi | 3. Ammónia nit- | P2O5 | mennyisége leg- | foszfátot, részben feltárt fosz- | csony klór tar- | ||||||||
| eredetű szer- | rogén | alább 1 tömeg%, fel | fátércet és őrölt lágy foszfátér- | talmú" megje- | |||||||||
| ves tápanya- | 3. Semleges ammó- | kell tüntetni | cet, a foszfortartalmat az 1-es, | lölés legfel- | |||||||||
| gok hozzá- | 4. Karbamid nit- | nium-citrátban és vízben | 2-es és 3-as típusú oldhatóság | jebb 2% klór- | |||||||||
| adása nélkül | rogén | oldódó P2O5 | szerint kell feltüntetni: | tartalomra vo- | |||||||||
| — ha a vízben oldódó P2O5 | natkozhat | ||||||||||||
| 5. Ciánamidnitro- | 4. Kizárólag ásványi sa- | nem éri el a 2%-ot, csak a 2-es | |||||||||||
| gén | vakban oldódó P2O5 | típusú oldhatóságot kell feltün- | 3. A klórtarta- | ||||||||||
| tetni, | lom feltüntet- | ||||||||||||
| 5. Lúgos ammónium- | — ha a vízben oldódó P2O5 | hető | |||||||||||
| citrátban oldódó P2O5 | meghaladja a 2%-ot, a 3-as tí- | ||||||||||||
| (Petermann) | pusú oldhatóságot kell feltün- | ||||||||||||
| tetni és a vízben oldódó P2O5 | |||||||||||||
| 6. a) Ásványi savakban | tartalmat jelölni kell (1-es típu- | ||||||||||||
| oldódó P2O5, melynek | sú oldhatóság) | ||||||||||||
| legalább 75%-a oldódó | A kizárólag ásványi savakban | ||||||||||||
| 2%-os citromsavban | oldódó P2O5 tartalom nem le- | ||||||||||||
| het több mint 2% | |||||||||||||
| 6. b) 2%-os citromsav- | (Ezen 1-es típus 2-es és 3-as | ||||||||||||
| ban oldódó P2O5 | típusú oldhatóságának megha- | ||||||||||||
| tározására 1 g mintát kell be- | |||||||||||||
| 7. Ásványi savakban ol- | mérni.) | ||||||||||||
| dódó P2O5, melynek leg- | |||||||||||||
| alább 75%-a oldódó lú- | |||||||||||||
| gos ammónium-citrátban | |||||||||||||
| (Joulie) | |||||||||||||
| 8. Ásványi savakban ol- | 2. a) Az őrölt lágy foszfátér- | ||||||||||||
| dódó P2O5, melynek leg- | cet vagy részben feltárt foszfát- | ||||||||||||
| alább 55%-a oldódó | ércet tartalmazó NPK műtrá- | ||||||||||||
| 2%-os hangyasavban | gyánál, amely nem tartalmaz- | ||||||||||||
| hat Thomas salakot, kalcinált | |||||||||||||
| foszfátércet és alumínium-kal- | |||||||||||||
| cium-foszfátot, a foszfortartal- | |||||||||||||
| mat az 1-es, 3-as és 4-es típusú | |||||||||||||
| oldhatóság szerint kell feltün- | |||||||||||||
| tetni. Ezen műtrágya típusnak | |||||||||||||
| tartalmaznia kell: | |||||||||||||
| — legalább 2% csak ásványi | |||||||||||||
| savakban oldódó P2O5-t (4-es | |||||||||||||
| típusú oldhatóság), | |||||||||||||
| — legalább 5% semleges | |||||||||||||
| ammónium-citrátban és vízben | |||||||||||||
| oldódó P2O5-t (3-as típusú oldha- | |||||||||||||
| tóság), | |||||||||||||
| — legalább 2,5% vízben oldó- | |||||||||||||
| dó P2O5-t (1-es típusú oldható- | |||||||||||||
| ság) | |||||||||||||
| Ezt a típusú műtrágyát a követ- | |||||||||||||
| kező megjelöléssel kell forga- | |||||||||||||
| lomba hozni: „NPK műtrágya, | |||||||||||||
| őrölt lágy foszfátérc tartalmú' ' | |||||||||||||
| vagy „NPK műtrágya, részben | |||||||||||||
| feltárt foszfátérc tartalmú" | |||||||||||||
| [Ezen 2. a) típus 3-as típusú | |||||||||||||
| oldhatóságának meghatározá- | |||||||||||||
| sára 3 g mintát kell bemérni.] | |||||||||||||
| 2. b) Az alumínium-kalcium- | |||||||||||||
| foszfátot-tartalmazó NPK mű- | |||||||||||||
| trágyánál, amely nem tartal- | |||||||||||||
| mazhat Thomas salakot, kalci- | |||||||||||||
| nált foszfátércet, őrölt lágy | |||||||||||||
| foszfátércet és részben feltárt | |||||||||||||
| foszfátércet, a foszfortartalmat | |||||||||||||
| az 1-es és 7-es típusú oldható- | |||||||||||||
| ság szerint kell feltüntetni, az | |||||||||||||
| utóbbit a vízben oldhatóság le- | |||||||||||||
| vonásával alkalmazzuk | |||||||||||||
| Ezen műtrágya típusnak tartal- | |||||||||||||
| maznia kell: | |||||||||||||
| — legalább 2% vízben oldódó | |||||||||||||
| P2O5-t (1-es típusú oldhatóság), | |||||||||||||
| — legalább 5%, 7-es típusú | |||||||||||||
| oldhatóság szerinti P2O5-t | |||||||||||||
| Ezt a típusú műtrágyát a követ- | |||||||||||||
| kező megjelöléssel kell forga- | |||||||||||||
| lomba hozni: „NPK műtrágya, | |||||||||||||
| alumínium-kalcium-foszfát tar- | |||||||||||||
| talmú' ' | |||||||||||||
| - | 3. Azon NPK műtrágyák eseté- | ||||||||||||
| ben, melyek kizárólag egyet tar- | |||||||||||||
| talmaznak az alábbi foszfor mű- | |||||||||||||
| trágya típusok közül: Thomas | |||||||||||||
| salak, kalcinált foszfátérc, alumí- | |||||||||||||
| nium-kalcium-foszfát, őrölt lágy | |||||||||||||
| foszfátérc, a típus megjelölése | |||||||||||||
| után kell megadni a foszfát ható- | |||||||||||||
| anyag tartalmat. | |||||||||||||
| Az oldódó P2O5 tartalmat a kö- | |||||||||||||
| vetkező oldhatósági vizsgála- | |||||||||||||
| tok szerint kell megadni: | |||||||||||||
| — Thomas salak alapú műtrá- | |||||||||||||
| gyák [6. b) típusú oldhatóság | |||||||||||||
| szerint], | |||||||||||||
| — kalcinált foszfát alapú mű- | |||||||||||||
| trágyák (5-ös típusú oldható- | |||||||||||||
| ság szerint), | |||||||||||||
| — alumínium-kalcium- | |||||||||||||
| foszfát alapú műtrágyák (7-es | |||||||||||||
| típusú oldhatóság szerint), | |||||||||||||
| — lágy őrölt foszfátérc alapú | |||||||||||||
| műtrágyák (8-as típusú oldha- | |||||||||||||
| tóság szerint). | |||||||||||||
| Bázikus foszfát hatóanyagok szemcsemérete: | |||||||||||||
| Thomas salak: | legalább 75%-a átesik a 0,160 mm-es szitán; | ||||||||||||
| Alumínium-kalcium-foszfát: | legalább 90%-a átesik a 0,160 mm-es szitán; | ||||||||||||
| Kalcinált foszfát: | legalább 75%-a átesik a 0,160 mm-es szitán; | ||||||||||||
| Lágy őrölt foszfátérc: | legalább 90%-a átesik a 0,063 mm-es szitán; | ||||||||||||
| Részben feltárt foszfátérc: | legalább 90%-a átesik a 0,160 mm-es szitán. | ||||||||||||
B/2. NP MŰTRÁGYÁK
| Típus | Az előállításra vonatkozó adatok | Minimális tápanyag tartalom (tömegszázalékban) | A műtrágyák 8., 9. és 10. oszlopban meghatározott formái, oldhatósága és a megadott tápanyag tartalom; szemcseméret eloszlás | A műtrágyák azonosítási adatai; egyéb követelmények | |||||||||||||
| Összes | Az egyes tápanyagok | N | P2O5 | K2O | N | P2O5 | K2O | ||||||||||
| 1. | 2. | 3. | 4. | 5. | 6. | 7. | 8. | 9. | 10. | ||||||||
| NP | Kémiai úton | 18% | 3% N | 1. Összes nitrogén | 1. Vízben oldódó P2O5 | 1. Összes nitrogén | 1. Az olyan NP műtrágyánál, | ||||||||||
| műtrágyák | vagy keverés- | (N + P2O5) | 5% P2O5 | amely nem tartalmaz Thomas | |||||||||||||
| sel előállított | 2. Nitrát nitrogén | 2. Semleges ammó- | 2. Habármelyiknit- | salakot, kalcinált foszfátércet, | |||||||||||||
| termék, állati | nium-citrátban oldódó | rogén forma (2 — 5) | alumínium-kalcium-foszfátot, | ||||||||||||||
| vagy növényi | 3. Ammónia nit- | P2O5 | mennyisége leg- | részben feltárt foszfátércet és | |||||||||||||
| eredetű szer- | rogén | alább 1 tömeg%, fel | őrölt lágy foszfátércet, a fosz- | ||||||||||||||
| ves tápanya- | 3. Semleges ammó- | kell tüntetni | fortartalmat az 1-es, 2-es és 3- | ||||||||||||||
| gok hozzá- | 4. Karbamid nit- | nium-citrátban és vízben | as típusú oldhatóság szerint | ||||||||||||||
| adása nélkül | rogén | oldódó P2O5 | kell feltüntetni: | ||||||||||||||
| — ha a vízben oldódó P2O5 | |||||||||||||||||
| 5. Ciánamidnitro- | 4. Kizárólag ásványi sa- | nem éri el a 2%-ot, csak a 2-es | |||||||||||||||
| gén | vakban oldódó P2O5 | típusú oldhatóságot kell feltün- | |||||||||||||||
| tetni, | |||||||||||||||||
| 5. Lúgos ammónium- | — ha a vízben oldódó P2O5 | ||||||||||||||||
| citrátban oldódó P2O5 | meghaladja a 2%-ot, a 3-as tí- | ||||||||||||||||
| (Petermann) | pusú oldhatóságot kell feltün- | ||||||||||||||||
| tetni és a vízben oldódó P2O5 | |||||||||||||||||
| 6. a) Ásványi savakban | tartalmat jelölni kell (1-es típu- | ||||||||||||||||
| oldódó P2O5, melynek | sú oldhatóság) | ||||||||||||||||
| legalább 75%-a oldódó | A kizárólag ásványi savakban | ||||||||||||||||
| 2%-os citromsavban | oldódó P2O5 tartalom nem le- | ||||||||||||||||
| het több mint 2% | |||||||||||||||||
| 6. b) 2%-os citromsav- | (Ezen 1-es típus 2-es és 3-as | ||||||||||||||||
| ban oldódó P2O5 | típusú oldhatóságának megha- | ||||||||||||||||
| tározására 1 g mintát kell be- | |||||||||||||||||
| 7. Ásványi savakban ol- | mérni.) | ||||||||||||||||
| dódó P2O5, melynek leg- | |||||||||||||||||
| alább 75%-a oldódó lú- | |||||||||||||||||
| gos ammónium-citrátban | |||||||||||||||||
| (Joulie) | - | ||||||||||||||||
| 8. Ásványi savakban oldódó P2O5, melynek legalább 55%-a oldódó 2%-os hangyasavban | 2. a) Az őrölt lágy foszfátércet vagy részben feltárt foszfátércet tartalmazó NP műtrágyánál, amely nem tartalmazhat Thomas salakot, kalcinált foszfátércet és alumínium-kalcium-foszfátot, a foszfortartalmat az 1 -es, 3-as és 4-es típusú oldhatóság szerint kell feltüntetni. Ezen műtrágya típusnak tartalmaznia kell: | ||||||||||||||||
| — legalább 2% csak ásványi savakban oldódó P2O5-t (4-es típusú oldhatóság), | |||||||||||||||||
| — legalább 5% semleges am-mónium-citrátban és vízben oldódó P2O5-t (3-as típusú oldhatóság), | |||||||||||||||||
| — legalább 2,5% vízben oldódó P2O5-t (1-es típusú oldhatóság) | |||||||||||||||||
| Ezt a típusú műtrágyát a következő megjelöléssel kell forgalomba hozni: „NP műtrágya, őrölt lágy foszfátérc tartalmú" vagy „NP műtrágya, részben feltárt foszfátérc tartalmú' ' [Ezen 2. a) típus 3-as típusú oldhatóságának meghatározására 3 g mintát kell bemérni.] | |||||||||||||||||
| 2. b) Az alumínium-kalcium-foszfátot tartalmazó NP műtrágyánál, amely nem tartalmazhat Thomas salakot, kalcinált foszfátércet, őrölt lágy foszfátércet és részben feltárt foszfátércet, a foszfortartalmat az 1-es és 7-es típusú oldhatóság szerint kell feltüntetni, az utóbbit a vízben oldhatóság levonásával alkalmazzuk | |||||||||||||||||
| Ezen műtrágya típusnak tartalmaznia kell: | |||||||||||||||||
| — legalább 2% vízben oldódó P2O5-t (1-es típusú oldhatóság), | |||||||||||||||||
| — legalább 5%, 7-es típusú oldhatóság szerinti P2O5-t | |||||||||||||||||
| Ezt a típusú műtrágyát a következő megjelöléssel kell forgalomba hozni: „NP műtrágya, alumínium-kalcium-foszfát tartalmú" | |||||||||||||||||
| 3. Azon NP műtrágyák esetében, amelyek kizárólag egyet tartalmaznak az alábbi foszfor műtrágya típusok közül: Thomas salak, kalcinált foszfátérc, alumínium-kalcium-foszfát, őrölt lágy foszfátérc, a típus megjelölése után kell megadni a foszfát hatóanyag tartalmat | |||||||||||||||||
| Az oldódó P2O5 tartalmat a következő oldhatósági vizsgálatok szerint kell megadni: | |||||||||||||||||
| — Thomas salak alapú műtrágyák [6. b) típusú oldhatóság szerint], | |||||||||||||||||
| — kalcinált foszfát alapú műtrágyák (5-ös típusú oldhatóság szerint), | |||||||||||||||||
| — alumínium-kalcium-foszfát alapú műtrágyák (7-es típusú oldhatóság szerint), | |||||||||||||||||
| — lágy őrölt foszfátérc alapú műtrágyák (8-as típusú oldhatóság) szerint. | |||||||||||||||||
| Bázikus foszfát hatóanyagok szemcsemérete: | |||||||||||||||||
| Thomas salak: | legalább 75%-a átesik a 0,160 mm-es szitán; | ||||||||||||||||
| Alumínium-kalcium-foszfát: | legalább 90%-a átesik a 0,160 mm-es szitán; | ||||||||||||||||
| ' | Kalcinált foszfát: | legalább 75%-a átesik a 0,160 mm-es szitán; | |||||||||||||||
| Lágy őrölt foszfátérc: | legalább 90%-a átesik a 0,063 mm-es szitán; | ||||||||||||||||
| Részben feltárt foszfátérc: | legalább 90%-a átesik a 0,160 mm-es szitán. | ||||||||||||||||
B/3. NK MŰTRÁGYÁK
| Típus | Az előállításra vonatkozó adatok | Minimális tápanyag tartalom (tömegszázalékban) | A műtrágyák 8., 9. és 10. oszlopban meghatározott formái, oldhatósága és a megadott tápanyag tartalom; szemcseméret eloszlás | A műtrágyák azonosítási adatai; egyéb követelmények | |||||||
| Összes | Az egyes tápanyagok | N | P2O5 | K2O | N | P2O5 | K2O | ||||
| 1. | 2. | 3. | 4. | 5. | 6. | 7. | 8. | 9. | 10. | ||
| NK | Kémiai úton | 18% | 3% N | 1. Összes nitrogén | Vízben | 1. Összes nitrogén | 1. Vízben ol- | ||||
| műtrágyák | vagy keverés- | (N + K2O) | 5% K2O | oldódó | dódó K2O | ||||||
| sel előállított | 2. Nitrát nitrogén | K2O | 2. Habármelyiknit- | ||||||||
| termék, állati | rogén forma (2 — 5) | 2. Az „ala- | |||||||||
| vagy növényi | 3. Ammónia nit- | mennyisége leg- | csony klór tar- | ||||||||
| eredetű szer- | rogén | alább 1 tömeg%, fel | talmú' ' megje- | ||||||||
| ves tápanya- | kell tüntetni | lölés legfel- | |||||||||
| gok hozzá- | 4. Karbamid nit- | jebb 2% klór- | |||||||||
| adása nélkül | rogén | tartalomra vo- | |||||||||
| natkozhat | |||||||||||
| 5. Ciánamidnitro- | |||||||||||
| . | gén | 3. A klórtarta- | |||||||||
| lom feltüntet- | |||||||||||
| hető | |||||||||||
B/4. PK MŰTRÁGYÁK
| B/4. PK MŰTRÁGYÁK |
| Típus | Az előállításra vonatkozó adatok | Minimális tápanyag tartalom (tömegszázalékban) | A műtrágyák 8., 9. és 10. oszlopban meghatározott tormái, oldhatósága és a megadott tápanyag tartalom; szemcséméret eloszlás | A műtrágyák azonosítási adatai; egyéb követelmények | |||||||
| Összes | Az egyes tápanyagok | N | P2O5 | K2O | N | P2O5 | K2O | ||||
| 1. | 2. | 3. | 4. | 5. | 6. | 7. | 8. | 9. | 10. | ||
| PK | Kémiai úton | 18% | 5% P2O5 | 1. Vízben oldódó P2O5 | Vízben | 1. Az olyan PK műtrágyánál, | 1. Vízben ol- | ||||
| műtrágyák | vagy keverés- | (P2O5 + | 5% K2O | oldódó | amely nem tartalmaz Thomas | dódó K2O | |||||
| sel előállított | K2O) | 2. Semleges ammó- | K2O | salakot, kalcinált foszfátércet, | |||||||
| termék, állati | nium-citrátban oldódó | alumínium-kalcium-foszfátot, | 2. Az „ala- | ||||||||
| vagy növényi | P2O5 | részben feltárt foszfátércet és | csony klór tar- | ||||||||
| eredetű szer- | őrölt lágy foszfátércet, a fosz- | talmú" megje- | |||||||||
| ves tápanya- | 3. Semleges ammó- | fortartalmat az 1-es, 2-es és 3- | lölés legfel- | ||||||||
| gok hozzá- | nium-citrátban és vízben | as típusú oldhatóság szerint | jebb 2% klór- | ||||||||
| adása nélkül | oldódó P2O5 | kell feltüntetni: | tartalomra vo- | ||||||||
| — ha a vízben oldódó P2O5 | natkozhat | ||||||||||
| 4. Kizárólag ásványi sa- | nem éri el a 2%-ot, csak a 2-es | ||||||||||
| vakban oldódó P2O5 | típusú oldhatóságot kell feltün- | 3. A klórtarta- | |||||||||
| tetni, | lom feltüntet- | ||||||||||
| 5. Lúgos ammónium- | — ha a vízben oldódó P2O5 | hető | |||||||||
| citrátban oldódó P2O5 | meghaladja a 2%-ot, a 3-as tí- | ||||||||||
| (Petermann) | pusú oldhatóságot kell feltün- | ||||||||||
| tetni és a vízben oldódó P2O5 | |||||||||||
| 6. a) Ásványi savakban | tartalmat jelölni kell (1-es típu- | ||||||||||
| oldódó P2O5, melynek | sú oldhatóság) | ||||||||||
| legalább 75%-a oldódó | A kizárólag ásványi savakban | ||||||||||
| 2%-os citromsavban | oldódó P2O5 tartalom nem le- | ||||||||||
| het több mint 2% | |||||||||||
| 6. b) 2%-os citromsav- | (Ezen 1-es típus 2-es és 3-as | ||||||||||
| ban oldódó P2O5 | típusú oldhatóságának megha- | ||||||||||
| tározására 1 g mintát kell be- | |||||||||||
| 7. Ásványi savakban ol- | mérni.) | ||||||||||
| dódó P2O5, melynek leg- | |||||||||||
| alább 75%-a oldódó lú- | |||||||||||
| gos ammónium-citrátban | |||||||||||
| (Joulie) | |||||||||||
| 8. Ásványi savakban oldódó P2O5, melynek legalább 55%-a oldódó 2%-os hangyasavban | 2. a) Az őrölt lágy foszfátércet vagy részben feltárt foszfátércet tartalmazó PK műtrágyánál, amely nem tartalmazhat Thomas salakot, kalcinált foszfátércet és alumínium-kalcium-foszfátot, a foszfortartalmat az 1-es, 3-as és 4-es típusú oldhatóság szerint kell feltüntetni. Ezen műtrágya típusnak tartalmaznia kell: | ||||||||||
| — legalább 2% csak ásványi savakban oldódó P2O5-t (4-es típusú oldhatóság), | |||||||||||
| — legalább 5% semleges am-mónium-citrátban és vízben oldódó P2O5-t (3-as típusú oldhatóság), | |||||||||||
| — legalább 2,5% vízben oldódó P2O5-t (1-es típusú oldhatóság) | |||||||||||
| Ezt a típusú műtrágyát a következő megjelöléssel kell forgalomba hozni: „PK műtrágya, őrölt lágy foszfátérc tartalmú' ' vagy „PK műtrágya, részben feltárt foszfátérc tartalmú" [Ezen 2. a) típus 3-as típusú oldhatóságának meghatározására 3 g mintát kell bemérni.], | |||||||||||
| 2. b) Az alumínium-kalciumfoszfátot tartalmazó PK műtrágyánál, amely nem tartalmazhat Thomas salakot, kalcinált foszfátércet, őrölt lágy foszfátércet és részben feltárt foszfátércet, a foszfortartalmat az 1-es és 7-es típusú oldhatóság szerint kell feltüntetni, az utóbbit a vízben oldhatóság levonásával alkalmazzuk | |||||||||||
| Ezen műtrágya típusnak tartalmaznia kell: | |||||||||||
| — legalább 2% vízben oldódó P2O5-t (1-es típusú oldhatóság), | |||||||||||
| — legalább 5%, 7-es típusú oldhatóság szerinti P2O5-t | |||||||||||
| Ezt a típusú műtrágyát a következő megjelöléssel kell forgalomba hozni: „PK műtrágya, alumínium-kalcium-foszfát tartalmú' ' | |||||||||||
| - | 3. Azon PK műtrágyák esetében, amelyek kizárólag egyet tartalmaznak az alábbi foszfor műtrágya típusok közül: Thomas salak, kalcinált foszfátérc, alumínium-kalcium-foszfát, őrölt lágy foszfátérc, atípus megjelölése után kell megadni a foszfát hatóanyag tartalmat | ||||||||||
| Az oldódó P2O5 tartalmat a következő oldhatósági vizsgálatok szerint kell megadni: | |||||||||||
| — Thomas salak alapú műtrágyák [6. b) típusú oldhatóság szerint], | |||||||||||
| — kalcinált foszfát alapú műtrágyák (5-ös típusú oldhatóság szerint), | |||||||||||
| — alumínium-kalcium-foszfát alapú műtrágyák (7-es típusú oldhatóság szerint), | |||||||||||
| — lágy őrölt foszfátérc alapú műtrágyák (8-as típusú oldhatóság) szerint | |||||||||||
| Bázikus foszfát hatóanyagok szemcsemérete: | |||||||||||
| Thomas salak: | legalább 75%-a átesik a 0,160 mm-es szitán; | ||||||||||
| Alumínium-kalcium-foszfát: | legalább 90%-a átesik a 0,160 mm-es szitán; | ||||||||||
| Kalcinált foszfát: | legalább 75%-a átesik a 0,160 mm-es szitán; | ||||||||||
| Lágy őrölt foszfátérc: | legalább 90%-a átesik a 0,063 mm-es szitán; | ||||||||||
| Részben feltárt foszfátérc: | legalább 90%-a átesik a 0,160 mm-es szitán. | ||||||||||
B/5. NPK MŰTRÁGYÁK KROTONILIDÉN-DIKARBAMID IZOBUTILIDÉN-DIKARB AMID
VAGY KARBAMID-FORMALDEHID TARTALOMMAL
| B/5. NPK MŰTRÁGYÁK KROTONILIDÉN-DIKARBAMID |
| IZOBUTILIDÉN-DIKARB AMID VAGY KARBAMID-FORMALDEHID TARTALOMMAL |
| Típus | Az előállításra vonatkozó adatok | Minimális tápanyag tartalom (tömegszázalékban) | A műtrágyák 8., 9. és 10. oszlopban meghatározott formái, oldhatósága és a megadott tápanyag tartalom; szemcseméret eloszlás | A műtrágyák azonosítási adatai; egyéb követelmények | |||||
| Összes | Az egyes tápanyagok | N | P2O5 | K2O | N | P2O5 | K2O | ||
| 1. | 2. | 3. | 4. | 5. | 6. | 7. | 8. | 9. | 10. |
| NPK | Kémiai úton | 20%(N | 5% N | 1. Összes nitrogén | 1. Vízben oldódó P2O5 | Vízben | 1. Összes nitrogén | 1. Az olyan NPK műtrágyá- | 1. Vízben ol- |
| műtrágyák | vagy keverés- | + P2O5 + | A meg- | oldódó | nál, amely nem tartalmaz Tho- | dódó K2O | |||
| krotonili- | sel előállított | K2O) | adott ösz- | 2. Nitrát nitrogén | 2. Semleges | K2O | 2. Habármelyiknit- | mas salakot, kalcinált fosz- | |
| dén-dikar- | termék, állati | szes nitro- | ammónium-citrátban | rogén forma (2 — 4) | fátércet, alumíniumkalcium- | 2. Az „ala- | |||
| bamid, | vagy növényi | gén tarta- | 3. Ammónia nit- | oldódó P2O5 | mennyisége leg- | foszfátot, részben feltárt fosz- | csony klór tar- | ||
| izobutili- | eredetű szer- | lom leg- | rogén | alább 1 tömeg%, fel | fátércet és őrölt lágy foszfátér- | talmú" megje- | |||
| dén-dikar- | ves tápanya- | alább | 3. Semleges | kell tüntetni | cet, a foszfor tartalmat az 1-es, | lölés legfel- | |||
| bamid | gok hozzá- | 25%-a az | 4. Karbamid nit- | ammónium-citrátban és | 2-es és 3-as típusú oldhatóság | jebb 2% klór | |||
| vagy kar- | adása nélkül, | 5., 6. vagy | rogén | vízben oldódó P2O5 | 3. Az 5., 6. és 7. tí- | szerint kell feltüntetni: | tartalomra vo- | ||
| bamid — | krotonilidén- | 7. típusú | pusú nitrogén for- | — ha a vízben oldódó P2O5 | natkozhat | ||||
| formaidé | dikarbamid, | nitrogén | 5. Krotonilidén- | mák egyike | nem éri el a 2%-ot csak a 2-es | ||||
| hid tarta- | izobutilidén- | forma le- | dikarbamid nitro- | A 7. típusú nitrogén | típusú oldhatóságot kell feltün- | 3. A klór tar- | |||
| lommal | dikarbamid | gyen | gén | forma mennyiségét | tetni, | talom feltün- | |||
| vagy karba- | A meg- | a 8. és 9. típusú nit- | — ha a vízben oldódó P2O5 | tethető | |||||
| mid — for- | adott 7. tí- | 6. Izobutilidén- | rogén formában is ki | meghaladja a 2%-ot, a 3-as tí- | |||||
| maldehid tar- | pusú nitro- | dikarbamid nitro- | kell fejezni | pusú oldhatóságot kell feltün- | |||||
| talommal | gén tarta- | gén | tetni és a vízben oldódó P2O5 | ||||||
| lom leg- | tartalmat jelölni kell (1-es típu- | ||||||||
| alább | 7. Karbamid | sú oldhatóság) | |||||||
| 60%-a ol- | formaldehid nitro- | A kizárólag ásványi savakban | |||||||
| dódjon | gén | oldódó P2O5 tartalom nem le- | |||||||
| forró víz- | het több mint 2% | ||||||||
| ben | 8. Karbamid ' | (Ezen 1-es típus 2-es és 3-as | |||||||
| 5% P2O5 | formaldehid nitro- | típusú oldhatóságának megha- | |||||||
| 5% K2O | gén csak forró víz- | tározására 1 g mintát kell be- | |||||||
| ben oldódó | mérni.) | ||||||||
| 9. Karbamid | |||||||||
| formaldehid nitro- | |||||||||
| gén hideg vízben | |||||||||
| oldódó | |||||||||
B/6. NP MŰTRÁGYÁK KROTONILIDÉN-DIKARBAMID,IZOBUTILIDÉN-DIKARBAMID VAGY KARBAMID - FORMALDEHID TARTALOMMAL
| B/6. NP MŰTRÁGYÁK KROTONILIDÉN-DIKARBAMID,IZOBUTILIDÉN-DIKARBAMID VAGY KARBAMID — FORMALDEHID TARTALOMMAL |
| Típus | Az előállításra vonatkozó adatok | Minimális tápanyag tartalom (tömegszázalékban) | A műtrágyák 8., 9. és 10. oszlopban meghatározott tonnái, oldhatósága és a megadott tápanyag tartalom; szemcseméret eloszlás | A műtrágyák azonosítási adatai; egyéb követelmények | |||||
| összes | Az egyes tápanyagok | N | P2O5 | K2O | N | P2O5 | K2O | ||
| 1. | 2. | 3. | 4. | 5. | 6. | 7. | 8. | 9. | 10. |
| NP műtrá- | Kémiai úton | 18% | 5% N | 1. Összes nitrogén | 1. Vízben oldódó P2O5 | 1. Összes nitrogén | 1. Az olyan NP műtrágyánál, | ||
| gyák kro- | vagy keverés- | (N + P2O5) | A meg- | amely nem tartalmaz Thomas | |||||
| tonilidén- | sel előállított | adott ösz- | 2. Nitrát nitrogén | 2. Semleges | 2. Habármelyiknit- | salakot, kalcinált foszfátércet, | |||
| dikarba- | termék, állati | szes nitro- | ammónium-citrátban | rogén forma (2 — 4) | - alumíniumkalcium-foszfátot, | ||||
| mid, izo- | vagy növényi | gén tarta- | 3. Ammónia nit- | oldódó P2O5 | mennyisége leg- | részben feltárt foszfátércet és | |||
| butilidén- | eredetű szer- | lom leg- | rogén | alább 1 tömeg%, fel | őrölt lágy foszfátércet, a fosz- | ||||
| dikar- | ves tápanya- | alább | 3. Semleges | kell tüntetni | for tartalmat az 1-es, 2-es és | ||||
| bamid | gok hozzá- | 25%-a az | 4. Karbamid nit- | ammónium-citrátban és | 3-as típusú oldhatóság szerint | ||||
| vagy kar- | adása nélkül, | 5., 6. vagy | rogén | vízben oldódó P2O5 | 3. Az 5., 6. és 7. tí- | kell feltüntetni: | |||
| bamid | krotonilidén- | 7. típusú | pusú nitrogén for- | — ha a vízben oldódó P2O5 | |||||
| — formal- | dikarbamid, | nitrogén | 5. Krotonilidén- | mák egyike | nem éri el a 2%-ot, csak a 2-es | ||||
| dehid tar- | izobutilidén- | forma le- | dikarbamid nitro- | A 7. típusú nitrogén | típusú oldhatóságot kell feltün- | ||||
| talommal | dikarbamid | gyen | gén | forma mennyiségét | tetni, | ||||
| vagy | A meg- | a 8. és 9. típusú nit- | — ha a vízben oldódó P2O5 | ||||||
| karbamid- | adott 7. tí- | 6. Izobutilidén- | rogén formában is ki | meghaladja a 2%-ot, a 3-as tí- | |||||
| formaldehid | pusú nitro- | dikarbamid nitro- | kell fejezni | pusú oldhatóságot kell feltün- | |||||
| tartalommal | gén tarta- | gén | tetni és a vízben oldódó P2O5 | ||||||
| lom leg- | tartalmat jelölni kell (1-es típu- | ||||||||
| alább | 7. Karbamid | sú oldhatóság) | |||||||
| 60%-a ol- | formaldehid nitro- | A kizárólag ásványi savakban | |||||||
| dódjon | gén | oldódó P2O5 tartalom nem le- | |||||||
| forró víz- | het több mint 2% | ||||||||
| ben | 8. Karbamid | (Ezen 1-es típus 2-es és 3-as | |||||||
| 5% P2O5 | formaldehid nitro- | típusú oldhatóságának megha- | |||||||
| gén csak forró víz- | tározására 1 g mintát kell be- | ||||||||
| ben oldódó | mérni.) | ||||||||
| 9. Karbamid | |||||||||
| formaldehid nitro- | |||||||||
| gén hideg vízben | |||||||||
| oldódó | |||||||||
B/7. NK MŰTRÁGYÁK KROTONILIDÉN-DIKARBAMID IZOBUTILIDÉN-DIKARBAMID VAGY KARBAMID-FORMALDEHID TARTALOMMAL
| B/7. NK MŰTRÁGYÁK KROTONILIDÉN-DIKARBAMID IZOBUTILIDÉN-DIKARBAMID VAGY KARBAMID-FORMALDEHID TARTALOMMAL |
| Típus | Az előállításra vonatkozó adatok | Minimális tápanyag tartalom (tömegszázalékban) | A műtrágyák 8., 9. és 10. oszlopban meghatározott tonnái, oldhatósága és a megadott tápanyag tartalom; szemcseméret eloszlás | A műtrágyák azonosítási adatai; egyéb követelmények | |||||
| Összes | Az egyes tápanyagok | N | P2O5 | K2O | N | P2O5 | K2O | ||
| 1. | 2. | 3. | 4. | 5. | 6. | 7. | 8. | 9. | 10. |
| NK műtrá- | Kémiai úton | 18% | 5% N | 1. Összes nitrogén | Vízben | 1. Összes nitrogén | 1. Vízben ol- | ||
| gyák kro- | vagy keverés- | (N + K2O) | A meg- | oldódó | dódó K2O | ||||
| tonilidén- | sel előállított | adott ösz- | 2. Nitrát nitrogén | K2O | 2. Habármelyiknit- | ||||
| dikarba- | termék, állati | szes nitro- | rogén forma (2 — 4) | 2. Az „ala- | |||||
| mid, izo- | vagy növényi | gén tarta- | 3. Ammónia nit- | mennyisége leg- | csony klór tar- | ||||
| butilidén- | eredetű szer- | lom lég | rogén | alább 1 tömeg%, fel | talmú" megje- | ||||
| dikar- | ves tápanya- | alább | kell tüntetni | lölés legfel- | |||||
| 3amid | gok hozzá- | 25%-a az | 4. Karbamid nit- | jebb 2% klór | |||||
| vagy kar- | adása nélkül, | 5., 6. vagy | rogén | 3. Az 5., 6. és 7. tí- | tartalomra vo- | ||||
| bamid | krotonilidén- | 7. típusú | pusú nitrogén for- | natkozhat | |||||
| — formal- | dikarbamid, | nitrogén | 5. Krotonilidén- | mák egyike | |||||
| dehid tar- | izobutilidén- | forma le- | dikarbamid nitro- | A 7. típusú nitrogén | 3. A klór tar- | ||||
| talommal | dikarbamid | gyen | gén | forma mennyiségét | talom feltün- | ||||
| vagy karba- | A meg- | a 8. és 9. típusú nit- | tethető | ||||||
| mid-formal- | adott 7. tí- | 6. Izobutilidén- | rogén formában is ki | ||||||
| dehid tarta- | pusú nitro- | dikarbamid nitro- | kell fejezni | ||||||
| lommal | gén tarta- | gén | |||||||
| lom leg- | |||||||||
| alább | 7. Karbamid | ||||||||
| 60%-a ol- | formaldehid nitro- | ||||||||
| dódjon | gén | ||||||||
| forró víz- | |||||||||
| ben | 8. Karbamid | ||||||||
| 5% K2O | formaldehid nitro- | ||||||||
| gén csak forró víz- | |||||||||
| ben oldódó | |||||||||
| 9. Karbamid | |||||||||
| formaldehid nitro- | |||||||||
| gén hideg vízben | |||||||||
| oldódó | |||||||||
C. FOLYÉKONY MŰTRÁGYÁK
C/1. EGYSZERŰ FOLYÉKONY MŰTRÁGYÁK
| C/1. EGYSZERŰ FOLYÉKONY MŰTRÁGYÁK |
| Szám | Típus | Az előállításra és az alapvető összetevőkre vonatkozó adatok | Előírt minimális tápanyag tartalom (tömegszázalék); a tápanyagok kifejezésére vonatkozó adatok; egyéb követelmények | A típus megjelölésére vonatkozó egyéb adatok | A deklarált tápanyag tartalom; a tápanyagok formái és oldhatósága; egyéb követelmények | ||||
| 1. | 2 | 3. | 4. | 5. | 6. | ||||
| 1. | Nitrogén műtrágya oldat | Kémiai úton és vízben való oldással nyert termék, atmoszférikus nyomáson állandó | 15% N Összes nitrogénben kifejezve vagy, hacsak | Összes nitrogén, és ha bármelyik nitrogén forma mennyisége legalább tömeg 1%: | |||||
| formában, növényi vagy állati eredetű szer- | egyféle formában van, akkor nitrát, ammó- | — nitrát nitrogén | |||||||
| ves tápanyag hozzáadása nélkül. | nia vagy karbamid nitrogén formában kife- | — ammónia nitrogén | |||||||
| jezve | — karbamid nitrogén | ||||||||
| - | Biuret tartalom: legfeljebb a karbamid-nitrogén x 0,026 | Ha a biuret tartalom kisebb mint 0,2% az „alacsony biuret-tartalmú" jelölést fel lehet tüntetni | |||||||
| 2. | Ammónium-nitrát-karbamid műtrágya oldat | Kémiai úton és vízben való oldással nyert termék, amely ammónium-nitrátot és kar-bamidot tartalmaz. | 26% N Összes nitrogénben kifejezve, ahol a karbamid nitrogén mennyisége az összes nitrogénnek körülbelül a fele Biuret tartalom: legfeljebb 0,5% | Összes nitrogén Nitrát nitrogén Ammónia nitrogén Karbamid nitrogén Ha a biuret tartalom kisebb mint 0,2% az „alacsony biuret-tartalmú" jelölést fel lehet tüntetni | |||||
| 3. | Kalcium-nitrát oldat | A terméket kalcium-nitrát vízben való oldásával nyerik. | 8% N Nitrát nitrogénben kifejezve, az ammónia | A típus megjelölés a következő megjegy- | Összes nitrogén Megadható: | ||||
| nitrogén tartalom legfeljebb 1 % | zésekkel kiegészít- | — nitrát nitrogén | |||||||
| hető: | — ammónia nitrogén | ||||||||
| — levéltrágyázás | — vízben oldódó kalcium oxid az 5. osz- | ||||||||
| céljára | lopban felsorolt felhasználás esetén | ||||||||
| — tápoldatos öntö- | |||||||||
| zés céljára | |||||||||
| 4. | Magnézium-nitrát oldat | Kémiai úton és magnézium-nitrát vízben való oldásával nyert termék. | 6% N Nitrát nitrogénben kifejezve 9% MgO Vízben oldódó magnézium-oxidban kifejezve | Nitrát nitrogén Vízben oldódó magnézium-oxid | |||||
| A pH: legalább 4. | |||||||||
| 5. | Kalcium-nitrát | A terméket kalcium-nitrát vízben való | 8% N | A típus megjelölés a | Összes nitrogén | ||||
| szuszpenzió | szuszpendálásával nyerik. | Összes vagy nitrát és ammónia nitrogén- | következő megjegy- | Nitrát nitrogén | |||||
| ben kifejezve. Az ammónia nitrogén tarta- | zésekkel kiegészít- | Vízben oldódó kalcium-oxid | |||||||
| lom legfeljebb 1 % | hető: | ||||||||
| 14% CaO | — levéltrágyázás | ||||||||
| ' | Vízben oldódó kalcium-oxidban kifejezve | céljára | |||||||
| — oldat és szusz- | |||||||||
| penzió készítés cél- | |||||||||
| jára | |||||||||
| — tápoldatos öntö- | |||||||||
| zés céljára | |||||||||
| 6. | Karbarrüd- formaid | Kémiai úton vagy karbamid-formaldehid | 18% N | Összes nitrogén | |||||
| ehid tartalmú nitro- | és nitrogén műtrágya (A/1 . táblázat a 3. a), | Összes nitrogénben kifejezve | Az a nitrogén forma, amelyik legalább | ||||||
| gén műtrágya oldat | 3. b) és 5. termékek kivételével) vízben | A deklarált összes nitrogén tartalom leg- | 1 tömeg%: | ||||||
| való oldásával keletkezik. | alább 1/3-a karbamid-formaldehid formá- | Nitrát nitrogén | |||||||
| ban van | Ammónia nitrogén | ||||||||
| Biuret tartalom: legfeljebb (karbamid nit- | Karbamid nitrogén | ||||||||
| rogén + karbamid-formaldehid nitrogén) x | Karbamid-formaldehidben kötött nitrogén | ||||||||
| 0,026 | |||||||||
| 7. | Karbamid- formaid | Kémiai úton vagy karbamid-formaldehid | 18% N | Összes nitrogén | |||||
| ehid tartalmú nitro- | és nitrogén műtrágya (A/1. táblázat a 3. a), | Összes nitrogénben kifejezve | Az a nitrogén forma, amelyik legalább | ||||||
| gén műtrágya szusz- | 3. b) és 5. termékek kivételével) vízben | A deklarált összes nitrogén tartalom leg- | 1 tömeg%: | ||||||
| penzió | való szuszpendálásával keletkezik. | alább 1/3-a karbamid-formaldehid formá- | Nitrát nitrogén | ||||||
| ban van | Ammónia nitrogén | ||||||||
| A deklarált érték legalább 60%-a oldódjon | Karbamid nitrogén | ||||||||
| forró vízben | Karbamid-formaldehidben kötött nitrogén | ||||||||
| Biuret tartalom: legfeljebb (karbamid nit- | Hideg vízben oldódó karbamid-formalde- | ||||||||
| rogén + karbamid-formaldehid nitrogén) x | hidben kötött nitrogén | ||||||||
| 0,026 | Csak forró vízben oldódó karbamid-for- | ||||||||
| maldehidben kötött nitrogén | |||||||||
C/2. ÖSSZETETT FOLYÉKONY MŰTRÁGYÁK
C/2. 1. NPK MŰTRÁGYA OLDAT
| Típus | Az előállításra vonatkozó adatok | Minimális tápanyag tanalom (tömegszázalékban) | A műtrágyák 8., 9. és 10. oszlopban meghatározott formái, oldhatósága és a megadott tápanyag tartalom; szemcseméret eloszlás | A műtrágyák azonosítási adatai; egyéb követelmények | |||||
| Összes | Az egyes tápanyagok | N | P2O5 | K2O | N | P2O5 | K2O | ||
| 1. | 2. | 3. | 4. | 5. | 6. | 7. | 8. | 9. | 10. |
| NPK mű- | Kémiai úton | 15% | 2% N | 1. Összes nitrogén | Vízben oldódó P2O5 | Vízben | 1. Összes nitrogén | Vízben oldódó P2O5 | 1. Vízben ol- |
| trágya ol- | és vízben való | (N + | 3% P2O5 | oldódó | dódó K2O | ||||
| dat | oldással nyert | P2O5 + | 3% K2O | 2. Nitrát nitrogén | K2O | 2. Habármelyiknit- | |||
| termék, at- | K2O) | Biuret tar- | rogénforma (2 — 4) | 2. Az „ala- | |||||
| moszférikus | Biuret | talom: leg- | 3. Ammónia nit- | mennyisége leg- | csony klór tar- | ||||
| nyomáson ál- | tartalom: | feljebb | rogén | alább 1 tömeg%, fel | talmú" megje- | ||||
| landó formá- | legfeljebb | karbamid | kell tüntetni | lölés legfel- | |||||
| ban, növényi | karbamid | nitrogén x | 4. Karbamid nit- | Ha a biuret tartalom | jebb 2% klór | ||||
| vagy állati | nitrogén x | 0,026 | rogén | kisebb, mint 0,2%, | tartalomra vo- | ||||
| eredetű szer- | 0,026 | az „alacsony biuret- | natkozhat | ||||||
| ves tápanyag | tartalmú' ' jelölést | ||||||||
| hozzáadása | fel lehet tüntetni | 3. A klór tar- | |||||||
| nélkül | talom feltün- | ||||||||
| tethető | |||||||||
C/2.2. NPK MŰTRÁGYA SZUSZPENZIÓ
| Típus | Az előállításra vonatkozó adatok | Minimális tápanyag tartalom (tömegszázalékban) | A műtrágyák 8., 9. és 10. oszlopban meghatározott tonnái, oldhatósága és a megadott tápanyag tartalom; szemcseméret eloszlás | A műtrágyák azonosítási adatai; egyéb követelmények | |||||
| Összes | Az egyes tápanyagok | N | P2O5 | K2O | N | P2O5 | K2O | ||
| 1. | 2. | 3. | 4. | 5. | 6. | 7. | 8. | 9. | 10. |
| NPK mű- | Folyékony | 20% | 3% N | 1. Összes nitrogén | 1. Vízben oldódó P2O5 | Vízben | 1. Összes nitrogén | A műtrágya nem tartalmazhat | 1. Vízben ol- |
| trágya | termék, mely- | (N + | 4% P2O5 | oldódó | Thomas salakot, alumínium-kal- | dódó K2O | |||
| szuszpen- | ben a táp- | P2O5 + | 4% K2O | 2. Nitrát nitrogén | 2. Semleges | K2O | 2. Habármelyiknit- | ciumfoszfátot, kalcinált foszfá- | |
| zió | anyagok víz- | K2O) | Biuret tar- | ammónium- | rogénforma (2 — 4) | tot, részben oldott foszfátot vagy | 2. Az „ala- | ||
| ben szuszpen- | Biuret | talom: leg- | 3. Ammónia nit- | citrátban oldódó P2O5 | mennyisége leg- | természetes foszfátokat | csony klór tar- | ||
| dált és oldott | tartalom: | feljebb | rogén | alább 1 tömeg%, fel | talmú" megje- | ||||
| formában | legfeljebb | karbamid | 3. Vízben és semleges | kell tüntetni | 1. Ha a vízben oldódó P2O5 tar- | lölés legfel- | |||
| vannak, növé- | a karbamid | nitrogén x | 4. Karbamid nit- | ammónium-citrátban ol- | Ha a biuret tartalom | talom kisebb, mint 2%, csak a | jebb 2% klór | ||
| nyi vagy állati | nitrogén x | 0,026 | rogén | dódó P2O5 | kisebb, mint 0,2%, | 2-es oldhatóságot kell megadni | tartalomra vo- | ||
| eredetű szer- | 0,026 | az „alacsony biuret- | natkozhat | ||||||
| ves tápanyag | tartalmú" jelölést | 2. Ha a vízben oldódó P2O5 | |||||||
| hozzáadása | fel lehet tüntetni | tartalom legalább 2%, a 3-as | 3. A klór tar- | ||||||
| nélkül | típusú oldhatóságot és a vízben | talom feltün- | |||||||
| oldódó P2O5 tartalmat kell | tethető | ||||||||
| megadni | |||||||||
C/2.3. NP MŰTRÁGYA OLDAT
| Típus | Az előállításra vonatkozó adatok | Minimális tápanyag tartalom (tömegszázalékban) | A műtrágyák 8., 9. és 10. oszlopban meghatározott formái, oldhatósága és a megadott tápanyag tartalom; szemcseméret eloszlás | A műtrágyák azonosítási adatai; egyéb követelmények | |||||
| Összes | Az egyes tápanyagok | N | P2O5 | K2O | N | P2O5 | K2O | ||
| 1. | 2. | 3. | 4. | 5. | 6. | 7. | 8. | 9. | 10. |
| NP műtrá- | Kémiai úton | 18% | 3% N | 1. Összes nitrogén | Vízben | 1. Összes nitrogén | Vízben oldódó P2O5 | ||
| gya oldat | és vízben való | (N + P2O5) | 5% P2O5 | oldódó | |||||
| oldással nyert | Biuret | Biuret tar- | 2. Nitrát nitrogén | P2O5 | 2. Habármelyiknit- | ||||
| termék, at- | tartalom: | talom: leg- | rogén forma (2 — 4) | ||||||
| moszférikus | legfeljebb | feljebb | 3. Ammónia nit- | mennyisége leg- | |||||
| nyomáson ál- | karbamid | karbamid | rogén | alább 1 tömeg%, fel | |||||
| landó formá- | nitrogén x | nitrogén x | kell tüntetni | ||||||
| ban, növényi | 0,026 | 0,026 | 4. Karbamid nit- | Ha a biuret tartalom | |||||
| vagy állati | rogén | kisebb mint 0,2%, az | |||||||
| eredetű szer- | „alacsony biuret- | ||||||||
| ves tápanyag | tartalmú' ' jelölést | ||||||||
| hozzáadása | fel lehet tüntetni | ||||||||
| nélkül | |||||||||
C/2.4. NP MŰTRÁGYA SZUSZPENZIÓ
| Típus | Az előállításra vonatkozó adatok | Minimális tápanyag tartalom (tömegszázalékban) | A műtrágyák 8., 9. és 10. oszlopban meghatározott formái, oldhatósága és a megadott tápanyag tartalom; szemcseméret eloszlás | A műtrágyák azonosítási adatai; egyéb követelmények | |||||
| Összes | Az egyes tápanyagok | N | P2O5 | K2O | N | P2O5 | K2O | ||
| 1. | 2. | 3. | 4. | 5. | 6. | 7. | 8. | 9. | 10. |
| NP műtrá- | Folyékony | 18% | 3% N | 1. Összes nitrogén | 1. Vízben oldódó P2O5 | 1. Összes nitrogén | 1. Ha a vízben oldódó P2O5 tar- | ||
| gya szusz- | termék, mely- | (N + P2O5) | 5% P2O5 | talom kisebb, mint 2%, csak a | |||||
| penzió | ben a táp- | Biuret | Biuret tar- | 2. Nitrát nitrogén | 2. Semleges ammóni- | 2. Habármelyiknit- | 2-es oldhatóságot kell megadni | ||
| anyagok víz- | tartalom: | talom: leg- | um-citrátban oldódó | rogén forma (2 — 4) | |||||
| ben szuszpen- | legfeljebb | feljebb | 3. Ammónia nit- | P2O5 | mennyisége leg- | 2. Ha a vízben oldódó P2O5 | |||
| dált és oldott | karbamid | karbamid | rogén | alább 1 tömeg%, fel | tartalom legalább 2%, a 3-as | ||||
| formában | nitrogén x | nitrogén x | 3. Vízben és semleges | kell tüntetni | típusú oldhatóságot és a vízben | ||||
| vannak, növé- | 0,026 | 0,026 | 4. Karbamid nit- | ammónium-citrátban ol- | Ha a biuret tartalom | oldódó P2O5 tartalmat kell | |||
| nyi vagy állati | rogén | dódó P2O5 | kisebb mint 0,2%, az | megadni | |||||
| eredetű szer- | „alacsony biuret- | A műtrágya nem tartalmazhat | |||||||
| ves tápanyag | tartalmú" jelölést | Thomas salakot, alumínium-kal- | |||||||
| hozzáadása | fel lehet tüntetni | cium-foszfátot, kalcinált foszfá- | |||||||
| nélkül | tot, részben oldott foszfátot vagy | ||||||||
| természetes foszfátokat | |||||||||
C/2.5. NK MŰTRÁGYA OLDAT
| Típus | Az előállításra vonatkozó adatok | Minimális tápanyag tartalom (tömegszázalékban) | A műtrágyák 8., 9. és 10. oszlophan meghatározott formái, oldhatósága és a megadott tápanyag tartalom; szemcseméret eloszlás | A műtrágyák azonosítási adatai; egyéb követelmények | |||||
| Összes | Az egyes tápanyagok | N | P2O5 | K2O | N | P2O5 | K2O | ||
| 1. | 2. | 3. | 4. | 5. | 6. | 7. | 8. | 9. | 10. |
| NK műtrá- | Kémiai úton | 15% | 3% N | 1. Összes nitrogén | Vízben | 1. Összes nitrogén | 1. Vízben ol- | ||
| gya oldat | és vízben való | (N + K2O) | 5% K2O | oldódó | dódó K2O | ||||
| oldással nyert | Biuret | Biuret tar- | 2. Nitrát nitrogén | K2O | 2. Habármelyiknit- | ||||
| termék, at- | tartalom: | talom: leg- | rogén forma (2 — 4) | 2. Az „ala- | |||||
| moszférikus | legfeljebb | feljebb | 3. Ammónia nit- | mennyisége leg- | csony klór tar- | ||||
| nyomáson ál- | a karbamid | karbamid | rogén | alább 1 tömeg%, fel | talmú" megje- | ||||
| landó formá- | nitrogén x | nitrogén x | kell tüntetni | lölés legfel- | |||||
| ban, növényi | 0,026 | 0,026 | 4. Karbamid nit- | Ha a biuret tartalom | jebb 2% klór | ||||
| vagy állati | rogén | kisebb mint 0,2%, az | tartalomra vo- | ||||||
| eredetű szer- | „alacsony biuret- | natkozhat | |||||||
| ves tápanyag | tartalmú' ' jelölést | ||||||||
| hozzáadása | fel lehet tüntetni | 3. A klór tar- | |||||||
| nélkül | talom feltün- | ||||||||
| tethető | |||||||||
C/2.6. NK MŰTRÁGYA SZUSZPENZIÓ
| Típus | Az előállításra vonatkozó adatok | Minimális tápanyag tartalom (tömegszázalékban) | A műtrágyák 8., 9. és 10. oszlopban meghatározott formái, oldhatósága és a megadott tápanyag tartalom; szemcseméret eloszlás | A műtrágyák azonosítási adatai; egyéb követelmények | |||||
| Összes | Az egyes tápanyagok | N | P2O5 | K2O | N | P2O5 | K2O | ||
| 1. | 2. | 3. | 4. | 5. | 6 | 7. | 8. | 9. | 10. |
| NK műtrá- | Folyékony | 18% | 3% N | 1. Összes nitrogén | Vízben | 1. Összes nitrogén | 1. Vízben ol- | ||
| gya szusz- | termék, mely- | (N + K2O) | 5% K2O | oldódó | dódó K2O | ||||
| penzió | ben a táp- | Biuret | Biuret tar- | 2. Nitrát nitrogén | K2O | 2. Habármelyiknit- | |||
| anyagok víz- | tartalom: | talom: leg- | rogén forma (2 — 4) | 2. Az „ala- | |||||
| ben szuszpen- | legfeljebb | feljebb | 3. Ammónia nit- | mennyisége leg- | csony klór tar- | ||||
| dált és oldott | a karbamid | karbamid | rogén | alább 1 tömeg%, fel | talmú" megje- | ||||
| formában | nitrogén x | nitrogén x | kell tüntetni | lölés legfel- | |||||
| vannak, növé- | 0,026 | 0,026 | 4. Karbamid nit- | Ha a biuret tartalom | jebb 2% klór | ||||
| nyi vagy állati | rogén | kisebb mint 0,2%, az | tartalomra vo- | ||||||
| eredetű szer- | „alacsony biuret- | natkozhat | |||||||
| ves tápanyag | tartalmú" jelölést | ||||||||
| hozzáadása | fel lehet tüntetni | 3. A klór tar- | |||||||
| nélkül | talom feltün- | ||||||||
| tethető | |||||||||
C/2.7. PK MŰTRÁGYA OLDAT
| Típus | Az előállításra vonatkozó adatok | Minimális tápanyag tartalom (tömegszázalékban) | A műtrágyák 8., 9. és 10. oszlopban meghatározott formái, oldhatósága és a megadott tápanyag tartalom; szemcseméret eloszlás | A műtrágyák azonosítási adatai; egyéb követelmények | |||||
| összes | Az egyes tápanyagok | N | P2O5 | K2O | N | P2O5 | K2O | ||
| 1. | 2. | 3. | 4. | 5. | 6. | 1. | 8. | 9. | 10. |
| PK műtrá- | Kémiai úton | 18% | 5% P2O5 | Vízben oldódó P2O5 | Vízben | Vízben oldódó P2O5 | 1. Vízben ol- | ||
| gya oldat | és vízben való | (P2O5 + | 5% K2O | oldódó | dódó K2O | ||||
| oldással nyert | K2O) | K2O | |||||||
| termék, növé- | 2. Az „ala- | ||||||||
| nyi vagy állati | csony klór tar- | ||||||||
| eredetű szer- | talmú" megje- | ||||||||
| ves tápanyag | lölés legfel- | ||||||||
| hozzáadása | jebb 2% klór | ||||||||
| nélkül | tartalomra vonatkozhat | ||||||||
| 3. A klór tartalom feltüntethető | |||||||||
C/2.8. PK MŰTRÁGYA SZUSZPENZIÓ
| Típus | Az előállításra vonatkozó adatok | Minimális tápanyag tartalom (tömegszázalékban) | A műtrágyák 8., 9. és 10. oszlopban meghatározott formái, oldhatósága és a megadott tápanyag tartalom; szemcseméret eloszlás | A műtrágyák azonosítási adatai; egyéb követelmények | |||||
| összes | Az egyes tápanyagok | N | P2O5 | K2O | N | P2O5 | K2O | ||
| 1. | 2. | 3. | 4. | 5. | 6. | 7. | 8. | 9. | 10. |
| PK műtrá- | Folyékony | 18% | 5% P2O5 | 1. Vízben oldódó P2O5 | Vízben | 1. Ha a vízben oldódó P2O5 tar- | 1. Vízben ol- | ||
| gya szusz- | termék, mely- | (P2O5 + | 5% K2O | oldódó | talom kisebb, mint 2% csak a | dódó K2O | |||
| penzió | ben a táp- | K2O) | 2. Semleges ammó- | K2O | 2-es oldhatóságot kell megadni | ||||
| anyagok víz- | nium-citrátban oldódó | 2. Az „ala- | |||||||
| ben szuszpen- | P2O5 | 2. Ha a vízben oldódó P2O5 | csony klór tar- | ||||||
| dált és oldott | tartalom legalább 2%, a 3-as | talmú" megje- | |||||||
| formában | 3. Vízben és semleges | típusú oldhatóságot és a vízben | lölés legfel- | ||||||
| vannak, növé- | ammónium-citrátban ol- | oldódó P2O5 tartalmat kell | jebb 2% klór | ||||||
| nyi vagy állati | dódó P2O5 | megadni | tartalomra vo- | ||||||
| eredetű szer- | A műtrágya nem tartalmazhat | natkozhat | |||||||
| ves tápanyag | Thomas salakot, alumínium-kal- | ||||||||
| hozzáadása | ciumfoszfátot, kalcinált foszfá- | 3. A klór tar- | |||||||
| nélkül | tot, részben oldott foszfátot vagy | talom feltün- | |||||||
| természetes foszfátokat. | tethető | ||||||||
D. MEZOELEM TARTALMÚ MŰTRÁGYÁK
| Szám | Típus | Az előállításra és az alapvető összetevőkre vonatkozó adatok | Előírt minimális tápanyag tartalom (tömegszázalék); a tápanyagok kifejezésére vonatkozó adatok; egyéb követelmények | A típus megjelölésére vonatkozó egyéb adatok | A deklarált tápanyag tartalom; a tápanyagok formái és oldhatósága; egyéb követelmények |
| 1. | 2 | 3. | 4. | 5. | 6. |
| 1. | Kalcium-szulfát | Természetes vagy ipari eredetű termék, mely különböző kristályvíz tartalmú kalcium-szulfátot tartalmaz | 25% CaO 35% SO3 A kalcium és kén tartalom összes CaO -ban és SO3-ban van kifejezve Szemcseméret: — legalább 80% átesik a 2 mm-es szitán — legalább 99% átesik a 10 mm-es szitán | A használatos kereskedelmi nevek megadhatók | Összes kén-trioxid Az összes kalcium-oxid tartalom is megadható |
| 2. | Kalcium-klorid oldat | Ipari eredetű kalcium-klorid oldat | 12% CaO A kalcium vízben oldódó CaO-ban van kifejezve. | Kalcium-oxid Feltüntethető: kijuttatás levéltrágyaként | |
| 3. | Elemi kén | Többé-kevésbé tisztított, természetes vagy ipari eredetű termék. | 98% S (245% SO3) A kén tartalom összes SO3-ban van kifejezve. | - | Összes kén-trioxid |
| 4. | Kieserit | Ásványi eredetű termék, mely fő alkotóként magnézium-szulfát- monohidrátot tartalmaz. | 24% MgO 45% SO3 A magnézium és kén tartalom vízben oldódó MgO-ban és SO3-ban van kifejezve. | A használatos kereskedelmi nevek megadhatók | Vízben oldódó magnézium-oxid A vízben oldódó kén-trioxid tartalom is megadható |
| 5. | Magnézium-szulfát | A termék fő alkotóként magnézium-szul-fát-heptahidrátot tartalmaz. | 15% MgO 28% SO3 A magnézium és kén tartalom vízben oldódó MgO-ban és SO3-ban van kifejezve. | A használatos kereskedelmi nevek megadhatók | Vízben oldódó magnézium-oxid A vízben oldódó kén-trioxid tartalom is megadható |
| 5.1. | Magnézium-szulfát oldat | A terméket ipari eredetű jnagnézium-szul-fát vízben való oldásával nyerik. | 5% MgO 10% SO3 A magnézium és kén tartalom vízben oldódó MgO-ban és SO3-ban van kifejezve. | A használatos kereskedelmi nevek megadhatók | Vízben oldódó magnézium-oxid A vízben oldódó kén-trioxid tartalom is megadható |
| 5.2. | Magnéziumhidroxid | Kémiai úton nyert termék, amely magnézium-hidroxidot tartalmaz alapvető összetevőként. | 60% MgO Szemcseméret: legalább 99% átesik a 0,063 mm-es szitán | Összes magnézium-oxid | |
| 5.3. | Magnéziumhidroxid szuszpenzió | A terméket az 5.2.-es típus szuszpendálá-sával nyerik | 24% MgO | Összes magnézium-oxid | |
| 6. | Magnézium-klorid oldat | A terméket ipari eredetű magnézium-klorid oldásával nyerik. | 13% MgO A magnéziumot magnézium-oxidban fejezik ki Kalcium tartalom: legfeljebb 3% CaO | Magnézium-oxid |
| A mezoelemek tartalma abban az esetben tüntethető fel, ha a tápelem tartalom eléri legalább az alábbi értékeket: |
| Tápelem | Minimális érték tömegszázalékban (m/m%) |
| Kalcium | 1,4 Ca |
| — Kalcium-oxid | 2,0 CaO |
| Magnézium | 1,2 Mg |
| — Magnézium-oxid | 2,0 MgO |
| Nátrium | 2,2 Na |
| — Nátrium-oxid | 3,0 Na20 |
| Kén | 2,0 S |
| — Kén-trioxid | 5,0 SO3 |
| A kalcium tartalom a folyékony műtrágyáknál akkor tüntethető fel, ha eléri legalább az alábbi értéket: |
| Kalcium | 5,7 Ca |
| — Kalcium-oxid | 8,0 CaO |
E. MIKROELEM TARTALMÚ MŰTRÁGYÁK
E/1. EGYETLEN MIKROELEMET TARTALMAZÓ MŰTRÁGYÁK
| Szám | Típus | Az előállításra és az alapvető összetevőkre vonatkozó adatok | Előírt minimalis tápanyag tartalom (tömegszázalék); a tápanyagok kifejezésére vonatkozó adatok; egyéb követelmények | A típus megjelölésére vonatkozó egyéb adatok | A deklarált tápanyag tartalom; a tápanyagok formai és oldhatósága; egyéb követelmények | ||||||||||
| 1. | 2. | 3. | 4. | 5. | 6. | ||||||||||
| BÓR | |||||||||||||||
| 1. a) | Bórsav | A terméket borát savval történő reakciójával nyerik. | 14% vízben oldódó B | A használatos kereskedelmi nevek megadhatók. | Vízben oldódó bór (B) | ||||||||||
| 1. b) | Nátrium-borát | Kémiai úton nyert termék, amely nátrium-borátot tartalmaz alapvető összetevőként. | 10% vízben oldódó B | A használatos kereskedelmi nevek megadhatók. | Vízben oldódó bór (B) | ||||||||||
| 1. c) | Kalcium-borát | A terméket colemanitból vagy pandermit-ből nyerik, melyek kalcium-borátot tartalmaznak alapvető összetevőként. | 7% összes B Szemcseméret: legalább 98% átesik a 0,063 mm-es szitán | A használatos kereskedelmi nevek megadhatók. | Összes bór (B) | ||||||||||
| 1. d) | Bór-etanol-amin | A terméket bórsav és etanol-amin reakciójával nyerik. | 8% vízben oldódó B | Vízben oldódó bór (B) | |||||||||||
| 1. e) | Bór alapú műtrágya oldat | A terméket az 1. a) és/vagy 1. b) és/vagy 1. d) oldásával nyerik. | 2% vízben oldódó B | A jelölésnek tartalmaznia kell az alkotórészek nevét. | Vízben oldódó bór (B) | ||||||||||
| 1. f) | Bór alapú műtrágya szuszpenzió | A terméket az 1. a) és/vagy 1. b) és/vagy 1. d) vízben való szuszpendálásával nyerik. | 2% vízben oldódó B | A jelölésnek tartalmaznia kell az alkotórészek nevét. | Vízben oldódó bór (B) | ||||||||||
| KOBALT | |||||||||||||||
| 2. a) | Kobalt só | Kémiai úton nyert termék, amely a kobalt ásványi sóját tartalmazza alapvető összetevőként. | 19% vízben oldódó Co | A címkén fel kell tüntetni az ásványi anion nevét. | Vízben oldódó kobalt (Co) | ||||||||||
| 2. b) | Kobalt kelát | Vízben oldódó termék, melyet kobalt és egy kelátképző kémiai reakciójával nyernek. | 2% vízben oldódó Co, a deklarált érték legalább 80%-a kelát kötésben | A kelátképző reagens neve. | Vízben oldódó kobalt (Co) Kelát kötésben levő kobalt (Co) | ||||||||||
| 2. c) | Kobalt műtrágya oldat | A terméket a 2. a) és/vagy a 2. b) típusú műtrágyák vízben való oldásával nyerik. | 2% vízben oldódó kobalt (Co) | A címkének tartalmaznia kell: a) az ásványi ani-on(ok) nevét; b) a kelátképző re a-gens(ek) nevét. | Vízben oldódó kobalt (Co) Kelát kötésben levő kobalt (Co), ha van benne. | ||||||||||
| RÉZ | |||||||||||||||
| 3; a) | Réz só | Kémiai úton nyert termék, amely a réz ásványi sóját tartalmazza alapvető összetevőként | 20% vízben oldódó Cu | A címkén fel kell tüntetni az ásványi anion nevét. | Vízben oldódó réz (Cu) | ||||||||||
| 3. b) | Réz-oxid | Kémiai úton nyert termék, amely réz-oxidot tartalmaz alapvető összetevőként | 70% összes Cu Szemcseméret: legalább 98% átesik a 0,063 mm-es szitán | Összes réz (Cu) | |||||||||||
| 3. c) | Réz-hidroxid | Kémiai úton nyert termék, amely a réz-hidroxidot tartalmaz alapvető összetevőként | 45% összes Cu Szemcseméret: legalább 98%-a átesik a 0,063 mm-es szitán | Összes réz (Cu) | |||||||||||
| 3. d) | Réz-kelát | Vízben oldódó termék, melyet réz és egy kelátképző kémiai reakciójával nyernek | 9% vízben oldódó Cu, a deklarált érték legalább 80%-a kelát kötésben | A kel átképző reagens neve. | Vízben oldódó réz (Cu) Kelát kötésben levő réz (Cu) | ||||||||||
| 3. e) | Réz alapú műtrágya | A terméket a 3. a) és/vagy a 3. b) és/vagy a 3. c) és/vagy egy egyedüli 3. d) típus, és ha szükséges egy sem tápanyagot sem toxikus anyagot nem tartalmazó adalékanyagból, keveréssel nyerik | 5% összes Cu | A címkének tartalmaznia kell: a) az ásványi ani-on(ok) nevét; b) a kelátképző re a-gens(ek) nevét, ha van benne. | Összes réz (Cu) Vízben oldódó réz (Cu), ha ennek mennyisége legalább az összes réz L' -e Kelát kötésben levő réz (Cu), ha van benne | ||||||||||
| 3. f) | Réz oldat műtrágya | A terméket a 3. a) és/vagy a 3. d) típus vízben való oldásával nyerik | 3% vízben oldódó Cu | A címkének tartalmaznia kell: a) az ásványi ani-on(ok) nevét; b) a kelátképző rea-gens(ek) nevét, ha van benne. | Víz-oldódó réz (Cu) Kelát kötésben levő réz (Cu), ha van benne | ||||||||||
| 3. g) | Réz-oxiklorid | Kémiai úton nyert termék, amely réz-oxi-kloridot tartalmaz alapvető összetevőként | 50% összes Cu Szemcseméret: legalább 98%-a átesik a 0,063 mm-es szitán | Összes réz (Cu) | |||||||||||
| 3. h) | Réz-oxiklorid szuszpenzió | A terméket a 3. g) típus vízben való szusz-pendálásával nyerik | 17% összes Cu | Összes réz (Cu) | |||||||||||
| VAS | |||||||||||||||
| 4. a) | Vas só | Kémiai úton nyert termék, amely a vas ásványi sóját tartalmazza alapvető összetevőként. | 12% vízben oldódó Fe | A címkén fel kell tüntetni az ásványi anion nevét. | Vízben oldódó vas (Fe) | ||||||||||
| 4. b) | Vas ke lát | Vízben oldódó termék, melyet vas és egy kelátképző kémiai reakciójával nyernek. | 5% vízben oldódó Fe, a deklarált érték legalább 80%-a kelát kötésben. | A kelátképző reagens neve. | Vízben oldódó vas (Fe) Kelát kötésben levő vas (Fe) | ||||||||||
| 4. c) | Vas műtrágya oldat | A terméket a 4. a) és/vagy a 4. b) típus vízben való oldásával nyerik. | 2% vízben oldódó Fe | A címkének tartalmaznia kell: a) az ásványi ani-on(ok) nevét; b) a kelátképző rea-gens(ek) nevét, ha van benne. | Vízben oldódó vas (Fe) Kelát kötésben levő vas (Fe), ha van benne. | ||||||||||
| MANGÁN | |||||||||||||||
| 5. a) | Mangán só | Kémiai úton nyert termék, amely a mangán (Mn ) ásványi sóját tartalmazza alapvető összetevőként. | 17% vízben oldódó Mn | A címkén fel kell tüntetni az ásványi anion nevét. | Víz-oldódó mangán (Mn) | ||||||||||
| 5. b) | Mangán kelát | Vízben oldódó termék, melyet mangán és egy kelátképző kémiai reakciójával nyernek. | 5% vízben oldódó Mn, a deklarált érték legalább 8/10-e kelát kötésben. | A kelátképző reagens neve. | Víz-oldódó mangán (Mn) Kelát kötésben levő mangán (Mn) | ||||||||||
| 5. c) | Mangán-oxid | Kémiai úton nyert termék, amely a mangán-oxidokat tartalmazza alapvető összetevőként. | 40% összes Mn Szemcseméret: legalább 80%-a átesik a 0,063 mm-es szitán. | Összes mangán (Mn) | |||||||||||
| 5. d) | Mangán alapú műtrágya | A terméket az 5. a) és az 5. c) típusból keveréssel nyerik. | 17% összes Mn | A címkének tartalmaznia kell a mangán komponensek nevét. | Összes mangán (Mn) Víz-oldódó mangán (Mn), ha ennek mennyisége legalább az összes mangán 1/4-e | ||||||||||
| 5. e) | Mangán alapú műtrágya oldat | A terméket az 5. aj és/vagy az 5. b) típus vízben való oldásával nyerik. | 3% vízben oldódó Mn | A címkének tartalmaznia kell: a) az ásványi ani-on(ok) nevét; b) a kelátképző rea-gens(ek) nevét, ha van benne. | Vízben oldódó mangán (Mn) Kelát kötésben levő mangán (Mn) ha van benne. | ||||||||||
| MOLIBDÉN | |||||||||||||||
| 6. a) | Nátrium-molibden át | Kémiai úton nyert termék, amely nátrium-molibdenátot tartalmaz alapvető összetevőként. | 35% vízben oldódó Mo | Vízben oldódó molibdén (Mo) | |||||||||||
| 6. b) | Ammónium-molib denát | Kémiai úton nyert termék, amely ammó-nium-molibdenátot tartalmaz alapvető összetevőként. | 50% vízben oldódó Mo | Vízben oldódó molibdén (Mo) | |||||||||||
| 6. c) | Molibdén alapú műtrágya | A terméket a 6. a) és a 6. b) típusból keveréssel nyerik | 35% vízben oldódó Mo | A címkének tartalmaznia kell a molib-dén komponens(ek) nevét. | Vízben oldódó molibdén (Mo) | ||||||||||
| 6. d) | Molibdén alapú műtrágya oldat | A terméket a 6. aj és/vagy a 6. b) típus vízben való oldásával nyerik | 3% vízben oldódó Mo | A címkének tartalmaznia kell a molib-dén komponens(ek) nevét. | Vízben oldódó molibdén (Mo) | ||||||||||
| CINK | |||||||||||||||
| 7. a) | Cink só | Kémiai úton nyert termék, amely a cink ásványi sóját tartalmazza alapvető összetevőként | 15% vízben oldódó Zn | A címkén fel kell tüntetni az ásványi anion nevét. | Vízben oldódó cink (Zn) | ||||||||||
| 7. b) | Cink kelát | Vízben oldódó termék, melyet cink és egy kelátképző kémiai reakciójával nyernek. | 5% vízben oldódó Zn, a deklarált érték legalább 8/10-e kelát kötésben van | A kelátképző reagens neve. | Vízben oldódó cink (Zn) Kelát kötésben levő cink (Zn) | ||||||||||
| 7. c) | Cink-oxid | Vegyi úton nyert termék, amely cink-oxidot tartalmaz alapvető összetevőként | 70% összes Zn Szemcseméret: legalább 80%-a átesik a 0,063 mm-es szitán | Összes cink (Zn) | |||||||||||
| 7. d) | Cink alapú műtrágya | A terméket a 7. a) és a 7. c) típusból keveréssel nyerik | 30% összes Zn | A címkének tartalmaznia kell a cink komponens(ek) nevét. | Összes Cink (Zn) Vízben oldódó cink (Zn), ha ennek mennyisége legalább az összes cink 1/4-e | ||||||||||
| 7. e) | Cink alapú műtrágya oldat | A terméket a 7. a) és/vagy a 7. b) típus vízben való oldásával nyerik | 3% vízben oldódó Zn | A címkének tartalmaznia kell: a) az ásványi ani-on(ok) nevét; b) a kelátképző rea-gens(ek) nevét, ha van benne. | Vízben oldódó cink (Zn) Kelát kötésben levő cink (Zn), ha van jelen | ||||||||||
E/2. ÖSSZETETT MIKROELEM TARTALMÚ MŰTRÁGYÁK HATÓANYAGTARTALMÁNAK ELŐÍRÁSAI
E/2.1. SZILÁRD ÉS FOLYÉKONY MIKROELEM MŰTRÁGYÁK KEVERÉKEINEK LEGKISEBB
MIKROELEM TARTALMA
| Mikroelemek | Kizárólag szervetlen formában levő mikroelem (tömegszázalék) | Kelát vagy komplex formában levő mikroelem (tömegszázalék) |
| Bór (B) | 0,2 | 0,2 |
| Kobalt (Co) | 0,02 | 0,02 |
| Réz (Cu) | 0,5 | 0,1 |
| Vas (Fe) | 2,0 | 0,3 |
| Mangán (Mn) | 0,5 | 0,1 |
| Molibdén (Mo) | 0,02 | — |
| Cink (Zn) | 0,5 | 0,1 |
A szilárd műtrágya keverék mikroelem tápanyag tartalma: legalább 5 tömeg%. A folyékony műtrágya keverék mikroelem tartalma: legalább 2 tömeg%.
E/2.2. TALAJTRÁGYÁZÁSRA SZOLGÁLÓ MAKRO- ÉS/VAGY MEZOELEMEKET IS TARTALMAZÓ ÖSSZETETT MIKROELEM TARTALMÚ MŰTRÁGYÁK LEGKISEBB MIKROELEM TARTALMA
| Mikroelemek | Szántó és rét-legelő (tömegszázalék) | Kertészeti kultúrák (tömegszázalék) |
| Bór (B) | 0,01 | 0,01 |
| Kobalt (Co) | 0,002 | — |
| Réz (Cu) | 0,01 | 0,002 |
| Vas (Fe) | 0,5 | 0,02 |
| Mangán (Mn) | 0,1 | 0,01 |
| Molibdén (Mo) | 0,001 | 0;001 |
| Cink (Zn) | 0,01 | 0,002 |
E/2.3. LEVÉLTRÁGYÁZÁSRA SZOLGÁLÓ MAKRO- ÉS/VAGY MEZELEMEKET IS TARTALMAZÓ ÖSSZETETT MIKROELEM TARTALMÚ MŰTRÁGYÁK LEGKISEBB MIKROELEM TARTALMA
| Mikroelemek | Tömegszázalék . |
| Bór (B) | 0,01 |
| Kobalt (Co) | 0,002 |
| Réz (Cu) | 0,002 |
| Vas (Fe) | 0,02 |
| Mangán (Mn) | 0,01 |
| Molibdén (Mo) | 0,001 |
| Cink (Zn) | 0,002 |
E/3. MAGYARORSZÁGON FELHASZNÁLHATÓ KELÁTKÉPZŐK
A következő savak, illetve azok nátrium, kálium vagy ammónium sói:
| Etiléndiamin-tetraecetsav | EDTA | C10H16O8N2 |
| Dietiléntriamin-pentaecetsav | DPTA | C14H23O10N3 |
| Etiléndiamin-di(o-hiroxifenil)ecetsav | EDDHÁ | C18H26O6N2 |
| Hidroxi-2-etiléndiamin-tetraecetsav | HEEDTA | C10H18O7N2 |
| Etiléndiamin-di (o-hidroxi-p-metil-fenil)ecetsav | EDDHMA | C20H24O6N2 |
| Etiléndiamin-di (5-karboxi-2-hidroxi-fenil)ecetsav | EDDCHA | C20H20O10N2 |
| Parafenol-szulfonsav K- vagy Mg sója | KPSHA" |
3. számú melléklet 2. számú függeléke
A műtrágyákra vonatkozó toleranciák
- A táblázatokban megadott toleranciák a tápanyag értékének a címkén, műbizonylaton feltüntetett értéktől való megengedett eltérését jelentik.
- A toleranciák célja a gyártás, a mintavétel és az elemzés során jelentkező eltérések figyelembevétele.
- A 3. számú melléklet 1. számú függelékében meghatározott minimális és maximális értékek tekintetében semmiféle tűrés figyelembevétele nem engedélyezett.
- Ha maximális érték nincs megadva, a tápanyagokat tekintve a feltüntetett érték túllépése - a mikroelemeket tartalmazó műtrágyák kivételével - nem korlátozott.[70]
- A különböző nitrogén formák vagy a foszfor különböző oldhatósági formák esetében a feltüntetett tartalomra vonatkozó tolerancia az adott tápanyag összes tartalmának 0,1-e, de legfeljebb 2 tömegszázalék, feltéve, hogy az adott tápanyag összes tartalma a 3. számú melléklet 1. számú függelékében meghatározott határokon és a táblázatokban meghatározott toleranciákon belül marad.
A. EGYSZERŰ MŰTRÁGYÁK
A/1. NITROGÉN MŰTRÁGYÁK
| Műtrágya típus | Tolerancia (m/m%) |
| Kalcium-nitrát | 0,4 N |
| Kalcium-magnézium-nitrát | 0,4 N |
| Magnézium-nitrát | 0,4 N |
| Nátrium-nitrát | 0,4 N |
| Chilei salétrom | 0,4 N |
| Kalcium-ciánamid | 1,0 N |
| Nitrát tartalmú kalcium-ciánamid | 1,0 N |
| Ammónium-szulfát | 0,3 N |
| Ammónium-nitrát vagy kalcium-ammónium-nitrát – 32 tömegszázalék N tartalom alatt – 32 tömegszázalék N tartalom felett | 0,8 N 0,6 N |
| Ammónium-szulfát-nitrát | 0,8 N |
| Magnézium-szulfo-nitrát | 0,8 N |
| Magnézium-ammónium-nitrát | 0,8 N |
| Karbamid | 0,4 N |
| Krotonilidén-dikarbamid | 0,5 N |
| Izobutilidén-dikarbamid | 0,5 N |
| Karbamid-formaldehid | 0,5 N |
| Krotonilidén-dikarbamid tartalmú nitrogén műtrágya | 0,5 N |
| Izobutilidén-dikarbamid tartalmú nitrogén műtrágya | 0,5 N |
| Karbamid-formaldehid tartalmú nitrogén műtrágya | 0,5 N |
| Ammónium-szulfát nitrifikáció gátlóval (diciándiamid) | 0,5 N |
| Ammónium-szulfo-nitrát nitrifikáció gátlóval (diciándiamid) | 0,5 N |
| Karbamid-ammónium-szulfát | 0,5 N |
A/2. FOSZFOR MŰTRÁGYÁK
| Műtrágya típus | Tolerancia (m/m%) |
| Bázikus salak, Thomas foszfátok, Thomas salak – 2 tömegszázaléknyi határok között kifejezett feltüntetés esetén – egyetlen számmal kifejezett feltüntetés esetén | – 1,0 P2O5 |
| Szuperfoszfát – Semleges ammónium-citrátban oldható – Vízoldható | 0,8 P2O5 0,9 P2O5 |
| Koncentrált szuperfoszfát – Semleges ammónium-citrátban oldható – Vízoldható | 0,8 P2O5 0,9 P2O5 |
| Triplaszuperfoszfát – Semleges ammónium-citrátban oldható – Vízoldható | 0,8 P2O5 1,3 P2O5 |
| Részlegesen feltárt foszfátérc – Ásványi savban oldható – Vízoldható | ' 0,8 P2O5 0,9 P2O5 |
| Dikalcium-foszfát – Lúgos ammónium-citrátban oldható | 0,8 P2O5 |
| Kalcinált foszfát – Lúgos ammónium-citrátban oldható | 0,8 P2O5 |
| Alumínium-kalcium-foszfát – Ásványi savban oldható – Lúgos ammónium-citrátban oldható | 0,8 P2O5 0,8 P2O5 |
| ínium-kalcium-foszfát – Ásványi savban oldható – Hangyasavban oldható | 0,8 P2O5 0,8 P2O5 |
A/3. KÁLIUM MŰTRÁGYÁK
| Műtrágya típus | Tolerancia (m/m%) |
| Kamit | 1,5 K2O |
| Dúsított kainit só | 1,0 K2O |
| Kálium-klorid – 55 tömegszázalék K2O alatt – 55 tömegszázalék K2O felett | 1,0 K2O 0,5 K2O |
| Magnéziumsó tartalmú kálium-klorid | 1,5 K2O |
| Kálium-szulfát | 0,5 K2O |
| Magnéziumsó tartalmú kálium-szulfát | 1,5 K2O |
| Kieserit kálium szulfáttal | 1,0 K2O |
| K2OMagnéziumsó – Magnézium – Klór | 0,9 Mg0 vagy 0,55 Mg 0,2 Cl |
B. ÖSSZETETT MŰTRÁGYÁK
| Műtrágya típus | Tolerancia (m/m%) |
| NPK műtrágyák – összes hatóanyagra vonatkoztatva – N-re vonatkoztatva – P2O5-re vonatkoztatva – K2O-ra vonatkoztatva | 1,9 1,1 N 1,1 P2O5 1,1 K2O |
| NP műtrágyák – összes hatóanyagra vonatkoztatva – N-re vonatkoztatva – P2O5-re vonatkoztatva | 1,5 1,1 N 1,1 P2O5 |
| NK műtrágyák – összes hatóanyagra vonatkoztatva – N-re vonatkoztatva – K2O-ra vonatkoztatva | 1,5 1,1 N 1,1 K2O |
| PK műtrágyák – összes hatóanyagra vonatkoztatva – P2O5-re vonatkoztatva – K2O-ra vonatkoztatva | 1,5 1,1 P2O5 1,1 K2O |
C. FOLYÉKONY MŰTRÁGYÁK
C/1. EGYSZERŰ FOLYÉKONY MŰTRÁGYÁK
| Műtrágya típus | Tolerancia (m/m%) |
| Nitrogén műtrágya oldat | 0,6 |
| Ammónium-nitrát-karbamid műtrágya oldat | 0,6 |
| Kalcium-nitrát oldat | |
| Magnézium-nitrát oldat | |
| Kalcium-nitrát szuszpenzió | 0,4 |
| Karbamid-formaldehid tartalmú nitrogén műtrágya oldat | 0,4 |
| Karbamid-formaldehid tartalmú nitrogén műtrágya szuszpenzió | 0,4 |
C/2. ÖSSZETETT FOLYÉKONY MŰTRÁGYÁK
| Műtrágya típus | Tolerancia (m/m%) |
| NPK műtrágya oldat és műtrágya szuszpenzió | |
| – összes hatóanyagra vonatkoztatva – N-re vonatkoztatva – P2O5-re vonatkoztatva – K2O-ra vonatkoztatva | 1,9 1,1 N 1,1 P2O5 1,1 K2O |
| NP műtrágya oldat és műtrágya szuszpenzió | |
| – összes hatóanyagra vonatkoztatva – N-re vonatkoztatva – P2O5-re vonatkoztatva | 1,5 1,1 N 1,1 P2O5 |
| NK műtrágya oldat és műtrágya szuszpenzió | |
| – összes hatóanyagra vonatkoztatva – N-re vonatkoztatva – K2O-ra vonatkoztatva | 1,5 1,1 N 1,1 K2O |
| Műtrágya típus | Tolerancia |
| PK műtrágya oldat és műtrágya szuszpenzió – összes hatóanyagra vonatkoztatva – P2O5-re vonatkoztatva – K2O-ra vonatkoztatva | 1,5 1,1 P2O5 1,1 K2O |
D. MEZOELEM TARTALMÚ MŰTRÁGYÁK
A tolerancia a deklarált érték egynegyede, de legfeljebb 0,9%
| Tápelem típus | Tolerancia (m/m%) |
| Kalcium – Kalcium-oxid | 0,64 Ca 0,9 CaO |
| Magnézium – Magnézium-oxid | 0,55 Mg 0,9 MgO |
| Nátrium – Nátrium-oxid | 0,67 Na 0,9 Na2O |
| Kén – Kén-trioxid | 0,36 S 0,9 S03 |
E. MIKROELEM TARTALMÚ MŰTRÁGYÁK[71]
| Mikroelem tartalom (m/m%) | Tolerancia (m/m%) |
| 2 tömegszázalék felett | +/- 0,4 |
| 2 tömegszázalék alatt | +/ - a feltüntetett tartalom 20%-a |
F. EGYÉB MŰTRÁGYÁK[72]
| Hatóanyag tartalom (m/m%) | Tolerancia (m/m%) |
| 2 tömegszázalék alatt | +/- a feltüntetett tartalom 20%-a |
| 2 - 10 tömegszázalék között | +/- 0,4 |
| 10 tömegszázalék felett | +/- 1,1 |
4. számú melléklet a 8/2001. (1. 26.) FVMrendelethez
Az engedélyezett termésnövelő anyag csomagolóeszközén, valamint a 100 kg nettó tömeget meghaladó áru és az ömlesztett áru kísérő okmányán feltüntetendő adatok
1. Az engedély száma és érvényesség ideje.
2. A termésnövelő anyag kereskedelmi neve (feltűnő helyen, egyértelműen, olvashatóan).
3. A termésnövelő anyag engedélyben meghatározott típusa.
4. A gyártó neve, címe és statisztikai számjele.
5. Az engedélyes neve, címe és statisztikai számjele.
6. Az engedélyben előírt tápanyag tartalom és oldhatóság a 4. számú melléklet függelékében meghatározott módon.
7. Felhasználási előírás.
8. Veszélyességi besorolás, veszély jelek, a kockázatokra (R) és a biztonságra (S) utaló mondatok, az alkalmazás során előírt védőeszközök, elsősegélynyújtási eljárás.
9. Környezetvédelmi előírások.
10. Tűzveszélyességi osztályba sorolás.
11. Tárolási előírások.*
12. Gyártási idő (év, hónap, nap), eltarthatóság.
13. A termésnövelő anyag - kérelmező által megadott - vámtarifaszáma hat számjegyig.[73]
14. Garantált nettó tömeg.
15. Ha a termésnövelő anyag a veszélyes áruk szállításáról szóló (ADR/RID) szabályzatok hatálya alá esik, a termék UN száma és ADR/RlD osztálya.
16. Egyéb előírások.
* Folyékony műtrágyáknál külön ki kell emelni a tárolási hőmérsékletre és a tárolás során bekövetkező balesetek megelőzésére vonatkozó elbírásokat.
4. számú melléklet függeléke
A tápelemek feltüntetésére vonatkozó előírások
1. A csomagoláson, címkén, illetve a kísérőokmányon a tápelemeket vegyjellel és szavakkal is, a táblázat szerint kell feltüntetni. A táblázat tartalmazza az elemi és oxidos forma közötti átváltás vonatkozó szorzószámait. A tápelemek sorrendje egyberi a felsorolás kötelező sorrendjét is jelenti.
2. A szilárd műtrágyák tápelem tartalmát tömegszázalékban (m/m%), a folyékony műtrágyákét vegyes százalékban (m/v°o) kell feltüntetni. A makro- és mezoelemeket egész számokban, adott esetben egy tizedes helyig kell megadni. A mikroelemeket a 3. számú melléklet 1. számú függelékének előírásai szerinti minimális mikroelem tartalomnak megfelelő tizedes helyig kell megadni.
3. Az oldhatóságot a műtrágya tömegszázaléka szerint kell kifejezni. Amennyiben az elem teljes mértékben vízben oldható, akkor csak a vízben oldható tartalmat kell feltüntetni, ha a vízben oldható mennyiség eléri a teljes mennyiség egynegyedét, a teljes és vízben oldható mennyiséget egyidejűleg kell feltüntetni.
4. A műtrágya mikroelem tartalmát akkor lehet feltüntetni, ha a 3. számú melléklet 1. számú függelékének a minimális mikroelem tartalomra vonatkozó előírása teljesül, és ha a műtrágya egyébként is megfelel a 3. számú melléklet 1. számú függeléke előírásainak.
5. A csak mikroelemet tartalmazó műtrágyánál a "mikroelem keverék" és az elemek nevét kell feltüntetni, ha a mikroelemeket más műtrágyához adagolják, akkor a műtrágya típus meghatározását a "mikroelemek hozzáadásával" és az elemek nevének kell követnie.
6. Amikor a mikroelemek kelőt vagy komplex formában vannak kötve, akkor a mikroelem nevének megadása után a ........ kelátja/komplexe pontosítást kell alkalmazni és fel kell tüntetni a kelátképző vegyület nevét.
7. A mikroelemeket tartalmazó műtrágyák csomagolásán, címkéjén, illetve a kísérő okmányon fel kell tüntetni az alábbi szöveget is:
"Kizárólag tényleges szükség esetén alkalmazandó, a túladagolás kerülendő!"
| Tápelem neve | Vegyjel | Átszámítás | |
| Makroelemek | Nitrogén | N | |
| Foszfor – Foszfor-pentoxid | P P2O5 | P = P2O5 × 0,436 | |
| Kálium – Kálium-oxid | K K2O | K = K2O × 0,830 | |
| Mezoelemek | Kalcium – Kalcium-oxid | Ca CaO | Ca = CaO × 0,715 |
| Magnézium – Magnézium-oxid | Mg MgO | Mg = MgO × 0,6 | |
| Nátrium – Nátrium-oxid | Na Na2O | Na = Na2O × 0,742 | |
| Kén – Kén-tűoxid Klór | S SO3O C1 | S = SO3 × 0,400 | |
| Mikroelemek | Bór Kobalt Réz Vas Mangán Molibdén Cink | B Co Cu Fe Mn Mo Zn |
5. számú melléklet a 8/2001. (I. 26.) FVM rendelethez
Bejelentő lap mintája
BEJELENTŐ LAP TERMÉSNÖVELŐ ANYAG IMPORTÁLÁSÁRÓL
A termésnövelő anyag kereskedelmi neve:
A termésnövelő anyag engedélyokiratának száma:
A behozandó termésnövelő anyag mennyisége:
Az importőr neve, címe és statisztikai számjele:
Dátum
importőr aláírása
6. számú melléklet a 8/2001. (I. 26.) FVM rendelethez
Kísérleti felhasználás iránti kérelem
1. Kérelmező neve és címe
2. Kísérleti készítmény
a) kereskedelmi neve,
b) termésnövelő anyag típusa az 1. számú melléklet függeléke szerint,
c) gyártó neve, címe és statisztikai számjele,
d) összetétele, hatóanyagok, alapanyagok, szennyező anyagok megnevezése és részaránya,
e) a gyártó által szavatolt minőségi jellemzők,
f) a gyártó által megadott felhasználási lehetőségek és alkalmazási dózisok,
g) tűzveszélyességi osztályba sorolás,
h) a gyártó által megadott tárolási és eltarthatósági feltételek.
3. Kísérlet
a) helyszíne (megye, helység, gazdaság, földhasználó),
b) az összes kísérleti terület nagysága ha-ban kifejezve,
c) kísérleti növény (faj, fajta, alany),
d) kísérlet elrendezése (parcellaméret, kezelésszám, ismétlések száma),
e) kezelések adatai (dózis, koncentráció, fenofázis, kezelés módja, felhasznált vízmennyiség),
f) értékelési szempontok.
7. számú melléklet a 8/2001. (I. 26.) FVM rendelethez[74]
MINTAVÉTELI ÉS ELEMZÉSI MÓDSZEREK
I. MINTAVÉTELI ELJÁRÁS MŰTRÁGYÁK ELLENŐRZÉSÉHEZ
1. CÉL ÉS ALKALMAZÁSI TERÜLET
A műtrágyák minőségének és összetételének hatósági ellenőrzése céljára szánt mintákat az alábbi módszerek szerint kell venni. Az így nyert mintákat a megmintázott tételekre nézve reprezentatívnak kell tekinteni.
2. HATÓSÁGI MINTAVEVŐK
A mintákat a tagállamok által e célra felhatalmazott szakképzett hatósági mintavevőnek kell vennie.
3. FOGALOMMEGHATÁROZÁSOK
Megmintázott tétel: a termék egy egységet képező, feltételezetten egységes tulajdonságokat mutató mennyisége.
Elemi minta: a megmintázott tétel egy pontjából vett mennyiség.
Átlagminta: azonos tételből vett elemi minták egyesítéséből származó minta.
Redukált minta: az átlagminta reprezentatív része, amelyet az átlagmintából csökkentési eljárással nyertek.
Végső (laboratóriumi) minta: a redukált minta reprezentatív része.
4. FELSZERELÉS
4.1. A mintavételi felszerelésnek olyan anyagból kell készülnie, amely a mintázandó termék tulajdonságait nem változtatja . meg. E felszerelés kizárólag a tagállamok által hivatalosan jóváhagyott lehet.
4.2. Szilárd műtrágyák mintavételére javasolt eszközök
4.2.1. Kézi mintavétel
4.2.1.1. Lapos fenekű kézi lapát függőleges oldalfalakkal.
4.2.1.2. Mintavevő szonda hosszú nyílással vagy kamrákkal. A mintavevő szonda mérete alkalmazkodjon a mintázandó tétel jellemzőihez (tartály mélysége, zsákméret stb.) és a műtrágya szemcsenagyságához.
4.2.2. Automata mintavétel
Jóváhagyott automata mintavevő készüléket lehet használni a mozgásban lévő műtrágya mintázásakor (a tétel átrakásakor vagy ki- és betárolása esetén).
4.2.3. Mintaosztó
Olyan berendezés, amelyet a minta egyenlő méretű részekre való osztására készítettek, alkalmazható elemi minták vételezésére és csökkentett, illetve végső minták készítésére.
4.3. Folyékony műtrágyák mintavételezésére javasolt készülékek
4.3.1. Kézi mintavételezés
Nyitott cső, szonda, üveg vagy más megfelelő eszköz, amely alkalmas a mintavételezésre kiválasztott részből véletlenszerűen mintát venni.
4.3.2. Mechanikus mintavételezés
Mozgó folyékony műtrágyák mintavételezésére jóváhagyott mechanikus készülékeket kell alkalmazni.
5. MENNYISÉGI KÖVETELMÉNYEK
5.1. Megmintázott tétel
A megmintázott tétel mérete olyan legyen, hogy minden alkotórészt lehessen mintázni.
5.2. Elemi minták
5.2.1. Ömlesztett szilárd műtrágyák vagy folyékony műtrágyák 100 kg-ot meghaladó méretű tartályokban
| Elemi minták száma minimum | |
| 5.2.1.1. 2,5 tonnát meg nem haladó tételnél | hét |
| 5.2.1.2. 2,5 tonnát meghaladó tételnél 80 tonnáig | 20 x a tétel tonnáinak száma1 |
| 5.2.1.3. 80 tonnát meghaladó tételnél | negyven |
| 5.2.2. Csomagolt szilárd műtrágyák vagy folyékony műtrágyák olyan tartályokban (= csomagok), amelyek egyike sem | |
| haladja meg a 100 kg-ot | |
| Megmintázandó csomagok száma, minimum2 | |
| 5.2.2.1. 1 kg fölötti csomagok | |
| 5.2.2.1.1. 5 csomagnál kevesebb csomagból álló tételek" | minden csomag |
| 5.2.2.1.2. 5 — 16 csomagból álló tételek. | négy |
| 5.2.2.1.3. 17 — 400 csomagból álló tételek | V tételben levő csomagok száma |
| 5.2.2.1.4. 400 csomagot meghaladó méretű tételek | húsz |
| 5.2.2.2. 1 kg-ot meg nem haladó csomagok | négy |
| 5.3. Átlagminta | |
| Minden megmintázott tételből egy átlagmintát kell képezni. Az elemi minták teljes mennyisége alkotja az átlagmintát, ennek | |
| Összes súlya nem lehet kevesebb, mint az alábbi értékek: | |
| 5.3.1. Ömlesztett szilárd műtrágyák vagy folyékony műtrágyák 100 kg-ot | |
| meghaladó méretű tartályokban: | 4 kg |
| 5.3.2. Csomagolt szilárd műtrágyák vagy folyékony műtrágyák olyan tartá- | |
| lyokban (= csomagok), amelyek egyike sem haladja meg a 100 kg-ot: | |
| 5.3.2.1. 1 kg-nál nagyobb csomagok | 4 kg |
| 5.3.2.2. 1 kg-nál nem nagyobb | négy eredeti csomag tartalmának súlya |
| 5.3.3. Ammónium-nitrát műtrágya minta vizsgálat céljára, | |
| a III. FÜGGELÉK 2. pontjában meghatározott vizsgálatokhoz: | 75 kg |
| 5.4. Végső laboratóriumi minták | |
| A végső mintát az átlagminta csökkentésével nyerjük, ha szükséges. Legalább egy végső minta vizsgálatát el kell végezni. | |
| A vizsgálatra küldött minta súlya 500 g-nál nem lehet kevesebb. | |
| 5.4.1. Szilárd és folyékony műtrágyák | |
| 5.4.2. Ammónium-nitrát műtrágya minták vizsgálat céljára | |
| Az egyesített minta adja a végső mintákat, szükség esetén csökkentéssel. | |
| 5.4.2.1. A III. FÜGGELÉK 1. pontja szerinti vizsgálatokhoz szükséges legki- | |
| sebb mintatömeg | 1 kg |
| 5.4.2.2. A III. FÜGGELÉK 2. pontja szerinti vizsgálatokhoz szükséges legki- | |
| sebb mintatömeg | 25 kg |
| 1 Ha a kapott szám törtszám, a következő egész számra kerekítsük. |
| 2 1 kg-ot meg nem haladó méretű csomagoknál egy csomagot tekintünk elemi mintának. |
6. A MINTÁK VÉTELÉRE, ELKÉSZÍTÉSÉRE ES CSOMAGOLÁSÁRA VONATKOZÓ UTASÍTÁSOK
6.1. Általános rész
A mintákat a lehető leggyorsabban kell venni és elkészíteni, figyelembe véve azokat az óvórendszabályokat, amelyek biztosítják, hogy a minta reprezentatív maradjon a megmintázott műtrágyára nézve. A minta vételére szolgáló eszközök éppúgy, mint a felületek és a mintatároló edények és zacskók tiszták és szárazak legyenek.
Folyékony műtrágya esetén a mintavételezésre kiválasztott részt a mintavétel előtt lehetőség szerint meg kell keverni.
6.2. Elemi minták
Az elemi mintákat a mintázandó tétel egészéből véletlenszerűen kell venni. Méretük megközelítőleg egyforma legyen.
6.2.1. Ömlesztett szilárd műtrágyák vagy folyékony műtrágyák 100-kg-ot meghaladó méretű tartályokban
A megmintázandó tételt képzeletben több egyenlő részre kell felosztani. Az 5.2. pont szerinti elemi minták számával megegyező számú részt véletlenszerűen ki kell választani és minden ilyen részből legalább egy mintát kell venni.
Ha nincs lehetőség az 5.1. szerinti követelményeknek való megfelelésre ömlesztett műtrágya vagy 100 kg-ot meghaladó méretű tartályokban tárolt folyékony műtrágya mintavételezésének esetében, akkor a mintavételt a mintavételezésre kiválasztott rész mozgatása (betöltése vagy kitöltése) közben kell végezni. Ilyen esetben a mintákat a fenti véletlenszerűen kiválasztott képzeletbeli részekből akkor kell venni, amikor azok mozgásban vannak.
6.2.2. Csomagolt szilárd vagy folyékony műtrágyák olyan tartályokban (= csomagok), amelyek egyike sem haladja meg a 100 kg-ot
Az 5.2. pont szerint megkívánt számú minta kiválasztását követően minden egyes ilyen csomag tartalmának egy részét ki kell venni. Ha szükséges, a mintát úgy kell venni, hogy a csomagokat egyenként kiürítik.
6.3. Átlagminta készítése
Az elemi mintákat össze kell keverni, hogy egyetlen átlagmintát képezzenek.
6.4. A végső (laboratóriumi) minták elkészítése Az átlagminta anyagát alaposan össze kell keverni.[75]
Ha szükséges, az átlagmintát először legalább 2 kg-ra kell csökkenteni (redukált minta) mintaosztóval vagy negyedelési módszerrel.
Ezután legalább három, körülbelül egyforma, az 5.4. pont követelményeit kielégítő mennyiségű végső mintát kell képezni. Minden mintát megfelelő légzáró tárolóedénybe vagy zacskóba kell helyezni. Minden szükséges óvóintézkedést meg kell tenni, hogy a minta tulajdonságaiban beálló bárminemű változást elkerüljük.
A III. FÜGGELÉK 1. és 2. pontja szerinti vizsgálatokhoz a végső mintákat 0 és 25 °C között kell tárolni.
7. A VÉGSŐ (LABORATÓRIUMI) MINTÁK CSOMAGOLÁSA
A tárolóedényeket vagy zacskókat le kell pecsételni és címkével ellátni olyan módon, hogy a zárás megsértése nélkül ne lehessen kinyitni (a teljes címkét be kell fogni a lezárásba).
8. MINTAVÉTELI JEGYZŐKÖNYV
Minden mintavételről jegyzőkönyvet kell felvenni, hogy minden egyes mintát kétséget kizáróan azonosítani lehessen.
9. A MINTÁK RENDELTETÉSI HELYE
Minden tételből legalább egy végső mintát a lehető leggyorsabban el kell juttatni a megbízott analitikai laboratóriumba, a vizsgáló személyzet számára szükséges információkkal együtt.
II. MŰTRÁGYÁK VIZSGÁLATI MÓDSZEREI
A) A MŰTRÁGYÁK ELEMZÉSI MÓDSZEREIRE VONATKOZÓ ÁLTALÁNOS RENDELKEZÉSEK
1. REAGENSEK
Amennyiben az elemző módszerben más rendelkezés nincsen, minden reagens analitikailag tiszta (a. t.) minőségű legyen. Mikroelemek vizsgálatakor a reagensek tisztaságát vakpróbával kell ellenőrizni. A kapott eredménytől függően további tisztítás elvégzése válhat szükségessé.
2. VÍZ
Az elemző módszerekben említett oldási, hígítási, öblítési és mosási műveletek esetében, ha nincs pontos meghatározás az oldószer vagy a hígítószer természetére vonatkozóan, víz alkalmazását kell feltételezni. Általános esetben a vizet ioncserélőn kell átvezetni vagy desztillálni kell. Ezt a vizet meghatározott esetekben - amikor azt az elemző módszerek előírják - különleges tisztítási eljárásoknak kell alávetni.
3. LABORATÓRIUMI FELSZERELÉS
A módszerek leírásai az általános laboratóriumi felszerelések pontos felsorolását - a lombikok és pipetták mérete kivételével - nem tartalmazzák. Figyelembe véve az ellenőrző laboratóriumokban általánosan használt felszerelést, az elemző módszerekben a felszerelések leírása a különleges eszközök és berendezések részletes megadására korlátozódik vagy azokéra, amelyeket valamilyen különleges elvárás követel meg. Valamennyi módszernél a laboratóriumi felszerelést előzetesen gondosan meg kell tisztítani, különösen azokban az esetekben, ahol kis mennyiségű elemeket kell meghatározni. A laboratóriumnak biztosítania kell minden osztással ellátott üvegedény pontosságát a szabványok figyelembevételével.
4. KONTROLL VIZSGÁLATOK
A vizsgálat előtt, ahol szükséges, ismert összetételű vegyületek (ammónium-szulfát, káliumdihidrogén-foszfát) segítségével meg kell győződni arról, hogy valamennyi berendezés jól működik, és az analitikai eljárást pontosan hajtják végre. Mindazonáltal a megvizsgált műtrágyákra kapott adatok a kémiai összetétel tekintetében félrevezetők lehetnek, ha az analitikai eljárást nem követik pontosan. Másrészt a meghatározások egy része empirikus és komplex kémiai összetételű termékekre vonatkozik. Ajánlatos, hogy amennyiben ilyenek rendelkezésre állnak, a laboratóriumnak használjanak jól definiált összetételű standard referencia műtrágyákat.
B) A MINTA ELŐKÉSZÍTÉSE VIZSGÁLATHOZ
1. TÁRGYKÖR
A jelen fejezet a végső (laboratóriumi) mintából vett minta vizsgálathoz való előkészítését írja le.
2. A MÓDSZER ELVE
A laboratórium által átvett, végső (laboratóriumi) minta előkészítése műveletek sorozatából, rendszerint szitálásból, darálásból és keverésből áll, melyeket oly módon visznek véghez, hogy
- egyrészt az egyes módszerek által előírt legkisebb bemért mintamennyiség is reprezentálja a laboratóriumi mintát,
- másrészt a műtrágya szemcseméretét (finomságát) az előkészítéssel nem szabad olyan mértékben megváltoztatni, hogy annak a különböző kivonószerekben való oldhatósága észrevehetően megváltozzon.
3. FELSZERELÉS
Mintamegosztó (nem kötelező).
Sziták 0,2 és 0,5 mm lyukbőséggel.
250 ml-es lombikok dugóval.
Porcelán mozsár és törő vagy daráló.
4. VÁLASZTHATÓ ELJÁRÁSOK
Bevezető megjegyzés
Ha a termék megfelelő, a végső (laboratóriumi) mintának csak egy reprezentatív részét kell megőrizni.
4.1. Olyan végső (laboratóriumi) minták, amelyeket nem szabad darálni.
Kalcium-nitrát, kalcium magnézium-nitrát, nátrium-nitrát, chilei salétrom, kalcium-ciánamid, nitráttartalmú kalcium-ciánamid, ammónium-szulfát, ammónium-nitrát 30% feletti N-tartalommal, karbamid, bázikus salak, részlegesen oldhatóvá feltárt természetes foszfátok, kicsapott dikalcium-foszfát dihidrát, kalcinált foszfát, alumíniumkalcium-foszfát, lágy őrölt ásványi foszfát.
4.2. Olyan végső (laboratóriumi) minták, amelyeket darálni kell.
Ezek olyan termékek, amelyek esetében a vizsgálatok egy részét előzetes darálás nélkül kell elvégezni (pl. őrlési finomság meghatározását), a vizsgálatok másik részét pedig őrlés után. Valamennyi olyan összetett műtrágya ebbe a csoportba tartozik, ' amelyek a következő foszfát összetevőket tartalmazzák: bázikus salak, alumíniumkalcium-foszfát, kalcinált-foszfát, lágy őrölt ásványi foszfát és részlegesen oldhatóvá feltárt természetes foszfátok. Ezért a végső (laboratóriumi) mintát két, lehetőség szerint azonos részre kell osztani, minta-osztóval vagy negyedeléssel.
4.3. Olyan végső (laboratóriumi) minták, amelyek esetében valamennyi vizsgálatot az őrölt termékből kell elvégezni.
A mintának csak egy reprezentatív részét kell megdarálni. Ide tartozik valamennyi, a listán felsorolt műtrágya, amely a 4.1. vagy 4.2. szakaszban nincs megemlítve.
5. MÓDSZER
A végső (laboratóriumi) minta 4.2., ill. 4.3. pont szerinti részét 0,5 mm-es szitán gyorsan átszitáljuk. A szitamaradékot durván megdaráljuk úgy, hogy a finomszemcsés részecskék aránya minimális legyen és újra átszitáljuk. A darálást olyan feltételek mellett kell elvégezni, hogy az anyag észlelhetően ne melegedjék fel. Ezt a műveletet addig ismételjük, amíg a szitán már nem marad fenn semmi. A kivitelezés olyan gyors legyen, hogy anyagok felvételét vagy leadását (víz, ammónia) megelőzzük. A megdarált és átszitált minta teljes mennyiségét dugóval lezárható tiszta üvegbe visszük át.
Vizsgálatra történő bemérés előtt az üveg tartalmát alaposan össze kell keverni.
6. SPECIÁLIS ESETEK
a) Különböző kristály kategóriák keverékét tartalmazó műtrágyák.
Ilyen esetben gyakran előfordul fajtázódás. Ezért elengedhetetlen a mintát megőrölni és 0,2 mm-es szitán átszitálni. Például: a foszfát és kálium-nitrát keveréke. Ezeknél a termékeknél a végső (laboratóriumi) minta teljes mennyiségét le kell darálni.
b) Olyan szitamaradványok, amelyeket nehéz megdarálni és nem tartalmaznak műtrágya anyagokat. Mérjük le a szitamaradékot és vegyük korrekcióba a tömegét a végeredmény kiszámításánál.
c) Hőre bomló termékek.
A darálást a felmelegedés elkerülésével kell elvégezni. Ilyen esetben előnyös dörzsmozsarat használni aprításra. Például: kalciumot és ciánamidot tartalmazó keverék műtrágyák.
d) Olyan termékek, amelyek a szokásosnál nedvesebbek és daráláskor kenődnek.
A homogenitás biztosítására olyan szitát kell választani, amelyen a csomókat a porcelánmozsár dörzsölőjével át tudjuk nyomni. Ilyen eset akkor állhat elő, ha a keverék bizonyos összetevői kristályvizet tartalmaznak.
C) NITROGÉN
C/1. AMMÓNIA NITROGÉN MEGHATÁROZÁSA
1. TÁRGY
A jelen fejezet az ammónia-kötésben lévő nitrogén meghatározásának eljárását írja le.
2. ALKALMAZÁSI TERÜLET
A módszer alkalmazható minden nitrogéntartalmú műtrágyára, beleértve a keverék műtrágyákat is, amelyben a nitrogén kizárólag ammónium sók formájában vagy ammónium sók és nitrátok formájában együttesen fordul elő. Nem alkalmazható karbamidot, ciánamidot és más szerves nitrogén vegyületet tartalmazó műtrágyákra.
3. A MÓDSZER ELVE
Az ammóniát fölös nátrium hidroxiddal felszabadítjuk; desztilláljuk, a fejlődött ammóniát meghatározott koncentrációjú kénsavban nyeletjük el, és a savfelesleget meghatározott koncentrációjú nátrium- vagy kálium-hidroxiddal titráljuk.
4. VEGYSZEREK
Desztillált vagy ioncserélt víz, amely széndioxidtól és nitrogénvegyületektől mentes.
| 4.1. Higított sósav: egy térfogategység HCl (d20 =1,18 g/ml), plusz egy térfogategység víz. | |
| 4.2. Kénsav: 0,1 mol/l. | az a) változathoz |
| 4.3. Nátrium- vagy kálium-hidroxid oldat, karbonátmentes: 0,1 mol/l. | |
| 4.4. Kénsav: 0,2 mol/l. | a b) változathoz, lásd a 2. megjegyzést |
| 4.5. Nátrium- vagy kálium-hidroxid oldat, karbonátmentes: 0,2 mol/l. | |
| 4.6. Kénsav: 0,5 mol/l. | a c) változathoz, lásd a 2. megjegyzést |
| 4.7. Nátrium- vagy kálium-hidroxid oldat, karbonátmentes: 0,5 mol/l. | |
| 4.8. Nátrium-hidroxid oldat, kb. 30% NaOH (d20 = 1,33 g/ml), ammóniamentes. | |
| 4.9. Indikátor oldatok. | |
4.9.1. Keverék indikátor
A oldat: oldjunk fel 1 g metilvörös indikátort 37 ml 0,1 mol/l nátrium-hidroxid oldatban és töltsük fel 1 literre vízzel. B oldat: oldjunk fel 1 g metilénkék indikátort vízben, és egészítsük ki egy 1 literre.
Egy térfogatrész A oldatot keverjünk össze két térfogatrész B oldattal. Ez az indikátor savas oldatban ibolyaszínű, semleges oldatban szürke és lúgos oldatban zöld színű. Használjunk 0,5 ml-t (10 cseppet) ebből az indikátor oldatból.
4.9.2. Metilvörös indikátor oldat
Oldjunk fel 0,1 g metilvörös indikátort 5 ml 95%-os etanolban. Egészítsük ki 100 ml-re vízzel, és ha szükséges, szűrjük meg. Ezt az indikátor oldatot (négy-öt cseppjét) az előző indikátor helyett használhatjuk.
4.10. Horzsakő forráskönnyítő granulátum, sósavval mosva és kiizzítva.
4.11. Ammónium-szulfát analitikai célra.
5. FELSZERELÉS
5.1. Megfelelő térfogatú gömblombikból és desztilláló feltéten át hozzákapcsolt hűtőből álló desztilláló berendezés.
Megjegyzés;
Az ehhez a meghatározáshoz jóváhagyott és ajánlott készülékek típusait valamennyi konstrukciós paraméter feltüntetésével az 1., 2., 3. és 4. számú ábrák mutatják.
5.2. Pipetták, 10, 20, 25, 50, 100 és 200 ml-es.
5.3. 500 ml-es mérőlombik, 1 db.
5.4. Körkörös rázógép (35-40 fordulat percenként).
6. A MINTA ELŐKÉSZÍTÉSE Lásd az A) fejezetet.
7. VIZSGÁLATI MÓDSZER
7.1. Az oldat elkészítése
Végezzük el a minta oldhatósági próbáját vízben, szobahőmérsékleten, 2 (m/v)%-os koncentrációban. Mérjünk le az előkészített mintából 5 vagy 7 vagy 10 g-ot, az 1. táblázat szerint, 0,001 g pontossággal és vigyük 500 ml-es mérőlombikba.
Az oldhatósági próba eredményétől függően a következőképpen járjunk el:
a) Vízben teljesen oldódó termékek
Adjunk a lombikban lévő mintához annyi vizet, amennyi a teljes oldódásához szükséges. Rázogassuk, majd a teljes oldódás után töltsük jelig, és gondosan keverjük össze.
b) Vízben nem teljesen oldódó termékek
Adjunk a lombikba 50 ml vizet, majd 20 ml sósavat (4.1.). Rázzuk össze. Hagyjuk állni, amíg a széndioxid fejlődés befejeződik. Adjunk hozzá 400 ml vizet, rázzuk fél óra hosszat a rázógépen (5.4). Töltsük jelig vízzel, keverjük össze, és szűrjük át száraz szűrőn tiszta edénybe.
7.2. Az oldat elemzése
A választott változattól függően vigyük a szedőlombikba az ismert koncentrációjú kénsav mért mennyiségét az 1. táblázat szerint. Adjuk hozzá a választott indikátor (4.9.1. vagy 4.9.2.) megfelelő mennyiségét, és ha szükséges, annyi vizet, hogy a térfogata legalább 50 ml legyen. A hűtő kivezető csövének az oldat felszíne alá be kell nyúlnia.
A táblázatban megadottak szerint a tiszta oldat egy alikvot részét <- vigyük hitelesített pipettával a berendezés desztilláló lombikjába. Adjunk hozzá annyi vizet, hogy az összes térfogat kb. 350 ml legyen, és néhány szem horzsakövet az egyenletes forrás biztosítására.
Állítsuk össze a desztilláló készüléket, ügyelve arra, hogy ne legyen ammóniaveszteség, adjunk a desztillál lombikba 10 ml koncentrált nátrium-hidroxid oldatot (4.8.) vagy pedig 20 ml-t azokban az esetekben, ha 20 ml sósavat (4.1.) használtunk a vizsgálati minta feloldásához. Melegítsük a lombikot óvatosan, a heves forrást elkerülve. Amikor a forrás megindul, 10-15 perc alatt kb. 100 ml-t desztilláljunk át; a desztillátum teljes térfogata 250 ml körül legyen ↑. Ha úgy látjuk, hogy már nem fejlődik ammónia, engedjük lejjebb a szedőlombikot úgy, hogy a hűtő kifolyócsöve a folyadék felszíne fölé kerüljön.
<- Az 1. táblázat szerint vett alikvot részben az ammóniakötésben lévő nitrogén mennyisége kb.:
- 0,05 g az a) változat szerint,
- 0,10 g a b) változat szerint,
- 0,20 g a c) változat szerint,
↑ A hűtést úgy állítsuk be, hogy a kondenzátum a hűtőben folyamatosan keletkezzék. A desztillációnak 30-40 percen belül be kell fejeződnie.
Alkalmas reagenssel vizsgáljuk meg az ezt követően kondenzálódó desztillátumot és így bizonyosodjunk meg arról, hogy az ammónia desztillációja már befejeződött. Mossuk le a hűtő kifolyócsövének végét egy kis vízzel a szedőlombikba, majd titráljuk meg a sav felesleget ismert koncentrációjú nátrium vagy kálium mérőoldattal a kiválasztott változatnak megfelelően (lásd a 2. megjegyzést).
Megjegyzés:
Különféle erősségű, ismert koncentrációjú mérőoldatok használhatók a visszatitrálásra, azzal a feltétellel, hogy a fogyás lehetőleg ne haladja meg a 40-45 ml-t.
7.3. Vakpróba
Végezzünk vakpróba meghatározást a fentiekkel megegyező körülmények között és vegyük azt figyelembe a végeredmény kiszámításakor.
7.4. Ellenőrző vizsgálat
Mielőtt a vizsgálatot megkezdenénk, ellenőrizni kell, hogy a berendezés helyesen működik-e, illetve, hogy a módszereket korrekt módon alkalmazzuk-e. Erre a célra ammónium-szulfát (4.11.) frissen készített oldatát kell használni, amely a választott változat által előírt maximális nitrogén mennyiséget tartalmazza.
8. AZ EREDMÉNY KIFEJEZÉSE
A vizsgálat eredményét az ammónia-kötésben lévő nitrogén százalékos arányában fejezzük ki a beérkezett állapotú műtrágyára vonatkoztatva.
9. MELLÉKLETEK
Az 5.1 "Felszerelések" pont 1. megjegyzésében leírtak szerint az 1., 2., 3. és 4. ábra a jelen fejezetben leírt készülék három különféle típusának konstrukciós paramétereit adja meg.
1. táblázat
Ammónia nitrogén és nitrát nitrogén meghatározása műtrágyában
A módszer a), b) és c) változata végrehajtásakor a bemérés, a hígítás és a számítás elvégzéséhez szükséges táblázatok
a) változat
Átdesztillálandó maximális nitrogén mennyiség kb. 50 mg.
A szedőlombikba bemért 0,1 mol/l kénsav térfogata: 50 ml.
Visszatitrálás: 0,1 mol/l nátrium-hidroxiddal vagy kálium-hidroxiddal.
| Deklarált tartalom (N%) | Minta bemérés (g) | Hígítás (ml) | Alikvot minta térfogata (ml) | Eredmény kiszámítása [N% = (50— A) F]* |
| 0—5 | 10 | 500 | 50 | (50— A) x 0,14 |
| 5—10 | 10 | 500 | 25 | (50— A) x 0,28 |
| 10—15 | 7 | 500 | 25 | (50— A) x 0,40 |
| 15—20 | 5 | 500 | 25 | (50— A) x 0,56 |
| 20—40 | 7 | 500 | 10 | (50— A) x 1,00 |
b) változat
Átdesztillálandó maximális nitrogén mennyiség kb. 100 mg.
A szedőlombikba bemért 0,1 mol/l kénsav térfogata: 50 ml.
Visszatitrálás: 0,2 mol/l nátrium-hidroxiddal vagy kálium-hidroxiddal.
| Deklarált tartalom (N%) | Minta bemérés (g) | Hígítás (ml) | Alikvot minta térfogata (ml) | Eredmény kiszámítása [N% = (50— A) F]* |
| 0—5 | 10 | 500 | 100 | (50— A) x 0,14 |
| 5—10 | 10 | 500 | 50 | (50— A) x 0,28 |
| 10—15 | 7 | 500 | 50 | (50— A) x 0,40 |
| 15—20 | 5 | 500 | 50 | (50— A) x 0,56 |
| 20—40 | 7 | 500 | 20 | (50— A) x 1,00 |
c) változat
Átdesztillálandó maximális nitrogén mennyiség kb. 200 mg.
A szedőlombikba bemért 0,1 mol/l kénsav térfogata: 35 ml.
Visszatitrálás: 0,5 mol/l nátrium-hidroxiddal vagy kálium-hidroxiddal.
| Deklarált tartalom (N%) | Minta bemérés (g) | Hígítás (ml) | Alikvot minta térfogata (ml) | Eredmény kiszámítása [N% = (50— A) F]* | ||
| 0—5 | 10 | 500 | 200 | (35— A) x 0,175 | ||
| 5—10 | 10 | 500 | 100 | (35— A) x 0,350 | ||
| 10—15 | 7 | 500 | 100 | (35— A) x 0,500 | ||
| 15—20 | 5 | 500 | 100 | (35— A) x 0,700 | ||
| 20—40 | 5 | 500 | 50 | (35— A) x 1,400 | ||
| <— Az eredmények kiszámítására megadott képletben: | ||||||
| 50 vagy 35 | — a szedőlombikba bemért ismert koncentrációjú kénsav oldat térfogata (ml). | |||||
| A = | — a visszatitrálásra fogyott nátrium-hidroxid vagy kálium-hidroxid mérőoldat térfogata (ml). | |||||
| F = | — szorzószám, amely figyelembe veszi a bemérést, a hígítást, a kivett alikvot részt és a | |||||
| mérőoldat faktorát. | ||||||
1. ábra
2. ábra
3. ábra
4. ábra
Az 1., 2., 3. és 4. ábra magyarázata
1. ábra
a) Hosszúnyakú, 1000 ml-es gömblombik.
b) Cseppfogóval ellátott desztillálófeltét, amely gömbcsiszolattal (18-as) csatlakozik a hűtőhöz. (A gömbcsiszolatot megfelelő gumicsatlakozással is lehet helyettesíteni.)
c) Teflon csappal ellátott tölcsér a nátrium-hidroxid adagolásához (a csapot szorítóval ellátott gumicsatlakozással is lehet helyettesíteni).
d) Hat golyóból álló golyós hűtő gömbcsiszolata csatlakozással (18-as) a bevezetésnél és a kivezetésnél egy kis gumicsatlakozóval rögzített üvegcső toldással. (Ha a desztillálófeltéthez a csatlakozást gumicsővel oldjuk meg, akkor a gömbcsiszolatot megfelelő gumi dugóval helyettesíthetjük.)
e) 500 ml-es szedőlombik.
A készülék boroszilikát üvegből készül.
2. ábra
a) Rövid nyakú 1000 ml-es gömblombik, 35-ös gömbcsiszolattal.
b) Desztillálófeltét cseppfogóval, 35-ös gömbcsiszolattal a bemenetelnél és 18-as gömbcsiszolattal a kimenetelnél. Oldalról egy tölcsér csatlakozik be a Nátrium-hidroxid oldat hozzáadására.
c) Hat golyóból álló golyós hűtő, bevezetésénél 18-as gömbcsiszolattal, a kivezetésnél kis gumicsatlakozóval rögzített üvegcső toldással.
d) 500 ml-es lombik, amelybe a desztillátumot gyűjtik. A készülék boroszilikát üvegből készül.
3. ábra
a) Hosszúnyakú gömblombik, 750 vagy 1000 ml-es, kúpos csatlakozással.
b) Desztillálófeltét cseppfogóval ellátva a kimenetelnél 18-as csiszolattal.
c) Könyökcső a bemenetnél 18-as csiszolattal és kónikus kivezetéssel (a desztillálófeltéthez való csatlakozást a gömbcsiszolat helyett gumicsővel is meg lehet oldani).
d) Hat golyóból álló golyós hűtő, kivezetésénél kis gumi csatlakozóval rögzített üvegcső toldással.
e) 500 ml-es lombik, amelybe a desztillátumot gyűjtik.
A készülék boroszilikát üvegből készül.
4. ábra
a) Hosszúnyakú gömblombik, 1000 ml-es, kúpos bevezetőnyílással.
b) Desztillálófeltét cseppfogóval, a kivezetésnél 18-as gömbcsiszolattal ellátva. Oldalról tefloncsappal ellátott tölcsér csatlakozik be a nátrium-hidroxid bevezetésére (a gömbcsiszolatot' gumicsöves csatlakozással, a tefloncsapot szorítóval ellátott gumicsővel helyettesíthetjük).
c) Hat golyóból álló golyós hűtő, a bemenetelnél 18-as gömbcsiszolattal, kivezetésénél kis gumicsatlakozással rögzített üvegcső toldással (ha a desztillálófeltéthez a csatlakozást gumicsővel oldjuk meg, a gömbcsiszolatot megfelelő gumidugóval helyettesíthetjük).
d) 500 ml-es lombik, amelybe a desztillátumot gyűjtik. A készülék boroszilikát üvegből készül.
C/2. NITRÁT ÉS AMMÓNIA NITROGÉN MEGHATÁROZÁSA
C/2.1. NITRÁT ÉS AMMÓNIA NITROGÉN MEGHATÁROZÁSA ULSCH SZERINT
1. TÁRGYA
A jelen fejezet eljárást ír le a nitrát és ammónium kötésben lévő nitrogén redukció útján, Ulsch módszer szerint történő meghatározására.
2. ALKALMAZÁSI TERÜLET
A módszer alkalmazható valamennyi nitrogéntartalmú műtrágyára, beleértve a keverék műtrágyákat, amelyekben a nitrogén csak nitrát kötésben vagy pedig ammónium és nitrát kötésben található.
3. A MÓDSZER ELVE
A nitrátokat és nitriteket fémvassal savas közegben ammóniává redukáljuk, az így képződött ammóniát fölös nátriumhidroxiddal felszabadítjuk, az ammóniát kidesztilláljuk és meghatározott koncentrációjú kénsav ismert térfogatában nyeletjük el. A fölös kénsavat meghatározott koncentrációjú nátrium- vagy kálium-hidroxiddal titráljuk.
4. VEGYSZEREK
Desztillált vagy ioncserélt, széndioxid és nitrogéntartalmú vegyületektől mentes víz.
4.1. Hígított sósav: egy térfogat koncentrált sósav (d20 =1,18 g/ml) plusz egy térfogat víz.
4.2. Kénsav: 0,1 mol/l.
4.3. Nátrium- vagy kálium-hidroxid oldat, karbonátmentes: 0,1 mol/l.
4.4. Kénsav oldat, kb. 30%-os H2SO4 (m/v), ammóniamentes.
4.5. Hidrogénben redukált vaspor (az előírt vas mennyiségnek legalább 0,05 g nitrát-kötésű nitrogén redukciójára legyen képes).
4.6. Nátrium-hidroxid, kb. 30% NaOH tartalmú (d20 = 1,33 g/ml) ammóniamentes.
4.7. Indikátor oldat.
4.7.1. Keverék indikátor
A oldat: Oldjunk fel 1 g metilvöröst 37 ml 0,1 mol/l nátrium-hidroxid oldatban és vízzel töltsük fel 1 literre.
B oldat: Oldjunk fel 1 g metilénkéket vízben egészítsük ki az oldat térfogatát 1 literre.
Keverjünk össze egy térfogategység A oldatot két térfogategység B oldattal.
Ez az indikátor savas oldatban ibolyaszínű, semleges oldatban szürke és lúgos oldatban zöld. Használjuk 0,5 ml-t (10 csepp).
4.7.2. Metilvörös indikátor oldat
Oldjunk fel 0,1 g metilvöröst 50 ml 95%-os etanolban. Egészítsük ki vízzel 100 ml-re, és ha szükséges, szűrjük meg. Ezt az indikátort (4-5 cseppjét) az előző indikátor helyett használhatjuk.
4.8. Horzsakő sósavban mosott és izzított.
4.9. Nátrium-nitrát analitikai célra.
5. FELSZERELÉS
Lásd a C/1. "Ammónia nitrogén meghatározása" című fejezetet.
6. A MINTA ELŐKÉSZÍTÉSE
Lásd a B) "A minta előkészítése vizsgálathoz" című fejezetet.
7. VIZSGÁLATI MÓDSZER
7.1. Az oldat elkészítése
Lásd a C/1. "Ammónia nitrogén meghatározása" című fejezetet.
7.2. Eljárás
Ontsuk a szedő lombikba meghatározott koncentrációjú kénsav oldat pontos mennyiségét, a 2.1 módszer 1. táblázata [a) változat] szerint. Adjuk hozzá a 4.7.1. vagy 4.7.2. pont szerinti indikátor mennyiséget. A hűtő kivezetéséhez toldott cső vége a szedőben a meghatározott koncentrációjú sav felszíne alá érjen.
Hiteles pipettát használva vigyük a tiszta oldat 2.1. módszer 1. táblázata [a) változat] szerint megválasztott alikvot mennyiségét a berendezés desztilláló lombikjába. Adjunk hozzá 350 ml vizet, 20 ml 30%-os kénsav oldatot (4.4.) keverjük össze, majd adjunk hozzá 5 g redukált vasat (4.5.). Mossuk a lombik nyakát néhány ml vízzel és helyezzünk a lombik nyakába egy kisméretű, hosszú szárú tölcsért. Egy óráig melegítsük forró vízfürdőben, majd mossuk be a tölcsér végét néhány ml vízzel.
Vigyázva, hogy ne legyen ammóniaveszteség, adjunk a desztilláló lombik tartalmához 50 ml koncentrált nátrium-hidroxid oldatot (4.6.) vagy ha 20 ml 1 + 1 arányú sósav-víz elegyet használtunk a feltáráskor, akkor 60 ml koncentrált nátrium-hidroxid oldatot (4.6.). Állítsuk össze a desztilláló készüléket. Desztilláljuk ki az ammóniát 2.1 módszer szerint.
7.3. Vakpróba
Végezzünk vakpróbát (minta nélkül) a vizsgálattal azonos körülmények között, és vegyük figyelembe az eredmény kiszámításánál.
7.4. Kontroll vizsgálat
Vizsgálat előtt nátrium-nitrát (4.9.) frissen készített oldatának 0,045-0,050 g nitrogént tartalmazó alikvot részével ellenőrizzük, hogy a berendezés jól működik, és a módszert helyesen alkalmazzuk.
8. AZ EREDMÉNY KIFEJEZÉSE
Az eredményt a nitrát-kötésű nitrogén vagy a kombinált ammónia- és nitrát-kötésű nitrogén százalékában fejezzük ki a vizsgálatra beérkezett állapotú műtrágyára vonatkoztatva.
C/2.2. A NITRÁT- ÉS AMMÓNIA-KÖTÉSBEN LÉVŐ NITROGÉN MEGHATÁROZÁSA ARND SZERINT
1. TÁRGY
A jelen fejezet eljárást ír le a nitrát- és ammónia-kötésben lévő nitrogén meghatározására redukció útján, Arnd módszere szerint [az a), b) és c) változatokra egyenként módosítva].
2. ALKALMAZÁSI TERÜLET Lásd a C/2.1. fejezetet.
3. A MÓDSZER ELVE
A nitrátokat és nitriteket semleges vizes oldatban 60% Cu és 40% Mg tartalmú fémötvözettel (Arnd-féle ötvözet), magnézium klorid jelenlétében ammóniává redukáljuk.
Az ammóniát kidesztilláljuk, és meghatározott koncentrációjú kénsav ismert térfogatú oldatában nyeletjük el. A fölös savat nátrium- vagy kálium-hidroxid meghatározott koncentrációjú oldatával titráljuk vissza.
4. VEGYSZEREK
Desztillált vagy ioncserélt, széndioxid és nitrogéntartalmú vegyületektől mentes víz.
| 4.1. Hígított sósav: egy térfogat HCl (d20 =1,18 g/ml) plusz egy térfogat víz | |
| 4.2. Kénsav: 0,1 mol/l. | az a) változathoz |
| 4.3. Nátrium- vagy kálium-hidroxid oldat, karbonátmentes: 0,1 mol/l. | |
| 4.4. Kénsav: 0,2 mol/l. | a b) változathoz, lásd a C/1. módszer |
| 4.5. Nátrium- vagy kálium-hidroxid oldat, karbonátmentes: 0,2 mol/l. | 2. megjegyzését |
| 4.6. Kénsav: 0,5 mol/l. | a c) változathoz, lásd a C/1. módszer |
| 4.7. Nátrium- vagy kálium-hidroxid oldat, karbonátmentes: 0,5 mol/l. | 2. megjegyzését |
4.8. Nátrium-hidroxid oldat: kb. 2 mol/l.
4.9. Arnd-féle ötvözet vizsgálati célra: az 1 mm-nél kisebb lyukbőségü szitán áteső, poralakú készítmény.
4.10. 20%-os magnézium-klorid oldat.
Oldjunk fel 200 g magnézium-kloridot (MgCl2.6H2O) kb. 600-700 ml vízben egy literes álló lombikban. A habzás megelőzésére adjunk hozzá 15 g magnézium-szulfátot (MgSO4.7H2O).
Oldódás után adjunk hozzá 2 g magnézium-oxidot és néhány szem horzsakövet. Forralással pároljuk be az oldatot 200 ml-re, így elűzve a reagensből az esetleges ammónia nyomokat. Hűtsük le, egészítsük ki a térfogatát 1 l-re és szűrjük le.
4.11. Indikátor oldatok
4.11.1. Keverék indikátor
A oldat: Oldjunk fel 1 g metilvöröst 37 ml 0,1 mol/l nátrium-hidroxid oldatban és vízzel töltsük fel egy literre.
B oldat: Oldjunk fel 1 g metilénkéket vízben és egészítsük ki az oldat térfogatát 1 literre.
Keverjünk össze egy térfogategység A oldatot két térfogategység B oldattal.
Ez az indikátor savas oldatban ibolyaszínű, semlegesben szürke és lúgos oldatban zöld. Használjunk 0,5 ml-t (10 csepp).
4.11.2. Metilvörös indikátor oldat
Oldjunk fel 0,1 g metilvöröst 50 ml 95%-os etanolban. Egészítsük ki vízzel 100 ml-re, és ha szükséges, szűrjük meg. Ezt az indikátort (4-5 cseppjét) az előző indikátor helyett használhatjuk..
4.11.3. Kongó vörös indikátor oldat
Oldjunk fel 3 g Kongó vörös indikátort egy liter meleg vízben. Lehűlés után szűrjük meg, ha szükséges. Ezt az indikátort a fent leírt két indikátor helyett használhatjuk, az extraktum desztilláció előtti semlegesítése során 100 ml semlegesítendő oldatra 0,5 ml indikátort számítva.
4.12. Horzsakő sósavban mosott és izzított.
4.13. Nátrium-nitrát analitikai célra.
5. KÉSZÜLÉKEK
Lásd a C/1. "Ammónia nitrogén meghatározása" című fejezetet.
6. A MINTA ELŐKÉSZÍTÉSE
Lásd a B) "A minta előkészítése vizsgálathoz" című fejezetet.
7. VIZSGÁLATI MÓDSZER
7.1. Az oldat elkészítése
Lásd a C/1. "Ammónia nitrogén meghatározása'' című fejezetet.
7.2. Az oldat vizsgálata
A kiválasztott változatnak megfelelően, pipettázzuk a szedőlombikba a meghatározott koncentrációjú kénsav pontos mennyiségét a C/1. módszer 1. táblázata szerint. Adjuk hozzá a kiválasztott indikátor oldat (4.11.1. vagy 4.11.2.) megfelelő mennyiségét, és végül annyi vizet, hogy a térfogat legalább 50 ml legyen. A hűtő kivezetéséhez toldott cső végének a folyadékfelszín alá kell érnie.
Hiteles pipettával vegyünk ki az oldatból az 1. táblázat szerinti alikvot mennyiséget, és tegyük a desztilláló lombikba.
Adjunk hozzá annyi vizet, hogy az összes térfogat kb. 350 ml legyen (lásd az 1. megjegyzést), 10 g Arnd-féle ötvözetet (4.9.), 50 ml magnézium-klorid oldatot (4.10.) és néhány szem horzsakő granulátumot (4.12.). Gyorsan csatlakoztassuk a lombikot a desztilláló készülékhez. Óvatosan melegítsük kb. 30 percig. Azután emeljük a hőfokot, hogy az ammóniát kidesztillálhassuk. Kb. egy óra hosszat folytassuk a desztillációt. Ezt követően a desztilláló lombikban maradt folyadéknak szirupszerűnek kell lennie. Amikor a desztilláció befejeződött, titráljuk meg a szedőben lévő fölös sav mennyiségét a C/1. módszerben leírt eljárás szerint:
Megjegyzés:
Ha a minta oldata savas [feloldáskor 20 ml hígított sósavat (4.1.) adtunk hozzá], akkor a vizsgálatra kivett alikvot részt a következőképpen kell semlegesíteni: a desztilláló lombikba vitt alikvot részhez adjunk kb. 250 ml vizet, valamelyik indikátor (4.11.1., 4.11.2., 4.11.3.) szükséges mennyiségét, és gondosan rázzuk össze.
Semlegesítsük 2N nátrium-hidroxiddal (4.8.), és egy csepp (l + 1) arányú sósavval (4.1.) savanyítsuk ismét vissza. Utána a 7.2. pont második sora szerint járjunk el.
7.3. Vakpróba
Végezzünk vakpróbát a vizsgálattal azonos körülmények között, és azt vegyük figyelembe az eredmény kiszámításánál.
7.4. Kontroll vizsgálat
Vizsgálat előtt nátrium-nitrát frissen készült, a kiválasztott változattól függően 0,050-0,150 g nitrát-kötésű nitrogént tartalmazó oldatával ellenőrizzük, hogy a berendezés jól működik-e, és a módszert helyesen alkalmazzuk.
8. AZ EREDMÉNY KIFEJEZÉSE Lásd a C/1. fejezetet.
C/2.3. A NITRÁT- ÉS AMMÓNIA-KÖTÉSBEN LÉVŐ NITROGÉN MEGHATÁROZÁSA DEVARDA SZERINT
1. TÁRGY
A jelen fejezet eljárást ír le a nitrát- és ammónia-kötésben lévő nitrogén meghatározására redukció útján, Devarda módszere szerint [az a), b) és c) változatokra egyenként módosítva].
2. ALKALMAZÁSI TERÜLET Lásd a C/2.1. fejezetet.
3. A MÓDSZER ELVE
A nitrátokat és nitriteket ammóniává redukáljuk erősen lúgos közegben Devarda ötvözettel (45% Al, 5% Zn és 50% Cu). Az ammóniát kidesztilláljuk, és meghatározott koncentrációjú kénsav ismert térfogatú oldatában nyeletjük el. A fölös savat nátrium- vagy kálium-hidroxid meghatározott koncentrációjú oldatával titráljuk vissza.
4. VEGYSZEREK
Desztillált vagy ioncserélt, széndioxid és nitrogéntartalmú vegyületektől mentes víz.
4.1. Hígított sósav: egy térfogat HCl (d20 =1,18 g/ml) plusz egy térfogat víz.
| 4.2. Kénsav: 0,1 mol/l. | az a) változathoz |
| 4.3. Nátrium- vagy kálium-hidroxid oldat, karbonátmentes: 0,1 mol/l. | |
| 4.4. Kénsav: 0,2 mol/l. | a b) változathoz, lásd a C/1. módszer |
| 4.5. Nátrium- vagy kálium-hidroxid oldat, karbonátmentes: 0,2 mol/l. | 2. megjegyzését |
| 4.6. Kénsav: 0,5 mol/l. | a c) változathoz, lásd a C/1. módszer |
| 4.7. Nátrium- vagy kálium-hidroxid oldat, karbonátmentes: 0,5 mol/l. | 2. megjegyzését |
4.8. Devarda ötvözet analitikai célra.
Poralakú; 90-100%-a át essen a 0,25 mm-nél kisebb lyukbőségü szitán, 50-75%-a át essen a 0,075 mm-nél kisebb lyukbőségü szitán.
Legfeljebb 100 g-os előrecsomagolt üvegek használata javasolt.
4.9. Nátrium-hidroxid oldat, kb. 30% NaOH (d20 = 1,33 g/ml), ammóniamentes.
4.10. Indikátor oldatok
4.10.1. Keverék indikátor
A oldat: Oldjunk fel 1 g metilvöröst 37 ml 0,1 mol/l nátrium-hidroxid oldatban és vízzel töltsük fel egy literre.
B oldat: Oldjunk fel 1 g metilénkéket vízben, egészítsük ki az oldat térfogatát 1 literre.
Keverjünk össze egy térfogategység A oldatot két térfogategység B oldattal.
Ez az indikátor savas oldatban ibolyaszínű, semleges oldatban szürke és lúgos oldatban zöld. Használjunk 0,5 ml-t (10 csepp).
4.10.2. Metilvörös indikátor oldat
Oldjunk fel 0,1 g metilvöröst 50 ml 95%-os etanolban. Egészítsük ki vízzel 100 ml-re, és ha szükséges, szűrjük meg. Ezt az indikátort (4-5 cseppjét) az előző indikátor helyett használhatjuk.
4.11. Etanol, 95-96%-os.
4.12. Nátrium-nitrát analitikai célra.
5. KÉSZÜLÉKEK Lásd a C/1. fejezetet.
5.1. Desztilláló berendezés megfelelő térfogatú gömblombikkal, cseppfogóval ellátott desztillálófeltéten keresztül csatlakozik a hűtőhöz. A szedőlombik buborékcsapdával van felszerelve az ammóniaveszteség elkerülésére.
A meghatározáshoz jóváhagyott készülék rajzát, az összes szerkezeti jellemző feltüntetésével az 5. ábra tartalmazza.
5.2. Pipetták, 10, 20, 25, 50, 100 és 200 ml-es.
5.3. 500 ml-es mérőlombik.
5.4. Körkörös rázógép (percenként 35-40 fordulattal).
6. A MINTA ELŐKÉSZÍTÉSE
Lásd a B) "A minta előkészítése vizsgálathoz" című fejezetet.
7. VIZSGÁLATI MÓDSZER
7.1. Az oldat elkészítése
Lásd a C/1. "Ammónia nitrogén meghatározása" című fejezetet.
7.2. Az oldat vizsgálata
Az oldat alikvot részében lévő nitrát-kötésben lévő nitrogén mennyisége nem haladhatja meg az 1. táblázatban megadott maximális értéket.
A kiválasztott változatnak megfelelően vigyük a lombikba a meghatározott koncentrációjú kénsav 1. táblázat szerinti, . pontosan kimért mennyiségét. Adjuk hozzá a kiválasztott indikátor (4.10.1., 4.10.2.) megfelelő mennyiségét, végül annyi vizet, hogy térfogat 50 ml legyen. A hűtő végéhez toldott üvegcsőnek a folyadékfelszín alá kell érnie. A buborékcsapdát töltsük fel desztillált vízzel.
Hiteles pipettával vegyünk ki egy alikvot részt a módszer 1. táblázata szerint. Vigyük át a desztilláló lombikba.
Adjunk hozzá annyi vizet, hogy a térfogat 250-300 ml között legyen, 5 ml etanolt (4.11.) és 4 g Devarda ötvözetet (4.8.).
Megtéve minden szükséges óvóintézkedést, hogy az ammóniaveszteséget elkerüljük, adjunk a lombikba 30 ml a 30%-os nátrium-hidroxid oldatot (4.9.) és végül, a savban oldható minták esetében még annyi lúgot, amennyi vizsgálatra kivett alikvot mennyiségben jelenlévő sósav semlegesítéséhez szükséges. Csatlakoztassuk a desztilláló lombikot a készülékhez, ügyelve a csatlakozások tömítettségére. Óvatosan rázzuk meg a lombikot, hogy a tartalmát összekeverjük. '
Óvatosan melegítsük, hogy a hidrogénfejlődés kb. fél óra alatt észrevehetően csökkenjen, és a folyadék forrásba jöjjön. Folytassuk a desztillációt, növelve a fűtést úgy, hogy kb. 30 perc alatt legalább 200 ml desztilláljon át (ne növeljük a desztillációs időt 45 percen túl).
Amikor a desztilláció befejeződött, vegyük le a szedőlombikot, óvatosan öblítsük le az üvegcső toldást. A szedő tartalmát ontsuk át titráló lombikba, és titráljuk meg a savfelesleget a C/1. módszerben leírt eljárás szerint.
Megjegyzés:
Kalcium sók, mint kalcium-nitrát és kalciumammónium-nitrát jelenlétében a desztilláció előtt az alikvot részben levő minta minden grammjára, 0,700 g dinátriumhidrogén-foszfátot (Na2HPO4.2H20) kell adni a Ca(OH)2 képződés megakadályozására.
7.3. Vakpróba
Végezzünk vakpróbát a vizsgálattal azonos körülmények között, és azt vegyük figyelembe a végeredmény kiszámításánál.
7.4. Kontroll vizsgálat
Vizsgálat előtt nátrium-nitrát frissen készült, a kiválasztott változattól függően 0,050-0,150 g nitrát-kötésű nitrogént tartalmazó oldatával ellenőrizzük, hogy a berendezés jól működik-e, és a módszert helyesen alkalmazzuk.
8. AZ EREDMÉNY KIFEJEZÉSE Lásd a C/2.1. fejezetet.
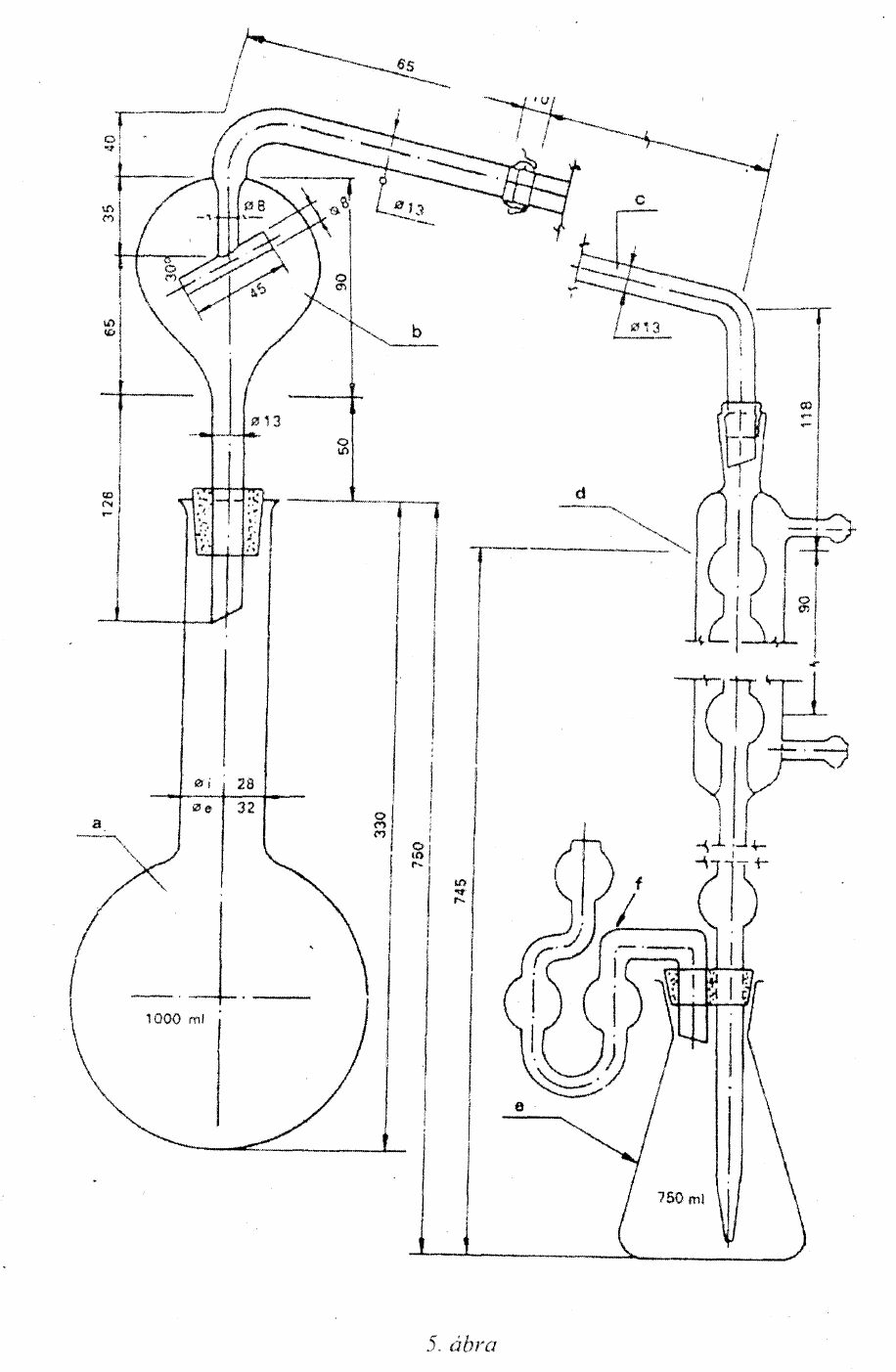
5. ábra
Az 5. ábra magyarázata
a) 750 ml-s (1000 ml-s) hosszúnyakú gömblombik, kónikus nyakkal.
b) Desztillációs feltét cseppfogóval, és kivezetésénél 18-as gömbcsiszolattal.
c) Könyökcső a bevezetésnél 18-as gömbcsiszolattal, kónikus csepegtető véggel.
d) Hatgolyós golyóshűtő, üvegcső toldással, amely átvezet egy gőzcsapdával felszerelt gumidugón.
e) 750 ml-es szedő lombik.
f) Gőzcsapda az ammóniaveszteség megelőzésére.
A berendezés boroszilikát üvegből készült.
C/3. ÖSSZES NITROGÉNTARTALOM MEGHATÁROZÁSA
C/3.1. NITRÁTMENTES KALCIUM CIÁNAMID ÖSSZES NITROGÉNTARTALMÁNAK MEGHATÁROZÁSA
1. TÁRGYKÖR
A jelen fejezet eljárást ír le a nitrát mentes kalcium ciánamid összes nitrogéntartalmának meghatározására.
2. ALKALMAZÁSI TERÜLET
Kizárólag kalcium ciánamid (nitrát mentes) műtrágya.
3. A MÓDSZER ELVE
Kjeldahl roncsolást követően az ammónia-kötésben levő nitrogént nátrium-hidroxiddal felszabadítjuk, és ismert térfogatú, meghatározott koncentrációjú kénsav oldatban nyeletjük el, és a kénsav felesleget visszatitrálva határozzuk meg.
4. VEGYSZEREK
Desztillált vagy ioncserélt víz, széndioxidtól és nitrogén vegyületektől mentes.
4.1. Hígított kénsav (d20 = 1,54 g/ml): egy térfogatrész kénsav (d20 = 1,84 g/ml) + egy térfogatrész víz.
4.2. Kálium szulfát analitikai célra. - '
4.3. Rézoxid (CuO) 0,3-0,4 g minden meghatározáshoz vagy rézszulfát pentahidrát (CuSO4.5H2O) egyenértékű mennyisége, 0,95-1,25 g minden meghatározáshoz.
4.4. Nátrium-hidroxid oldat, kb. 30% NaOH (d20 = 1,33 g/ml), ammóniamentes.
| 4.5. Kénsav: 0,1 mol/l. | az a) változathoz |
| 4.6. Nátrium- vagy kálium-hidroxid oldat, karbonátmentes: 0,1 mol/l. | |
| 4.7. Kénsav: 0,2 mol/l. | a b) változathoz, lásd a C/1. módszer |
| 4.8. Nátrium- vagy kálium-hidroxid oldat, karbonátmentes: 0,2 mol/l. | 2. megjegyzését |
| 4.9. Kénsav: 0,5 mol/l. | a c) változathoz, lásd a C/1. módszer |
| 4.10. Nátrium- vagy kálium-hidroxid oldat, karbonátmentes: 0,5 mol/l. | 2. megjegyzését |
4.11. Indikátor oldatok
4.11.1. Keverék indikátor
A oldat: Oldjunk fel 1 g metilvöröst 37 ml 0,1 mol/l nátrium-hidroxid oldatban, és vízzel töltsük fel egy literre.
B oldat: Oldjunk fel 1 g mefilénkéket vízben, és egészítsük ki az oldat térfogatát 1 literre.
Keverjünk össze egy térfogatrész A oldatot két térfogatrész B oldattal.
Ez az indikátor savas oldatban ibolyaszínű, semlegesben szürke és lúgos oldatban zöld színű. Használjunk 0,5 ml-t (10 csepp).
4.11.2. Metilvörös indikátor oldat
Oldjunk fel 0,1 g metilvöröst 50 ml 95%-os etanolban. Egészítsük ki vízzel 100 ml-re és ha szükséges, szűrjük meg. Ezt az indikátort (4-5 cseppjét) az előző indikátor helyett használhatjuk.
4.12. Horzsakő forráskönnyítő granulátum, sósavban mosott, izzított.
4.13. Kálium-rodanid, analitikai célra
5. FELSZERELÉS
5.1. Desztilláló berendezés, lásd C/1. módszer "Ammónia nitrogén meghatározása."
5.2. Hosszúnyakú Kjeldahl lombik, megfelelő térfogatú.
5.3. Pipetták, 50, 100 és 200 ml-es.
5.4. 250 ml-es mérőlombik.
6. A MINTA ELŐKÉSZÍTÉSE Lásd a B) fejezetet.
7. ELJÁRÁS
7.1. Az oldat elkészítése
Mérjünk be 0,001 g pontossággal 1 g mintát Kjeldahl lombikba. Adjunk hozzá 50 ml hígított kénsavat (4.1.), 10 és 15 g közötti mennyiségű kálium-szulfátot (4.2.) és az előírt katalizátort (4.3.). Melegítsük lassan a víz elpárologtatására, majd 2 óra hosszat forraljuk óvatosan. Hagyjuk lehűlni, és hígítsuk 100-150 ml vízzel. Hűtsük le újra. Vigyük a szuszpenziót veszteség nélkül 250 ml-es lombikba, töltsük jelig vízzel, rázzuk össze, és száraz szűrőn szűrjük száraz lombikba.
7.2. Az oldat vizsgálata
A fentiek szerint kapott oldatból a kiválasztott változatnak megfelelően (lásd C/1. módszer) 50, 100 vagy 200 ml-t veszünk ki, kidesztilláljuk az ammóniát a C/1. módszernél leírtak szerint, és elegendő NaOH oldat (4.4.) hozzáadásával biztosítjuk, hogy a lúg jelentős feleslegben legyen,
7.3. Vakpróba
Végezzünk vakpróbát (minta hozzáadása nélkül) a vizsgálattal azonos körülmények között, és vegyük figyelembe az eredmények kiszámításánál.
7.4. Kontroll vizsgálat
Vizsgálat előtt kálium-rodanid (4.13.) meghatározott koncentrációjú oldatából kivett, a minta nitrogéntartalmával közelítőleg azonos nitrogéntartalmú alikvot részével ellenőrizzük, hogy a berendezés jól működik-e, és a módszert helyesen alkalmazzuk.
8. AZ EREDMÉNY SZÁMÍTÁSA
Az eredményt a nitrogéntartalom (N) százalékában fejezzük ki, a beérkezési állapotú műtrágyára vonatkoztatva.
a) változat: N% = (50-A) x 0,7
b) változat: N% = (50-A) x 0,7
c) változat: N% = (35-A) x 0,875
C/3.2. NITRÁTTARTALMÚ KALCIUM CIÁNAMID ÖSSZES NITROGÉN TARTALMÁNAK MEGHATÁROZÁSA
1. TÁRGY
A jelen fejezet eljárást ír le a kalcium-ciánamid összes nitrogéntartalom meghatározására.
2. ALKALMAZÁSI TERÜLET
A módszer alkalmazható nitráttartalmú kalcium-ciánamid vizsgálatára.
3. A MÓDSZER ELVE
A Kjeldahl módszert közvetlenül nem lehet alkalmazni a nitráttartalmú kalcium-ciánamid vizsgálatára. Ezért a nitrát-kötésben lévő nitrogént ammóniává kell redukálni fém vassal és/vagy ón-kloriddal a Kjeldahl roncsolást megelőzően.
4. VEGYSZEREK
Desztillált vagy ioncserélt víz, széndioxidtól és nitrogénvegyületektől mentes.
| 4.1. Kénsav (d20 = 1,84 g/ml). | |
| 4.2. Vaspor, hidrogénben redukált. | |
| 4.3. Kálium-szulfát, finoman porított, analitikai célra. | |
| 4.4. Kénsav: 0,1 mol/l. | az a) változathoz |
| 4.5. Nátrium- vagy kálium-hidroxid oldat, karbonátmentes: 0,1 mol/l. | |
| 4.6. Kénsav: 0,2 mol/l. | a b) változathoz, lásd a C/1. módszer |
| 4.7. Nátrium- vagy kálium-hidroxid oldat, karbonátmentes: 0,2 mol/l. | 2. megjegyzését |
| 4.8. Kénsav: 0,5 mol/l. | a c) változathoz, lásd a C/1 módszer |
| 4.9. Nátrium- vagy kálium-hidroxid oldat, karbonátmentes: 0,5 mol/l. | 2. megjegyzését |
4.10. Indikátor oldatok
4.10.1. Keverék indikátor
A oldat: Oldjunk fel 1 g metilvöröst 37 ml 0,1 mol/l nátrium-hidroxid oldatban és vízzel töltsük fel egy literre.
B oldat: Oldjunk fel 1 g metilénkéket vízben, és egészítsük ki az oldat térfogatát 1 literre.
Keverjünk össze egy térfogatrész A oldatot két térfogatrész B oldattal.
Ez az indikátor savas oldatban ibolyaszínű, semlegesben szürke, és lúgos oldatban zöld színű. Használjuk 0,5 ml-t (10 csepp).
4.10.2. Metilvörös indikátor oldat
Oldjunk fel 0,1 g metilvöröst 50 ml 95%-os etanolban. Egészítsük ki vízzel 100 ml-re, és ha szükséges, szűrjük meg. Ezt az indikátort (4-5 cseppjét) az előző indikátor helyett használhatjuk.
4.11. Ón-kloridoldat
Oldjunk fel 120 g SnCl2.2H2O-t 400 ml tömény sósavban (d20 =1,18 g/ml), és töltsük fel vízzel egy literre. Az oldatnak teljesen tisztának kell lennie. Közvetlenül felhasznál előtt kell készíteni. Elengedhetetlen, hogy az ón-klorid redukáló tulajdonságát ellenőrizzük.
Megjegyzés:
Oldjunk fel 0,5 g SnCl2.2H2O-t 2 ml tömény sósavban (d20 =1,18 g/ml) és egészítsük ki vízzel 50 ml-re. Adjunk hozzá 5 g Rochelle sót (káliumnátrium-tartarátot) és annyi analitikai tisztaságú nátrium bikarbonátból készült oldatot, hogy az oldat lakmusz papírral lúgos kémhatást mutasson.
Titráljuk meg 0,1 mol/l jódoldattal keményítő indikátor mellett, 1 ml 0,1 mol/l jódoldat 0,01128 g SnCl2.2H2O-nak felel meg.
Az így készített oldatban jelenlévő összes ón minimum 80%-a kétértékű formában legyen. A titrálásnál legalább 35 ml 0,1 mol/l jódoldatnak kell fogynia.
4.12. Nátrium-hidroxid oldat, 30%-os NaOH (d20 = 1,33 g/ml), ammóniamentes.
4.13. Nitrát-ammónia standard oldat
Mérjünk le 2,5 g analitikai tisztaságú kálium-nitrátot és 10,16 g analitikai tisztaságú ammónium-szulfátot egy 250 ml-es mérőlombikba. Oldjuk fel vízzel, töltsük fel 250 ml-re. Ennek az oldatnak 1 ml-e 0,01 g nitrogénnek felel meg.
4.14. Horzsakő forráskönnyítő granulátum, sósavban mosott, izzított.
5. FELSZERELÉS Lásd C/3.1. fejezetet.
6. A MINTA ELŐKÉSZÍTÉSE Lásd a B) fejezetet.
7. ELJÁRÁS
7.1. Az oldat elkészítése
Mérjünk le 1 g mintát 0,001 g pontossággal és vigyük a Kjeldahl lombikba. Adjunk hozzá 0,5 g vasport (4.2.) és 50 ml ón-klorid oldatot (4.11.), keverjük össze, és hagyjuk állni fél óráig. Ezalatt a 10. és a 20. percben újra keverjük össze. Ezután adjunk hozzá 10 g kálium-szulfátot (4.3.) és 30 ml kénsavat (4.1.). Forraljuk fel, és folytassuk a forralást a fehér gőzök megjelenésétől számított egy óra hosszat. Hagyjuk hűlni, és hígítsuk 100 és 150 ml közötti térfogatú vízzel. Vigyük át a szuszpenziót veszteség nélkül egy 250 ml-es mérőlombikba, hűtsük le, töltsük jelig, és szűrjük le száraz szűrőn át egy száraz lombikba. Ahelyett, hogy szuszpenziót leszivornyáznánk a C/1. módszer a), b) vagy c) változata szerinti meghatározáshoz, ebből az oldatból az ammónia kötésben lévő nitrogént is meghatározhatjuk, azután, hogy elegendő nátrium-hidroxidot adtunk a lúgfelesleg biztosításához (4.12.).
7.2. Az oldat vizsgálata
A fentiek szerint nyert oldatból a 2.1. módszer kiválasztott a), b) vagy c) változatának megfelelően 50, 100 vagy 200 ml-t veszünk ki. A C/1. módszer szerint kidesztilláljuk az ammóniát, vigyázva arra, hogy desztilláló lombikba elegendő Nátrium-hidroxid oldatot (4.12.) adjunk a nagy lúgfelesleg biztosítására.
7.3. Vakpróba
Végezzünk vakpróbát (minta hozzáadása nélkül) a vizsgálattal azonos körülmények között, és vegyük figyelembe a végeredmény kiszámításánál.
7.4. Kontroll vizsgálat
A vizsgálat elvégzése előtt ellenőrizzük a berendezés működésének helyességét és a módszer megfelelő alkalmazását olyan oldattal, amely az ammónia-kötésű és a nitrát-kötésű nitrogént meghatározott, a nitráttartalmú kalcium-ciánamid műtrágya nitrát-nitrogén és ciánamid-nitrogén tartalmával összemérhető mennyiségben tartalmazza.
Erre a célra mérjük a standard oldat (4.13.) 20 ml-ét a Kjeldahl lombikba.
Végezzük el a vizsgálatot a 7.1. és 7.2. pontban leírtak szerint.
8. AZ EREDMÉNY SZÁMÍTÁS A
Az eredményt a nitrogéntartalom (N) százalékában fejezzük ki, a beérkezési állapotú műtrágyára vonatkoztatva.
a) változat: N% = (50-A) x 0,7
b) változat: N% = (50-A) x 0,7
c) változat: N% = (35-A) x 0,875
C/3.3. ÖSSZES NITROGÉNTARTALOM MEGHATÁROZÁSA KARBAMIDBAN
1. TÁRGY
A jelen fejezet eljárást ír le a karbamid összes nitrogéntartalmának meghatározására.
2. ALKALMAZÁSI TERÜLET
A módszert kizárólag nitrátmentes karbamid műtrágyákra lehet alkalmazni.
3. A MÓDSZER ELVE
A karbamidot kénsav jelenlétében történő forralással ammóniává alakítjuk. Az így kapott ammóniát a meglúgosított oldatból kidesztilláljuk, a desztillátumot meghatározott koncentrációjú, ismert mennyiségű kénsavban fogjuk fel. A sav felesleget meghatározott koncentrációjú lúgoldattal visszatitráljuk.
4. VEGYSZEREK
| Desztillált vagy ioncserélt, széndioxid és nitrogén vegyületektől mentes víz. | |
| 4.1. Koncentrált kénsav (d20 = 1,84 g/ml). | |
| 4.2. Nátrium-hidroxid oldat, kb. 30% NaOH (d20 = 1,33 g/ml), mentes. | |
| 4.3. Kénsav: 0,1 mol/l. | az a) változathoz |
| 4.4. Nátrium- vagy kálium-hidroxid oldat, karbonátmentes: 0,1 mol/l. | |
| 4.5. Kénsav: 0,2 mol/l. | a b) változathoz, lásd a C/1. módszer |
| 4.6. Nátrium- vagy kálium-hidroxid oldat, karbonátmentes: 0,2 mol/l. | 2. megjegyzését |
| 4.7. Kénsav: 0,5 mol/l. | a c) változathoz, lásd a C/1. módszer |
| 4.8. Nátrium- vagy kálium-hidroxid oldat, karbonátmentes: 0,5 mol/l. | 2. megjegyzését |
4.9. Indikátor oldatok
4.9.1. Keverék indikátor
A oldat: Oldjunk fel 1 g metilvöröst 37 ml 0,1 mol/l nátrium-hidroxid oldatban, és vízzel töltsük fel egy literre. B oldat: Oldjunk fel 1 g metilénkéket vízben, és egészítsük ki az oldat térfogatát 1 literre. Keverjünk össze egy térfogatrész A oldatot két térfogatrész B oldattal.
Ez az indikátor savas oldatban ibolyaszínű, semlegesben szürke, és lúgos oldatban zöld színű. Használjunk 0,5 ml-t (10 csepp).
4.9.2. Metilvörös indikátor oldat
Oldjunk fel 0,1 g metil vöröst 50 ml 95%-os etanolban. Egészítsük ki vízzel 100 ml-re, és ha szükséges, szűrjük meg. Ezt az indikátort (4-5 cseppjét) az előző indikátor helyett használhatjuk.
4.10. Horzsakő forráskönnyítő granulátum, sósavban mosott, izzított.
4.11. Karbamid, analitikai célra.
5. FELSZERELÉS
5.1. Desztilláló berendezés, lásd C/1. módszert, "Ammónia nitrogén meghatározása".
5.2. 500 ml-es mérőlombik.
5.3. Pipetták, 25, 50 és 100 ml.
6. A MINTA ELŐKÉSZÍTÉSE Lásd a B) fejezetet.
7. ELJÁRÁS
7.1. Az oldat elkészítése
Mérjük az előkészített minta 2,5 g-ját 0,001 pontossággal egy 300 ml-es Kjeldahl lombikba, és nedvesítsük meg 20 ml vízzel.Adjunk hozzá 20 ml tömény kénsavat (4.1.) és néhány szem üveggyöngyöt az egyenletes forrás biztosítására. A kifröcsögés megakadályozására tegyünk lombik nyakába hosszúszárú üvegtölcsért. Melegítsük kezdetben lassan, majd növelve a hőt mindaddig, amíg fehér füst keletkezése válik megfigyelhetővé (30-40 perc).
Hűtsük le, és hígítsuk 100-150 ml vízzel. Veszteség nélkül vigyük át 500 ml-es mérőlombikba, az üledékeket is átöntve. Hagyjuk szobahőmérsékletre hűlni, töltsük jelig vízzel, keverjük össze, és ha szükséges, szűrjük át száraz szűrőn száraz lombikba.
7.2. Az oldat vizsgálata
A fentiek szerint nyert oldatból hiteles pipettával vigyünk a C/1. módszer kiválasztott változatának megfelelően 25, 50 vagy 100 ml-t a desztilláló lombikba. Desztilláljuk ki az ammóniát a C/1. módszerben leírtak szerint, elegendő NaOH-t (d2o = 1,33 g/ml) (4.2.) adva a desztilláló lombikba jelentős lúgfölösleg biztosításához.
7.3. Vakpróba
Végezzünk vakpróbát (minta hozzáadása nélkül) a vizsgálattal azonos körülmények között, és ezt vegyük figyelembe a végeredmény kiszámításánál.
7.4. Kontroll vizsgálat
A vizsgálat előtt frissen készített karbamid oldat (4.11.) alikvot részével ellenőrizzük, hogy a berendezés jól működik-e, és a módszert helyesen alkalmazzuk.
8. AZ EREDMÉNY KISZÁMÍTÁSA
Az eredményt a beérkezési állapotú műtrágya százalékos nitrogén (N) tartalmában adjuk meg:
a) változat: N% = (50-A) x 1,12
b) változat: N% = (50-A) X 1,12
c) változat: N% = (35-A) x 1,40.
C/4. A CIÁNAMID KÖTÉSBEN LÉVŐ NITROGÉN MEGHATÁROZÁSA
1. TÁRGY
A jelen fejezet eljárást ír le a ciánamid kötésben lévő nitrogén meghatározására.
2. ALKALMAZÁSI TERÜLET Kalcium-ciánamid és kalcium-ciánamid /nitrát keveréke.
3. A MÓDSZER ELVE
A ciánamidot lecsapjuk ezüst komplex formájában, és csapadékból határozzuk meg Kjeldahl módszerrel.
4. VEGYSZEREK
Desztillált vagy ioncserélt, széndioxid és nitrogén vegyületektől mentes víz.
4.1. Jégecet.
4.2. Ammóniaoldat, 10 súly% ammónia gáz tartalommal (d2o=0,96 g/ml).
4.3. Tollens reagens (ezüst-ammónia oldat).
Elegyítsünk 10%-os vizes ezüst-nitrát (AgNO3) oldatot 500 ml 10%-os ammónia oldattal (4.2.).
Óvjuk fénytől, hőtől és levegőtől. Az oldatot rendes körülmények között évekig el lehet tartani. A reagens jó minőségűnek minősül mindaddig, amíg az oldat tiszta.
4.4. Tömény kénsav (d20= 1,84 g/ml).
4.5. Kálium-szulfát analitikai célra.
4.6. Rézoxid (CuO), 0,3-0,4 g minden meghatározáshoz vagy pedig rézszulfát-pentahidrát (CuSO4.5H2O) ekvivalens mennyisége, 0,95-1,25 g minden meghatározáshoz.
4.7. Nátrium-hidroxid oldat, ammóniamentes kb. 30% NaOH (d20 = 1,33 g/ml).
4.8. Kénsav: 0,1 mol/l.
4.9. Nátrium vagy kálium hidroxid: 0,1 mol/l.
4.10. Indikátor oldatok
4.10.1. Keverék indikátor
A oldat: Oldjunk fel 1 g metilvöröst 37 ml 0,1 mol/l nátrium-hidroxid oldatban és vízzel töltsük fel egy literre.
B oldat: Oldjunk fel 1 g metilénkéket vízben, és egészítsük ki az oldat térfogatát 1 literre.
Keverjünk össze egy térfogatrész A oldatot két térfogatrész B oldattal.
Ez az indikátor savas oldatban ibolyaszínű, semlegesben szürke és lúgos oldatban zöld színű. Használjunk 0,5 ml-t (10 csepp).
4.10.2. Metilvörös indikátor oldat
Oldjunk fel 0,1 g metilvöröst 50 ml 95%-os etanolban. Egészítsük ki vízzel 100 ml-re és ha szükséges, szűrjük meg. Ezt az indikátort (4-5 cseppjét) az előző indikátor helyett használhatjuk.
4.11. Horzsakő forráskönnyítő granulátum, sósavban mosott, izzított.
4.12. Kálium-tiocianát analitikai célra.
5. FELSZERELÉS
5.1. Desztillációs berendezés, lásd C/1. módszer "ammónia kötésben lévő nitrogén meghatározása".
5.2. 500 ml-es kalibrált lombik (pl. Stohman lombik).
5.3. Hosszúnyakú Kjeldahl lombik megfelelő térfogattal (300-500 ml).
5.4. Pipetta, 50 ml-es.
5.5. Körkörös rázógép (35-40 fordulat/perc).
6. A MINTA ELŐKÉSZÍTÉSE Lásd a B) fejezetet.
7. ELJÁRÁS
7.1. Biztonsági intézkedések
Ezüst-ammónia tartalmú oldatokkal való munka közben védőszemüveget kell viselni. Ha a folyadék felszínén vékony hártya képződik, rázkódás hatására robbanás következhet be, a legnagyobb óvatosság indokolt.
7.2. A vizsgálati oldat elkészítése
Mérjünk le 2,5 g mintát 0,001 g pontossággal, és vigyük kis porcelán mozsárba. Dörzsöljük el a mintát háromszor vízzel, a vizet minden dörzsölés után az 500 ml-re kalibrált Stohman lombikba (5.2.) töltve. Végül vigyük át a mintát veszteség nélkül a Stohman lombikba, belemosva a dörzsmozsarat, a dörzsölőt és a tölcsért is. Adjunk hozzá annyi vizet, hogy kb. 400 ml legyen. Adjunk hozzá 15 ml ecetsavat (4.1.). Rázassuk a rázógépen (5.5.) 2 óra hosszat.
Töltsük fel a lombikot 500 ml vízzel, keverjük össze, és szűrjük meg.
A vizsgálatot a lehető leggyorsabban kell elvégezni.
7.3. Az oldat vizsgálata
A szűrlet 50 ml-ét vigyük 250 ml-es főzőpohárba.
Adjunk hozzá ammónia oldatot (4.2.), amíg enyhén lúgos lesz, majd adjunk hozzá 30 ml meleg ammóniás ezüst-nitrát oldatot (4.3.) a ciánamid sárga színű ezüst-komplexének leválasztása céljából.
Hagyjuk egy éjszakán át állni, szűrjük le a csapadékot, és mossuk hideg vízzel, amíg teljesen ammóniamentes lesz.
A szűrőt és a csapadékot még nedvesen helyezzük Kjeldahl lombikba, adjunk hozzá 10-16 g kálium-szulfátot (4.5.), az előírt mennyiségű katalizátort (4.6.), majd 50 ml vizet és 25 ml tömény kénsavat (4.4.).
Melegítsük lassan a lombikot óvatos rázogatás mellett, amíg az elegy forrni kezd. Fokozzuk a melegítést, forraljuk az oldatot, amíg átlátszó színtelen vagy világoszöld lesz. Ezután még egy óra hosszat forraljuk, majd hagyjuk lehűlni.
Vigyük át az oldatot veszteség nélkül a desztilláló lombikba, adjunk hozzá néhány forráskönnyítő horzsakő granulátumot (4.11.) és töltsük fel vízzel kb. 350 ml-re Keverjük össze és hűtsük le.
Desztilláljuk ki az ammóniát a C/1. módszer aj változata szerint, elegendő mennyiségű NaOH oldatot (4.7.) adagolva ahhoz, hogy jelentős lúgfölösleg legyen.
7.4. Vakpróba
Végezzünk vakpróbát (minta hozzáadása nélkül) a vizsgálattal azonos körülmények között, és vegyük figyelembe a végeredmény kiszámításánál.
7.5. Kontroll vizsgálat
Vizsgálat előtt kálium-rodanid (4.12.) standard oldatának kb. 0,05 g nitrogénnek megfelelő alikvot részével ellenőrizzük, hogy a berendezés jól működik-e és a módszert helyesen alkalmazzák-e.
8. AZ EREDMÉNYEK KISZÁMÍTÁS A
Az eredményt a ciánamid kötésben lévő nitrogén tömegszázalékában adjuk meg, a beérkezési állapotú műtrágyára vonatkoztatva.
N% = (50-A) x 0,56
C/5. KARBAMID BIURET TARTALMÁNAK SPEKTROFOTOMETRIÁS MEGHATÁROZÁSA
1. TÁRGY
A jelen fejezet eljárást ír le karbamid biuret tartalmának meghatározására.
2. ALKALMAZÁSI TERÜLET
A módszert kizárólag karbamidra lehet alkalmazni.
3. A MÓDSZER ELVE
Lúgos közegben, kálium nátrium tartarát jelenlétében a biuret és a kétértékű réz ion ibolyaszínű rézvegyületet képeznek. Az oldat optikai sűrűségét kb. 546 nm (nanométer) hullámhosszon kell meghatározni.
4. VEGYSZEREK
Desztillált vagy ioncserélt víz, széndioxidtól és nitrogén vegyületektől mentes. A víz minősége ennél a vizsgálatnál különösen fontos.
4.1. Metanol.
4.2. Kénsav oldat, kb. 0,1 mol/l.
4.3. Nátrium-hidroxid oldat, kb. 0,1 mol/l.
4.4. Káliumnátrium-tartarát lúgos oldata
Egy literes mérőlombikban oldjunk fel 40 g nátrium-hidroxidot 500 ml vízben, és hagyjuk lehűlni. Adjunk hozzá 50 g káliumnátrium-tartarátot (NaKC4H4O6.H2O). Töltsük fel jelig. Hagyjuk 24 órát állni használat előtt.
4.5. Rézszulfát oldat
Egy literes mérőlombikban oldjunk fel 15 g rézszulfátot (CuSO4.4H2O) 500 ml vízben. Töltsük fel jelig.
4.6. Frissen készített biuret oldat
250 ml-es mérőlombikban oldjunk fel vízben 0,25 g tiszta biuretet. Töltsük fel 250 ml-re. Ennek az oldatnak 1 ml-e 0,001 g biuretet tartalmaz.[76]
4.7. Indikátor oldat
100 ml-es mérőlombikban oldjunk fel 0,1 g metilvörös indikátort 50 ml 95%-os etanolban. Töltsük fel vízzel 100 ml-re. Ha oldhatatlan részek vannak benne, szűrjük le.
5. KÉSZÜLÉK
5.1. Spektrofotométer vagy fotométer szűrőkkel, amelynek érzékenysége és pontossága lehetővé teszi a 0,5% T alatti méréseket[77]
5.2. Mérőlombik, 100, 250 és 1000 ml-es.
5.3. Osztott pipetták, 2, 5, 10, 20, 25 és 50 ml-es vagy 25 ml-es 0,05 ml beosztású büretta.
5.4. Főzőpohár, 250 ml-es.
6. A MINTA ELŐKÉSZÍTÉSE Lásd a B) fejezetet.
7. ELJÁRÁS
7.1. A standard görbe elkészítése
Vigyük a biuret standard (4.6.) oldat 0, 2, 5, 10, 20, 25 és 50 ml-es részleteit egy 7 db 100 ml mérőlombikból álló mérőlombik-sorozatba. A térfogatot egészítsük ki vízzel kb. 50 ml-re, semlegesítsük 0,1 mol/l kénsavval (4.2.). Adjunk hozzá 20 ml-t a lúgos tartarát oldatból (4.4.), majd 20 ml-t a réz-szulfát oldatból (4.5.).
Megjegyzés:
Ezeket az oldatokat két pontos bürettával kell bemérni vagy még jobb pipettával.
Töltsük fel a mérőlombikokban lévő oldatokat vízzel 100 ml-re, keverjük össze, és hagyjuk állni 15 percig 30 ± 2 "C-on. A "0" biuret tartalmú oldattal szemben mérjük a többi oldat abszorbanciáját kb. 546 nm hullámhosszon megfelelő hosszúságú küvettában.
Rajzoljuk fel a kalibrációs görbét, az abszorbanciát az ordinátán, a biuret mg-ban megadott megfelelő mennyiségeit pedig az abszcisszán.
7.2. A vizsgálandó oldat elkészítése
Mérjük be az előkészített minta 10 g-ját 0,001 pontossággal; oldjuk fel kb. 150 ml vízben egy 250 ml mérőlombikban, majd töltsük jelig. Szűrjük le, ha szükséges.
1. észrevétel
Ha a vizsgálandó minta több, mint 0,015 g ammónia-kötésben lévő nitrogént tartalmaz, akkor 250 ml főzőpohárban, 50 ml metanolban (4.1.) oldjuk fel. Pároljuk be a metanolt, míg a térfogat kb. 25 ml-re csökken. Ezt vigyük veszteség nélkül 250 ml-es mérőlombikba, töltsük jelig vízzel. Ha szükséges, száraz redős szűrőpapíron száraz edénybe szűrjük le.
2. észrevétel
Az opaleszcencia kiküszöbölése: ha kolloid részecskék vannak jelen, szűréskor nehézségek támadhatnak. Ilyen esetben az oldatot a következőképpen kell elkészíteni: oldjuk fel a mintát 150 ml vízben, adjunk hozzá 2 ml 1 mol/l sósavat, és szűrjük át az oldatot sima, dupla, nagyon finom szűrőpapíron 250 ml-es mérőlombikba. Mossuk a szűrőket vízzel, majd töltsük fel a lombikot. Folytassuk az eljárást a 7.3. "Meghatározás" pontban előírt eljárás szerint.
7.3. Meghatározás
A feltételezett biuret koncentrációnak megfelelően pipettázzunk ki a 7.2. szerinti oldatból 25 vagy 50 ml-t és vigyük ezt a mennyiséget 100 ml-es mérőlombikba, és ha szükséges, semlegesítsük 0,1 mol/l kénsav vagy nátronlúg reagenssel (4.2. vagy 4.3.), metilvörös indikátor mellett, és ugyanolyan pontossággal, mint ahogy a standard görbe felvételénél eljártunk, adjuk hozzá a káliumnátrium-tartarát (4.4.) lúgos oldatának 20 ml-ét, és a réz oldatnak (4.5.) ugyancsak 20 ml-ét. Töltsük jelig, alaposan keverjük össze, és hagyjuk állni 15 percig 30 ± 2 °C-n.
Ezután végezzük el a fotometriás méréseket, és számítsuk ki a karbamid biuret tartalmát.
8. AZ EREDMÉNY KISZÁMÍTÁSA
Ha a "C" a biuret tömege milligrammban a kalibrációs görbéről leolvasva, "V" pedig az alikvot rész térfogata, akkor:
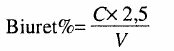
9. FÜGGELÉK
Ha "JO'' a monokromatikus fénysugarak intenzitása (a mérési hullámhosszon) a fényáteresztő közegbe belépéskor, "J'' pedig a kilépéskor, akkor:

| ahol: | ||
| s | = a rétegvastagság cm-ben, | |
| c | = a koncentráció mg/l-ben, | |
| k | = az anyagra jellemző tényező a Lambert-Beer törvény szerint. |
C/6. KÜLÖNFÉLE FORMÁBAN KÖTÖTT NITROGÉN MEGHATÁROZÁSA UGYANABBAN A MINTÁBAN
C/6.1. KÜLÖNFÉLE FORMÁBAN KÖTÖTT NITROGÉN MEGHATÁROZÁSA NITRÁT, AMMÓNIA, KARBAMID ÉS CIÁNAMID NITROGÉNT TARTALMAZÓ MŰTRÁGYA MINTÁBAN
1. TÁRGY
A jelen fejezet eljárást ír le a valamely formában kötött nitrogén meghatározására bármely fenti formában kötött nitrogén jelenlétében.
2. ALKALMAZÁSI TERÜLET
A módszer alkalmazható valamennyi olyan, a rendelet hatálya alá tartozó műtrágyára, amely különféle formában kötött nitrogént tartalmaz.
3. A MÓDSZER ELVE
3.1. Az összes oldható és nem oldható nitrogén
A standard műtrágyáknak a 3. számú melléklet 1. függeléke szerinti listája alapján a jelen meghatározás kalcium ciánamidot tartalmazó termékekre alkalmazható.
3.1.1. Nitrát vegyületek távollétében a vizsgálati mintát közvetlen Kjeldahl módszerrel kell elroncsolni.
3.1.2. Nitrát vegyületek jelenlétében a vizsgálati mintát vassal és ón-kloriddal végzett redukciót követően Kjeldahl módszerrel leroncsolni.
Mindkét esetben a C/1. módszerrel kell meghatározni az ammóniát.
Megjegyzés:
Ha a vizsgálat 0,5-nél magasabb oldhatatlan nitrogén jelenlétére utal, akkor arra a következtetésre lehet jutni, hogy a műtrágya olyan oldhatatlan nitrogén vegyületeket tartalmaz, amelyek nem tartoznak a 8/2001. (I. 26.) FVM rendelet hatálya alá.
3.2. Oldható nitrogén formák
A következőket ugyanabból a mintaoldatból vett, különböző alikvot részekből határozzuk meg.
3.2.1. Összes oldható nitrogén
3.2.1.1. - nitrátok távollétében közvetlen Kjeldahl roncsolással.
3.2.1.2. - nitrátok jelenlétében az oldat Ulsch szerinti redukcióját követően kivett alikvot részének Kjeldahl szerinti roncsolásával.
Az ammóniát mindkét esetben a C/1. fejezet szerint határozzuk meg.
3.2.2. Összes oldható nitrogén a nitrát-kötésű nitrogén kivételével Kjeldahl roncsolással, a nitrát-kötésű nitrogén vasszulfáttal történt előzetes eltávolítása után.
Az ammóniát a C/1. fejezet szerint határozzuk meg.
3.2.3. nitrát-kötésű nitrogén különbség alapján:
3.2.3.1. -kalcium-ciánamid távollétében a 3.2.1.2. és 3.2.2. közötti különbség, vagy az összes oldható nitrogén (3.2.1.2.) és az ammóniakötésű és karbamid-kötésű szerves nitrogén (3.2.4 + 3.2.5.) közötti különbség alapján.
3.2.3.2. - kalcium ciánamid jelenlétében a 3.2.1.2. és a 3.2.2. közötti különbség, vagy a 3.2.1.2. és a 3.2.4. + 3.2.5. + 3.2.6. összege közötti különbség alapján.
3.2.4. ammóniakötésű nitrogén:
3.2.4.1. ha csak ammónia vagy ammónia + nitrát-kötésű nitrogén van jelen, az 1. módszer alkalmazásával.
3.2.4.2. karbamid-kötésű nitrogén és/vagy ciánamid nitrogén jelenlétében enyhe lúgosítást követő hideg desztillációval, az ammóniát ismert koncentrációjú kénsavban elnyeletve, és a C/1. módszer szerint meghatározva.
3.2.5. karbamid kötésű nitrogén:
3.2.5.1. ureáz segítségével ammóniává alakítva, amelyet ismert koncentrációjú sósavval titrálunk meg, vagy
3.2.5.2. gravimetriás módszerrel xanthidrollal: az együtt leváló biuretet nagyobb hiba nélkül karbamidként számolhatjuk, mivel a biuret koncentrációja ezekben az összetett műtrágyákban abszolút értékben alacsony,
vagy
3.2.5.3. különbség alapján számítással az alábbi táblázat szerint:
| Eset | Nitrát nitrogén | Ammónia nitrogén | Ciánamid nitrogén | Különbség számítása |
| 1. | nincs | van | van | (3.2.1.1.)— [(3.2.4.2.) + (3.2.6.)] |
| 2. | van | van | van | (3.2.2.)— [(3.2.4.2.) + (3.2.6.)] |
| 3. | nincs | van | nincs | (3.2.1.1.) — (3.2.4.2.) |
| 4. | van | van | nincs | (3.2.2.)— (3.2.4.2.) |
3.2.6. ciánamid nitrogén ezüstvegyületként történő lecsapását követően a csapadékból Kjeldahl módszerrel.
4. VEGYSZEREK
Desztillált vagy ioncserélt víz, széndioxidtól és nitrogén vegyületektől mentes.
4.1. Kálium-szulfát, analitikai célra.
4.2. Vaspor, redukált.
(Az előírt vaspor mennyisége legalább 50 mg nitrát nitrogén redukálására legyen alkalmas.)
4.3. Káliurn-rodanid analitikai célra.
4.4. Kálium-nitrát analitikai célra.
4.5. Ammónium-szulfát analitikai célra.
4.6. Karbamid analitikai célra.
4.7. Hígított kénsav, 1:1 térfogatarányú egy rész kénsav (d2o = 1,84 g/ml) egy rész víz.
4.8. Meghatározott koncentrációjú kénsav oldat: 0,2 mol/l.
4.9. Tömény nátrium-hidroxid oldat. Kb. 30 vegyes%-os NaOH oldat, ammóniamentes.
4.10. Meghatározott koncentrációjú nátrium- vagy kálium-hidroxid: 0,2 mol/l, karbonát mentes.
4.11. Ón-kloridoldat.
Oldjunk fel 120 g SnCl2-2H2O-t 400 ml tömény sósavban (d20 =1,18 g/ml), és töltsük fel vízzel egy literre. Az oldatnak teljesen tisztának kell lennie. Közvetlenül felhasználás előtt kell készíteni. Elengedhetetlen, hogy az ón-klorid redukáló tulajdonságát ellenőrizzük.
Megjegyzés:
Oldjunk fel 0,5 g SnCl2.2H2O-t + 2 ml tömény sósavban (d20 = 1,18 g/ml) és egészítsük ki vízzel 50 ml-re. Adjunk hozzá 5 g Rochelle sót (káliumnátrium-tartarátot) és annyi analitikai tisztaságú nátriumhidrogén-karbonátból készült oldatot, hogy az oldat lakmusz papírral lúgos kémhatást mutasson.
Titráljuk meg 0, 1 mol/l jódoldattal keményítő indikátor mellett, 1 ml 0,1 mól/l mérőoldat megfelel 0,001128 g SnCl2.2H2O-nak. Az így készített oldatban jelenlévő összes ón minimum 80%-a kétértékű formában legyen. A titrálásra legalább 35 ml 0,1 mol/l jódoldat fogyjon.
4.12. Kénsav (d20 = 1,84 g/ml).
4.13. Hígított sósav: 1:1 térfogatarányú, egy rész sósav (d20 =1,18 g/ml) egy rész víz.
4.14. Ecetsav: 96- 100%-os.
4.15. Kénsavoldat kb. 30% (m/v) kénsavtartalommal.
4.16. Vasszulfát, kristályos: FeSO4.7H2O.
4.17. Meghatározott koncentrációjú kénsav oldat: 0,1 mol/l.
4.18. Oktilalkohol.
4.19. Kálium-karbonát telített oldata.
4.20. Nátrium- vagy kálium-hidroxid meghatározott koncentrációjú oldata: 0,1 mol/l (karbonátmentes).
4.21. Bárium-hidroxid telített oldata.
4.22. Nátrium-karbonát oldat: 10% (m/v).
4.23. Sósav: 2 mol/l.
4.24. Meghatározott koncentrációjú sósav oldat: 0,1 mol/l.
4.25. Ureáz oldat
Szuszpendáljunk 0,5 g aktív ureázt 100 ml desztillál vízben. 0,1 mol/l sósavat (4.24.) használva, pH-mérő segítségével állítsuk a pH-t 5,4-re.
4.26. Xanthidrol
5%-os etanolos vagy metanolos (4.31.) oldat. (Ne használjunk olyan terméket, amelyben sok az oldhatatlan rész.) Az oldatot jól zárható edényben fénytől védve 3 hónapig el lehet tartani.
4.27. Rézoxid CuO:0,3-0,4 g vizsgálatonként, vagy rézszulfát pentahidrát (CuSO4.5H2O) ekvivalens mennyisége: 0,95-1,25 g vizsgálatonként.
4.28. Horzsakő forráskönnyítő granulátum, sósavban mosott, izzított.
4.29. Indikátor oldatok
4.29.1. Keverék indikátor
A oldat: Oldjunk fel 1 g metilvöröst 37 ml 0,1 mol/l nátrium-hidroxid oldatban és vízzel töltsük fel egy literre.
B oldat: Oldjunk fel 1 g metilénkéket vízben, és egészítsük ki az oldat térfogatát 1 literre.
Keverjünk össze egy térfogatrész A oldatot két térfogatrész B oldattal.
Ez az indikátor savas oldatban ibolya-, semlegesben szürke és lúgos oldatban zöld színű. Használjuk 0,5 ml-t (10 csepp).
4.29.2. Metilvörös indikátor oldat
Oldjunk fel 0,1 g metil vöröst 50 ml 95%-os etanolban. Egészítsük ki vízzel 100 ml-re és ha szükséges, szűrjük meg. Ezt az indikátort (4-5 cseppjét) az előző indikátor helyett használhatjuk.
4.30. Indikátor papírok
Lakmusz, brómtimolkék (vagy más, pH 6 és 8 között i változásra érzékeny papírok).
4.31. Etanol vagy metanol: 95%-os oldat
5. FELSZERELÉS
5.1. Desztillációs berendezés: lásd a C/1. módszert.
5.2. Az ammóniakötésű nitrogén meghatározására szolgáló készülék a 7.2.5.3. módszernek megfelelően (lásd a C/6.1. fejezetet és a 6. ábrát).
A berendezés speciálisan kiképzett, gömbölyű üveg fejrésszel ellátott szedőből áll, amelynek egy oldal kivezető nyaka is van. A fejen keresztül egy cseppfogóval ellátott cső nyúlik ki, valamint egy merőlegesen hajlított cső a levegő bevezetésére. A csöveket egyszerű gumi csatlakozással lehet a szedőhöz kapcsolni. Fontos, hogy a levegőt bevezető cső végének kiképzése megfelelő formájú legyen, mivel gázbuborékoknak tökéletesen kell eloszolniuk a szedőben és az gázmosóban lévő oldatokban. A legjobb erre a kis, gombaformájú gázbevezető fej, amelynek átmérője 20 mm, és közben a kerületén 6 db 1 mm-es nyílása van.
5.3. A karbamid-kötésű nitrogén ureáz technikával történő meghatározására szolgáló készülék (7.2.6.1.). Egy 300 ml-es Erlenmeyer lombikból, egy csepegtetőtölcsérből és egy kis gázmosóból áll (lásd a 7. ábra).
5.4. Körkörös rázógép (35-40 fordulat/perc).
5.5. pH-méter.
5.6. Állítható hőfokú szárítószekrény.
5.7. Üvegáru
- pipetták, 2, 5, 10, 20, 25, 50 és 100 ml-es,
- hosszúnyakú Kjeldahl lombikok, 300 és 500 ml-es,
- mérőlombikok 100, 250, 500 és 1000 ml-es,
- zsugorított üvegszűrő tégelyek, 5-15 pm pórusátmérőjű,
- mozsarak.
6. A MINTA ELŐKÉSZÍTÉSE Lásd a B) fejezetet.
7. ANALITIKAI TECHNIKÁK
7.1. Összes oldható és oldhatatlan nitrogén. 7.1.1. Nitrátok távollétében
7.1.1.1. Roncsolás
Mérjünk be 0,001 g pontossággal olyan mennyiségű mintát, amely maximum 100 mg nitrogént tartalmaz. Vigyük a desztilláló berendezés lombikjába (5.1.). Adjunk hozzá 10 és 15 g közötti mennyiségű kálium-szulfátot (4.1.), a katalizátort (4.27.) és pár szem forráskönnyítő granulátumot (4.28.). Ezután adjunk hozzá 50 ml hígított kénsavat (4.7.), és keverjük gondosan össze. Először óvatosan melegítsük, időnként rázogatva, amíg a habzás megszűnik, azután úgy állítsuk be a melegítést, hogy az oldat egyenletesen forrjon. Az oldat kitisztulása után még egy órát forraljuk, ügyelve arra, hogy a lombik nyakára vagy falára ne tapadjon fel szerves anyag. Hagyjuk lehűlni. Óvatosan hígítsuk meg 350 ml vízzel rázogatás közben. Biztosítsuk, hogy az oldódás a lehető legteljesebb legyen. Hagyjuk lehűlni, és csatlakoztassuk a lombikot a desztilláló készülékhez (5.1.).
1.1.1.2. Az ammónia desztillációja
Vigyünk a berendezés szedőjébe hiteles pipettával 50 ml meghatározott koncentrációjú 0,2 mol/l kénsav oldatot (4.8.). Adjuk hozzá az indikátort (4.29.1. vagy 4.29.2.). Ügyeljünk rá, hogy a hűtő vége legalább 1 cm-rel az oldat felszíne alá nyúljon.
Ügyelve, hogy ammóniaveszteség ne léphessen fel, óvatosan adjunk annyi tömény nátrium-hidroxid oldatot (4.9.) a desztilláló lombikba, hogy a folyadékelegy erősen lúgos legyen, 120 ml általában elegendő; győződjünk meg erről néhány csepp fenolftalein oldat adagolásával. A lombik tartalmának a desztilláció befejezésekor is határozottan lúgosnak kell lennie. Állítsuk be a lombik melegítését úgy, hogy 150 ml kb. fél óra alatt desztilláljon le. Vizsgáljuk meg indikátor papírral (4.30.), hogy a desztilláció végbement-e. Ha ez lenne a helyzet, desztilláljunk át további 50 ml-t, és ismételjük meg az indikátorpapíros ellenőrzést, amíg a desztillátum újabb részlete már semleges kémhatást mutat az indikátor papírral (4.30.). Engedjük le a szedőt, desztilláljunk még pár ml-t, és öblítsük le a hűtő végét. A savfelesleget kálium- vagy nátrium-hidroxid meghatározott koncentrációjú 0,2 mol/l oldatával (4.10.) titráljuk az indikátor színének megváltozásáig.
7.1.1.3. Vakpróba
Végezzünk vakpróbát a vizsgálattal azonos körülmények között, és azt vegyük figyelembe a végeredmény kiszámításánál.
7.1.1.4. Az eredmény kiszámítása
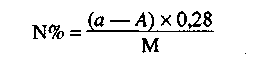
| ahol: | ||
| a = | 0,2 mol/l nátrium- vagy kálium-hidroxid meghatározott koncentrációjú oldata, ml, amely a vakpróbánál fogyott, amikor a készülék (5.1.) szedőjébe 50 ml meghatározott koncentrációjú 0,2 mol/l kénsavat (4.8.) adtunk, | |
| A = | 0,2 mol/l nátrium- vagy kálium-hidroxid meghatározott koncentrációjú oldata, ml, amely a minta vizsgálatára fogyott, | |
| M = | a bemérés tömege, g. |
7.1.2. Nitrát jelenlétében
7.1.2.1. Vizsgálati minta
Mérjünk be 0,001 g pontossággal annyi mintát, amely 40 mg nitrát nitrogénnél nem tartalmaz többet.
7.1.2.2. A nitrát redukciója
Elegyítsük a bemért mintát 50 ml vízzel egy kis mozsárban. Minimális vízmennyiséggel mossuk át egy 500 ml-es Kjeldahl lombikba. Adjunk hozzá 5 g redukált vasat (4.2.) és 50 ml ón-klorid oldatot (4.11.). Rázzuk össze, és hagyjuk állni fél óra hosszat. Állás közben 10, illetve 20 perc elteltével újra keverjük össze.
7.1.2.3. Kjeldahl roncsolás
Adjunk 30 ml kénsavat (4.12.), 5 g kálium szulfátot (4.1.), a katalizátor előírt mennyiségét (4.27.), néhány horzsakövet (4.28.) a Kjeldahl lombikba. Az enyhén ferdén befogott lombikot kíméletesen melegítsük. Lassan növeljük a hőt, és időnként rázzuk meg a lombikot az elegy szuszpenzióban tartása végett. A folyadék megsötétedik, majd kivilágosodik sárgászöld vízmentes vasszulfát szuszpenzió keletkezése közben. Folytassuk egy óra hosszat a melegítést, az oldatot éppen a forráspontján tartva. Hagyjuk lehűlni. Óvatosan vegyük fel a lombik tartalmát kevés vízben, majd folyamatosan adjunk hozzá 100 ml vizet, és a lombik tartalmát mossuk át egy 500 ml-es mérőlombikba. Töltsük jelig vízzel. Elegyítsük. Szűrjük száraz edénybe száraz szűrőn keresztül.
7.1.2.4. Az oldat vizsgálata
Pipettával vigyük a desztilláló berendezés (5.1.) szedőjébe az oldat alikvot részét, amely legfeljebb 100 mg nitrogént tartalmaz. Hígítsuk 350 ml-re desztillált vízzel, adjunk hozzá pár horzsakövet (4.28.), csatlakoztassuk a lombikot a desztilláló berendezéshez és folytassuk a meghatározást a 7.1.1.2. leírás szerint.
7.1.2.5. Vakpróba Lásd a7.1.1.3. fejezetet.
7.1.2.6. Az eredmény kiszámítása
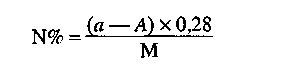
| ahol: | ||
| a = | 0,2 mol/l Nátrium- vagy kálium-hidroxid meghatározott koncentrációjú oldata, ml, amely a vakpróbánál fogyott, amikor a készülék (5.1.) szedőjébe 50 ml meghatározott koncentrációjú 0,2 mol/l kénsavat (4.8.) adtunk, | |
| A = | 0,2 mol/l Nátrium- vagy kálium-hidroxid meghatározott koncentrációjú oldata, ml, amely a minta vizsgálatára fogyott, | |
| M = | a 7.1.2.4. pont szerint vett alikvot oldatrészben jelenlévő minta mennyisége, g. |
7.2. Oldható nitrogénvegyületek
7.2.1. A vizsgálandó oldat készítése
Mérjünk 10 g mintát 1 mg pontossággal 500 ml-es mérőlombikba.
1.2.1.1. Ciánamidot nem tartalmazó műtrágyák esetében
Adjunk a lombikba 50ml vizet, majd 20 ml higított sósavat (4.13.). Rázzuk össze, és hagyjuk állni, amíg széndioxid fejlődése befejeződik. Azután adjunk hozzá 400 ml vizet, és fél óra hosszat rázassuk körkörös rázógépen (5.4.). Töltsük jelig vízzel, keverjük össze, és szűrjük száraz szűrőn át száraz edénybe.
7.2.1.2. Ciánamidot tartalmazó műtrágyák esetében
Adjunk a lombikba 400 ml vizet és pár csepp metilvörös indikátort (4.29.2.). Ha szükséges, savanyítsuk az oldatot ecetsavval (4.14.). Adjunk hozzá 15 ml ecetsavat (4.14.). Rázzuk a körkörös rázógépen 2 óra hosszat (5.4.). Ha szükséges, ecetsavval újra (4.14.) savanyítsuk meg az oldatot. Töltsük jelig vízzel, keverjük össze, és szűrjük haladéktalanul száraz szűrőn át száraz lombikba. Azonnal végezzük el a ciánamid nitrogén meghatározását.
Mindkét esetben ugyanaznap végezzük el az oldható nitrogénvegyületek meghatározását, mint amikor az oldatot készítettük, kezdve a ciánamid-kötésű és karbamid-kötésű nitrogénnel, ha ilyen jelen van.
7.2.2. Összes oldható nitrogén
7.2.2.1. Nitrátok távollétében
Pipettázzuk 300 ml-es Kjeldahl lombikba a szűrlet (7.2.1.1. vagy 7.2.1.2.) legfeljebb 100 mg nitrogént tartalmazó alikvot részét. Adjunk hozzá 15 ml koncentrált kénsavat (4.12.), 0,4 g rézoxidot vagy 1,25 g rézszulfátot (4.27.) és néhány szem horzsakövet (.4.28.). Először gyengén melegítsük a roncsolás megkezdésére, majd vigyük magasabb hőmérsékletre, és tartsuk ott, amíg az oldat színtelen vagy enyhén zöldes lesz és a fehér füstképződés jól láthatóvá válik. Lehűlés után veszteség nélkül vigyük az oldatot a desztilláló lombikba, hígítsuk kb. 500 ml-re vízzel, és adjunk hozzá néhány szem horzsakövet (4.28.). Csatlakoztassuk a lombikot a desztilláló készülékhez (5.1.), és folytassuk a meghatározást a 7.1.1.2. pont leírása szerint.
7.2.2.2. Nitrát jelenlétében
Hiteles pipettával vigyük 500 ml-es Erlemeyer lombikba a 7.2.1.1. vagy 7.2.1.2. pont szerinti szűrlet legfeljebb 40 mg nitrát-kötésű nitrogént tartalmazó alikvot mennyiségét. A vizsgálatnak ezen a fokán az összes nitrogén mennyisége nem lényeges. Adjunk hozzá 10 ml 30%-os kénsavat (4.15.), 5 g redukált vasport (4.2.), és azonnal takarjuk le az Erlenmeyer lombikot óraüveggel. Melegítsük kíméletesen, amíg a reakció állandóvá, de nem túl hevessé válik. Ekkor állítsuk le a melegítést, és hagyjuk a lombikot legalább három óra hosszat szobahőmérsékleten állni. Veszteség nélkül mossuk át vízzel a lombik tartalmát egy 250 ml-es mérőlombikba, dekantálva (az oldhatatlan vasat visszahagyva a reakciólombikban). Töltsük jelig vízzel a mérőlombikot. Gondosan keverjük össze a mérőlombik tartalmát, majd hiteles pipettával vigyük egy 300 ml-es Kjeldahl lombikba az oldat legfeljebb 100 mg nitrogént tartalmazó alikvot részét. Adjunk hozzá 15 ml koncentrált kénsavat (4.12.), 0,4 g rézoxidot vagy 1,25 g rézszulfátot (4.27.) és néhány horzsakövet (4.28.). Először óvatosan melegítsük a forrás megindítására, majd emeljük a hőmérsékletet magasabbra, amíg az oldat színtelenné vagy zöldessé, és a fehér füstképződés jól láthatóvá válik. Lehűlés után kvantitatív vigyük át az oldatot a desztilláló lombikba, hígítsuk kb. 500 ml-re vízzel, és adjunk hozzá néhány szem horzsakövet (4.28.). Csatlakoztassuk a lombikot a desztilláló berendezéshez (5.1.), és folytassuk a meghatározást a 7.1.1.2. szakasz szerint.
7.2.2.3. Vakpróba Lásd 7.1.1.3.
7.2.2.4. Az eredmény kiszámítása
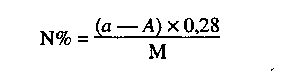
| ahol: | ||
| a = | 0,2 mol/l nátrium- vagy kálium-hidroxid meghatározott koncentrációjú oldata, ml, amely a vakpróbánál fogyott, amikor a készülék (5.1.) szedőjébe 50 ml meghatározott koncentrációjú 0,2 mol/l kénsavat (4.8.) adtunk, | |
| A = | 0,2 mol/l nátrium- vagy kálium-hidroxid meghatározott koncentrációjú oldata, ml, amely a minta vizsgálatára fogyott, | |
| M = | a 7.2.2.1 . vagy a 7.2.2.2. szakasz szerint vett alikvotban jelenlévő minta mennyisége, g. |
7.2.3. Összes oldható nitrogén, a nitrát nitrogén kivételével.
Hiteles pipettával vigyük a 7.2.1.1. vagy 7.2.1.2. pont szerinti szűrlet legfeljebb 50 mg meghatározandó nitrogént tartalmazó alikvot részét egy 300 ml-es Kjeldahl lombikba. Hígítsuk 100 ml-re vízzel, adjunk hozzá 5 g vasszulfátot (4.16.), 20 ml tömény kénsavat (4.1.) és néhány horzsakövet (4.28.) Először enyhén melegítsük, majd fokozzuk a hőt a fehér gőzök megjelenéséig. 15 percig folytassuk a roncsolást. Állítsuk le a melegítést, adjunk hozzá rézoxid (4.27.) katalizátort és tartsuk további 10-15 percen át azon a hőmérsékleten, ahol a fehér gőzök fejlődése megkezdődött. Lehűlés után a Kjeldahl lombik teljes tartalmát ontsuk át a desztilláló berendezés (5.1.) lombikjába. Hígítsuk vízzel kb. 500 ml-re, és adjunk hozzá néhány szem horzsakövet. Csatlakoztassuk a lombikot a desztilláló berendezéshez, és folytassuk le a meghatározást a 7.1.1.2. szakasz szerint.
7.2.3.1. Vakpróba Lásd 7.1.1.3.
7.2.3.2. Az eredmény kiszámítása
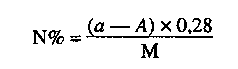
| ahol: | ||
| a = | 0,2 mol/l nátrium- vagy kálium-hidroxid meghatározott koncentrációjú oldata, ml, amely a vakpróbánál fogyott, amikor a készülék (5.1.) szedőjébe 50 ml meghatározott koncentrációjú 0,2 mol/l kénsavat (4.8.) adtunk, | |
| A = | 0,2 mol/l nátrium- vagy kálium-hidroxid meghatározott koncentrációjú oldata, ml, amely a minta vizsgálatára fogyott, | |
| M = | a vizsgálatra kivett alikvot részben jelenlévő minta mennyisége g-ban. |
7.2.4. Nitrát nitrogén
7.2.4.1. Kalcium ciánamid távollétében
A nitrát-kötésű nitrogént a 7.2.2.4. és 7.2.3.2. pont szerinti meghatározások különbségéből, illetve a 7.2.2.4. pont szerinti eredmény és a (7.2.5.2. vagy 7.2.5.5.) és/vagy a (7.2.6.3. vagy 7.2.6.5. vagy 7.2.6.6.) összegzett eredmény különbségéből számítjuk.
7.2.4.2. Kalcium ciánamid jelenlétében
A nitrát-kötésű nitrogént a 7.2.2.4. és 7.2.3.2. pontok szerinti meghatározások különbségéből, illetve 7.2.2.4. pont szerinti eredmény és a (7.2.5.5.), (7.2.6.3. vagy 7.2.6.5. vagy 7.2.6.6.) és (7.2.7.) pont szerinti eredmények összegének különbségéből számítjuk.
7.2.5. Ammónia kötésű nitrogén
7.2.5.1. Ha csak ammónia nitrogén vagy ammónia és nitrát nitrogén van jelen
Vigyük a desztilláló készülék (5.1.) lombikjába a. 7.2.1.1. szakasz szerinti szűrlet legfeljebb 100 mg ammónia nitrogént tartalmazó alikvot részletét. Adjunk hozzá annyi vizet, hogy térfogata kb. 350 ml legyen, és néhány szem horzsakövet (4.28.). Csatlakoztassuk a lombikot a desztilláló berendezéshez, adjunk hozzá 20 ml Nátrium-hidroxid oldatot (4.9.) és desztilláljuk le a 7.1.1.2. szakasz szerint.
7.2.5.2. Az eredmények kiszámítása:
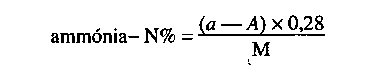
| ahol: | ||
| a = | 0,2 mol/l nátrium- vagy kálium-hidroxid meghatározott koncentrációjú oldata, ml, amely a vakpróbánál fogyott, amikor a készülék (5.1.) szedőjébe 50 ml meghatározott koncentrációjú 0,2 mol/l kénsavat (4.8.) adtunk, | |
| A = | 0,2 mol/l nátrium- vagy kálium-hidroxid meghatározott koncentrációjú oldata, ml, amely a minta vizsgálatára fogyott, | |
| M = | a vizsgálatra kivett alikvot részben jelenlévő minta mennyisége g-ban. |
7.2.5.3. Karbamid és/vagy ciánamid nitrogén jelenlétében
Hiteles pipettával vigyük a berendezés (5.2.) száraz lombikjába a 7.2.1.1. vagy 7.2.1.2. szakasz szerinti szűrlet legfeljebb 20 mg ammóniakötésű nitrogént tartalmazó alikvot részét. Állítsuk össze a készüléket. Pipettázzuk a 0,1 mol/l meghatározott koncentrációjú kénsavoldat (4.17.) 50 ml-ét egy 300 ml-es Erlenmeyer lombikba, és adjunk hozzá annyi vizet, hogy a folyadékszint kb. 5 cm-rel a bevezető cső nyílása fölött legyen. A reakcióedény oldalsó nyakán keresztül adjunk annyi desztillált vizet, hogy a térfogat 50 ml körül legyen. Keverjük össze. A levegőztetés alatti habzási elkerülendő, adjunk hozzá pár csepp oktil-alkoholt (4.18.). Lúgosítsuk meg az oldatot 50 ml telített kálium-karbonát oldattal (4.19.), és azonnal kezdjük meg a felszabadított ammónia kiűzését a hideg szuszpenzióból. Az ehhez szükséges erős levegőáramot (kb. 3 l/perc), előzetesen híg kénsavval, illetve híg nátrium-hidroxiddal töltött gázmosó palackokon vezetjük át. Sűrített levegő alkalmazása helyett vákuumban (vízsugár szivattyúval) is dolgozhatunk, feltéve, hogy a bevezető cső kielégítően légzáró módon kapcsolódik
az ammóniát felfogó gyűjtőedényhez. Az ammónia kiűzése általában három óra alatt fejeződik be. Azonban erről célszerű meggyőződni a gyűjtőedény kicserélésével. Amikor a műveletnek vége, válasszuk le a gyűjtőedényt, öblítsük le a cső végét és a lombik oldalát kevés desztillált vízzel. Titráljuk a savfelesleget 0,1 mol/l nátrium-hidroxid ismert koncentrációjú oldatával, amíg az indikátor (4.29.1.) színe szürkére változik.
7.2.4.3. Vakpróba Lásd 7.1.1.3.
7.2.5.5. Az eredmény kiszámítása
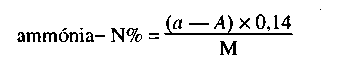
| ahol: | ||
| a = | 0,1 mol/l nátrium- vagy kálium-hidroxid meghatározott koncentrációjú oldata, ml, amely a vakpróbánál fogyott, amikor a készülék (5.2.) szedőjébe 50 ml meghatározott koncentrációjú 0,1 mol/l kénsavat (4.17.) adtunk, | |
| A = | 0,1 mol/l nátrium- vagy kálium-hidroxid meghatározott koncentrációjú oldata, ml, amely a minta vizsgálatára fogyott, | |
| M = | a vizsgálatra kivett alikvot részben jelenlévő minta mennyisége g-ban. |
7.2.6. Karbamid nitrogén
7.2.6.1. Ureáz módszer
Hiteles pipettával vigyük egy 500 ml-es mérőlombikba a 7.2.1.1. vagy 7.2.1.2. szakasz szerinti szűrlet legfeljebb 250 mg karbamid-nitrogént tartalmazó alikvot részét. A foszfátok lecsapására adjunk az oldathoz telített bárium-hidroxid oldatot (4.21.), amíg már nem figyelhető meg csapadékleválás. Ezután távolítsuk el a bárium ionokat (és az esetleg feloldódott kalcium ionokat) 10%-os nátrium-karbonát oldat (4.22.) segítségével. Hagyjuk a lombikban lévő oldatot ülepedni, ellenőrizzük, hogy a csapadékleválás teljesen végbement-e.
Ezután töltsük jelig, elegyítsük és szűrjük meg redős szűrőpapíron. A szűrlet 50 ml-ét pipettázzuk a készülék (5.3.) 300 ml-es Erlenmeyer lombikjába. Savanyítsuk meg a szűrletet 2N sósavval (4.23.), amíg a pH mérő 3-as pH-t mutat. Azután lúgosítsuk vissza az oldatot 5,4-re 0,1 mol/l nátrium-hidroxiddal.
Hogy az ureáz reakció alatti ammóniaveszteséget elkerüljük, zárjuk le az Erlenmeyer lombikot dugóval, amelybe egy csepegtető tölcsér, és egy 2 ml meghatározott koncentrációjú 0,1 mol/l sósavat tartalmazó buborékcsapda csatlakozik. A választótölcséren adjunk hozzá 20 ml ureáz oldatot (4.25.), és hagyjuk állni 20-25 C-on egy óra hosszat. Ezután pipettázzuk a 0,1 mol/l sósav mérőoldat (4.2.4.) 25 ml-ét az adagoló-tölcsérbe, engedjük, hogy átfusson rajta oldatba, majd öblítsük át egy kis vízzel. Hasonlóan mossuk át a védő gázmosók tartalmát is az Erlenmeyer lombikban lévő oldathoz. Titráljuk meg a savfölösleget 0,1 mol/l Nátrium-hidroxid mérőoldattal (4.20.) amíg a pH mérőn mérve 5,4 pH értéket kapunk.
7.2.6.2. Vakpróba Lásd 7.1.1.3.
7.2.6.3. Az eredmény kiszámítása
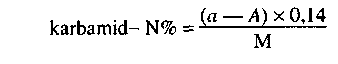
| ahol: | ||
| a = | 0,1 mol/l nátrium- vagy kálium-hidroxid mérőoldata, ml, amely a vizsgálattal pontosan azonos körülmények között elvégzett vakpróba során fogyott, | |
| A = | 0,1 mol/l nátrium- vagy kálium-hidroxid mérőoldata, ml, amely a minta vizsgálatára fogyott. | |
| M = | a vizsgálatra kivett alikvot részben jelenlévő minta mennyisége g-ban. |
Megjegyzések:
1. A báriumhidroxiddal és nátrium karbonáttal végzett kicsapás után a lehető leggyorsabban végezzük a lombik jelig töltését, szűrését és semlegesítést.
2. A titrálást indikátorral (4.29.2.) is el lehet végezni, de akkor nehezebb a végpont észlelése.
7.2.6.4. Xanthidrolos gravimetriás módszer
Hiteles pipettával vigyük a 7.2.1.1. vagy 7.2.1.2. szakasz szerinti szűrlet legfeljebb 20 mg karbamidot tartalmazó alikvot részét egy 250 ml-es főzőpohárba. Adjunk hozzá 40 ml ecetsavat (4.14.). Egy percig keverjük üvegbottal, a kivált csapadékot hagyjuk 5 percig ülepedni. Szűrjük egy 100 ml-es főzőpohárba, és mossuk át néhány ml ecetsavval (4.14.), majd cseppenként adjunk a szűrlethez 10 ml xanthidrolt (4.26.), közben üvegbottal folyamatosan kevergetve. A csapadék megjelenéséig hagyjuk állni. Amikor a csapadék kiválás megkezdődik, 1-2 percig újra kevergessük. Másfél óra hosszat hagyjuk ismét állni. Előzetesen kiszárított és lemért üvegszűrő tégelyen szűrjük meg, enyhén tömörítve. Háromszor mossuk át 5 ml etanollal (4.31.) anélkül, hogy az összes ecetsav kimosására törekednénk. Tegyük szárítószekrénybe, és szárítsuk 130 °C-on egy óra hosszat (a hőmérséklet ne emelkedjék 145 °C fölé). Exszikkátorban hagyjuk lehűlni és mérjük.
7.2.6.5. Az eredmény kiszámítása
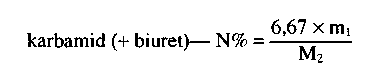
| ahol: | ||
| m1 = | a kapott csapadék tömege, g, | |
| M2 = | a vizsgálatra kivett alikvot részben lévő minta tömege, g. |
Az eredményt korrigáljuk a vakértékkel. Általában a biuretet nagyobb hiba elkövetése nélkül belemerhetjük a karbamid értékbe, mivel az összetett műtrágyákban a biuret tartalom abszolút értékben jelentéktelen.
7.2.6.6. Különbségen alapuló módszer
A karbamid nitrogént az alábbi táblázat szerint is kiszámíthatjuk:
| Eset | Nitrát nitrogén | Ammónia nitrogén | Ciánamid nitrogén | Különbség számítása |
| 1. | nincs | van | van | (7.2.2.4.)— [(7.2.5.5.) + (7.2.7.)] |
| 2. | van | van | van | (7.2.3.2.)— ( (7.2.5.5.) + (7.2.7.)] |
| 3. | nincs | van | nincs | (7.2.2.4.)— (7.2.5.5.) |
| 4. | van | van | nincs | (7.2.3.2.)— (7.2.5.5.) |
7.2.7. Ciánamid nitrogén
Vegyünk a 7.2.1.2. szakasz szerinti szűrletből 10-30 mg ciánamid tartalmú alikvot részt, vigyük át egy 250 ml-es főzőpohárba. Folytassuk le a vizsgálatot a 2.4. szakasz szerint.
8. AZ EREDMÉNYEK HITELESSÉGÉNEK ELLENŐRZÉSE
8.1. Bizonyos esetekben a bemért mintából közvetlenül meghatározott összes nitrogén tartalom (7.1.) és az összes oldható nitrogén tartalom (7.2.2.) különbözhet egymástól, de a különbség 0,5%-nál nem lehet nagyobb. Ha a különbség ennél nagyobb, akkor a műtrágya tartalmazhat olyan oldhatatlan nitrogén-tartalmú formákat, amelyek nem szerepelnek a 3. számú melléklet 1. függelékében közölt listában.
Minden vizsgálat előtt olyan standard oldattal ellenőrizzük a berendezések szabályos működését és a módszer helyes alkalmazását, amely oldat a különböző kötésben lévő nitrogént olyan vagy hasonló arányban tartalmazza, mint az a vizsgálandó mintából készült oldatban előfordul. Ezt a standard oldatot a következő vegyületek pontosan ismert koncentrációjú standard oldatainak felhasználásával készítjük: kálium-rodanid (4.3.), kálium-nitrát (4.4.), ammónium-szulfát (4.5.) és karbamid (4.6.).
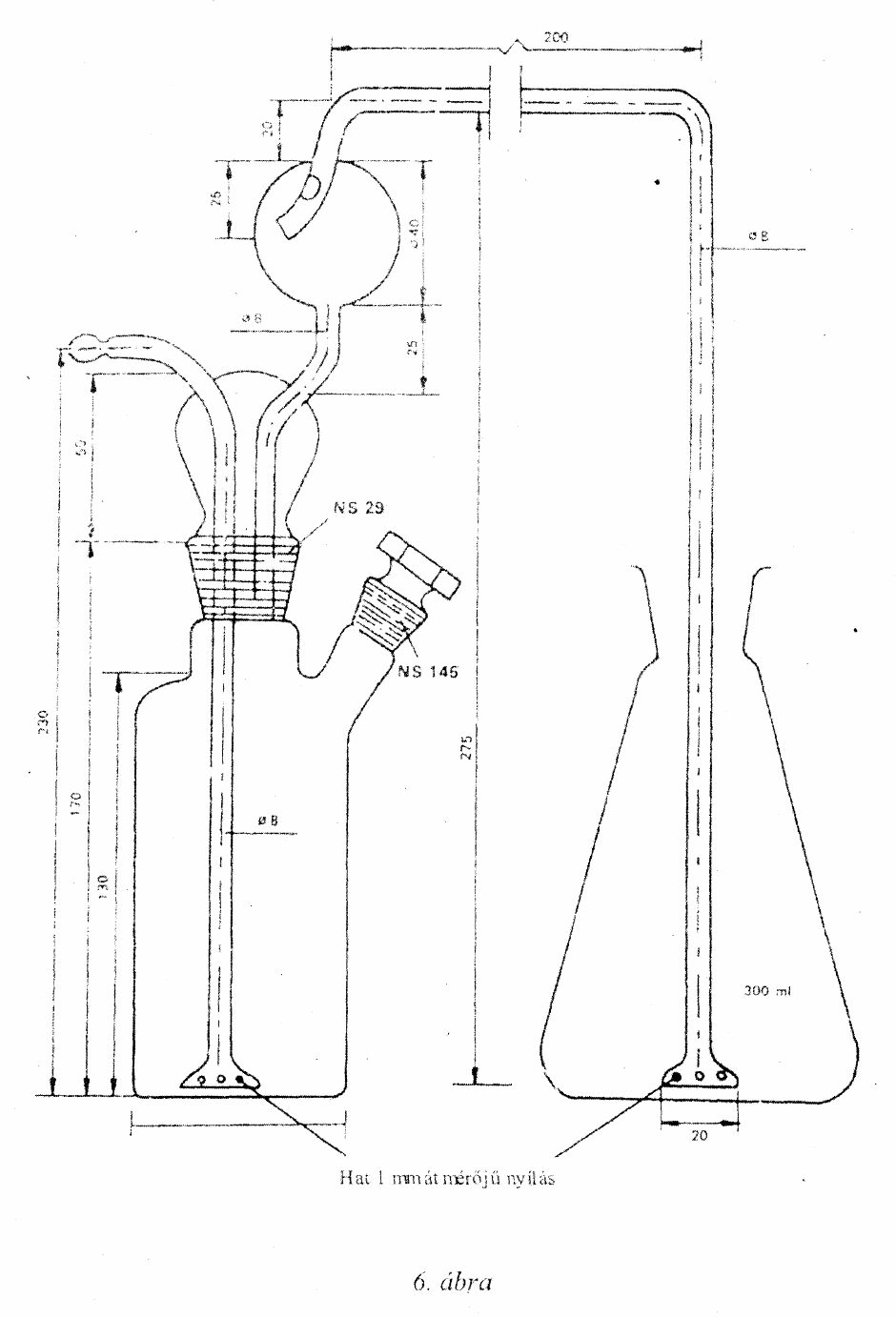
6. ábra
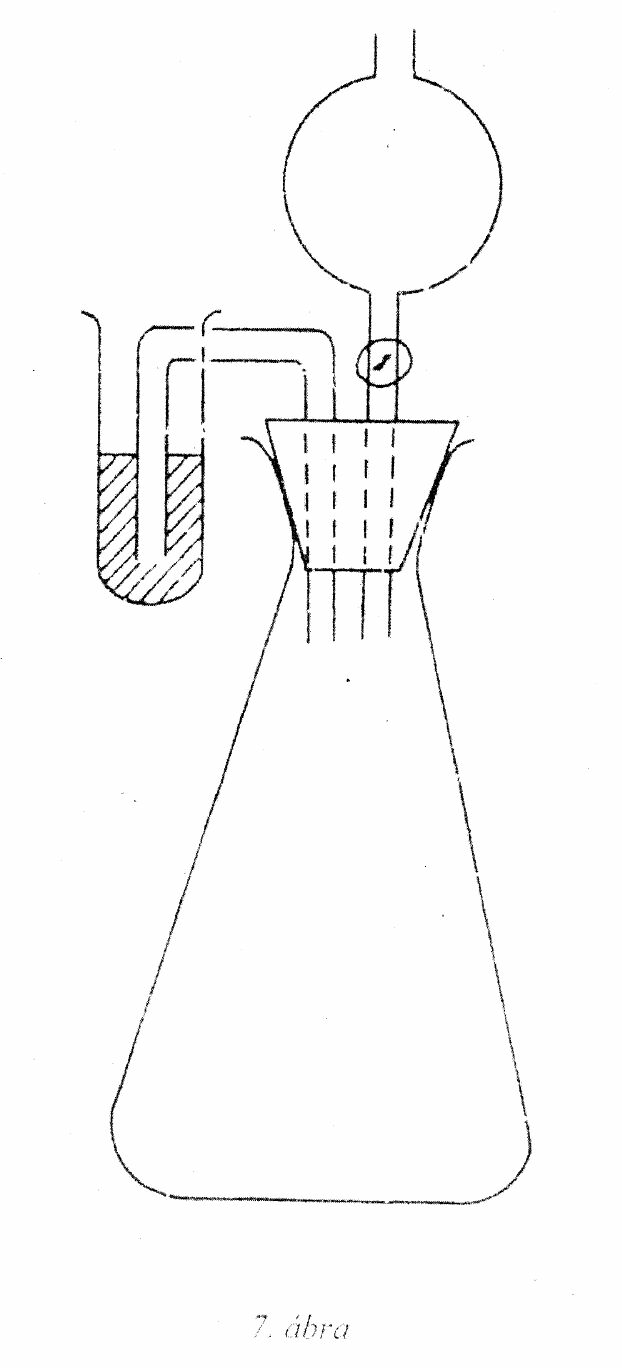
7. ábra
C/6.2. A NITROGÉN KÜLÖNBÖZŐ FORMÁINAK MEGHATÁROZÁSA OLYAN MŰTRÁGYÁKBAN, AMELYEK CSAK NITRÁT-, AMMÓNIA ÉS KARBAMID NITROGÉNT TARTALMAZNAK
1. TÁRGY
A jelen fejezet célja, hogy egyszerűsített módszert írjon le a nitrogén különféle formáinak meghatározására olyan műtrágyákban, amelyek a nitrogént csak nitrát-, ammónia- és karbamid-kötésben tartalmazzák.
2. ALKALMAZÁSI TERÜLET
A jelen módszer alkalmazható a 3. számú melléklet 1. függelékében említett minden olyan műtrágyára, amely kizárólag nitrát, ammónia vagy karbamid nitrogént tartalmaz.
3. A MÓDSZER ELVE
Ugyanazon mintaoldat különféle részleteiből az alábbi meghatározásokat végezzük el.
3.1. Összes oldható nitrogén
3.1.1. - nitrátok távollétében, az oldat Kjeldahl roncsolásával;
3.1.2. - nitrátok jelenlétében, az oldat egy részének Ulsch módszerrel történő redukcióját követő Kjeldahl roncsolással; az ammóniát mindkét esetben (3.1.1. és 3.1.2.) a C/1. módszer szerint határozzuk meg;
3.2. - nitrát kötésben levő nitrogén kivételével az összes oldható nitrogén meghatározása Kjeldahl roncsolással, miután a nitrát kötésű nitrogént savas közegben vasszulfáttal kicsaptuk, az ammóniát a C/1. módszer szerint meghatározzuk;
3.3. - nitrát nitrogén tartalom, a 3.1.2. és a 3.2. módszerek eredményei közötti eltérésből, vagy az összes oldható nitrogén (3.1.2.) és az ammónia vagy a karbamid kötésű nitrogén (3.4. + 3.5.) különbségéből;
3.4. - ammónia-kötésű nitrogén tartalom, enyhe lúgosítást követő hideg desztillációval, az ammóniát kénsavban elnyeletve a C/1. módszer szerint határozzuk meg;
3.5. - karbamid kötésű nitrogén;
3.5.1. - vagy ureáz segítségével ammóniává alakítva, és azt ismert koncentrációjú sósavval megtitrálva;
3.5.2. - vagy gravimetriás módszerrel xanthidrollal; - a biuretet a karbamiddal együtt mérjük, azonban ez csak elhanyagolható hibát okoz, mivel a biuret rendszerint igen kis mennyiségben fordul elő az összetett műtrágyákban;
3.5.3. - vagy különbség alapján az alábbi táblázat szerint:
| Eset | Nitrát kötésű nitrogén | Ammónia kötésű nitrogén | Karbamid nitrogén |
| 1. | nincs | van | (3.1.1.)— (3.4.) |
| 2. | van | van | (3.2.)— (3.4.) |
4. VEGYSZEREK Desztillált vagy ioncserélt víz.
4.1. Kálium-szulfát analitikai célra.
4.2. Vas, hidrogénnel redukálva, analitikai célra.
(A vas megadott mennyisége legalább 50 mg nitrát kötésű nitrogén redukciójára legyen képes.)
4.3. Kálium-nitrát analitikai célra.
4.4. Ammónium-szulfát analitikai célra.
4.5. Karbamid analitikai célra.
4.6. Kénsav oldat: 0,2 mol/l.
4.7. Tömény Nátrium-hidroxid oldat: közelítőleg 30% (m/v)-os vizes NaOH oldat, ammónia mentes.
4.8. Nátrium- vagy kálium-hidroxid oldat, 0,2 mol/l, ammónia mentes.
4.9. Kénsav, (d20 = 1,84 g/ml).
4.10. Hígított sósav, 1:1 térfogatarány, egy térfogat sósav (d20 =1,18 g/ml) egy térfogat víz.
4.11. Ecetsav, 96%-100%
4.12. Kénsav oldat, kb. 30% (m/v) H2SO4 tartalmú, ammóniamentes.
4.13. Vasszulfát; kristályos FeSO4.7H2O.
4.14. Faktorozott kénsavas oldat, 0,1 mol/l.
4.15. Oktil alkohol.
4.16. Telített kálium-karbonát oldat.
4.17. Nátrium- vagy kálium-hidroxid oldat: 0,1 mol/l.
4.18. Telített bárium-hidroxid oldat.
4.19. Nátrium-karbonát oldat: 10% (m/v).
4.20. Sósav oldat 2 mol/l.
4.21. Sósav oldat 0,1 mol/l.
4.22. Ureáz oldat
Készítsünk szuszpenziót 0,5 g aktív ureázból 100 ml vízben; 0,1 mol/l sósavval, pH-mérő segítségével állítsuk be a pH-ját 5,4-re.
4.23. Xanthidrol
5%-osetanolos vagy metanolos oldat (4.28.). (Ne használjunk olyan anyagot, amelyben magas az oldhatatlan részek aránya.) Gondosan lezárva, sötétben tárolva 3 hónapig eltartható.
4.24. Katalizátor
Rézoxid (CuO):0,3-0,4 g meghatározásonként, vagy rézszulfát CuSO4.5H2O, 0,95-1,25 g meghatározásonként.
4.25. Horzsakő forráskönnyítő granulátum, sósavval mosott, izzított.
4.26. Indikátor oldatok
A oldat: Oldjunk fel 1 g metilvöröst 37 ml 0,1 nátrium-hidroxid oldatban, és vízzel töltsük fel egy literre.
B oldat: Oldjunk fel 1 g metilénkéket vízben, és egészítsük ki az oldat térfogatát 1 literre.
Keverjünk össze egy térfogategység A oldatot két térfogategység B oldattal.
Ez az indikátor savas oldatban ibolyaszínű, semlegesben szürke, és lúgos oldatban zöld. Használjunk 0,5 ml-t (10 csepp).
4.26.2. Metilvörös indikátor oldat
Oldjunk fel 0,1 g metilvöröst 50 ml 95%-os etanolban. Egészítsük ki vízzel 100 ml-re és ha szükséges, szürjük meg.
Ezt az indikátort (4-5 cseppjét) az előző indikátor helyett használhatjuk.
4.27. Indikátorpapírok
Lakmusz, brómtimol kék (vagy más, pH 6-8 között érzékeny indikátorpapír).
4.28. Etanol vagy metanol: 95%-os (m/v).
5. FELSZERELÉS
5.1. Desztillációs berendezés: lásd a C/1. fejezetet.
5.2. Az ammóniakötésű nitrogén meghatározására szolgáló készülék a 7.5.1. fejezetnek megfelelően (lásd a C/6.1. fejezetet és a 6. ábrát).
A berendezés speciálisan kiképzett, gömbölyű üveg fejrésszel ellátott szedőből áll, amelynek egy oldal kivezető nyaka is van. A fejen keresztül egy cseppfogóval ellátott cső nyúlik ki, valamint egy merőlegesen hajlított cső a levegő bevezetésére. A csöveket egyszerű gumi csatlakozással lehet a szedőhöz kapcsolni. Fontos, hogy a levegőt bevezető cső végének kiképzése megfelelő formájú legyen, mivel gázbuborékoknak tökéletesen kell eloszolniuk a szedőben és az elnyeletőben lévő oldatokban. A legjobb erre a kis, gombaformájú gázbevezető fej, amelynek átmérője 20 mm, és közben a kerületén 6 db 1 mm-es nyílása van.
5.3. A karbamid-kötésű nitrogén ureáz módszerrel történő meghatározására szolgáló készülék (7.6.1.). Egy 300 ml-es Erlenmeyer lombikból, egy csepegtetőtölcsérből és egy kis gázmosóból áll (lásd a 7. ábra).
5.4. Körkörös rázógép (35-40 fordulat/perc).
5.5. pH-méter.
5.6. Üvegáru
- pipetták, 2, 5, 10, 20, 25, 50 és 100 ml-es,
- hosszúnyakú Kjeldahl lombikok, 300 és 500 ml-es,
- mérőlombikok 100, 250, 500 és 1000 ml-es,
- zsugorított üvegszűrő tégelyek, 5-15 μm pórusátmérőjű,
- mozsarak.
6. A MINTA ELŐKÉSZÍTÉSE Lásd a B) fejezetet.
7. ANALITIKAI MÓDSZEREK
7.1. Az oldatok elkészítése az analízishez
Mérjünk be 10 g mintát 1 mg pontossággal egy 500 ml-es Stohmann lombikba. Adjunk hozzá 50 ml vizet, azután 20 ml hígított sósavat (4.10.). Rázzuk össze. Hagyjuk állni amíg a széndioxid felszabadulás befejeződik. Adjunk hozzá 400 ml vizet; és rázassuk fél órán keresztül; töltsük fel jelig vízzel, homogenizáljuk, és szűrjük le egy száraz szűrőpapíron keresztül egy száraz üvegedénybe.
7.2. Összes nitrogén
7.2.1. Nitrátok jelenléte nélkül
Pipettázzuk a szűrlet (7.1.) legfeljebb 100 mg nitrogént tartalmazó részletét egy 300 ml-es Kjeldahl lombikba. Adjunk hozzá 15 ml koncentrált kénsavat (4.9.), 0,4 g rézoxidot vagy 1,25 g rézszulfátot (4.24.) és néhány szem üveggyöngyöt forráskönnyítőnek. Először óvatosan kezdjük hevíteni, hogy a reakció beinduljon, azután erősebben, amíg a folyadék színtelenné, vagy enyhén zöldessé válik, és a kéntrioxid fehér füstje félreismerhetetlenül megjelenik. Lehűlés után vigyük át az oldatot egy desztilláló lombikba, hígítsuk kb. 500 ml-re vízzel, és adjunk hozzá néhány szem horzsakövet (4.25.). Csatlakoztassuk a lombikot a desztilláló készülékhez (5.1.) és végezzük el a meghatározást a C/6.1. módszer 7.1.1.2. pontjában leírtak szerint.
7.2.2. Nitrátok jelenlétében
A 7.1. pont szerinti szűrlet legfeljebb 40 mg nitrát-kötésben lévő nitrogént tartalmazó részletét pipettázzuk egy 500 ml-es Erlenmeyer lombikba. A vizsgálatnak ezen a fokán az összes N mennyisége nem fontos. Adjunk hozzá 10 ml 30%-os kénsavat (4.12.), 5 g redukált vasat (4.2.) és azonnal fedjük le az Erlenmeyer lombik száját óraüveggel. Óvatosan melegítsük, amíg a reakció erőteljesen megindul, de nem válik túl hevessé. Állítsuk le a fűtést, és hagyjuk állni legalább 3 órát szobahőmérsékleten. A folyadékot veszteség nélkül ontsuk át egy 250 ml-es mérőlombikba, a fel nem oldott vasat az eredeti lombikban visszahagyva. Töltsük a lombikot jelig vízzel. Óvatosan elegyítsük. Pipettázzuk egy legfeljebb 100 mg N-t tartalmazó részletét 300 ml-es Kjeldahl lombikba. Adjunk hozzá 15 ml tömény kénsavat (4.9.) meg 0,4 rézoxidot vagy 1,25 g rézszulfátot (4.24.) és pár üveggyöngyöt a forrás szabályozására. Először óvatosan melegítsük a reakció megindítására, azután erősebben, amíg a folyadék színtelenné vagy halvány zöldessé válik, és fehér gőzök megjelenése egyértelműen észlelhető. Ha lehűlt, vigyük az oldatot veszteség nélkül a desztilláló lombikba, hígítsuk kb. 500 ml-re vízzel, adjunk hozzá néhány szem horzsakövet (4.25.). Csatlakoztassuk a lombikot a desztilláló készülékhez (5.1.) és folytassuk a meghatározást a C/6.1. módszer 7.1.1.2. pontja szerint.
7.2.3. Vakpróba
Végezzünk vakpróbát a vizsgálattal megegyező körülmények között, és eredményét vegyük figyelembe a végeredmény kiszámításánál.
7.2.4. Az eredmény kiszámítása
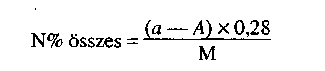
| ahol: | ||
| a = | faktorozott 0,2 mol/l nátrium- vagy kálium-hidroxid (4.8.) fogyása a vakpróba során, amikor a szedőbe 50 ml faktorozott 0,2 mol/l kénsavoldatot (4.6.) mértünk, ml, | |
| A = | faktorozott 0,2 mol/l nátrium- vagy kálium-hidroxid (4.8.) fogyása a minta vizsgálatakor, ml, | |
| M = | az oldat alikvot részében (7.2.1. vagy 7.2.2.) jelenlévő minta mennyisége, g. |
7.3. Összes nitrogén, a nitrát-kötésű nitrogén kivételével
7.3.1. Vizsgálat
A 7.1. szakasz szerinti szűrlet legfeljebb 50 mg meghatározandó nitrogént tartalmazó alikvot részletét 300 ml-es Erlenmeyer lombikba pipettázzuk. 100 ml vízzel hígítjuk. 5 g vasszulfátot (4.13.), 20 ml tömény kénsavat (4.9.) és néhány forráskönnyítő üveggyöngyöt adunk hozzá. Először óvatosan, majd erősebben melegítjük, a kéntrioxid fehér füstjének megjelenéséig. Folytassuk a melegítést 15 percig, majd állítsuk le a fűtést, tegyünk a lombikba 0,4 g rézoxidot vagy 1,25 g rézszulfátot (4.24.) a folyamat katalizálására. Ismét kezdjük meg a melegítést, és 10-15 percig fehér füstképződés közben forraljuk az oldatot. Lehűlés után vigyük át a Kjeldahl lombik tartalmát veszteség nélkül a desztilláló lombikba (5.1.). Hígítsuk kb. 500 ml vízzel, adjunk hozzá, néhány szem horzsakő granulátumot (4.25.). Csatlakoztassuk a lombikot a desztilláló készülékhez, és folytassuk a meghatározást a 2.6. módszer 7.1.1.2. pontjában leírtak szerint.
7.3.2. Vakpróba Lásd 7.2.3.
7.3.3. Az eredmény kiszámítása
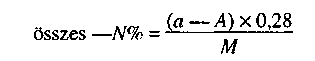
| ahol: | ||
| a = | faktorozott 0,2 mol/l nátrium- vagy kálium-hidroxid (4.8.) fogyása a vakpróba során, amikor a szedőbe 50 ml faktorozott 0,2 mol/l kénsavoldatot (4.6.) mértünk, ml, | |
| A = | faktorozott 0,2 mol/l nátrium- vagy kálium-hidroxid (4.8.) fogyása a minta vizsgálatakor, ml, | |
| M = | az oldatok vizsgálathoz felhasznált alikvot részében jelenlévő minta mennyisége, g. |
7.4. Nitrát nitrogén
A következő különbségek alapján:
7.2.4. -(7.5.3.+ 7.6.3.)
vagy
7.2.4. -(7.5.3.+ 7.6.5.)
vagy
7.2.4. - (7.5.3.+ 7.6.6.).
7.5. Ammónia kötésben lévő nitrogén
7.5.1. Vizsgálat
A berendezés (5.2.) száraz lombikjába vigyük a szűrlet (7.1.) legfeljebb 20 mg ammónia kötésben lévő nitrogént tartalmazó mennyiségét. Csatlakoztassuk a lombikot a készülékhez. Pipettázzunk a 300 ml-es Erlenmeyer lombikba pontosan 50 ml faktorozott 0,1 mol/l kénsavat (4.14.), és adjunk hozzá annyi desztillált vizet, hogy a folyadékszint kb. 5 cm-rel a bemeneti cső nyílása felett legyen. A reakció lombik oldalcsövén keresztül vigyünk be annyi desztillált vizet, hogy a térfogat kb. 50 ml legyen. Rázzuk össze. Adjunk hozzá néhány csepp oktil-alkoholt (4.15.), hogy megelőzzük a gázáram megindításakor fellépő habzást. Adjunk hozzá 50 ml telített kálium-karbonát oldatot (4.16.), és azonnal indítsuk meg az így felszabadított ammónia • kiűzését a hideg szuszpenzióból Az ehhez szükséges intenzív levegőáramot (áramlási sebesség kb. 3 liter/perc) előzetesen híg kénsavat és híg nátrium-hidroxidot tartalmazó gázmosó palackokon való átvezettetéssel tisztítjuk. Nyomás alatti levegő használata helyett vákuumot (vízsugárszivattyú) használhatunk, ha a készülék tömítettsége megfelelő.
Az ammónia kinyerése általában 3 óra alatt teljessé válik. Azonban erről kívánatos megbizonyosodni úgy, hogy az Erlenmeyer lombikot kicseréljük. Amikor a folyamat befejeződött, vegyük le az Erlenmeyer lombikot a készülékről, öblítsük bele a bevezető cső végét és az Erlenmeyer lombik belső falát egy kis desztillált vízzel, és titráljuk meg a savfelesleget az ismert faktorú 0,1 mol/l nátrium-hidroxid oldattal (4.17.).
7.5.2. Vakpróba Lásd 7.2.3.
7.5.3. Az eredmény kiszámítása
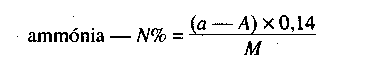
| ahol: | ||
| a = | faktorozott 0,1 mol/l nátrium- vagy kálium-hidroxid oldat (4.17.) fogyása a vakpróba során, amikor a berendezés (5.2.) 300 ml-es Erlenmeyer lombikjába 50 ml faktorozott 0,1 mol/l kénsav oldatot (4.14.) mértünk, ml, | |
| A = | faktorozott 0,1 mol/l nátrium- vagy kálium-hidroxid (4.17.) fogyása a minta vizsgálatakor, ml, | |
| M = | az oldatok vizsgálathoz felhasznált alikvot részében jelenlévő minta mennyisége, g. |
7.6. Karbamid-kötésű nitrogén
7.6.1. Ureázos módszer
A szűrlet (7.1.) legfeljebb 250 mg karbamidot tartalmazó mennyiségét vigyük 500 ml-es mérőlombikba. Adjunk hozzá megfelelő mennyiségű telített bárium-hidroxid oldatot (4.18.) addig, amíg az oldat további adagolására több csapadék már nem válik le. A fölös bárium ionokat (és az esetlegesen feloldódott kalcium ionokat) 10%-os nátrium karbonát oldattal (4.19.) távolítsuk el. Hagyjuk állni, majd ellenőrizzük, hogy a kicsapás teljes-e. Töltsük jelig a mérőlombikot, homogenizáljuk, majd redős szűrőpapíron szűrjük le a tartalmát. A szűrlet 50 ml-ét pipettázzuk a készülék (5.3.) 300 ml-es Erlenmeyer lombikjába. Savanyítsuk meg 2N sósavval (4.20.) pH 3 értékig, amelyet pH-méteren állítunk be. Emeljük a pH-t 5,4-re 0,1 mol/l nátrium hidroxiddal (4.17.). Hogy elkerüljük az ammónia veszteséget az ureáz által megindított hidrolízis során, az Erlenmeyer lombikot olyan dugóval zárjuk le, amelybe egy csepegtető tölcsér és egy pontosan 2 ml 0,1 mol/l sósavat (4.21.) tartalmazó gázcsapda tartály csatlakozik. A csepegtető tölcséren keresztül adjunk a lombikba 20 ml ureáz oldatot (4.22.). Hagyjuk a lombikot egy órán át 20-25 C-on állni. Pipettázzunk 25 ml meghatározott koncentrációjú 0,1 mol/l sósav oldatot (4.21.) a csepegtető tölcsérbe, engedjük bele a lombikba, majd öblítsük be egy kis vízzel. A gázcsapda tartályban lévő folyadékot szintén vigyük veszteség nélkül az Erlenmeyer lombikba. Titráljuk meg a savfelesleget meghatározott koncentrációjú 0,1 mol/l nátrium-hidroxid oldattal (4.17.), amíg a pH méteren mért pH érték eléri az 5,4-et.
Megjegyzések:
1. A bárium-hidroxid és a nátrium-karbonát oldatok hozzáadása után a jelig töltést, szűrést és semlegesítést a lehető leggyorsabban végezzük el.
2. A titrálást indikátor (4.26.) mellett is el lehet végezni, de a színátcsapás nehezebben észlelhető.
7.6.2. Vakpróba Lásd 7.2.3.
7.6.3. Az eredmény kiszámítása
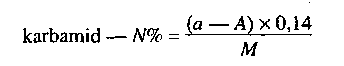
| ahol: | ||
| a = | faktorozott 0,1 mol/l nátrium- vagy káliumhidroxid oldat (4.17.) fogyása a vakpróba során, amelyet pontosan a vizsgálattal megegyező körülmények között végeztük el, ml, | |
| A = | faktorozott 0,1 mol/l nátrium- vagy káliumhidroxid (4.17.) fogyása a minta vizsgálatakor, ml, | |
| M = | az oldat vizsgálathoz felhasznált alikvot részében jelenlévő minta mennyisége, g. |
7.6.4. Xanthidrollal végzett gravimetriás meghatározás.
Pipettázzuk a szűrlet (7.1.) legfeljebb 20 mg karbamidot tartalmazó mennyiségét 100 ml-es főzőpohárba. Adjunk hozzá 40 ml ecetsavat (4.11.). Keverjük üvegbottal 1 percig. A kiváló csapadékot hagyjuk 5 percig ülepedni. Szűrjük, mossuk pár ml ecetsavval (4.11.). Cseppenként adjunk az oldathoz 10 ml xanthidrolt (4.23.), az oldatot közben állandóan kevergetve üvegbottal. Hagyjuk állni, amíg csapadékleválást észlelünk, majd egy-két percig keverjük újra az üvegbottal. Hagyjuk állni másfél óra hosszat. Enyhe vákuum mellett szűrjük előre kiszárított és lemért üvegszűrő tégelyen, mossuk át háromszor 5 ml etanollal (4.28.), anélkül, hogy az ecetsav teljes eltávolítására törekednénk. Helyezzük a szűrőt szárítószekrénybe és szárítsuk 1 óra hosszat 130 C-on (a 145 °C hőmérsékletet nem szabad elérni.). Hagyjuk lehűlni exszikkátorban és mérjük le.
7.6.5. Az eredmény kiszámítása
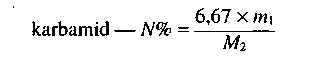
| ahol: | ||
| m = | a csapadék tömege, g, | |
| M = | az oldat vizsgálathoz felhasznált alikvot részében jelenlévő minta mennyisége, g. |
Az eredményt korrigáljuk a vakértékkel. Általában a biuretet nagyobb hiba elkövetése nélkül belemérhetjük a karbamid értékbe, mivel az összetett műtrágyákban a biuret tartalom abszolút értékben jelentéktelen.
7.6.6. Különbségen alapuló módszer
A karbamid nitrogént az alábbi táblázat szerint is kiszámíthatjuk:
| Eset | Nitrát kötésű nitrogén | Ammónia kötésű nitrogén | .Karbamid nitrogén |
| 1. | nincs | van | (7.2.4.)— (7.5.3.) |
| 2. | van | van | (7.3.3.)— (7.5.3.) |
8. AZ EREDMÉNYEK HITELESSÉGÉNEK ELLENŐRZÉSE
Minden vizsgálat előtt olyan standard oldattal ellenőrizzük a berendezések szabályos működését és a módszer helyes alkalmazását, amely oldat a különböző kötésben lévő nitrogént olyan vagy hasonló arányban tartalmazza, mint a vizsgálandó mintából készült oldatban előfordul. Ezt a standard oldatot a következő vegyületek pontosan ismert koncentrációjú standard oldatainak felhasználásával készítjük: kálium-nitrát (4.3.), ammónium-szulfát (4.4.) és karbamid (4.5.).
D) FOSZFOR
D/1. EXTRAKCIÓS MÓDSZEREK
D/1.1. ÁSVÁNYI SAVAKBAN OLDHATÓ FOSZFOR KIVONÁSA
1. TÁRGY
A jelen fejezet eljárást ír le az ásványi savakban oldható foszfor meghatározására.
2. ALKALMAZÁSI TERÜLET
A módszer kizárólag a 3. számú melléklet 1. függelékében felsorolt foszfát műtrágyákra alkalmazható.
3. A MÓDSZER ELVE
A foszfort salétromsav és kénsav elegyével vonjuk ki a műtrágyából.
4. VEGYSZEREK Desztillált vagy ioncserélt víz.
4.1. Kénsav (d20= l,84 g/ml).
4.2. Salétromsav (d20 = 1,40 g/ml).
5. FELSZERELÉS
Szokásos laboratóriumi felszerelés,
5.1. Kjeldahl lombik, legalább 500 ml-es, vagy 250 ml-es gömbölyű fenekű, visszafolyó hűtőként működő üvegcsővel.
5.2. 500 ml-es mérőlombik.
6. A MINTA ELŐKÉSZÍTÉSE Lásd a B) fejezetet.
7. ELJÁRÁS
7.1. Minta
Mérjünk be 2,5 g mintát 0,001 g pontossággal száraz Kjeldahl lombikba.
7.2. Extrakció
Adjunk hozzá 15 ml vizet, és összekeveréssel szuszpendáljuk az anyagot. Adjunk hozzá 20 ml salétromsavat (4.2.), majd óvatosan 30 ml kénsavat (4.1.).
Mikor a kezdetben heves reakció csillapodik, óvatosan hozzuk forrásba az oldatot, és forraljuk 30 percig. Hagyjuk lehűlni, és óvatos keverés közben adjunk hozzá 150 ml vizet. Folytassuk a forralást még 15 percig.
Hűtsük le teljesen, és vigyük 500 ml-es mérőlombikba. Töltsük jelig, homogenizáljuk, és száraz, foszfátmentes redős szűrőpapíron szűrjük, a szűrlet első részletét (kb. 20 ml) ontsuk el.
7.3. Meghatározás
A foszfor meghatározását az így nyert oldat alikvot részéből a D/2. módszer szerint végezzük el.
D/1.2. 2%-os (20 g/liter) HANGYASAVBAN OLDHATÓ FOSZFOR KIVONÁSA
1. TÁRGYKÖR
A jelen fejezet eljárást ír le a 2%-os (20 g/liter) hangyasavban oldható foszfor meghatározására.
2. ALKALMAZÁSI TERÜLET
A módszer kizárólag lágy természetes foszfátok kivonására alkalmazható.
3. A MÓDSZER ELVE
A kemény és lágy természetes foszfátok megkülönböztetése céljából a hangyasavban oldható foszfort meghatározott körülmények között vonjuk ki.
4. VEGYSZEREK
4.1. Hangyasav, 2%-os (20 g/liter).
Megjegyzés
82 ml hangyasavat (koncentráció: 98-100%; d20 = 1,22 g/ml) oldjunk 5 literre desztillált vízzel.
5. FELSZERELÉS
Szokásos laboratóriumi felszerelés.
5.1. 500 ml-es kalibrált lombik (pl. Stohmann lombik).
5.2. Körkörös rázógép (35-40 fordulat/perc).
6. A MINTA ELŐKÉSZÍTÉSE Lásd a B) módszert.
7. ELJÁRÁS
7.1. Minta
Mérjük az előkészített minta 5 g-ját 0,001 g pontossággal egy bő nyakú, száraz, 500 ml-re kalibrált Stohmann lombikba (5.1.).
7.2. Extrakció
Miközben a lombikot kézzel rázogatjuk, adjuk hozzá a 2%-os 20±l°C-os hangyasavat (4.1.), amíg a kalibrációs jelnél kb. 1 cm-rel alacsonyabb folyadékszintet nem érünk el. Töltsük jelig a lombikot. Zárjuk le gumidugóval, és 20±2 C-on rázassuk 30 percig a rázógépen (5.2.)
Az oldatot száraz, redős, foszfátmentes szűrőpapíron át száraz edénybe szűrjük. A szűrlet első (kb. 20 ml-es) részletét ontsuk el.
7.3. Meghatározás
A foszfort a teljesen tiszta szűrlet alikvot részéből a D/2. módszer szerint határozzuk meg.
D/1.3. 2%-os (20 g/liter) CITROMSAVBAN OLDHATÓ FOSZFOR KIVONÁSA
1. TÁRGY
A jelen fejezet eljárást ír le a 2%-os (20 g/liter) citromsavban oldható foszfor meghatározására.
2. ALKALMAZÁSI TERÜLET
A módszer kizárólag bázikus salak típusokra (lásd a 3. számú melléklet 1. függeléke) alkalmazható.
3. A MÓDSZER ELVE
A foszfort 2%-os (20 g/liter) citromsav oldattal, meghatározott körülmények között vonjuk ki a műtrágyából.
4. VEGYSZEREK Desztillált vagy ioncserélt víz.
4.1. 2%-os (20 g/liter) citromsav oldat, kristályos citromsavból (C6H8O7.H2O) készítve.
Megjegyzés:
A citromsav oldat koncentrációját ellenőrizzük úgy, hogy 10 ml-ét ismert faktorú 0,1 mol/l nátrium-hidroxid oldattal megtitráljuk, fenolftalein indikátor mellett. Ha a citromsav oldat megfelelő, a nátrium-hidroxid oldat fogyása 28,55 ml lesz.
5. FELSZERELÉS
5.1. Körkörös rázógép (35-40 fordulat/perc).
6. A MINTA ELŐKÉSZÍTÉSE
A vizsgálatot a beérkezési állapotú mintán végezzük el. A mintát gondosan össze kell keverni, hogy homogenitását biztosítsuk. Lásd az 1. módszert.
7. ELJÁRÁS
7.1. Minta
Mérjük az előkészített minta 5 g-ját 0,001 pontossággal, és vigyük olyan 600 ml térfogatú megfelelően széles nyakú száraz lombikba, amelyben a minta alapos rázatására lehetőség van.
7.2. Extrakció
Adjunk a mintához 500±1 ml, 20±1 °C hőmérsékletű citromsav oldatot (4.1.). Amikor az első millilitereket adagoljuk, erősen rázzuk a lombikot, így megelőzve, hogy csomók képződjenek, vagy az anyag a lombik falára tapadjon. Gumidugóval zárjuk le a lombikot, és pontosan 30 percig, 20±2 °C-on rázassuk körforgó rázógépen (5.1.).
Szűrjük késedelem nélkül száraz, foszfátmentes redős szűrőpapíron száraz üvegedénybe. Ontsuk el a szűrlet első kb. 20 ml-ét. Folytassuk a szűrést, amíg a foszfor meghatározáshoz elegendő mennyiségű szűrletet nem nyerünk.
7.3. Meghatározás
A foszfát meghatározást a D/2. módszerrel végezzük az így nyert oldat alikvot részéből.
D/1.4. SEMLEGES AMMÓNIUMCITRÁTBAN OLDHATÓ FOSZFOR KIVONÁSA
1. TÁRGY
A jelen fejezet módszert ír le semleges ammónium-citrátban oldható foszfor meghatározására.
2. ALKALMAZÁSI TERÜLET
A módszer minden olyan műtrágyára alkalmazható, amelynek semleges ammónium-citrát oldatban való oldhatóságát megállapították (lásd a 3. számú melléklet 1. függelékét).
3. A MÓDSZER ELVE
A foszfort 65 °C-on, semleges ammónium-citrát oldatta (pH 7.0) meghatározott körülmények között extraháljuk.
4. VEGYSZEREK Desztillált vagy ioncserélt víz.
4.1. Semleges ammónium citrát oldat (pH 7.0).
Az oldatnak 185 g kristályos citromsavat kell tartalmaznia literenként. pH 7-nél, 20 °C-on sűrűségének 1,09 g/ml-nek kell lennie.
A reagenst a következőképpen készítsük:
Oldjunk fel 370 g kristályos citromsavat (C6H8O7.H2O) kb. 1,5 l vízben, és állítsuk megközelítően semleges kémhatásúra 345 ml ammónium hidroxid oldat (28-29% közötti ammóniatartalommal) hozzáadásával. Ha az ammónia koncentráció alacsonyabb, mint 28%, akkor adagoljunk megfelelően több ammónium hidroxidot, és a citromsavat ennek megfelelően kevesebb víz hozzáadásával higítsuk.
Hűtsük le az oldatot, és pH méter elektródjainak az oldatba való mentésével állítsuk a pH-t pontosan semlegesre úgy, hogy mechanikus keverővel való folyamatos kevertetés közben cseppenként adagoljuk a 28 és 29% közé eső ammóniatartalmú oldatot mindaddig, amíg 20 °C hőmérsékleten pontosan 7,0 pH értéket kapunk. Ekkor egészítsük ki a térfogatot 2 l-re, és újra ellenőrizzük a pH-t. Tartsuk a reagenst zárt edényben, és pH-ját rendszeresen ellenőrizzük.
5. FELSZERELÉS
5.1. Főzőpohár, 2 l-es.
5.2. pH mérő.
5.3. Erlenmeyer lombik, 200 vagy 250 ml-es 5.4. Mérőlombikok, 500 ml-es és 200 ml-es.
5.4. 500 ml-es és 2000 ml-es mérőlombik.
5.5. Vízfürdő, termosztálható 65 °C-ra, megfelelő keverővel ellátva (lásd a 8. ábrát).
6. A MINTA ELŐKÉSZÍTÉSE Lásd a B) fejezetet.
7. ELJÁRÁS
7.1. Minta
A vizsgálandó műtrágya (lásd az Irányelv I. mellékletének A és B részét) 1 és 3 g közé eső mennyiségét 0,001 g pontossággal bemérve vigyük 200 ml-es vagy 250 ml-es Erlenmeyer lombikba, amelybe előzetesen 100 ml, 65 °C-ra melegített ammónium citrát oldatot töltöttünk.
7.2. Az oldat vizsgálata
Dugjuk be az Erlenmeyer lombikot, és rázzuk annak érdekében, hogy csomómentes szuszpenziót nyerjünk. Időnként egy pillanatra vegyük ki a dugót a nyomás kiegyenlítésére, majd zárjuk vissza az Erlenmeyer lombikot. Helyezzük a lombikot vízfürdőbe, hogy tartalmának hőmérsékletét pontosan 65 °C-on tarthassuk, és csatlakoztassuk a keverőhöz (8. ábra). Keverés közben a szuszpenzió szintjének állandóan a vízfürdőben lévő víz szintje alatt kell lennie. A mechanikai keverést úgy állítsuk be, hogy az anyag teljes szuszpendálását biztosítsuk.[78]
Pontosan 1 órai keverés után vegyük ki az Erlenmeyer lombikot a vízfürdőből.
Hűtsük le azonnal folyóvíz alatt szobahőmérsékletre, és tartalmát veszteség nélkül vigyük át egy 500 ml-es mérőlombikba fecskendővel ellátott desztillált vizes palackkal való bemosással. Töltsük jelig vízzel. Keverjük alaposan össze. Szűrjük át közepes sebességgel száraz, redős, közepes sebességű, foszfátmentes szűrőpapíron száraz edénybe. Ontsuk el a szűrlet első, kb. 50 ml-es részletét.
Ezután gyűjtsünk össze kb. 100 ml tiszta szűrletet.
7.3. Meghatározás
Az így nyert kivonat foszfortartalmát a D/2. módszer szerint határozzuk meg.
8. ábra
D/1.5. KIVONÁS LÚGOS AMMÓNIUMCITRÁTTAL D/1.5.1. OLDHATÓ FOSZFOR KIVONÁSA PETERMANN SZERINT 65 °C-on
1. TÁRGY
A jelen fejezet eljárást ír le a lúgos ammónium-citrátban oldható foszfor meghatározására.
2. ALKALMAZÁSI TERÜLET
A módszer kizárólag a lecsapott dikalcium-foszfor-dihidrátra (CaHPO4.2H2O) alkalmazható.
3. A MÓDSZER ELVE
A foszfort 65 °C-on ammónium citrát lúgos oldatával (Petermann szerint) meghatározott körülmények között vonjuk ki.
4. VEGYSZEREK
Desztillált víz vagy a desztillált vízzel azonos tulajdonságú ionmentes víz.
4.1. Petermann-féle oldat.
4.2. Jellemzők
Citromsav (C6H8O7.H2O): 173 g/liter.
Ammónia: 42 g/liter ammóniakötésben lévő nitrogén.
pH: 9,4 és 9,7 között.
Készítése diammónium-citrátból
Oldjunk fel 931 g diammónium-citrátot (mólsúly: 226,19) kb. 3500 ml vízben, 5 l-es kalibrált lombikban. Állítsuk folyóvizes fürdőbe, keverjük össze, hűtsük, majd kis részletekben adjuk hozzá az ammóniát. Pl. d20=0,906 g/ml sűrűségű, 20,81 súly%-os ammónia tartalmú oldatnál 502 ml ammónia oldatot kell használni. Állítsuk a hőmérsékletet 20 °C-ra, töltsük a lombikot jelig desztillált vízzel, és keverjük össze a lombik tartalmát.
Készítése citromsavból és ammóniából
Oldjunk fel 865 g citromsav monohidrátot kb. 2500 ml desztillált vízben egy kb. 5 l-es edényben. Helyezzük az edényt jeges fürdőbe, és folyamatos rázogatás közben adjunk hozzá ammónia oldatot egy olyan tölcséren keresztül, amelynek szára a citromsav oldatba merül. Pl. d20 = 0,906 g/ml sűrűségű, 20,81 súly%-os ammónia tartalmú oldatnál 1114 ml ammónia oldatot kell adagolni. Állítsuk a hőmérsékletet 20 °C-ra, töltsük át az oldatot egy 5 l-es mérőlombikba, töltsük jelig desztillált vízzel és keverjük össze.
Ellenőrizzük az ammónia-kötésű nitrogéntartalmat a következőképpen:
Vigyük az oldat 25 ml-ét egy 250 ml-es mérőlombikba, és töltsük jelig desztillált vízzel. Keverjük össze. Határozzuk meg ezen oldat 25 ml-ének ammónia nitrogén tartalmát a C/1. módszerrel. Megfelelő, ha a 0,5 mol/l H2SO4 fogyása 15 ml lesz.
Ha az ammónia nitrogéntartalom 42 g/liternél nagyobb, a felesleges ammóniát semleges gázárammal vagy enyhe melegítéssel kiűzhetjük, és a pH-t így 9,7 értékre hozhatjuk vissza. Végezzünk egy második meghatározást ennek ellenőrzésére.
Ha az ammónia nitrogéntartalom 42 g/liternél kevesebb (n), M vagy V mennyiségű ammónia oldatot szükséges hozzáadni
tömeg szerint:
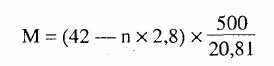
vagy térfogat szerint:
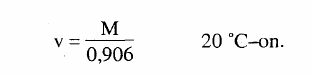
Ha V kevesebb, mint 25 ml, akkor adjuk közvetlenül az ammónia oldatot az 5 l-es lombikba, V x 0,173 g porított citromsavval együtt.
Ha V nagyobb, mint 25 ml, célszerű egy liter új reagenst készíteni az alábbi módon:
Mérjünk le 173 g citromsavat. Oldjuk fel 500 ml vízben. Betartva a jelzett óvatossági intézkedéseket, adjunk hozzá 225 + V x 1206 ml-t abból az ammónia oldatból, amelyet az 5 liter reagens elkészítéséhez használtunk. Töltsük jelig vízzel és keverjük össze.
Az így kapott 1 liter oldatot keverjük össze az előzőleg készített oldat 4975 ml-ével.
5. FELSZERELÉS
5.1. Vízfürdő, 65 ± 1 °C hőmérséklet beállítására alkalmas.
5.2. 500 ml-es kalibrált lombik (pl. Stohmann).
6. A MINTA ELŐKÉSZÍTÉSE Lásd a B) fejezetet.
7. ELJÁRÁS
7.1. Minta
Mérjük le az előkészített minta 1 g-ját 0,001 g pontossággal, és vigyük 500 ml-es kalibrált lombikba(5.2.).
7.2. Extrakció
Adjunk hozzá 200 ml lúgos ammónium-citrát oldatot (4.1.). Dugaszoljuk be a lombikot, és rázzuk erőteljesen kézzel, hogy elkerüljük a csomók képződését és az anyagnak a lombik falára való tapadását.
Helyezzük a lombikot 65 °C-os vízfürdőbe és rázzuk össze minden 5 percben az első fél óra alatt. Minden összezárást követően lazítsuk meg a dugót a nyomás kiegyenlítésére. A vízfürdőben a vízének szintje a lombikban lévő oldat szintje felett legyen. Hagyjuk a lombikot a vízfürdőben 65 °C-on egy további órára, és 10 percenként rázzuk össze. Vegyük ki a lombikot, hűtsük 20 °C körüli hőmérsékletre, és töltsük kb. 500 ml-re vízzel. Keverjük össze, és szűrjük száraz redős, foszfátmentes szűrőpapíron. A szűrlet első részletét ontsuk el.
7.3. Meghatározás
A kivont foszfortartalom meghatározását az így nyert oldat alikvot részletéből a D/2. módszerrel határozzuk meg.
D/1.5.2. OLDHATÓ FOSZFOR KIVONÁSA PETERMANN SZERINT SZOBAHŐMÉRSÉKLETEN
1. TÁRGY
A jelen fejezet eljárást ír le hidegen lúgos ammónium-citrátban oldható foszfor meghatározására.
2. ALKALMAZÁSI TERÜLET
A módszer kizárólag őrölt foszfátokra alkalmazható.
3. A MÓDSZER ELVE
A foszfort 20 °C körüli hőmérsékleten ammónium-citrát lúgos oldatával (Petermann-féle oldattal) meghatározott körülmények között vonjuk ki.
4. VEGYSZEREK Lásd a D/1.5. 1. fejezetet.
5. FELSZERELÉS
5.1. Szokásos laboratóriumi felszerelés, és egy 250 ml-es kalibrált lombik (pl. Stohmann lombik).
5.2. Körkörös rázógép (35-40 fordulat/perc).
6. A MINTA ELŐKÉSZÍTÉSE
Lásd a B) fejezetet.
7. ELJÁRÁS
7.1. Minta
Mérjük le az előkészített minta 2,5 g-ját 0,001 g pontossággal, és vigyük a 250 ml-es kalibrált lombikba (5.1.).
7.2. Extrakció
Adjunk hozzá egy kevés Petermann-féle oldatot 20 °C-on, rázzuk kézzel nagyon erősen, hogy gátat vessünk a csomóképződésnek, és megelőzzük az anyagnak a lombik oldalára való feltapadását. Töltsük a lombikot jelig Petermann-féle oldattal, és zárjuk le gumidugóval.
Rázzuk két óra hosszat a rázógépen (5.2.). Késedelem nélkül szűrjük le száraz, redős, foszfátmentes szűrőpapíron száraz edénybe. A szűrlet első részletét ontsuk el.
7.3. Meghatározás
A foszfor meghatározást az így nyert oldat alikvot részéből a D/2. módszer szerint végezzük el.
D/1.5.3. JOULIE SZERINTI LÚGOS AMMÓNIUM-CITRÁTBAN OLDHATÓ FOSZFOR KIVONÁSA
1. TÁRGY
A jelen fejezet eljárást ír le a Joulie szerinti lúgos ammónium-citrátban oldható foszfor meghatározására.
2. ALKALMAZÁSI TERÜLET
A módszer alkalmazható minden olyan egyszerű vagy összetett foszfát műtrágyára, amelyben a foszfor kalcinált alumínium-foszfát formában található.
3. A MÓDSZER ELVE
A kivonást az ammónium-citrát meghatározott összetételű lúgos oldatával történő erőteljes rázással végezzük (szükség szerint oxin jelenlétében) 20 °C-on.
4. VEGYSZEREK Desztillált vagy ionmentes víz.
4.1. Joulie-féle lúgos ammónium citrát oldat
Ez az oldat 400 g citromsavat és 153 g NH3-at tartalmaz literenként. Szabad ammóniumtartalma kb. 55 g/liter. A alább megadott módszerek egyikével készíthető:
4.1.1. Egy literes kalibrált lombikban oldjunk fel 400 g citromsavat (C6H8C7.H2O) kb. 600 ml ammónia oldatban (d20 = 0,925 g/ml, vagyis 200 g/l NH3). A citromsavat 50-80 g-os mennyiségekben folyamatosan adagoljuk, miközben a hőmérsékletet 50 °C alatt tartjuk. A térfogatot ammóniaoldattal 1 l-re egészítjük ki.
4.1.2. Oldjunk fel 1 literes kalibrált lombikban 432 g diammónium-citrátot (C6H14N2O7) 300 ml vízben. Adjunk hozzá 440 ml ammónia oldatot (d20 = 0,925 g/ml). A térfogatot vízzel 1 l-re egészítjük ki.
Megjegyzés:
Az összes ammóniatartalom ellenőrzése:
Vegyünk a citrát oldatból egy 10 ml-es mintát, és vigyük 250 ml-es mérőlombikba. Töltsük jelig desztillált vízzel. Határozzuk meg az így kapott oldat 25 ml-ének ammónia nitrogén tartalmát a C/1. módszer szerint.
1 ml 0,5 mol/l H2SO4 0,008516 g NH3-nak felel meg.
Ilyen körülmények között a reagenst akkor lehet megfelelőnek tekinteni, ha a titrálásnál fogyott ml-ek száma 17,7 és 18 ml között van.
Ha ez nem teljesül, akkor adjunk az oldathoz annyiszor 4, 25 ml ammóniát (d20 = 0,925 g/ml) ahányszor 0,1 ml-rel 18 ml alá esik a fentiek szerint fogyott ml-ek száma.
4.2. Porított 8-hidroxikinolin (oxin)
5. FELSZERELÉS
5.1. Szokásos laboratóriumi felszerelés, és egy kisméretű üveg vagy porcelán dörzsmozsár törővel.
5.2. 500 ml-es kalibrált lombikok.
5.3. 1000 ml-es kalibrált lombik.
5.4. Körkörös rázógép (35-40 fordulat/perc).
6. A MINTA ELŐKÉSZÍTÉSE Lásd a B) fejezetet.
7. ELJÁRÁS
7.1. Minta
Mérjük be az előkészített minta 1 g-ját 0,0005 pontossággal, és vigyük kisméretű dörzsmozsárba. Adjunk hozzá kb. 10 csepp citrátot (4.1.), hogy megnedvesítsük, és nagyon óvatosan dörzsöljük el a dörzsmozsár törőjével.
7.2. Extrakció
Adjunk hozzá 20 ml ammónium citrátot (4.1.) és keverjük péppé, majd hagyjuk kb. 1 percig állni.
Ontsuk le róla a folyadékot egy 500 ml-es kalibrált lombikba. Kaparjuk a mozsár aljára azokat a szemcséket is, amelyek az első nedves dörzsölést elkerülhették. Adjunk hozzá 20 ml citrát oldatot (4.1.), dörzsöljük el, mint fent, és a dekantált oldatot vigyük a kalibrált lombikba. Ezt az eljárást négyszer ismételjük meg. Ezt követően a mozsárban lévő összes anyagot áttölthetjük a kalibrált lombikba. A felhasznált citrát összes térfogatának megközelítőleg 100 ml-nek kell lennie.
Öblítsük a dörzsmozsarat és a törőt a kalibrált lombikba 40 ml desztillált vízzel.
A bedugaszolt lombikot rázógépen (5.4.) 3 óráig rázatjuk. Hagyjuk a lombikot 15-16 óráig állni, majd rázassuk ismét 3 óráig ugyanolyan feltételek mellett. A hőmérsékletet az egész eljárás alatt 20±2 °C hőmérsékleten kell tartani.
Töltsük a lombikot jelig desztillált vízzel. Szűrjük száraz szűrőn, ontsuk el a szűrlet első részét, a tiszta szűrletet gyűjtsük száraz edénybe.
7.3. Meghatározás
A kivont foszfor meghatározását az így nyert oldat alikvot részéből a D/2. módszer szerint végezzük el.
8. FÜGGELÉK
Az oxin alkalmazása lehetővé teszi, hogy ezt az eljárást magnéziumot tartalmazó műtrágyákra alkalmazzuk. Alkalmazása akkor javasolt, ha a magnézium és a foszforanhidrid aránya magasabb, mint 0,03 (Mg/P205>0,03). Ha ez a feltétel teljesül, adjunk a megnedvesített vizsgálati mintához 3 g oxint Az oxin magnézium távollétében feltételezhetően a továbbiakban nem zavarja a meghatározást. Azonban ha ismert, hogy a vizsgálandó anyag nem tartalmaz magnéziumot, az oxin használatától el lehet tekinteni.
D/1.6. VÍZOLDHATÓ FOSZFOR KIVONÁSA
1. TÁRGY
A jelen fejezet eljárást ír le a vízoldható foszfát kivonására.
2. ALKALMAZÁSI TERÜLET
A módszer alkalmazható minden műtrágyára, beleértve az összetett műtrágyákat is, ahol vízoldható foszfort kell meghatározni.
3. A MÓDSZER ELVE
A vízoldható foszfort vízzel extraháljuk meghatározott körülmények között történő rázatással.
4. VEGYSZER Desztillált vagy ionmentes víz.
5. FELSZERELÉS
5.1. 500 ml-es kalibrált lombik (pl. Stohmann).
5.2. Körkörös rázógép (35-40 fordulat/perc).
6. A MINTA ELŐKÉSZÍTÉSE Lásd a B) fejezetet.
7. ELJÁRÁS
7.1. Minta
Mérjük le a minta 5 g-ját 0,001 g pontossággal, és vigyük az 500 ml-es kalibrált lombikba (5.1.).
7.2. Extrakció
Adjunk a lombikba 450 ml vizet, amelynek hőmérséklete 20 és 25 °C között legyen.
Rázassuk a rázógépen (5.2.) 30 percig. Ezután a lombikot töltsük jelig vízzel, rázással alaposan keverjük össze, majd szűrjük száraz, foszfátmentes redős szűrőn egy száraz edénybe.
7.3. Meghatározás
A foszfor meghatározását az így nyert oldat egy alikvot részéből a D/2. módszerrel végezzük.
D/2. A KIVONT FOSZFOR MEGHATÁROZÁSA
(Gravimetriás módszer kinolin-foszfomolibdát alkalmazásával.)
1. TÁRGY
A jelen fejezet eljárást ír le a foszfor meghatározására műtrágya kivonatokban.
2. ALKALMAZÁSI TERÜLET
A módszer mindenfajta műtrágya kivonat7 esetében alkalmazható a foszfor különféle formáinak meghatározására.
3. A MÓDSZER ELVE
A foszfort egy esetleges hidrolízist követően savas közegben kinolin-foszfomolibdát formájában lecsapjuk. Szűrés és mosás után a csapadékot 250 °C-on szárítjuk és mérjük. A fenti körülmények között az oldatban valószínűen előforduló vegyületek (ásványi és szerves savak, ammónium ionok, oldható szilikátok stb.) részéről zavaró hatásra nem kell számítani, ha a lecsapáskor nátrium-molibdát vagy ammónium-molibdát reagenst használunk.
4. VEGYSZEREK
Desztillált vagy ionmentes víz.
4.1. Tömény salétromsav (d20 = 1,40 g/ml).
4.2. A reagens elkészítése
4.2.1. A nátrium-molibdát alapú reagens készítése.
A oldat: Oldjunk fel 70 g nátrium-molibdát. dihidrátot 100 ml desztillált vízben.
B oldat: Oldjunk fel 60 g citromsav monohidrátot 100 ml desztillált vízben és adjunk hozzá 85 ml tömény salétromsavat (4.1).
C oldat: A C oldatot az A és B oldatok elegyítésével nyerjük.
D oldat: 50 ml desztillált vízhez adjunk 35 ml tömény salétromsav (4.1) oldatot és 5 ml frissen desztillált kinolint. Ezt az oldatot adjuk a C oldathoz, alaposan keverjük össze és hagyjuk egy éjszakán át sötét helyen állni. Ezt követően töltsük fel desztillált vízzel 500 ml-re, ismételten keverjük össze, és zsugorított üveg-szűrőn (5.6) szűrjük le.
4.2.2. Az ammónium-molibdát alapú reagens készítése
A oldat: Oldjunk fel 100 g ammónium-molibdátot 300 ml desztillált vízben miközben gyengén melegítjük és kevergetjük időről időre.
B oldat: Oldjunk fel 120 g citromsav monohidrátot 200 ml desztillált vízben és adjunk hozzá 170 ml tömény salétromsavat (4.1).
C oldat: Adjunk 10 ml frissen desztillált kinolint 70 ml koncentrált salétromsavhoz (4.1.).
D oldat: Lassan ontsuk állandó keverés közben az A oldatot a B oldathoz. Azután alapos keverés közben adjuk a C oldatot ehhez az elegyhez, és egészítsük ki 1 literre. Hagyjuk állni két napig sötét helyen, és azután szűrjük át egy zsugorított üvegszűrőn (5.6.).
A 4.2.1. és a 4.2.2 szerint nyert reagenseket ugyanúgy kell használni; mindkettőt sötét helyen, lezárt polietilén üvegben kell eltartani.
5. FELSZERELÉS
5.1. Szokásos laboratóriumi felszerelés és egy széles nyakú 500 ml-es Erlenmeyer lombik.
5.2. Osztott pipetták, 10,25 és 50 ml-s.
5.3. Üvegszűró tégely, 5-20 μ pórusmérettel. 5.4. Szívópalack.
5.5. Szárítószekrény, 250±10 °C-ra állítható.
5.6. Zsugorított üveg szűrőtölcsér, 5-20 μ pórusmérettel.
6. ELJÁRÁS
6.1. Az oldat elkészítése
Vegyük ki pipettával a műtrágya kivonat kb. 0,01 g P2O5-ot tartalmazó részét (lásd a 2. táblázatot), és vigyük egy 500 ml-es Erlenmeyer lombikba. Adjunk hozzá 15 ml tömény salétromsavat (4.1.), és hígítsuk vízzel kb. 100 ml-re.[79][80]
2. táblázat
A foszfát kivonatok alikvot részének meghatározása
| P2O5% a műtrágyában | P% a műtrágyában | Vizsgálandó minta (g) | Hígítás (ml-re) | Minta (ml) | Hígítás (ml-re) | Alikvot rész a lecsapáshoz (ml) | Kinolin foszfo-molibdát átszámítási faktor (F) P2O5%-ra | Kinolin foszfo-molibdát átszámítási faktor (F) P%-ra |
| 5—10 | 2,2—4,4 | 1 | 500 | — | — | 50 | 32,074 | 13,984 |
| 5 | 500 | — | — | 10 | 32,074 | 13,984 | ||
| 10—25 | 4,4—11,0 | 1 | 500 | — | — | 25 | 64,148 | 27,968 |
| 5 | 500 | 50 | 500 | 50 | 64,148 | 27,968 | ||
| + 25 | + 11 | 1 | 500 | — | — | 10 | 160,370 | 69,921 |
| 5 | 500 | 50 | 500 | 25 | 128,296 | 55,937 |
6.2. Hidrolízis
Ha feltételezhető, hogy az oldatban metafoszfátok, pirofoszfátok és polifoszfátok vannak jelen, hidrolízist végzünk az alábbiak szerint:
Hozzuk lassú forrásba az Erlenmeyer lombik tartalmát és tartsuk ezen a hőmérsékleten, amíg a hidrolízis befejeződik (ez általában egy óra alatt következik be). Gondot kell fordítani arra, hogy elkerüljük az egyenetlen és túlzott bepárlódás okozta veszteségeket: visszafolyó hűtőt kell felszerelni, különben a kezdeti térfogat a felére is lecsökkenhet. A hidrolízis végeztével a térfogatot desztillált vízzel az eredeti térfogatra kell kiegészíteni.
6.3. A szűrőtégely lemérése
Szárítsuk a szűrőtégelyt (5.3.) legalább 15 percig 250±10 °C hőmérsékletű szárítószekrényben. Hagyjuk lehűlni exszikká-torban, majd mérjük le.
6.4. Lecsapás
Az Erlenmeyer lombikban lévő savas oldatot addig melegítjük, amíg forrásba jön. Ekkor megindítjuk a kinolin-foszfomo-libdát csapadék kiválását úgy, hogy 40 ml lecsapó reagenst (4.2.1. vagy 4.2.2. pont szerinti reagenst) adagolunk cseppenként, folyamatos keverés mellett. Helyezzük az Erlenmeyer lombikot forrásban lévő vízfürdőre, hagyjuk ott 15 percig, időnként megrázogatva. Az oldatot szűrhetjük azonnal, vagy pedig lehűlés után.[81]
6.5. Szűrés és mosás
Az oldatot vákuum segítségével dekantálva szűrjük. Az Erlenmeyer lombikban maradt csapadékot 30 ml vízzel mossuk. Dekantáljuk és szűrjük le az oldatot. Ezt az eljárást ötször megismételjük. Végül a csapadéknak a lombikban maradt részét is veszteség nélkül a szűrőre mossuk vízzel. Négyszer átmossuk 20-20 ml vízzel, minden alkalommal teljesen leszívatva a folyadékot a szűrőről. Alaposan szárítsuk meg a csapadékot.
6.6. Szárítás és mérés
Töröljük le a szűrőtégely külső oldalát szűrőpapírral. Helyezzük a szűrőtégelyt a szárítószekrénybe és szárítsuk 250 °C-n (5.5.) súlyállandóságig (általában 15 perc). Hagyjuk exszikkátorban lehűlni, és gyorsan mérjük le.
6.7. Vakpróba
Minden vizsgálatsorozathoz végezzünk vakpróbát úgy, hogy csak a reagenseket és oldószereket használjuk az extrakciónak megfelelő arányban. A kapott értéket vegyük figyelembe a végeredmény kiszámításánál.
6.8. Az eredmények helyességének igazolása
Végezzük el a meghatározást káliumdihidrogén-foszfát oldat 0,01 g P2O5-ot tartalmazó alikvot mennyiségével.
7. AZ EREDMÉNY KIFEJEZÉSE
Ha a 2, táblázat szerinti beméréseket és hígításokat alkalmazzuk, a következő képleteket kell használni:
P% a műtrágyában = (A-a) x F'
vagy
P2O5% a műtrágyában = (A-a) x F ahol:
A = a kinolin-foszfomolibdát tömege, g,
a = a vakpróba során kapott kinolin-foszfomolibdát tömege, g,
F és F' = a 2. táblázat utolsó két oszlopában megadott faktorok.
A 2. táblázattól eltérő bemérések és hígítások esetén a műtrágyában levő foszfortartalom kiszámítására a következő képleteket kell használni:
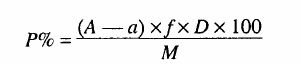
vagy
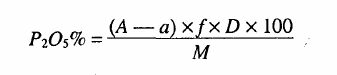
ahol:
f és f' = a kinolin-foszfomolibdát átszámítási faktorai: P2O5-ra 0,032074 (f), vagy P-ra 0,013984 (f),
D = hígítási faktor,
M = a vizsgált minta (bemérés) tömege, g-ban.
E) KÁLIUM
E/1. VÍZOLDHATÓ KÁLIUM TARTALOM MEGHATÁROZÁSA
1. TÁRGY
A jelen fejezet eljárást ír le a vízoldható kálium meghatározására.
2. ALKALMAZÁSI TERÜLET
A módszer valamennyi, a 3. számú melléklet 1. számú függelékében felsorolt kálium műtrágyára alkalmazható.
3. A MÓDSZER ELVE
A vizsgálandó mintában lévő káliumot vízben oldjuk. Eltávolítjuk vagy megkötjük azokat az anyagokat, amelyek a kálium mennyiségi meghatározását zavarhatnák, majd a káliumot enyhén lúgos közegben kálium-tetrafenilborát formájában lecsapjuk.
4. VEGYSZEREK Desztillált vagy ionmentes víz.
4.1. Formaldehid
Tiszta formaldehid oldat, 25 és 35% közötti formaldehid tartalommal.
4.2. Kálium klorid analitikai célra.
4.3. Nátrium-hidroxid oldat: 10 mol/l.
Ügyelni kell arra, hogy csak káliummentes nátrium-hidroxidot szabad használni.
4.4. Indikátor oldat
Oldjunk fel 0,5 g fenolftaleint 90%-os etanolban, és egészítsük ki az oldat térfogatát 100 ml-re.
4.5. EDTA oldat
Oldjunk fel 4 g-ot etiléndiamintetraecetsav dinátrium sójának dihidrátjából vízben, 100 ml-s mérő lombikban. Töltsük jelig és keverjük össze.
Ezt a reagenst műanyag edényben kell eltartani.
4.6. Nátriumtetrafenil-borát oldat (NaTPB oldat)
Oldjunk fel 32,5 g nátriumtetrafenil-borátot 980 ml vízben, adjunk hozzá 2 ml nátrium-hidroxid oldatot (4.3.) és 20 ml magnézium klorid oldatot (100 g MgCl2.6H20/liter). Keverjük 15 percig, és szűrjük finom, hamumentes szűrőpapíron. Ezt a reagenst műanyag edényben kell eltartani.
4.7. Mosófolyadék
20 ml STPB oldatot (4.6.) hígítsunk 1000 ml-re vízzel.
4.8. Brómos víz. Telített vizes brómoldat.
5. FELSZERELÉS
5.1. 1000 ml-s mérőlombik.
5.2. 250 ml-s főzőpohár.
5.3. Szűrőtégelyek 5 és 20 μm közötti porozitással.
5.4. Szárítószekrény 120±10 °C-ra beállítva.
5.5. Exszikkátor.
6. A MINTA ELŐKÉSZÍTÉSE
Lásd a B) fejezetet.
A kálium sók esetében a mintát elegendően finomra kell darálni ahhoz, hogy a vizsgálat céljára reprezentatív mintát kapjunk. Ezekhez a termékhez az 1. módszer (6) a) pontját kell alkalmazni.
7. ELJÁRÁS
7. 1 Minta
Mérjünk be az előkészített mintából 10 g-t 0,001 g pontossággal (50%-nál több káliumoxidot tartalmazó káliumsók esetében 5 g-t) és vigyük át 600 ml-s főzőpohárba kb. 400 ml vízzel.
Forraljuk fel és tartsuk forrásban 30 percig. Hűtsük le, vigyük át veszteség nélkül 1000 ml-s mérőlombikba, töltsük fel jelig, keverjük össze és szűrjük száraz edénybe. Ontsuk el a szűrlet első 50 ml-ét (lásd 7.6., megjegyzés az eljáráshoz).
7.2. Az alikvot rész elkészítése a lecsapáshoz
Pipettával vigyük a szűrlet 25-50 mg káliumot tartalmazó alikvot részét (lásd a 3. táblázat) 250 ml-es főzőpohárba. Ha szükséges, térfogatát vízzel egészítsük ki 50 ml-re.
A zavaró anyagok eltávolítása végett adjunk 10 ml EDTA oldatot (4.5.) és néhány csepp fenolftalein oldatot (4.4.) a főzőpohárba, keverjük el és adjunk hozzá cseppenként nátrium-hidroxid oldatot (4.3.), amíg az oldat meg nem pirosodik. Adjunk hozzá még pár csepp nátirum-hidroxidot a lúgfelesleg biztosítására. (Általában 1 ml nátrium-hidroxid elegendő a minta semlegesítésére és a lúgfelesleg biztosítására.)
Az ammónia nagy részének eltávolítására [lásd 7.6 b) megjegyzés az eljáráshoz] forraljuk kíméletesen 15 percig.
Ha szükséges, adjunk hozzá vizet, térfogatát 60 ml-re kiegészítve. Forraljuk fel az oldatot, majd vegyük le a főzőpoharat a rezsóról és adjunk hozzá 10 ml formaldehidet (4.1.). Adjunk hozzá néhány csepp fenolftaleint, és ha szükséges, még egy kevés nátrium-hidroxidot, amíg határozott piros szín jelenik meg. Takarjuk le a főzőpoharat óraüveggel és helyezzük forró vízfürdőre 15 percnyi időre.
7.3. A tégely lemérése
Szárítsuk a szűrőtégelyt (lásd 5. "Felszerelés") állandó tömegig (kb. 15 perc) a 120 °C-os szárítószekrényben. Hagyjuk a tégelyt exszikkátorban lehűlni, majd mérjük le.
7.4. Lecsapás
Vegyük le a főzőpoharat a forró vízfürdőről, cseppenként keverjünk hozzá 10 ml STPB oldatot (4.6.). Ez az adagolás kb. 2 percet vesz igénybe. Szűrés előtt legalább 10 percig hagyjuk állni.
7.5. Szűrés és mosás
Szűrjük vákuumban a lemért szűrőtégelybe, öblítsük be a főzőpoharat a mosófolyadékkal (4.7.). Mossuk a csapadékot háromszor a mosófolyadékkal (összesen 60 ml mosófolyadékkal) és kétszer 5-10 ml vízzel.
Gondosan szárítsuk meg a csapadékot.
7.6. Szárítás és mérés
Töröljük le a szűrőtégely külső oldalát szűrőpapírral. Helyezzük a szűrőtégelyt tartalmával együtt a szárítószekrénybe másfél órára, 120 °C hőmérsékleten. Hagyjuk a szűrőtégelyt exszikkátorban lehűlni, és gyorsan mérjük le.
Megjegyzés az eljáráshoz
a) Ha a szűrlet sötét színű, vegyünk ki belőle pipettával egy legfeljebb 100 mg K2O-ot tartalmazó alikvot részt, vigyük egy
100 ml-es mérőlombikba, adjunk hozzá brómos vizet és forraljuk fel, hogy a brómfelesleget eltávolítsuk. Hűtsük le, majd töltsük jelig, szűrjük le és a szűrlet alikvot határozzuk meg a kálium tartalmát mennyiségileg.
b) Ahol nincs vagy kevés az ammónia nitrogén tartalom, ott nem szükséges 15 percig forralni.
7.7. A kálium kivonatok alikvot részének meghatározása.
3. táblázat
| K2O% a műtrágyában | K% a műtrágyában | Vizsgálandó minta (g) | Extrakt oldat mintarészlete a hígításhoz (ml) | Hígítás (ml-re) | Alikvot rész a lecsapáshoz (ml) | Átszámítási faktor (F) K2O%-ra (g KTPB) | Átszámítási faktor (F') K%-ra (g KTPB) |
| 5—10 | 4,2—8,3 | 10 | — | — | 50 | 26,280 | 21,812 |
| 10—20 | 8,3—16,6 | 10 | — | — | 25 | 52,560 | 43,624 |
| 20—50 | 16,6—41,5 | 10 | vagy- | — | 10 | 131,400 | 109,060 |
| vagy 50 | 250 | 50 | 131,400 | 109,060 | |||
| >50 | >41,5 | 5 | vagy — | — | 10 | 262,800 | 218,120 |
| vagy 50 | 250 | 50 | 262,800 | 218,120 |
7.8. Vakpróba
Minden mérési sorozathoz végezzünk vakpróbát úgy, hogy csak a reagenseket használjuk az analízisnél használt mennyiségekben, és a végeredmény kiszámításakor vegyük figyelembe.
7.9. Ellenőrző vizsgálat
A vizsgálati módszer ellenőrzésére végezzük el a meghatározást kálium-klorid vizes oldatának legfeljebb 40 mg K2O-ot tartalmazó alikvot részével.
8. AZ EREDMÉNY KISZÁMÍTÁSA
Ha a 3. táblázat szerinti beméréseket és hígításokat használjuk, a műtrágyában levő káliumtartalom kiszámítására az alábbi képleteket kell alkalmazni:
K2O% a műtrágyában = (A-a) x F
vagy
K% a műtrágyában = (A-a) x F'
ahol:
A = a mintából nyert csapadék tömege, g, a = a vakpróbánál nyert csapadék tömege, g, F és F' = faktorok (lásd a 3. táblázatot).
Ha a 3. táblázatban megadottól eltérő beméréseket és hígításokat használjuk, a műtrágyában levő káliumtartalom kiszámítására tömegszázalékban az alábbi képleteket kell alkalmazni:
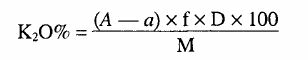
vagy
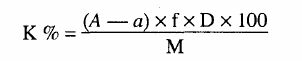
ahol:
f = átszámítási szorzószám, KTPB-ből K20-ra: 0,1314,
f = átszámítási szorzószám, KTPB-ből K-ra: 0,109,
D = hígítási faktor,
M = a bemért minta tömege, g.
F) KLORID
F/1. KLORID MEGHATÁROZÁSA SZERVES ANYAG TÁVOLLÉTÉBEN
1. TÁRGY
A jelen fejezet eljárást ír le a klorid meghatározására szervesanyag távollétében.
2. ALKALMAZÁSI TERÜLET
A módszer valamennyi szervesanyagot nem tartalmazó műtrágyára alkalmazható.
3. A MÓDSZER ELVE
A vízben feloldott kloridokat savas közegben ismert koncentrációjú ezüst-nitrát oldat feleslegével lecsapjuk. A felesleget ammónium-tiocianát oldattal vas(III)ammónium-szulfát jelenlétében visszatitráljuk (Volhard módszer).
3. VEGYSZEREK
Kloridmentes desztillált vagy ionmentes víz.
4.1. Nitrobenzol vagy dietiléter.
4.2. Salétromsav, 10 mol/l.
4.3. Indikátor oldat
Oldjunk fel vízben 40 g vas(III)ammónium szulfátot, Fe2(SO4)3.(NH4)2SO4.24H2O, és az oldat térfogatát egészítsük ki egy literre.
4.4. Ezüst-nitrát oldat, 0,1 mol/l, meghatározott koncentrációjú.
4.5. Ammónium-tiocianát mérőoldat, 0,1 mol/l. Készítése:
Mivel a só higroszkópos, és bomlás veszélye nélkül nem szárítható, ajánlatos kb. 9 g-ot lemérni, vízben feloldani, majd térfogatát 1 l-re kiegészíteni. Titrálással állítsuk be 0,1 mol/l-re a 0,1 mol/l-es AgNO3 literét.
5. FELSZERELÉS
5.1. Körkörös rázógép (35-40 fordulat/perc).
5.2. Büretták.
5.3. Mérőlombik, 500 ml-es.
5.4. Erlenmeyér lombik, 250 ml-es.
6. A MINTA ELŐKÉSZÍTÉSE Lásd a B) fejezetet.
7. ELJÁRÁS
7.1. Minta és az oldat előkészítése
A minta 5 g-ját mérjük le 0,001 g pontossággal és vigyük 500 ml-s mérőlombikba. Adjunk hozzá 450 ml vizet. Fél óra hosszat kevertessük a rázógépen (5.1). Töltsük fel 500 ml-re desztillált vízzel; keverjük össze és szűrjük főzőpohárba.
7.2. Meghatározás
Vegyük a szűrlet legfeljebb 0,150 g kloridot tartalmazó alikvot részét. Például 25 ml-t (0,25 g), 50 ml-t (0,5 g) vagy 100 ml-t (1 g). Ha a kivett rész térfogata 50 ml-nél kisebb, akkor egészítsük ki desztillált vízzel 50 ml-re.
Adjunk hozzá 5 ml 10 mol/l salétromsavat (4.2.), 20 ml indikátor oldatot (4.3.) és két csepp 0,1 mol/l ammónium-tiocianát mérőoldatot (4.5.). (Ez utóbbi reagensből e célra szolgáló, nullára állított bürettával mintát veszünk.)
Ezután bürettából adagoljunk ezüst-nitrát oldatot (4.4.) amíg 2-5 ml ezüst-nitrát felesleg keletkezik. Adjunk hozzá 5 ml nitrobenzolt vagy 5 ml dietilétert (4.1.) és rázzuk jól össze, hogy tömörítsük a csapadékot. Titráljuk meg a fölös ezüst-nitrátot 0, 1 mol/l ammónium-tiocianát oldattal (4.5.) amíg olyan vörösesbarna színt észlelünk, amely a lombik óvatos rázogatása közben . is megmarad.
Megjegyzés:
A nitrobenzol vagy dietiléter (de főleg a nitrobenzol) megakadályozza, hogy az ezüst-klorid reakcióba lépjen az ammónium-tiocianáttal. így tiszta színátcsapást kapunk.
7.3. Vakpróba
Készítsünk vakpróbát a vizsgálattal azonos körülmények között, és eredményét vegyük figyelembe a végeredmény kiszámításánál.
7.4. Kontrol vizsgálat
A vizsgálatot megelőzően ellenőrizzük a módszer pontosságát frissen készített kálium-klorid oldat olyan alikvot részével, amely 100 mg nagyságrendű, ismert mennyiségű kloridot tartalmaz.
8. AZ EREDMÉNY KIFEJEZÉSE
Az eredményt a beérkezési állapotú minta klorid tartalmának százalékos (m/m) értékében fejezzük ki. Számítsuk ki a klorid(Cl) százalékot az alábbi képlettel:
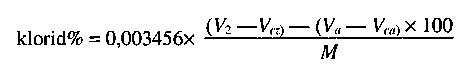
| ahol: |
| Vz = a 0,1 mol/l ezüst-nitrát ml-einek száma, |
| Vcz = a 0, 1 mol/l ezüst-nitrát vakpróbához fogyott ml-einek száma, |
| Va = a 0,1 mol/l ammónium-tiocianát ml-einek száma, |
| Vca = a 0,1 mol/l ammónium-tiocianát vakpróbához fogyott ml-einek száma, |
| M = a bemérés (7.2.) g-ban. |
G) AZ ŐRLÉS FINOMSÁGA
G/.1. AZ ŐRLÉS FINOMSÁGÁNAK MEGHATÁROZÁSA (SZÁRAZ SZITÁLÁS)
1. TÁRGY
A jelen fejezet száraz eljárást ír le az őrlés finomságának meghatározására.
2. ALKALMAZÁSI TERÜLET
A módszer alkalmazható az EC típusú műtrágyák közül mindazokra, amelyeknél 0,630 és 0,160 mm-es szitákra vonatkozó őrlési finomsági követelmény van előírva.
3. A MÓDSZER ELVE
Gépi szitálással meghatározzuk a terméknek azt a mennyiségét, amelynek szemcsemérete 0,630 fölött van, és azt a mennyiséget, amelynek szemcsemérete 0,160 és 0,630 mm között van. Az őrlés finomságának százalékos értékét kiszámítjuk.
4. FELSZERELÉS
4.1. Rázógép gépi szitáláshoz.
4.2. Szabványos méretű (20 cm átmérőjű, 5 cm magasságú) sziták 0,160-os és 0,630 nyílással.
5. ELJÁRÁS
Mérjük le a minta 50 g-ját 0,05 g pontossággal. Állítsuk össze a két szitát és a hozzájuk tartozó gyűjtő edényrészt és helyezzük a rázógépre (4.1.). A nagyobb nyílású szita kerüljön felülre. Tegyük a szitálandó mintát a felső szitára. Szitáljunk 10 percig, majd vegyük ki a gyűjtő edényrészben összegyűlt anyagot. Indítsuk újra a szitát, és egy perc múlva győződjünk meg arról, hogy a gyűjtő edényrészbe ez idő alatt nem hullott át 250 mg-ot meghaladó anyagmennyiség. Ismételjük az utóbbi eljárást mindaddig, amíg az 1 perces rázás alatt az összegyűlt anyag mennyisége kevesebb nem lesz 250 mg-nál. Mérjük le külön-külön a szitákon fennmaradt anyagot.
6. AZ EREDMÉNY KISZÁMÍTÁSA
A minta %-os finomsága a 0,630 mm-s szitán = (50 - M1)x 2.
A minta %-os finomsága a 0,160 mm-s szitán = (50 - (M1 + M2)x 2.
ahol:
M1: - a 0,630-as nyílású szitán fennmaradt szitamaradék tömege, g,
M2: - a 0,160-os nyílású szitán fennmaradt szitamaradék tömege, g.
A számítások eredményét a legközelebbi egész számra kell kerekíteni.
G/2. LÁGY TERMÉSZETES FOSZFÁTOK ŐRLÉSI FINOMSÁGÁNAK MEGHATÁROZÁSA
1. TÁRGY
A jelen módszer lágy természetes foszfátok őrlési finomságának meghatározására szolgál.
2. ALKALMAZÁSI TERÜLET
A módszer lágy természetes foszfátokra alkalmazható.
3. A MÓDSZER ELVE
Finom részecske eloszlású mintáknál az összetapadás miatt a száraz szitálás nehézkes, ezért általában nedves szitálást alkalmaznak.
4. VEGYSZEREK Nátrium-hexametafoszfát oldat, 1 %-os.
5. FELSZERELÉS
5.1. Szabványos méretű (20 mm átmérőjű, 5 cm magasságú) sziták 0,063 és 0,125 mm nyílással, gyűjtő edényrésszel.
5.2. Üvegtölcsér, 20 cm átmérővel, állványra szerelve.
5.3. 250 ml-s főzőpoharak.
5.4. Szárítószekrény.
6. VIZSGÁLATI MÓDSZER
6.1. Mintavétel
Mérjük le a minta 50 g-ját 0,05 g pontossággal. Mossuk a szita mindkét oldalát vízzel és helyezzük a 0,125 mm-s szitát a 0,063 mm-s szita tetejére.
6.2. Eljárás
Helyezzük a mintát a felső szitára. Szitáljunk gyenge hideg víz áram mellett (csapvizet lehet használni), amíg az átfolyó víz gyakorlatilag tiszta lesz. Ügyelni kell a folyadékáram beállítására, hogy az alsó szita sohase teljék meg vízzel.
Amikor úgy látjuk, hogy a felső szitán a maradék mennyisége többé-kevésbé állandóvá vált, vegyük le ezt a szitát és helyezzük addig egy gyűjtőedény-részre.
Folytassuk a nedves szitálást az alsó szitán pár percig, amíg az átfolyó víz közel tiszta lesz.
Tegyük vissza a 0,125 mm-s szitát a 0,063 mm-s szitára. A gyűjtő edényrészből vigyünk fel a felső szitára minden maradékot és kezdjük újra a szitálást gyenge vízáramlás mellett, amíg az átfolyó víz megint tisztává nem válik.
Mindegyik szitamaradékot mossuk veszteség nélkül külön-külön főzőpoharakba a tölcsér segítségével. Mindegyik szitamaradékot szuszpendáljuk fel vízzel a főzőpohárban, majd kb. 1 perces állás után dekantálunk annyi vizet, amennyit lehetséges.
A főzőpoharakat tegyük szárítószekrénybe és szárítsuk 150 °C-on 2 óra hosszat. Hagyjuk lehűlni, a szita maradékokat ecsettel nyerjük vissza, és mérjük le.
7. AZ EREDMÉNY KISZÁMÍTÁSA
A vizsgálatok eredményeit a következő egész számra kell kerekíteni.
%-os finomság a 0,125 mm-s szitán = (50 - M1)x 2
%-os finomság a 0,063 mm-s szitán = (50 - (M1 + M2) x 2,
ahol:
M1 - a 0,125 mm-s szitán fennmaradt maradék tömege, g,
M2 - a 0,063 mm-s szitán fennmaradt maradék tömege, g.
Megjegyzések:
Ha a szitálás után csomók jelenléte észlelhető, a vizsgálatot a következőképpen kell újra végezni:
Lassan, folyamatos keverés mellett ontsuk a minta 50 grammját egy literes lombikba, amelybe előzetesen 500 ml nátrium-hexametafoszfát oldatot öntöttünk. Dugaszoljuk be a lombikot, és kézzel erőteljesen rázzuk, hogy a csomók szétessenek. Az egész szuszpenziót vigyük a felső szitára, és gondosan mossuk át a lombikot is. Folytassuk a vizsgálatot a 6.2. pont előírásai szerint.
H) MEZOELEMEK
H/1. AZ ÖSSZES KALCIUM, ÖSSZES MAGNÉZIUM, ÖSSZES NÁTRIUM
ÉS A SZULFÁT FORMÁJÁBAN JELEN LÉVŐ ÖSSZES KÉN KIVONÁSA
1. TÁRGY
Ez a fejezet az összes kalcium, az összes magnézium, az összes nátrium és a szulfát formájában jelen lévő összes kén kivonásához határoz meg eljárást úgy, hogy ugyanaz a kivonat alkalmazható valamennyi megkövetelt tápelem meghatározásához.
2. ALKALMAZÁSI TERÜLET
Ez a módszer azokra a műtrágyákra alkalmazandó, amelyekre a 3. számú melléklet 1. függeléke megköveteli az összes kalcium, az összes magnézium, az összes nátrium és a szulfát formájában jelen lévő összes kén megadását.
3. A MÓDSZER ELVE Oldás híg sósavban forralva.
4. VEGYSZEREK
4.1. Hígított sósav
Egy térfogatrész sósav (d20 =1,18 g/ml) és egy térfogatrész víz.
5. FELSZERELÉS
Állítható hőmérsékletű elektromos főzőlap.
6. A MINTA ELŐKÉSZÍTÉSE Lásd a B) fejezetet.
7. ELJÁRÁS
7.1. Vizsgálati minta.
A kalciumot, a magnéziumot, a nátriumot és a szulfát formájában jelen lévő ként egy milligramm pontossággal bemért 5 g vizsgálati mintából vonjuk ki.
Ha azonban a műtrágya 15%-nál több ként (S), azaz 37,5%-nál több SO3-ot, és 18,8%-nál több kalciumot (Ca), azaz 26,3%-nál több CaO-t tartalmaz, akkor a kalcium és kén kivonását egy milligramm pontossággal bemért 1 g vizsgálati mintából végezzük. Tegyük a vizsgálati mintát 600 ml-es főzőpohárba.
7.2. Az oldat elkészítése
Adjunk hozzá kb. 400 ml vizet és kis adagokban - a minta magas karbonát tartalma esetén óvatosan - adjunk még hozzá kis adagokban 50 ml hígított sósavat (4.1.). Forraljuk fel és tartsuk forrásban 30 percig. Időnként megkeverve hagyjuk lehűlni. Veszteség nélkül, dekantálással vigyük át egy 500 ml-es mérőlombikba. Töltsük fel jelig vízzel, és keverjük össze. Száraz szűrőn keresztül szűrjük át egy száraz lombikba, és az első részletet ontsuk el. A szűrletnek teljesen átlátszónak kell lennie. Ha a szűrletet nem használjuk fel azonnal, akkor dugóval zárjuk le.
H/2. KÜLÖNBÖZŐ FORMÁBAN JELEN LÉVŐ ÖSSZES KÉN KIVONÁSA
1. TÁRGY
Ez a fejezet a műtrágyákban elemi formában és/vagy egyéb kémiai összetételben jelen lévő összes kén kivonásához határoz meg eljárást.
2. ALKALMAZÁSI TERÜLET
Ez a módszer azokra a műtrágyákra alkalmazandó, amelyekre a 3. számú melléklet 1. függeléke a különböző formában (elemi, tioszulfát, szulfit, szulfát) jelen lévő összes kén megadását követeli meg.
3. A MÓDSZER ELVE
Az elemi ként lúgos közegben átalakítjuk poliszulfidokká és tioszulfáttá; ezeket, továbbá az esetlegesen jelen lévő szulfitokat hidrogén-peroxiddal oxidáljuk. Ezáltal a kén különböző formáit szulfáttá alakítottuk, amelyet bárium-szulfátos kicsapással határozunk meg (8.9. módszer).
4. VEGYSZEREK
4.1. Hígított sósav:
Egy térfogatrész sósav (d20 =1,18 g/ml) és egy térfogatrész víz.
4.2. Nátrium-hidroxid oldat, NaOH, legalább 30% (d20 = 1,33 g/ml).
4.3. Hidrogén-peroxid oldat, 30% (m/m).
4.4. Bárium-klorid vizes oldata, BaCl2.2H2O, 122 g/l.
5. FELSZERELÉS
Elektromos főzőlap állítható hőmérséklettel.
6. A MINTA ELKÉSZÍTÉSE Lásd a B) fejezetet.
7. ELJÁRÁS
7.1. Vizsgálati minta
Egy milligramm pontossággal mérjünk be olyan mennyiségű műtrágyát, amely 80 és 350 mg közötti mennyiségű ként (S) vagy 200 és 875 mg közötti mennyiségű SO3-ot tartalmaz.
Rendszerint (ha S < 15%) 2,5 g-ot mérjünk be. Tegyük a vizsgálati mintát egy 400 ml-es főzőpohárba.
7.2. Oxidálás
Adjunk hozzá 20 ml nátrium-hidroxid oldatot (4.2.) és 20 ml vizet. Fedjük le óraüveggel. Forraljuk 5 percig a főzőlapon (5. 1.). Vegyük le a főzőlapról. Forró vízsugár segítségével gyűjtsük össze a főzőpohár falára ragadt ként és forraljuk 20 percig. Hagyjuk lehűlni.
Adjunk hozzá 2 ml-es adagokban hidrogén-peroxidot (4.3.) mindaddig, amíg már nem észlelünk további reakciót. 6-8 adag hidrogén-peroxidra lesz szükség. Hagyjuk tovább oxidálódni egy órát, aztán forraljuk fél órán keresztül. Hagyjuk lehűlni.
7.3. Az oldat elkészítése az elemzéshez
Adjunk hozzá kb. 50 ml vizet és 50 ml sósav oldatot (4.1.).
- Ha a kén (S) mennyisége kevesebb mint 5%:
szűrjük át egy 600 ml-es főzőpohárba. A szűrőn maradt csapadékot mossuk át többször hideg vízzel. Mosás után a szűrlet utolsó cseppjeiből ellenőrizzük a szulfát jelenlétének hiányát bárium-klorid oldat (4.4.) segítségével; a szűrletnek tökéletesen tisztának kell lennie. A szulfátot a teljes szűrletmennyiségben határozzuk meg a 8.9. módszer szerint.
- Ha a kén (S) mennyisége több mint 5%:
vigyük át veszteség nélkül egy 250 ml-es mérőlombikba, töltsük jelig vízzel és keverjük össze. Száraz szűrőn keresztül szűrjük át egy száraz tárolóedénybe; a szűrletnek teljesen tisztának kell lennie. Ha a szűrletet nem használjuk fel azonnal, akkor dugóval zárjuk le. Határozzuk meg a szulfátokat ennek az oldatnak egy alikvot részén bárium-szulfátos kicsapással (8.9. módszer).
H/3. A VÍZOLDHATÓ KALCIUM, MAGNÉZIUM, NÁTRIUM ÉS (A SZULFÁT FORMÁJÁBAN JELEN LÉVŐ) KÉN KIVONÁSA
1. TÁRGY
Ez a fejezet a vízben oldódó kalcium, magnézium, nátrium és (a szulfát formájában jelen lévő) kén kivonásához határoz meg eljárást úgy, hogy ugyanaz a kivonat alkalmazható legyen az összes megkövetelt tápelem meghatározásához.
2. ALKALMAZÁSI TERÜLET
Ez a módszer azokra az EGK műtrágyákra alkalmazandó, amelyekre a 3. számú melléklet 1. függeléke megköveteli a vízben oldódó kalcium, magnézium, nátrium és (a szulfát formájában jelen lévő) kén megadását.
3. A MÓDSZER ELVE
A tápelemeket forró vízben oldjuk.
4. VEGYSZEREK
Desztillált, vagy azonos minőségű ioncserélt víz.
5. FELSZERELÉS
Állítható hőmérsékletű elektromos főzőlap.
6. A MINTA ELKÉSZÍTÉSE Lásd a B) fejezetet.
7. ELJÁRÁS
7.1. Vizsgálati minta
a) Ha a műtrágyák nem tartalmaznak ként, vagy ha tartalmaznak, akkor nem több mint 3% ként (S), azaz 7,5% SO3-ot és ugyanakkor nem több mint 4% kalciumot (Ca), azaz 5,6% CaO-t, akkor mérjünk be 5 g műtrágyát egy milligramm pontossággal.
b) Ha a műtrágyák több mint 3% ként (S) és több mint 4% kalciumot (Ca) tartalmaznak, akkor mérjünk be 1 g műtrágyát egy milligramm pontossággal.
Tegyük a vizsgálati mintát 600 ml-es főzőpohárba.
7.2. Az oldat elkészítése
Adjunk hozzá kb. 400 ml vizet és tartsuk forrásban 30 percig. Időnként megkeverve hagyjuk lehűlni, és veszteség nélkül, dekantálással vigyük át egy 500 ml-es mérőlombikba. Töltsük fel jelig vízzel, és keverjük össze.
Száraz szűrőn keresztül szűrjük át egy száraz lombikba. A szűrlet első részleteit ontsuk el. A szűrletnek teljesen átlátszónak kell lennie.
Ha a szűrletet nem használjuk fel azonnal, akkor dugóval zárjuk le.
H/4. KÜLÖNBÖZŐ FORMÁBAN JELEN LÉVŐ, VÍZBEN OLDÓDÓ KÉN KIVONÁSA
1. TÁRGY
Ez a fejezet a műtrágyákban különböző, vízben oldódó formában jelen lévő kén kivonásához határoz meg eljárást.
2. ALKALMAZÁSI TERÜLET
Ez a módszer azokra az EC műtrágyákra alkalmazandó, amelyekre a 3. számú melléklet 1. függeléke megköveteli a vízben oldódó kéntrioxid megadását.
3. A MÓDSZER ELVE
A ként hideg vízben feloldjuk, majd lúgos közegben hidrogén-peroxidos oxidációval szulfáttá alakítjuk.
4. VEGYSZEREK
4.1. Hígított sósav:
Egy térfogatrész sósav (d20 =1,18 g/ml) és egy térfogatrész víz.
4.2. Nátrium-hidroxid oldat, NaOH, legalább 30% (d20 = 1,33 g/ml).
4.3. Hidrogén-peroxid oldat, 30% (w/w).
5. FELSZERELÉS
5.1. 500 ml-es Stohmann-lombik.
5.2. Forgó rázógép, 30-40 fordulat/perc.
5.3. Állítható hőmérsékletű elektromos főzőlap.
6. A MINTA ELŐKÉSZÍTÉSE Lásd a B) fejezetet.
7. ELJÁRÁS
7.1. Vizsgálati minta
a) Ha a műtrágyák legfeljebb 3% ként (S), azaz 7,5% SO3-ot és ugyanakkor legfeljebb 4% kalciumot (Ca), azaz 5,6% CaO-t tartalmaznak, akkor mérjünk be 5 g műtrágyát egy milligramm pontossággal.
b) Ha a műtrágyák több mint 3% ként (S) és ugyanakkor több mint 4% kalciumot (Ca) tartalmaznak, akkor mérjünk be 1 g műtrágyát milligrammos pontossággal.
Tegyük a vizsgálati mintát egy 500 ml-es lombikba (5.1.).
7.2. Az oldat elkészítése
Adjunk hozzá kb. 400 ml vizet. Zárjuk le dugóval. Rázassuk (5.2.) 30 percig. Töltsük fel jelig vízzel és keverjük össze. Száraz szűrőn keresztül szűrjük át egy száraz lombikba. Ha a szűrletet nem használjuk fel azonnal, akkor dugóval zárjuk le.
7.3. A vizsgálathoz használt alikvot rész oxidálása
A kivont oldatból mérjünk ki egy legfeljebb 50 ml-es alikvot részt, amely lehetőség szerint 20 mg és 100 mg közötti ként (S) tartalmazzon.
Ha szükséges, töltsük fel 50 ml-re vízzel. Adjunk hozzá 3 ml nátrium-hidroxid oldatot (4.2.) és 2 ml hidrogén-peroxid oldatot (4.3.). Takarjuk le óraüveggel és a főzőlapon (5.3.) óvatosan forraljuk egy órán keresztül. Milliliteres részletekbén folytassuk a hidrogén-peroxid oldat hozzáadását mindaddig, amíg a reakció tart (legfeljebb 5 ml).
Ezután hagyjuk lehűlni. Vegyük le az óraüveget és mossuk le az alsó oldalát a főzőpohárba. Adjunk hozzá kb. 20 ml hígított sósavat (4.1.). Töltsük fel vízzel kb. 300 ml-ig.
Határozzuk meg a szulfátok mennyiségét az oxidált oldat egészére vonatkoztatva a 8.9. módszer szerint.
H/5. AZ ELEMI KÉN KIVONÁSA ÉS MEGHATÁROZÁSA
FIGYELMEZTETÉS
Ez az elemzési módszer szén-diszulfid (CS2) használatát írja elő. Ezért különleges biztonsági intézkedéseket kell alkalmazni, különösen a következők tekintetében:
- a CS2 tárolása,
- a személyzet védőfelszerelése,
- a munkahelyi higiénia,
- a tűzesetek és a robbanások megelőzése,
- a használt vegyszer elhelyezése.
Ez a módszer jól képzett személyzetet és megfelelően felszerelt laboratóriumot kíván meg.
1. TÁRGY
Ez a fejezet a műtrágyákban jelen lévő elemi kén kivonásához és a mennyiségének megállapításához határoz meg eljárást.
2. ALKALMAZÁSI TERÜLET
Ez a módszer azokra az EC műtrágyákra alkalmazandó, amelyekre a 3. számú melléklet 1. függeléke megköveteli az elemi formában jelen lévő összes kén megadását.
3. A MÓDSZER ELVE
A vízben oldódó vegyületek eltávolítását követően az elemi ként szén-diszulfid segítségével kivonjuk, majd a kivont ként gravimetriásán meghatározzuk.
4. VEGYSZEREK Szén-diszulfid.
5. FELSZERELÉS
5.1. 100 ml-es extrakciós lombik, csiszolatos üvegdugóval ellátva.
5.2. Soxhlet-készülék, alkalmas szűrőelemekkel.
5.3. Vákuumos, forgó párologtató berendezés.
5.4. Ventilátorral ellátott elektromos kemence 90 ± 2 °C-ra beállítva.
5.5. 5-7 cm átmérőjű porcelán bepárló csészék, amelyek 5 cm-nél nem magasabbak.
5.6. Állítható hőmérsékletű elektromos főzőlap.
6. A MINTA ELŐKÉSZÍTÉSE Lásd a B) fejezetet.
7. ELJÁRÁS
7.1. Vizsgálati minta
Egy milligramm pontossággal mérjünk be 5 és 10 g közötti mennyiségű mintát, és tegyük a Soxhlet-készülék (5.2.) extrakciós hüvelyébe.
7.2. A kén kivonása
Alaposan mossuk át a tartalmát forró vízzel, hogy minden vízben oldható összetevőt eltávolítsunk. Szárítsuk a kemencében (5.4.) 90 °C-on legalább egy órán keresztül. Helyezzük bele a szűrőt a Soxhlet-készülékbe (5.2.).
Helyezzünk egy pár üveggyöngyöt a készülék extrakciós lombikjába (5.1.), mérjük le a tömegét (P0) és adjunk hozzá 50 ml szén-diszulfidot (4.1.).
Csatlakoztassuk a készüléket és hagyjuk, hogy az elemi kén kivonása folytatódjon hat órán keresztül. Kapcsoljuk ki a fűtést, és lehűlés után szedjük le a lombikot. Kössük a lombikot a forgó párologtató berendezésre (5.3.) és párologtassuk, amíg a lombik tartalma szivacsos masszává szilárdul.
Szárítsuk a lombikot a kemencében 90 °C-on (5.4.) (általában egy óra szükséges), amíg a tömege (P1) állandósul.
7.3. Az elemi kén tisztaságának ellenőrzése
Egyes anyagok az elemi kénnel együtt kivonódhattak a szén-diszulfidos eljárás során. Az elemi kén tisztaságának ellenőrzése a következőképpen zajlik:
homogenizáljuk a lombik tartalmát olyan alaposan, amennyire csak lehet, és vegyünk ki 2 vagy 3 g-ot milligrammos pontossággal (n). Helyezzük bepárló csészébe (5.5.). Mérjük meg a csészének és tartalmának együttes tömegét (P2). Helyezzük a főzőlapra (5.6.) és állítsuk azt be 220 °C-ot nem meghaladó hőmérsékletre, hogy a kén ne gyulladjon meg. Hagyjuk szublimálni 3 vagy 4 órán keresztül, amíg a tömege nem állandósul (P3).
Megjegyzés:
Egyes műtrágyák esetén nem feltétlenül szükséges a kén tisztaságát ellenőrizni. Ebben az esetben hagyjuk ki a 7.2. lépést.
AZ EREDMÉNYEK KIFEJEZÉSE
A műtrágya elemi kén (S) tartalmának százalékos aránya a következő:
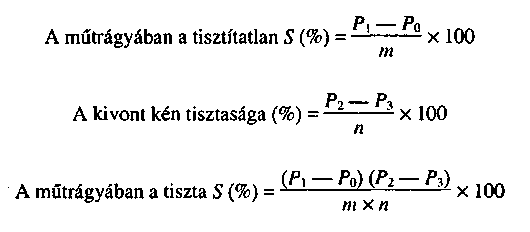
| ahol: | ||
| m = | a műtrágya vizsgálati mintájának tömege grammban kifejezve, | |
| Po = | a Soxhlet-lombik tömege grammban kifejezve, | |
| P1 .= | a Soxhlet-lombik és a tisztítatlan kén szárítás utáni együttes tömege, | |
| n = | a tisztításra szánt tisztítatlan kén tömege grammban kifejezve, | |
| P - | a bepárló-csésze tömege, | |
| P3 = | a bepárló-csésze tömege a kén szublimációja után. |
H/6. A KIVONT KALCIUM OXALÁT FORMÁJÁBAN TÖRTÉNŐ LECSAPÁST KÖVETŐ
PERMANGANOMETRIÁS MEGHATÁROZÁSA
1. TÁRGY
Ez a fejezet a műtrágya kivonatokban lévő kalcium mennyiségének megállapítására határoz meg eljárást.
2. ALKALMAZÁSI TERÜLET
Ez a módszer azokra az EGK műtrágyákra alkalmazandó, amelyekre a 3. számú melléklet l. függeléke megköveteli az összes és/vagy a vízben oldódó kalcium megadását.
3. A MÓDSZER ELVE
A kivont oldat alikvot részéből a jelen lévő kalciumot oxalát formájában lecsapjuk és azt kálium-permanganátos titrálással meghatározzuk.
4. VEGYSZEREK
4.1. Hígított sósav:
egy térfogatrész sósav (d20 =1,18 g/ml) és egy térfogatrész víz.
4.2. 1:10 arányban hígított kénsav:
egy térfogatrész kénsav (d20 = 1,84 g/ml) tíz térfogatrész vízben.
4.3. 1:1 arányban hígított ammónia oldat:
egy térfogatrész ammónia (d20 = 0,88 g/ml) és egy térfogatrész víz.
4.4. Ammónium-oxalát telített oldata [(NH4)2C2O4.H2O], szobahőmérsékleten (nagyjából 40 g/l).
4.5. Citromsav oldat, 30% (m/v).
4.6. Ammónium-klorid oldat, 5% (m/v).
4.7. Brómtimolkék oldat, 95%-os etanolban, 0,1% (m/v).
4.8. Brómkrezolzöld oldat, 95%-os etanolban, 0,04% (m/v).
4.9. Kálium-permanganát mérőoldat, 0,02 mol/l.
5. FELSZERELÉS
5.1. 5 és 20 μm közötti porozitású üveg szűrőtégely.
5.2. Forró vízfürdő.
6. AZ ALIKVOT RÉSZ ELŐKÉSZÍTÉSE
A 8.1. vagy a 8.2. módszer szerint készített kivont oldatból pipettával vegyünk ki egy 15 és 50 mg közötti mennyiségű Ca-ot (= 21 és 70 mg közötti mennyiségű CaO) tartalmazó alikvot részt. Legyen ennek az alikvot résznek a térfogata v2. Töltsük bele egy 400 ml-es főzőpohárba. Szükség esetén semlegesítsük egy pár csepp ammónia oldat (4.3.) segítségével [az indikátor (4.7.) zöldről kékre váltásáig].
Adjunk hozzá 1 ml citromsav oldatot (4.5.) és 5 ml ammónium-klorid oldatot (4.6.).
7. A KALCIUM-OXALÁT LECSAPÁSA
Adjunk hozzá kb. 100 ml vizet. Hozzuk forrásba, adjunk hozzá 8-10 csepp indikátor oldatot (4.8.) majd lassan 50 ml forró ammónium-oxalát oldatot (4.4.). Ha csapadék keletkezik, oldjuk fel néhány csepp sósav hozzáadásával (4.1.). Semlegesítsük folyamatos keverés közben, nagyon lassan ammónia oldattal (4.3.), amíg a pH 4,4 és 4,6 közé nem változik [az indikátor (4.8.) zöldről kékre változásáig]. Helyezzük a főzőpoharat forrásban lévő vízfürdőbe (5.2.) kb. 30 perc időtartamra. Vegyük ki a főzőpoharat a fürdőből, hagyjuk állni egy órán keresztül és szűrjük le a tégelyen keresztül (5.1.).
8. AZ OXALÁTOS CSAPADÉK TITRÁLÁSA
Mossuk a főzőpoharat és az üvegszűrőt addig, amíg az ammónium-oxalát felesleg teljes mértékben el nem távozik (ez a mosóvízben a klorid jelenlétének hiányával ellenőrizhető). Helyezzük a tégelyt egy 400 ml-es főzőpohárba és oldjuk fel a csapadékot 50 ml forró kénsavval (4.2.). Tegyünk annyi vizet a főzőpohárba, hogy kb. 100 ml legyen az oldat térfogata. Melegítsük fel 70-80 °C-os hőmérsékletre és cseppenként titráljuk kálium-permanganát oldattal (4.9.) mindaddig, amíg a rózsaszín elszíneződés 1 percig megmarad. Legyen ez a térfogat n.
9. AZ EREDMÉNYEK KISZÁMÍTÁSA
A műtrágya kalcium (Ca) tartalma tömegszázalékban a következő:
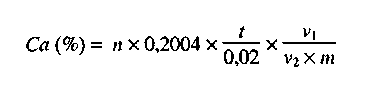
| ahol: | ||
| n = | a felhasznált kálium-permanganát mennyisége ml-ben kifejezve, | |
| m = | a vizsgálati minta tömege grammban kifejezve, | |
| V2 = | az alikvot rész térfogata milliliterben kifejezve, | |
| V1 = | a kivont oldat térfogata milliliterben kifejezve, | |
| t = | a kálium-permanganát oldat molaritása mol/l-ben kifejezve. |
CaO (%) = Ca (%) x 1,400.
H/7. MAGNÉZIUM MEGHATÁROZÁSA ATOMABSZORPCIÓS SPEKTROFOTOMETRIÁVAL
1. TÁRGY
Ez a fejezet a műtrágya kivonatokban lévő magnézium mennyiségének megállapítására határoz meg eljárást.
2. ALKALMAZÁSI TERÜLET
Ez a módszer azokra a 8.1. és 8.3. módszer szerint kivont műtrágya kivonatokra alkalmazandó, amelyekre az összes és/vagy a vízben oldódó magnézium megadása kötelező, a következő kivételekkel:
a mezoelemekről szóló 3. számú melléklet 1. függelékében felsorolt műtrágyák:
4. típus (kieserit),
5. típus (magnézium-szulfát) és
7. típus (kieserit kálium-szulfáttal) amelyekre a 8.8. módszer vonatkozik.
Az alább megadott módszer minden olyan műtrágya kivonatra alkalmazandó, amely olyan mennyiségben tartalmaz bizonyos elemeket, ami zavarhatja a magnézium komplexometriás meghatározását.
3. A MÓDSZER ELVE
A magnézium meghatározása atomabszorpciós spektrofotometriával, az oldat megfelelő hígítása után.
4. VEGYSZEREK
4.1. Sósav, 1 mol/l oldat.
4.2. Sósav, 0,5 mol/l oldat.
4.3. Magnézium törzsoldat, 1,00 mg/ml.
4.3.1. Oldjunk fel 10,141 g magnézium-szulfátot (MgSO4.7H2O) 500 ml 0,5 mol/l sósavoldatban (4.2.), 1000 ml-es főzőpohárban. Oldódás után mossuk át veszteség nélkül 1000 ml-es mérőlombikba, töltsük fel jelig, és keverjük össze.
4.3.2. Mérjünk be 1,658 g magnézium-oxidot (MgO), amelyet a karbonátosodás nyomainak eltüntetésére előzetesen kalcináltunk. Helyezzük főzőpohárba, amelybe 100 ml vizet és 120 ml 1 mol/l sósavat (4.1.) is teszünk. Amikor feloldódott, veszteség nélkül átmossuk egy 1000 ml-es mérőlombikba. Töltsük fel jelig, és keverjük össze.
vagy:
4.3.3. Kereskedelmi standard oldat
A laboratórium köteles az ilyen oldatokat ellenőrizni.
4.4. Stroncium-klorid oldat.
Oldjunk fel 75 g stroncium-kloridot (SrCl2.6H2O) sósavoldatban (4.2.), és egészítsük ki a térfogatát 500 ml-re ugyanazzal a sósavoldattal.
5. FELSZERELÉS
Atomabszorpciós mérésekre alkalmas spektrofotométer, magnézium lámpával, beállítva 285,2 nm-re. Levegő-acetilén láng.
6. A MINTA ELKÉSZÍTÉSE Lásd a H/1. és a H/3. fejezeteket.
7. ELJÁRÁS
7.1. Ha a műtrágya megadott magnéziumtartalma (Mg) több mint 6% (azaz 10% MgO formájában), akkor vegyünk 25 ml-t (v1) a kivont oldatból (6.). Vigyük át egy 100 ml-es mérőlombikba, töltsük jelig vízzel, és keverjük össze. A hígítási tényező D1 = 100/V1.
7.2. Pipettával vegyünk ki 10 ml-t a kivont oldatból (6.) vagy a (7.1.) oldatból. Vigyük át egy 200 ml-es mérőlombikba. Töltsük jelig a 0,5 mol/l sósavval (4.2.), és keverjük össze. A hígítási tényező 200/10.
7.3. Hígítsuk ezt az oldatot (7.2.) a 0,5 mol/l sósav oldattal (4.2.) úgy, hogy a koncentráció a spektrofotométer (5.1.) mérési tartományának optimális részébe essen. V2 a minta térfogata 100 ml-ben. A hígítási tényező D2 = 100/V2.
Az utolsó oldat 10 (térfogatszázalék) stroncium-klorid oldatot (4.4.) tartalmazzon.
7.4. Vakoldat készítése
Készítsünk vakoldatot a teljes eljárás megismétlésével a kivonástól kezdve (8.1. vagy 8.3. módszer), egyedül a műtrágya vizsgálati mintáját hagyjuk ki.
7.5. Kalibrációs oldatok készítése
A törzsoldat (4.3.) 0,5 mol/l sósavval történő hígításával készítsünk legalább öt növekvő koncentrációjú kalibrációs oldatot a készülék (5.1.) optimális mérési tartományán belül.
Ezek az oldatok 10% (m/v) stroncium-klorid oldatot (4.4.) tartalmazzanak.
7.6. Mérés
Állítsuk be a spektrofotométert (5.1.) 285,2 nm hullámhosszra.
Poriasszuk be egymás után a kalibrációs oldatokat (7.5.), a minta oldatot (7.3.) és a vakoldatot (7.4.), miközben mindig a következő mérendő oldattal mossuk át a készüléket. Ismételjük meg ezt a műveletet háromszor. Ábrázoljuk a kalibrációs görbét az egyes kalibrációs oldatok (7.5.) átlagos abszorbanciájának felhasználásával az ordinátán és a megfelelő magnéziumra vonatkoztatott koncentráció (μg/ml-ben) feltüntetésével az abszcisszán. Határozzuk meg a magnézium koncentrációját a mintában (7.3.), XS és a vakoldatban (7.4.), Xb, a kalibrációs görbe alapján.
8. AZ EREDMÉNYEK KISZÁMÍTÁSA
A magnézium (Mg) vagy magnézium-oxid (MgO) mennyiségét a mintában a kalibrációs oldatokhoz történő viszonyítás segítségével határozhatjuk meg, a vakpróba eredményének figyelembevételével. A magnézium (Mg) tömegszázalékos mennyisége a műtrágyában a következő:
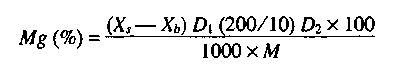
| ahol: | ||
| XS = | az elemzendő minta koncentrációja, amint azt felvettük a kalibrációs görbére, μg/ml-ben kifejezve, | |
| Xb = | a vakoldat koncentrációja, amint azt felvettünk a kalibrációs görbére, μg/ml-ben kifejezve, | |
| D1 = | a hígítási tényező, ha az oldatot hígítottuk (7.1.), | |
| egyenlő néggyel, ha 25 ml-t vettünk, | ||
| egyenlő eggyel, ha az oldatot nem hígítottuk, | ||
| D2 = | a 7.3.-nál szereplő hígítási tényező, | |
| M = | a vizsgálati minta tömege a kivonás során. |
MgO (%)=Mg (%)/0,6
H/8. MAGNÉZIUM MEGHATÁROZÁSA KOMPLEXOMETRIÁVAL
1. TÁRGY
Ez a fejezet a műtrágya kivonatokban lévő magnézium mennyiségének megállapítására határoz meg eljárást.
2. ALKALMAZÁSI TERÜLET
Ez a módszer azokra az EC műtrágya kivonatokra alkalmazandó, amelyekre az összes és/vagy a vízben oldódó magnézium megadása kötelező:
- a 3. számú melléklet 1. függelékében felsorolt műtrágyák: egyszerű nitrogénműtrágyák, 1 b. típus (kalcium-magnéziumnitrát), 7. típus (magnézium-szulfát-nitrát), 8. típus (nitrogénműtrágyák magnéziummal) és tiszta káliumműtrágyák, 2. típus (dúsított kainit), 4. típus (magnéziumot tartalmazó kálium-klorid), 6. típus (magnéziumsót tartalmazó kálium-szulfát),
- a függelék mezoelemekről szóló D táblázatában felsorolt műtrágyák.
3. A MÓDSZER ELVE
A magnéziumot a 8.1. és/vagy a 8.3. módszer szerint oldatba visszük. Első titrálás: a Ca és Mg titrálása EDTA segítségével, eriokrómfekete T jelenlétében. Második titrálás: a Ca titrálása EDTA segítségével, kalcein vagy kalkonkarbonsav jelenlétében. A magnéziumot a különbségből határozzuk meg.
4. VEGYSZEREK
4.1. Magnézium mérőoldat, 0,05 mol/l:
4.1.1. Oldjunk fel 1,232 g magnézium-szulfátot (MgSO4.7H2O) a 0,5 mol/l sósavoldatban (4.11.) és hígítsuk fel ugyanazzal a savval 100 ml-re.
vagy:
4.1.2. Mérjünk be 2,016 g magnézium-oxidot, amelyet a karbonátosodás nyomainak eltüntetésére előzetesen kaleináltunk. Helyezzük főzőpohárba, amelybe 100 ml vizet is teszünk.
Keverjünk bele kb. 120 ml kb. 1 mol/l sósavat (4.12.).
Amikor feloldódott, veszteség nélkül vigyük át egy 1000 ml-es mérőlombikba. Töltsük fel jelig, és keverjük össze.
Ezeknek az oldatoknak 1 ml-e 1,216 mg Mg-ot tartalmaz (= 2,016 mg MgO).
A laboratórium felelőssége a fenti mérőoldat koncentrációjának az ellenőrzése.
4.2. EDTA 0,05 mol/l oldata
Mérjünk be 18,61 g-ot a dinátrium-etilén-diamin-tetraacetát-dihidrátból (C10H14N2Na2O8.2H2O), tegyük egy 1000 ml-es főzőpohárba és oldjuk fel 600-800 ml vízben. Veszteség nélkül vigyük át egy 1000 ml-es mérőlombikba. Töltsük fel jelig, és keverjük össze. Ellenőrizzük ezt az oldatot a mérőoldattal (4.1.) úgy, hogy az utóbbiból kiveszünk egy 20 ml-es mintát, és megtitráljuk a (7.2.) módszer szerinti analitikai eljárással.
Egy ml EDTA oldat megfelel 1,216 mg Mg-nak (= 2,016 mg MgO) és 2,004 mg Ca-nak (= 2,804 mg CaO) (lásd 10.1. és 10.6. megjegyzés).
4.3. 0,05 mol/lkalcium mérőoldat
Mérjünk be 5,004 g száraz kalcium-karbonátot. Helyezzük főzőpohárba, amelybe 100 ml vizet is teszünk. Fokozatosan keverjünk bele 120 ml kb. 1 M sósavat (4.12.).
Forraljuk fel a széndioxid kiűzése céljából, hűtsük le és veszteség nélkül vigyük át egy 1000 ml-es mérőlombikba, majd töltsük fel jelig vízzel, és keverjük össze. Ellenőrizzük ezt az oldatot az EDTA oldattal (4.2.) a (7.3.) módszer szerinti analitikai eljárással. Ennek az oldatnak 1 ml-e 2,004 mg Ca-t tartalmaz (= 2,804 mg CaO), és megfelel 1 ml 0,05 mólos EDTA oldatnak (4.2.).
4.4. Kalcein indikátor
Alaposan keverjünk össze mozsárban 1 g kalceint 100 g nátrium-kloriddal. Ebből a keverékből 10 mg-ot használunk. Az indikátor zöldről narancsszínűre változik. A titrálást addig kell folytatni, amíg a szín narancsra változik, ami mentes a zöldes árnyalattól.
4.5. Kalkonkarbonsav indikátor
Oldjunk fel 400 mg kalkonkarbonsavat 100 ml metanolban. Ez az oldat legfeljebb csak kb. 4 hétig tárolható. Ebből az oldatból három cseppet kell használni. Az indikátor pirosról kék színűre változik. A titrálást addig kell folytatni, amíg a szín kékre változik, ami mentes a pirosas árnyalattól.
4.6. Eriokrómfekete-T indikátor
Oldjunk fel 300 mg eriokrómfekete-T indikátort 25 ml 1-propanol és 15 ml trietanol-amin keverékében. Ez az oldat legfeljebb csak kb. 4 hétig tárolható. Ebből az oldatból három cseppet kell felhasználni. Az indikátor pirosról kék színűre változik és a titrálást addig kell folytatni, amíg a szín kékre változik, ami mentes a pirosas árnyalattól. Csak akkor változtatja a színét, ha magnézium is jelen van. Szükség esetén adjunk hozzá 1 ml-t a mérőoldatból (4.1.).
Ha kalcium és magnézium egyaránt jelen van, akkor az EDTA először a kalciummal képez komplexet és azután a magnéziummal. Ebben az esetben a két elem meghatározása párhuzamosan történik.
4.7. Kálium-cianid oldat
KCN 2%-os vizes oldata. (Ne pipettázzuk szájjal és lásd 10.7.).
4.8. Kálium-hidroxid és kálium-cianid oldata
Oldjunk fel 280 g KOH-t és 66 g KCN-t vízben, egészítsük ki a térfogatát egy literre és keverjük össze.
4.9. pH 10,5-es puffer oldat
Egy 500 ml-es mérőlombikba tegyünk 33 g ammónium-kloridot és oldjuk fel 200 ml vízben, majd adjunk hozzá 250 ml ammóniát (d20 = 0,91 g/ml), végül töltsük fel jelig és keverjük össze. Rendszeresen ellenőrizzük az oldat pH-ját.
4.10. Hígított sósav: egy térfogat sósav (d20 = 1,18 g/ml) és egy térfogat víz.
4.11. Sósav oldat, kb. 0,5 mol/l.
4.12. Sósav oldat, kb. 1 mol/l.
4.13. Nátrium-hidroxid oldat, 5 mol/l.
5. FELSZERELÉS
5.1. Mágneses vagy mechanikus keverő.
5.2. pH-mérő.
6. ELLENŐRZŐ VIZSGÁLAT
Végezzünk el meghatározásokat az oldatok (4.1. és 4.3.) alikvot részein olyan módon, hogy a Ca/Mg arány nagyjából azonos legyen az elemzendő oldatban lévővel. Ebből a célból vegyünk (a) mennyiséget a (4.3.) mérőoldatból és (b-a) mennyiséget a (4.1.) mérőoldatból. Az (a) és (b) szám megfelel az EDTA oldatból az elemzendő minta két titrálása során felhasznált mennyiség ml-ben kifejezett értékének. Ez az eljárás csak akkor megfelelő, ha az EDTA a kalcium és a magnézium oldatok pontosan ekvivalensek. Ha ez nem áll fenn, akkor helyesbítéseket kell végezni.
7. A VIZSGÁLATI OLDAT ELŐKÉSZÍTÉSE Lásd a H/1. és a H/3. fejezeteket.
8. MEGHATÁROZÁS
8.1. Az alikvot rész kivétele.
Az alikvot rész lehetőleg 9 és 18 mg közötti mennyiségű magnéziumot tartalmazzon (= 15-30 mg MgO).
8.2. Titrálás eriokrómfekete-T jelenlétében
Pipettázzunk egy alikvot részt (8.1.) az elemzendő oldatból egy 400 ml-es főzőpohárba. Semlegesítsük a felesleges savat 5 M nátrium-hidroxid oldattal (4.12.), pH-mérő alkalmazása mellett. Hígítsuk fel vízzel kb. 100 ml-re. Adjunk hozzá 5 ml puffer oldatot (4.9.). A pH-mérő által mért pH 10,5 ±0,1 kell legyen. Adjunk hozzá 2 ml kálium-cianid oldatot (4.7.) és három csepp eriokrómfekete T indikátort (4.6.). Titráljuk EDTA oldattal (4.2.). Közben a keverővel (5.1.) lassan kevertetjük (lásd 10.2., 10.3. és 10.4.). Legyen "b" a 0,05 mólos EDTA oldat milliliteréinek száma.
8.3. Titrálás kalcein vagy kalkonkarbonsav jelenlétében
Pipettázzunk a fenti titrálás során alkalmazottal azonos alikvot részt az elemzendő oldatból egy 400 ml-es főzőpohárba. Semlegesítsük a felesleges savat 5 M nátrium-hidroxid oldattal (4.13.), pH-mérő alkalmazása mellett. Hígítsuk fel vízzel kb. 100 ml-re. Adjunk hozzá 10 ml KOH/KCN oldatot (4.8.) és az indikátort (4.4.) vagy (4.5.). A keverővel (5.1.) való lassú keverés közben titráljuk meg az EDTA oldattal (4.2.) (lásd 10.2., 10.3. és 10.4.). Legyen "a" a 0,05 mólos EDTA oldat milliliteréinek száma.
9. AZ EREDMÉNYEK KIFEJEZÉSE
Azokra az EGK műtrágyákra, amelyekre a módszer alkalmazható (5 g műtrágya 500 ml kivonatban), a műtrágya százalékos tartalma:
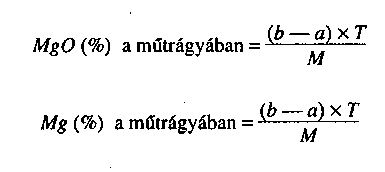
| ahol: | ||
| a = | a 0,05 mólos EDTA oldat milliliteréinek száma, amelyet a kalcein vagy kalkonkarbonsav jelenlétében | |
| végzett titrálás során használtunk fel, | ||
| b = | a 0,05 mólos EDTA oldat milliliteréinek száma, amelyet az eriokrómfekete-T jelenlétében végzett titrálás | |
| során használtunk fel, | ||
| M = | a vizsgálati minta tömege, amely az alikvot részben van (grammban kifejezve), | |
| T = | 0,2016 x az EDTA oldat molaritása/0,05 (lásd 4.2.), | |
| T' = | 0,1216 x az EDTA oldat molaritása/0,05 (lásd 4.2.). |
10. MEGJEGYZÉSEK
10.1. Komplexometriai elemzésekben a sztöchiometrikus EDTA: fém arány mindig 1:1, függetlenül a fém vegyértékétől és annak ellenére, hogy az EDTA négy vegyértékű. Az EDTA titráló oldat és a mérőoldatok ezért molárisak és nem normál oldatok.
10.2. A komplexometriás indikátorok gyakran érzékenyek a levegőre. Az oldat veszíthet a színéből a titrálás alatt. Ebben az esetben egy vagy két csepp indikátort kell hozzáadni. Ez elsősorban az eriokrómfekete-T és a kalkonkarbonsav esetében igaz.
10.3. A fém-indikátor komplexek gyakran relatíve stabilak, így esetenként valamennyi időt vehet igénybe, amíg a színváltozás megtörténik. Ezért az EDTA utolsó cseppjeit lassan kell hozzáadni és egy csepp 0,05 mólos magnézium oldatot (4.1.) vagy kalcium oldatot (4.3.) kell még hozzáadni, annak érdekében, hogy megbizonyosodjunk arról, hogy a színváltozás nem ment még végbe. Ez különösen igaz az eriokróm-magnézium komplex esetében.
10.4. Az indikátor színátcsapását nem függőlegesen, hanem vízszintesen, az oldat keresztmetszetében kell észlelni, és a főzőpoharat fehér háttér elé kell helyezni, jól megvilágított helyre. Az indikátor színátcsapása könnyen észlelhető, ha a főzőpoharat alulról gyengén megvilágított (25 wattos lámpa), homályos üvegre tesszük.
10.5. Ez az elemzés bizonyos fokú gyakorlatot követel meg. A feladat - többek között - tartalmazza a 4.1. és 4.3. mérőoldatok színváltozásainak megfigyelését. Ajánlatos, hogy a meghatározásokat ugyanaz a laborkémikus végezze.
10.6. Ha garantált titerű EDTA oldatot használunk (például Tritisol, Normex), akkor ez egyszerűsítheti a 4.1., 4.2. és 4.3. mérőoldatok egyenértékének ellenőrzését.
10.7. A kálium-cianidot tartalmazó oldatok nem önthetők el a lefolyóba a cianid ártalmatlanítása nélkül, például lúgosítást követő nátrium-hipokloritos oxidációval.
H/9. SZULFÁTOK MEGHATÁROZÁSA
1. TÁRGY
Ez a fejezet a műtrágya kivonatokban szulfát formájában jelen lévő kén mennyiségének megállapítására határoz meg eljárást.
2. ALKALMAZÁSI TERÜLET
Ez a módszer a 8.1, 8.2., 8.3. és 8.4. módszer során előállított kivonatokban jelen lévő szulfát mennyiségének megállapítására alkalmazható.
3. A MÓDSZER ELVE
Gravimetriás meghatározás bárium-szulfát formájában.
4. VEGYSZEREK
4.1. Hígított sósav:
Egy térfogatrész sósav (d20 =1,18 g/ml), és egy térfogatrész víz.
4.2. Bárium-klorid oldat BaCl22H2O: 122 g/l.
4.3. Ezüst-nitrátoldat: 5 g/l.
5. FELSZERELÉS
5.1. Porcelán olvasztótégelyek.
5.2. Forró vízfürdő.
5.3. Szárítószekrény 105 ± 1 °C-ra beállítva.
5.4. Elektromos kemence 800 ± 50 °C-ra beállítva.
6. A MINTA ELŐKÉSZÍTÉSE
6.1. Mintavétel az oldatból
Pipetta alkalmazásával vegyünk ki egy alikvot részt a 2. pontban megjelölt kivont oldatokból, amely 20-100 mg S-t vagy 50-350 mg SO3-ot tartalmaz.
Tegyük ezt az alikvot részt egy megfelelő térfogatú főzőpohárba. Adjunk hozzá 20 ml hígított sósavat (4.1.). Egészítsük ki kb. 300 ml-re vízzel.
6.2. A lecsapás
Forraljuk fel az oldatot. Cseppenként adjunk hozzá kb. 20 ml-t a bárium-klorid oldatból (4.2.), miközben élénken keverjük az oldatot. Forraljuk néhány percig.
A főzőpoharat fedjük le óraüveggel, és tegyük forrásban lévő vízfürdőbe (5.2.) egy órás időtartamra. Ezután hagyjuk forró (> 60 °C) állapotban állni, amíg a felülúszó folyadék kitisztul. Dekantáljuk a tiszta oldatot egy kis pórusú hamumentes szűrőn. Mossuk át a csapadékot néhányszor forró vízzel. Folytassuk a csapadék mosását a szűrőn, amíg a szűrlet klorid-mentes lesz. Ezt ezüst-nitrát oldat (4.3.) segítségével ellenőrizhetjük.
6.3. Elhamvasztás és a csapadék lemérése
Helyezzük a szűrőpapírt és a csapadékot egy előzőleg 0,1 mg pontossággal lemért porcelántégelybe (5.1.). Szárítsuk meg a szárítószekrényben (5.3.) és hamvasszuk kb. 800 °C-on fél órán keresztül (5.4.). Hagyjuk lehűlni exszikkátorban, és mérjük le 0,1 mg-os pontossággal.
7. AZ EREDMÉNYEK KIFEJEZÉSE
Egy mg bárium-szulfát megfelel 0,137 mg S-nek vagy 0,343 mg SO3-nak. A műtrágya tömegszázalékos kéntartalma a következő:
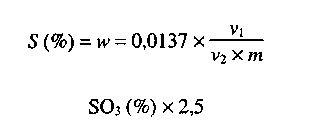
| Ahol: |
| w = a bárium-szulfát csapadék tömege grammban kifejezve, |
| v1 = a kivont oldat térfogata milliliterben kifejezve, |
| v2 = az alikvot térfogata milliliterben kifejezve, |
| m = a vizsgálati minta tömege grammban kifejezve. |
H/10. A KIVONT NÁTRIUM MEGHATÁROZÁSA
1. TÁRGY
Ez a fejezet a műtrágya kivonatokban jelen lévő nátrium mennyiségének megállapításához határoz meg eljárást.
2. ALKALMAZÁSI TERÜLET
Ez a módszer azokra a műtrágyákra alkalmazandó, amelyekre a 3. számú melléklet 1. függeléke megköveteli a nátrium megadását.
3. A MÓDSZER ELVE
Az I/2. és/vagy I/3. módszer szerint kapott kivonat megfelelő hígítása után az oldat nátrium-koncentrációját lángemissziós spektrofotometriával határozzuk meg.
4. VEGYSZEREK
4.1. Hígított sósav
Egy térfogatrész sósav (d20 =1,18 g/ml) és egy térfogatrész víz.
4.2. Alumínium-nitrát A1(NO3)3.9H2O.
4.3. Cézium-klorid, CsCl.
4.4. Vízmentes nátrium-klorid, NaCl.
4.5. Cézium-klorid és alumínium-nitrát oldat
Készítsünk vizes oldatot 50 g cézium-kloriddal (4.3.) és 250 g alumínium-nitráttal (4.2.) egy 1000 ml-es mérőlombikban. Töltsük fel jelig, és keverjük össze.
4.6. Nátrium törzsoldat, 1 mg/ml Na
Oldjunk fel vízben 2,542 g nátrium-kloridot (4.4.) egy 1000 ml-es mérőlombikban. Adjunk hozzá 10 ml sósavat (4.1.). Töltsük fel jelig, és keverjük össze.
5. FELSZERELÉS
Lángemisszióra alkalmas spektrofotométer, 589,3 nm-re beállítva.
6. KALIBRÁLÓ OLDATOK ELKÉSZÍTÉSE
6.1. Tegyünk 10 ml törzsoldatot (4.6.) egy 250 ml-es mérőlombikba. Töltsük fel jelig, és keverjük össze. Az oldat koncentrációja 40 μg/ml Na.
6.2. Tegyünk 0,5, 10, 15, 20, 25 ml közbenső oldatot (6.1.) 100 ml-es mérőlombikokba. Adjunk hozzá 10 ml (4.5.) oldatot. Töltsük fel jelig, és keverjük össze. Az oldatok koncentrációja: 0, 2, 4, 6, 8, 10 μg/ml Na.
7. A MÉRENDŐ OLDATOK ELŐKÉSZÍTÉSE
A kivont oldat várható nátriumtartalmától függően, az I/2. vagy I/3. módszer szerint (5 g műtrágya 500 ml-ben) végezzük el a hígításokat a következő táblázatnak megfelelően:
| Na2O (%) | Na (%) | Közbenső hígítás | + | Végső hígítás | + | A hígítás mértéke |
| Minta (ml) (V4) | Hígítás ml-re (v3) | Minta (ml) (V4) | Hígítás ml-re | |||
| 3—5 | 2,2—3,7 | 10 | 50 | 10 | 100 | 50 |
| 5—10 | 3,7—7,4 | 10 | 100 | 10 | 100 | 100 |
| 10—20 | 7,4—15 | 10 | 100 | 5 | 100 | 200 |
| 20—38 | 15—28 | 5 | 100 | 5 | 100 | 400 |
A közbenső oldat térfogatát vízzel egészítsük ki. A végső hígításhoz adjunk 10 ml-t a (4.5.) oldatból a 100 ml-es mérőlombikba.
Egy g-os vizsgálati mintához szorozzuk meg a végső hígítás térfogatát (v4) öttel.
8. MEGHATÁROZÁS
Készítsük elő a spektrofotométert (5.1.) az 589,3 nm-es mérésre. Kalibráljuk a műszert a kalibrációs oldatokkal (6.2.). Ezután állítsuk be a műszer érzékenységét úgy, hogy a teljes mérési tartományt kihasználjuk a legtöményebb kalibrációs oldat mérésénél. Ezután mérjük meg az elemzendő vizsgálati mintára (7.) adott választ. Ismételjük meg ezt a műveletet 3 alkalommal.
9. AZ EREDMÉNYEK KISZÁMÍTÁSA
Rajzoljuk fel a kalibrációs görbét az egyes kalibrációs oldatokra adott átlagos válaszok feltüntetésével az ordinátán, míg az abszcisszán a megfelelő koncentrációk szerepeljenek μg/ml mértékegységben kifejezve. Határozzuk meg ebből a vizsgálati minta nátrium-koncentrációját. Számítsuk ki a nátrium mennyiségét, a hígítások mértékének figyelembevételével. Az eredményt a minta százalékában fejezzük ki.
A műtrágya nátrium (Na) tartalma tömegszázalékban a következő:
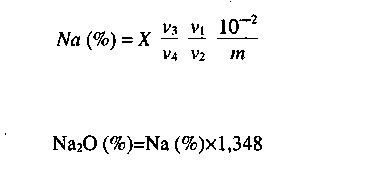
| Ahol: |
| x = a spektrofotométerbe bejuttatott oldat koncentrációja μg/ml-ben kifejezve, |
| v1 = a kivont oldat térfogata milliliterben kifejezve, |
| v2 = az alikvot térfogata a közbenső hígításnál milliliterben kifejezve, |
| v3 = a közbenső hígítás térfogata milliliterben kifejezve, |
| v 4 = az alikvot térfogata a végső hígításnál (100 milliliterben), |
| m = a vizsgálati minta tömege grammban kifejezve. |
I) MIKROELEMEK
I/1. AZ ÖSSZES MIKROELEM KIVONÁSA
1. TÁRGY
A módszer a következő mikroelemek kivonására vonatkozóan határozza meg az eljárást: összes bór, összes kobalt, összes réz, összes vas, összes mangán, összes molibdén és összes cink. A cél az, hogy minél kevesebb kivonatot kelljen előállítani, és ' amikor csak lehet, ugyanazt a kivonatot használjuk fel valamennyi fent felsorolt mikroelem teljes mennyiségének meghatározására.
2. ALKALMAZÁSI TERÜLET
Az eljárás azokra az EGK műtrágyákra vonatkozik, amelyek a 3. számú melléklet 1. függeléke hatálya alá tartoznak és a következő mikroelemek egyikét, vagy közülük többet tartalmaznak: bór, kobalt, réz, vas, mangán, molibdén és cink. Alkalmazható minden olyan mikroelemre, amelynek megadott koncentrációja 10% vagy annál kevesebb.
3. A MÓDSZER ELVE
Forrásban lévő hígított sósavban való oldás.
Megjegyzés:
A kivonás tapasztalati jellegű és ezért a terméktől vagy a műtrágya egyéb összetevőitől függően előfordulhat, hogy nem mindig veszteségmentes. Különösen egyes mangán-oxidok esetében a kivont mangán mennyisége a minta tényleges mangántartalmánál jelentősen kevesebb lehet. A műtrágya előállítók felelőssége, hogy a megadott érték ténylegesen annak a mennyiségnek feleljen meg, amit a vizsgálat körülményei között ki lehet vonni.
4. VEGYSZEREK
4.1. Hígított sósav (HCl) oldat, kb. 6 mol/l
Keverjünk össze 1 térfogatrész sósavat (d20 =1,18 g/ml) egy térfogatrész vízzel.
4.2. Tömény ammónia oldat (NH4OH, d20 = 0,9 g/ml).
5. FELSZERELÉS
Állítható hőmérsékletű elektromos főzőlap. Megjegyzés
Ha a kivonat bórtartalmát kell meghatározni, ne használjunk boroszilikát üveg felszerelést. Mivel a módszer forralást is tartalmaz, teflon vagy kvarcüveg alkalmazása javasolt. Ha a mosogatáshoz bórátokat tartalmazó mosogatószert használtak, akkor az üvegárut használat előtt gondosan át kell öblíteni.
6. A MINTA ELŐKÉSZÍTÉSE Lásd a B) fejezetet.
7. ELJÁRÁS
7.1. Vizsgálati minta
Mérjünk le a műtrágyából egy 2 és 10 g közé eső mennyiséget a minta adott elemre megadott koncentrációjától függően. Az alábbi táblázatot kell használni ahhoz, hogy megfelelő hígítás után a végső oldat az egyes módszereknek megfelelő mérési tartományon belül essen. A mintákat 1 mg-os pontossággal kell bemérni.
| A mikroelem megadott koncentrációja a műtrágyában (%) | < 0,01 | 0,01— < 5 | >5— 10 |
| A vizsgálati minta tömege (g) | 10 | 5 | 2 |
| Az adott elem tömege a mintában (mg) | 1 | 0,5—250 | 100—200 |
| A kivonat térfogata, V (ml) | 250 | 500 | 500 |
| Az adott elem koncentrációja a kivonatban (mg/l) | 4 | 1—500 | 200—400 |
Vigyük át a mintát egy 250 ml-es főzőpohárba.
7.2. Az oldat elkészítése
Ha szükséges, nedvesítsük meg a mintát egy kis vízzel, majd óvatosan, kis részletekben adjuk hozzá annyiszor 10 ml hígított sósavat (4.1.), ahány g-ot bemértünk a műtrágyából, és ezután adjunk hozzá kb. 50 ml vizet. Fedjük le a főzőpoharat óraüveggel és keverjük össze a tartalmát. A főzőlapon forraljuk fel, majd forraljuk 30 percen keresztül. Időnként megkeverve hagyjuk lehűlni. Vigyük át veszteség nélkül egy 250 vagy 500 ml-es mérőlombikba (lásd a táblázatot). Töltsük jelig vízzel, és gondosan keverjük össze. Szűrjük át egy száraz szűrőn, száraz edénybe. Ontsuk el a szűrlet első részét. A kivonatnak tökéletesen tisztának kell lennie.
Ajánlatos a meghatározásokat késedelem nélkül elvégezni a tiszta szűrlet alikvot részein. Ha nem azonnal végezzük a mérést, akkor a kivonatokat tartalmazó lombikokat dugóval zárjuk le.
Megjegyzés:
Az olyan kivonatoknál, amelyeknél a bórtartalmat kell meghatározni, a következőképpen járjunk el: állítsuk a pH-t koncentrált ammónia oldattal (4.2.) 4 és 6 közé.
8. MEGHATÁROZÁS
Az egyes mikroelemek meghatározását az azokra a mikroelemekre leírt módszerben megadott alikvot részekből kell elvégezni.
Ha szükséges, az I/3. módszerrel távolítsuk el a szerves kelát- vagy komplexképző anyagokat a kivonat egy alikvot részéből. Ha a meghatározást atomabszorpciós spektrometriával végezzük, akkor erre nincs feltétlenül szükség.
I/2. A VÍZBEN OLDHATÓ MIKROELEMEK KIVONÁSA
1. TÁRGY
A módszer eljárást ír le a következő mikroelemek vízben oldható formáinak meghatározásához: bór, kobalt, réz, vas, mangán, molibdén és cink. A cél az, hogy minél kevesebb kivonást kelljen elvégezni és amikor csak lehet, ugyanazt a kivonatot használjuk fel a fent felsorolt mikroelemek koncentrációjának meghatározásához.
2. ALKALMAZÁSI TERÜLET
Ez az eljárás azokra az EGK műtrágyákra vonatkozik, amelyek a 3. számú melléklet 1. függeléke hatálya alá tartoznak és a következő mikroelemek egyikét, vagy közülük többet tartalmaznak: bór, kobalt, réz, vas, mangán, molibdén és cink. Alkalmazható minden mikroelemre, amelynek megadott koncentrációja 10% vagy annál alacsonyabb.
3. A MÓDSZER ELVE
A mikroelemeket úgy vonjuk ki, hogy a műtrágyát 20 °C ± 2 °C hőmérsékleten vízzel rázatjuk.
Megjegyzés
A kivonás tapasztalati jellegű és előfordulhat, hogy nem veszteségmentes.
4. VEGYSZEREK
4.1. Hígított sósav (HCl) oldat, kb. 6 mol/l
Keverjünk össze 1 térfogatrész sósavat (d20 =1,18 g/ml) 1 térfogatrész vízzel.
5. FELSZERELÉS
5.1. Forgó rázógép, 35-40 fordulat/perc sebességre állítva.
5.2. pH-mérő.
Megjegyzés:
Ha a kivonat bórtartalmát kell meghatározni, ne használjunk boroszilikát üveg felszerelést. Teflon vagy kvarcüveg alkalmazása javasolt. Ha a mosogatáskor bórátokat tartalmazó mosogatószert használtak, az üvegárut használat előtt alaposan át kell öblíteni.
6. A MINTA ELŐKÉSZÍTÉSE Lásd a B) fejezetet
7. ELJÁRÁS
7.1. Vizsgálati minta
Mérjünk le a műtrágyából egy 2 és 10 g közé eső mennyiséget a minta adott elemre megadott koncentrációjától függően. Az alábbi táblázatot kell használni ahhoz, hogy a végső oldat megfelelő hígítás után az egyes módszereknek megfelelő mérési tartományon belül essen. A mintákat 1 mg-os pontossággal kell bemérni.
| A mikroelem megadott koncentrációja a műtrágyában (%) | <0,01 | 0,01— < 5 | >5— 10 |
| A vizsgálati minta tömege (g) | 10 | 5 | 2 |
| Az adott elem tömege a mintában (mg) | 1 | 0,5—250 | 100—200 |
| A kivonat térfogata, V (ml) | 250 | 500 | 500 |
| Az adott elem koncentrációja a kivonatban (mg/l) | 4 | 1—500 | 200—400 |
Vigyük a mintát egy 250 vagy 500 ml-es lombikba (a táblázat alapján).
7.2. Az oldat elkészítése
Adjunk kb. 200 ml vizet a 250 ml-es lombikba, vagy 400 ml vizet az 500 ml-es lombikba.
Zárjuk le jól a lombikot. Rázzuk meg erősen kézzel, hogy jól eloszlassuk a mintát, azután helyezzük a lombikot a rázógépre és 30 percig rázassuk.
Vízzel töltsük jelig és gondosan keverjük össze.
7.3. A vizsgálati oldat előkészítése
Szűrjük le késedelem nélkül száraz, tiszta lombikba. A lombikot zárjuk le dugóval. A meghatározást a szűrést követően azonnal végezzük el.
Megjegyzés:
Ha a szűrlet fokozatosan zavarossá válik, végezzünk új kivonást a 7.1. és 7.2. pont szerint, Ve térfogatú lombikot használva. Szűrjük át a kivonatot W térfogatú, előzetesen kiszárított mérőlombikba, amelybe már töltöttünk 5,00 ml hígított sósavat (4.1.). Állítsuk le a szűrést pontosan abban a pillanatban, amikor a folyadékszint eléri a kalibrációs jelet. Gondosan keverjük össze.
Ilyen körülmények között az eredmények kifejezésekor a V értéke:
V=Ve x W/(W-5)
Az eredmények kiszámításakor a hígítás ettől a V értéktől függ.
8. MEGHATÁROZÁS
Az egyes mikroelemek meghatározását az azokra a mikroelemekre leírt módszerben megadott alikvot részekből kell elvégezni.
Ha szükséges, az I/3. módszerrel távolítsuk el a szerves kelát- vagy komplexképző anyagokat a kivonat egy alikvot részéből. Ha a meghatározást atomabszorpciós spektrometriával végezzük, akkor erre nincs feltétlenül szükség.
I/3. SZERVES VEGYÜLETEK ELTÁVOLÍTÁSA MŰTRÁGYA KIVONATOKBÓL
1. TÁRGY
Ez a módszer a szerves vegyületek műtrágya kivonatokból való eltávolítására vonatkozó eljárást írja le.
2. ALKALMAZÁSI TERÜLET
Az eljárást azon műtrágyák az I/1. és az I/2. módszerrel kivont mintáinak elemzésére lehet alkalmazni, amelyekre a 3. számú melléklet 1. függeléke az összes és/vagy a vízben oldható mikroelemek megadását előírja.
Megjegyzés:
Kis mennyiségű szerves anyag jelenléte az atomabszorpciós spektrometriával történő meghatározást általában nem zavarja.
3. A MÓDSZER ELVE
Az alikvot részben jelen lévő szerves vegyületeket hidrogén-peroxiddal oxidáljuk.
4. VEGYSZEREK
4.1. Hígított sósav (HCl) oldat, kb. 0,5 mol/l
Keverjünk össze 1 térfogatrész sósavat (d20 =1,18 g/ml), 20 térfogatrész vízzel.
4.2. Hidrogén-peroxid oldat (30% H2O2 d20 =1,11 g/ml), mikroelemektől mentes.
5. FELSZERELÉS
Termosztálható hőmérsékletű elektromos főzőlap.
6. ELJÁRÁS
Vegyünk 25 ml-t az I/1. vagy az I/2. módszerrel nyert kivonatból és vigyük át egy 100 ml-es főzőpohárba. A 9.2. módszerrel nyert kivonat esetében adjunk hozzá 5 ml hígított sósav oldatot (4.1.). Azután adjunk hozzá 5 ml hidrogén-peroxid oldatot (4.2.). Takarjuk le óraüveggel. Hagyjuk kb. 1 órán keresztül szobahőmérsékleten oxidálódni, majd fokozatosan melegítsük forrásig és fél óráig forraljuk. Ha szükséges, lehűlés után még egyszer adjunk hozzá 5 ml hidrogén-peroxidot. Végül forraljuk fel a hidrogén-peroxid felesleg eltávolításához. Hagyjuk lehűlni és vigyük át veszteség nélkül egy 50 ml-es mérőlombikba, majd töltsük jelig. Szűrjük le, ha szükséges.
Amikor alikvot részeket veszünk ki, illetve a termékben a mikroelem százalékos koncentrációját kiszámítjuk, ezt a hígítást figyelembe kell venni.
I/4. MIKROELEMEK MEGHATÁROZÁSA MŰTRÁGYAKIVONATOKBAN ATOMABSZORPCIÓS SPEKTROMETRIÁVAL. (ÁLTALÁNOS ELJÁRÁS)
1. TÁRGY
A módszer bizonyos mikroelemek műtrágyakivonatokban jelen lévő mennyiségének atomabszorpciós spektrometriás módszerrel történő meghatározásához ad meg általános eljárást.
2. ALKALMAZÁSI TERÜLET
Az eljárást olyan műtrágyák az I/1. és az I/2. módszerrel kivont mintáinak vizsgálatára lehet alkalmazni, amelyekre a 3. számú melléklet 1. függeléke az összes és/vagy a vízben oldható elemek megadását előírja.
Ennek az eljárásnak az alkalmazása a különböző mikroelemekre az egyes elemek esetében meghatározott módszerek leírásában van részletezve.
Megjegyzés:
Kis mennyiségű szerves anyag jelenléte a legtöbb esetben nem zavarja az atomabszorpciós spektrometriával történő meghatározást.
3. A MÓDSZER ELVE
Miután a kivonatot szükség szerint kezeltük a zavaró kémiai anyagok mennyiségének csökkentésére vagy eltávolítására, a kivonatot meghígítjuk úgy, hogy a koncentráció a spektrofotométer optimális mérési tartományába essen a mikroelem meghatározásához alkalmas hullámhossznál.
4. VEGYSZEREK
4.1. Hígított sósav (HCl) oldat, kb. 6 mol/l
Keverjünk össze 1 térfogatrész sósavat (r = 1,18 g/ml) 1 térfogatrész vízzel.
4.2. Hígított sósav (HCI) oldat, kb. 0,5 mol/l
Keverjünk össze 1 térfogatrész sósavat (d20 =1,18 g/ml) 20 térfogatrész vízzel.
4.3. Lantánsó oldatok (10 g La/l).
Ezt a vegyszert a kobalt, vas, mangán és cink meghatározásához használjuk. Az alábbi módszerek egyikének alkalmazásával készíthető el:
a) Lantán-oxidot sósavban (4.1.) feloldunk. Tegyünk 11,73 g lantán-oxidot (La2O3) 150 ml vízzel egy literes mérőlombikba és adjunk hozzá 120 ml 6 mol/l sósavat (4.1.). Hagyjuk feloldódni, majd vízzel egészítsük ki egy literre és gondosan keverjük össze. Ez az oldat kb. 0,5 mol/l töménységű, sósavban oldva; vagy
b) Lantán-klorid, -szulfát vagy -nitrát oldattal. Oldjunk fel 26,7 g lantán-klorid-heptahidrátot (LaCl3.7H2O) vagy 31,2 g lantán-nitrát-hexahidrátot (La(NO3)3.6H2O) vagy 26,2 g lantán-szulfát-nonahidrátot (La2(SO4)3.9H2O) 150 ml vízben, azután adjunk hozzá 85 ml 6 mol/l sósavat (4.1.). Hagyjuk feloldódni, majd vízzel egészítsük ki térfogatát 1 literre. Gondosan keverjük össze. Ez az oldat kb. 0,5 mol/l töménységű, sósavban oldva.
4.4. Kalibrációs oldatok
Ezek készítésére vonatkozóan lásd az egyes mikroelemekre megadott egyedi vizsgálati módszereket.
5. FELSZERELÉS
Atomabszorpciós spektrométer a meghatározandó elemekre jellemző sugárzás kibocsátására alkalmas fényforrásokkal.
Az analitikusnak követnie kell a műszer használati utasítását és legyen járatos a műszer kezelésében. A műszer legyen alkalmas a háttérkorrekcióra, hogy azt szükség esetén (Co és Zn meghatározásokhoz) alkalmazni lehessen. A vizsgálatokhoz levegőt és acetilén gázt kell használni.
6. A VIZSGÁLATI OLDAT KÉSZÍTÉSE
6.1. A meghatározandó mikroelemek kivont oldatának készítése. Lásd az I/1. és/vagy I/2., illetve értelemszerűen a I/3. fejezeteket
6.2. A vizsgálati oldat kezelése
Hígítsuk meg az I/1., az I/2. vagy az I/3. módszerrel kapott kivonat alikvot részét vízzel és/vagy sósav oldattal (4.1.) vagy (4.2.) úgy, hogy a végső mérési oldatban a meghatározandó elem koncentrációja megfeleljen a használt kalibrációs tartománynak (7.2.) és a sósav koncentrációja legyen legalább 0,5 mol/l, és ne legyen több, mint 2,5 mol/l. Ehhez egy vagy több egymást követő hígítás elvégzésére van szükség.
A kivonat végső hígításával nyert oldat egy alikvot részét vegyük ki, a térfogatát nevezzük (a) ml-nek és vigyük át egy 100 ml-es mérőlombikba. Ha kobalt-, vas-, mangán- vagy cinktartalmat határozunk meg, akkor adjunk hozzá 10 ml lantánsó oldatot (4.3.). Töltsünk jelig a 0,5 mol/l sósavoldattal (4.2.) és gondosan keverjük össze. Ez a végső mérési oldat. A hígítási tényezőt nevezzük D-nek.
7. ELJÁRÁS
7.1. Vakoldat készítése
Készítsünk vakoldatot úgy, hogy a kivonástól kezdve megismételjük az egész eljárást, de nem adjuk hozzá a bemért műtrágyamintát.
7.2. Kalibrációs oldatok készítése
Az egyes mikroelemekre egyedileg megadott módszerek szerint készített kalibrációs munkaoldatok felhasználásával 100 ml-es mérőlombikokban készítsünk egy legalább 5 kalibrációs oldatból álló, növekvő koncentrációjú sorozatot, a spektrométer optimális méréstartományán belül. Ha szükséges, állítsuk be a sósav oldat koncentrációját úgy, hogy az a hígított vizsgálati oldatok (6.2.) sósav koncentrációjához a lehető legközelebb essen. Kobalt, vas, mangán vagy cink meghatározásakor adjunk az oldatokhoz 10 ml-t ugyanabból a lantánsó oldatból (4.3.), amit a 6.2. pontnál használtunk. Töltsük jelig a 0,5 mol/l sósav oldattal (4.2.) és keverjük alaposan össze.
7.3. Meghatározás
Készítsük elő a spektrofotométert (5.) a mérésekhez, és állítsuk be az érintett mikroelemhez a módszer által megadott hullámhosszra.
Poriasszuk be háromszor egymást követően a kalibrációs oldatokat (7.2.), a vizsgálati oldatot (6.2.) és a vakoldatot (7.1.), miközben feljegyzünk minden eredményt és kiöblítjük a készüléket az egyes porlasztások között desztillált vízzel.
Vegyük fel a kalibrációs görbét úgy, hogy minden kalibrációs oldatra (7.2.) nézve az átlagos spektrométer leolvasásokat tüntetjük fel az ordinátán és az elem μg/ml-ben kifejezett megfelelő koncentrációit pedig az abszcisszán.
Ebből a görbéből határozzuk meg az érintett mikroelem koncentrációját a minta oldatban (xS), (6.2.) és a vakoldatban (xb), (7.1.), a koncentrációkat μg/ml-ben kifejezve.
8. AZ EREDMÉNYEK KIFEJEZÉSE
A mikroelem %-os (m/m)koncentrációja (E) a műtrágyában egyenlő:
E(%)=[(xs-xb)xVxD]/(Mx104)
ha a 9.3. módszert használtuk:
E(%)=[(xs-xh)xVx2D]/(Mx104)
ahol:
E = a meghatározott mikroelem mennyisége, a műtrágya százalékában kifejezve,
xs = a vizsgálati oldat (6.2.) koncentrációja μg/ml-ben,
Xb = a vakoldat (7.1.) koncentrációja μg/ml-ben,
V = az I/1. vagy az I/2. módszerrel nyert kivonat térfogata ml-ben,
D = a végrehajtott hígításnak (6.2.) megfelelő tényező,
M = az I/1. vagy I/2. módszer szerint bemért minta tömege g-ban.
A D hígítási tényező számítása:
Ha az (a1), (a2), (a3), ... (ai) és (a) az alikvot részek és (vl), (v2), (v3), ... (vi) és (100) a hígításaiknak rendre megfelelő térfogatok, ml-ben kifejezve, akkor a D hígítási tényező:
D=(v1/a1)x(v2/a2)x(v3/a3)x.x.x.x.(vi/ai)x(100/a).
I/5. BÓR MEGHATÁROZÁSA MŰTRÁGYA-KIVONATOKBAN AZOMETIN-H SPEKTROFOTOMETRIÁS MÓDSZERREL
1. TÁRGY
A módszer a bór műtrágya-kivonatokban történő meghatározásának eljárását írja le.
2. ALKALMAZÁSI TERÜLET
Az eljárást olyan műtrágyák az I/1. és az I/2. módszerrel kivont mintáinak vizsgálatára lehet alkalmazni, amelyekre a 3. számú melléklet 1. függeléke az összes és/vagy a vízben oldható bórtartalom megadását előírja.
3. A MÓDSZER ELVE
A borátionok azometin-H oldatban sárga színű komplexet képeznek, amelynek koncentrációját molekuláris abszorpciós spektrometriával 410 nm-en határozzuk meg. A zavaró ionokat EDTA-val fedjük el.
4. VEGYSZEREK
4.1. EDTA puffer oldat
300 ml vizet tartalmazó 500 ml-es mérőlombikba tegyünk:
- 75 g ammónium-acetátot (NH4OOCCH3);
- 10 g dinátrium-etilén-diamin-tetraacetátot (Na2EDTA);
- 40 ml ecetsavat (CH3COOH, d20= 1,05 g/ml).
Töltsük jelig vízzel és gondosan keverjük össze. Az oldat pH-ját üvegelektróddal ellenőrizzük, az 4,8 + 0,1 legyen.
4.2. Azometin-H oldat
200 ml-es mérőlombikba tegyünk
- 10 ml puffer oldatot (4.1.);
- 400 mg azometin-H-t (C17H12NNaO8S2);
- 2 g aszkorbinsavat (C6H8O6).
Töltsük jelig és gondosan keverjük össze. Ebből a reagensből ne készítsünk nagy mennyiséget, mert csak pár napig stabil.
4.3. Bór kalibráló oldatok
4.3.1. Bór törzsoldat (l00 μg/ml)
Oldjunk fel 0,5719 g bórsavat (H3BO3) vízben, egy 1000 ml-es mérőlombikban. Töltsük jelig vízzel és gondosan keverjük össze. Töltsük műanyag edénybe és tároljuk hűtőszekrényben.
4.3.2. Bór munkaoldat (10 μg/ml)
Tegyünk 50 ml törzsoldatot (4.3.1.) egy 500 ml-es mérőlombikba. Töltsük jelig vízzel és gondosan keverjük össze.
5. FELSZERELÉS
Spektrométer molekuláris abszorpciós mérésekhez, 10 mm optikai úthosszú küvettákkal, 410 nm-re állítva.
6. AZ ELEMZENDŐ OLDAT ELŐKÉSZÍTÉSE
6.1. A bór oldat készítése
Lásd az I/1. és/vagy az I/2., illetve értelemszerűen az I/3. fejezetek.
6.2. A vizsgálati oldat előkészítése
A kivonat (6.1.) alikvot részét hígítsuk meg úgy, hogy a 7.2. pontban megadott koncentrációt kapjunk. Két egymást követő hígítás lehet szükséges.
Nevezzük a hígítási tényezőt D-nek.
6.3. A korrekciós oldat elkészítése
Ha a vizsgálati oldat (6.2.) színes, készítsünk megfelelő korrekciós oldatot olymódon, hogy műanyag edénybe teszünk 5 ml vizsgálati oldatot (6.2.), 5 ml EDTA puffer oldatot (4.1.) és 5 ml vizet, majd jól elkeverjük.
7. ELJÁRÁS
7.1. A vakoldat elkészítése
Készítsünk vakoldatot úgy, hogy az egész eljárást megismételjük a kivonástól kezdve, de nem mérünk be műtrágya mintát.
7.2. A kalibrációs oldatok elkészítése
Tegyünk 0, 5, 10, 15, 20 és 25 ml kalibrációs munkaoldatot (4.3.2.) 100 ml-es mérőlombikokba. Töltsük jelig vízzel és gondosan keverjük össze. Ezek az oldatok 0 és 2,5 μg/ml közé eső bórt tartalmaznak.
7.3. Színelőhívás
Tegyünk a kalibrációs oldatokból (7.2.), a vizsgálati oldatokból (6.2.) és a vakoldatból (7.1.) 5-5 ml-t műanyag edényekbe. Adjunk hozzájuk 5 ml EDTA puffer oldatot (4.1.). Adjunk hozzájuk 5 ml azometin-H oldatot (4.2.). Gondosan keverjük össze és 2,5-3 órára sötét helyre állítva hagyjuk a színt kialakulni.
7.4. Meghatározás
Mérjük meg a 7.3. pont szerint kapott oldatok, és ha szükség van rá, a korrekciós oldat (6.3.) abszorbanciáját 410 nm-en, vízzel szemben. Minden leolvasás előtt vízzel öblítsük át a küvettákat.
8. AZ EREDMÉNYEK KIFEJEZÉSE
Ábrázoljuk a kalibrációs görbét úgy, hogy a kalibrációs oldatok (7.2.) koncentrációját az abszcisszára, a spektrofotométerről leolvasott abszorbanciát (7.4.) az ordinátára mérjük fel.
Olvassuk le a kalibrációs görbéről a bórkoncentrációt a vakpróbában (7.1.) a bórkoncentrációt a vizsgálati oldatban (6.2.) és ha a vizsgálati oldatunk színes volt, akkor a vizsgálati oldat korrigált koncentrációját is. Az utóbbi kiszámításához vonjuk ki a korrekciós oldat abszorbanciáját a vizsgálati oldat abszorbanciájából (6.2.) és így határozzuk meg a vizsgálati oldat korrigált koncentrációját. Jegyezzük fel a vizsgálati oldat korrigált (6.2.) vagy korrekció nélküli, X(xS) koncentrációját és a vakoldat (xb) koncentrációját.
A műtrágya tömegszázalékban kifejezett bórtartalmát az alábbi képlet adja meg:
B%=[(xs-xb)xVxD]/(M x104)
Ha az I/3. módszert használtuk:
B%=[(xs-xh)xVx2D]/(M x104)
ahol:
B = a bórtartalom a műtrágya százalékában kifejezve,
xs = a vizsgálati oldat (6.2.) koncentrációja (μg/ml), korrekcióval vagy anélkül,
xb = a vakoldat (7.1.) koncentrációja (μg/ml),
V = az I/1. vagy az I/2. módszer szerint nyert kivonat térfogata,
D = a 6.2. pont szerint végzett hígítás tényezője,
M = az I/1. vagy az I/2. módszer szerint bemért minta tömege g-ban.
A D hígítási tényező kiszámítása: ha (a1) és (a2) egymást követő alikvot részek és (v1) és (v2) a megfelelő higítások szerinti térfogatok, a D higítási tényező a következő:
D=(v1/a1)x(v2/a2).
I/6. KOBALT MEGHATÁROZÁSA MŰTRÁGYÁKBAN ATOMABSZORPCIÓS SPEKTROMETRIÁVAL
1. TÁRGY
Ez a módszer a kobalt műtrágyakivonatokban történő meghatározásának eljárását írja le.
2. ALKALMAZÁSI TERÜLET
Ezt az eljárást azon műtrágyák az I/1. és az I/2. módszerrel kivont mintáinak vizsgálatára lehet alkalmazni, amelyekre a 3. számú melléklet 1. függeléke az összes és/vagy a vízben oldható kobalttartalom megadását előírja.
3. A MÓDSZER ELVE
A kivonatok megfelelő kezelése és hígítása után a kobalttartalmat atomabszorpciós spektrometriával határozzuk meg.
4. VEGYSZEREK
4.1. Sósav oldat, kb. 6 mol/l Lásd az I/4. fejezet (4.1.).
4.2. Sósav oldat, kb. 0,5 mol/l Lásd az I/4. fejezet (4.2.).
4.3. Lantánsó oldatok (10 g La/l) Lásd az I/4. fejezet (4.3.).
4.4. Kobalt kalibrációs oldatok
4.4.1. Kobalt törzsoldat (l000 μg/ml
250 ml-es főzőpohárba mérjünk be 1 g kobaltot 0,1 mg-os pontossággal, adjunk hozzá 25 ml 6 mol/l sósavat (4.1.) és melegítsük főzőlapon, amíg a kobalt teljesen fel nem oldódik. Amikor lehűlt, vigyük át veszteség nélkül egy 1000 ml-es mérőlombikba. Töltsük jelig vízzel és gondosan keverjük össze.
4.4.2. Kobalt munkaoldat (100 μg/ml).
Vigyünk 10 ml törzsoldatot (4.4.1.) egy 100 ml-es mérőlombikba. Töltsük jelig 0,5 mol/l sósav oldattal (4.2.) és gondosan keverjük össze.
5. FELSZERELÉS
Atomabszorpciós spektrométer: lásd az I/4. módszert (5.). A műszernek a kobalthoz megfelelő sugárforrással (240,7 nm) kell rendelkeznie. A spektrométernek alkalmasnak kell lennie háttérkorrekció elvégzésére.
6. A VIZSGÁLATI OLDAT ELŐKÉSZÍTÉSE
6.1. Kivont kobalt-oldat
Lásd az I/1. és/vagy az I/2., illetve értelemszerűen az I/3. fejezeteket.
6.2. A vizsgálati oldat előkészítése
Lásd az I/4. fejezet (6.2.). A vizsgálati oldat 10% (v/v) lantánsó oldatot (4.3.) tartalmazzon.
7. ELJÁRÁS
7.1. A vakoldat készítése
Lásd az I/4. fejezet (7.1.). A vakoldat 10% (v/v) lantánsó oldatot (4.3.) tartalmazzon.
7.2. A kalibrációs oldatok elkészítése Lásd az I/4. fejezet (7.2.).
A kobalt számára optimális 0-5 μg/ml meghatározási tartomány beállításához tegyünk rendre 0, 0,5, l, 2, 3, 4 és 5 ml-t a munkaoldatból (4.4.2.) 100 ml-es mérőlombikokba. Ha szükséges, a sósav koncentrációt állítsuk be úgy, hogy a vizsgálati oldatok sósav-koncentrációjához a lehető legközelebb essen. Adjunk mindegyik mérőlombikba 10 ml-t a 6.2. pontban alkalmazott lantánsó oldatból. Töltsük fel 100 ml-re 0,5 M sósav oldattal (4.2.) és gondosan keverjük össze. Ezek az oldatok rendre 0, 0,5, l, 2, 3, 4 és 5 μg/ml kobaltot tartalmaznak.
7.3. Meghatározás
Lásd az I/4. fejezet (7.3.). Készítsük elő a spektrométert (5.) 240,7 nm-en történő mérésre.
8. AZ EREDMÉNYEK KIFEJEZÉSE
Lásd az 1.4. fejezet (8.).
A műtrágya tömegszázalékban kifejezett kobalttartalmát az alábbi egyenlet adja meg:
Co% = [(xs-xh)xVxD]/(Mx104
Ha az I/3. fejezetet használtuk:
Co% = [(xs-xb)xVx2D]/(Mxl04
ahol:
Co = a műtrágya százalékban kifejezett kobalttartalma,
xs = a vizsgálati oldat (6.2.) koncentrációja (μg/ml),
xb = a vakoldat (7.1.) koncentrációja (μg/ml),
V = az I/1. vagy az I/2. fejezet szerint nyert kivonat térfogata (ml),
D = a 6.2. pont szerint végzett hígítás tényezője,
M = az I/1. fejezet vagy az I/2. fejezet szerint bemért minta tömege (g).
A D hígítási tényező kiszámítása: Ha az (a1), (a2), (a3), ... (ai) és (a) az alikvot részek, (v1), (v2), (v3), ... (vi) és (100) a hígításoknak rendre megfelelő térfogatok, akkor a D hígítási tényező:
D = (v1/a1) x (v2/a2) x (v3/a3) x.x.x.x. (vi/ai) x (100/a).
I/7. RÉZ MEGHATÁROZÁSA MŰTRÁGYAKIVONATOKBAN ATOMABSZORPCIÓS SPEKTROMETRIÁVAL
1. TÁRGY
Ez a módszer a réz műtrágyakivonatokban való meghatározásának eljárását írja le.
2. ALKALMAZÁSI TERÜLET
Ezt az eljárást azon műtrágyák az I/1. és az I/2. módszerrel kivont mintáinak vizsgálatára lehet alkalmazni, amelyekre a 3. számú melléklet 1. függeléke az összes és/vagy a vízben oldható réztartalom megadását előírja.
3. A MÓDSZER ELVE
A kivonatok megfelelő kezelését és hígítását követően a réztartalmat atomabszorpciós spektrometriával határozzuk meg.
4. VEGYSZEREK
4.1. Sósav oldat, kb. 6 mol/l Lásd az I/4. fejezet (4.1.).
4.2. Sósav oldat, kb. 0,5 mol/l Lásd az I/4. fejezet (4.2.).
4.3. Hidrogén-peroxid oldat (30% H2O2, d20 = 1,11 g/ml), mikroelemektől mentes.
4.4. Réz kalibrációs oldatok
4.4.1. Réz törzsoldat (1000 μg/ml)
Mérjünk be 250 ml-es főzőpohárba 1 g rezet 0,1 mg-os pontossággal, és adjunk hozzá 25 ml 6 mol/l sósavat (4.1.), majd adjunk még hozzá 5 ml hidrogén-peroxid oldatot (4.3.) végül melegítsük főzőlapon, amíg a réz teljesen fel nem oldódik. Vigyük át veszteség nélkül egy 1000 ml-es mérőlombikba. Töltsük jelig vízzel és gondosan keverjük össze.
4.4.2. Réz munkaoldat (100 μg/ml)
Helyezzünk 20 ml törzsoldatot (4.4.1.) egy 200 ml-es mérőlombikba. Töltsük fel jelig 0,5 mol/l sósav oldattal (4.2.) és gondosan keverjük össze.
5. FELSZERELÉS
Atomabszorpciós spektrométer: lásd az I/4. módszert (5.). A műszer legyen felszerelve rézre jellemző sugarak (324,8 nm) kibocsátására alkalmas fényforrással.
6. A VIZSGÁLATI OLDAT ELŐKÉSZÍTÉSE
6.1. Kivont réz-oldat
Lásdaz I/1. és/vagy az I/2., illetve értelemszerűen az I/3. fejezetet.
6.2. A vizsgálati oldat előkészítése Lásd az I/4. fejezetet (6.2.).
7. ELJÁRÁS
7.1. Vakoldat készítése Lásd az I/4. fejezetet (7.1.).
7.2. A kalibrációs oldatok elkészítése Lásd az I/4. fejezetet (7.2.).
A réz számára optimális 0-5 μg/ml meghatározási tartomány beállításához vigyünk át rendre 0, 0,5, l, 2, 3, 4 és 5 ml munkaoldatot (4.4.2.) 100 ml-es mérőlombikokba. Ha szükséges, a sósav-koncentrációt állítsuk be úgy, hogy az a vizsgálati oldat (6.2.) sósav-koncentrációjához a lehető legközelebb essen. Töltsük fel 100 ml-re 0,5 mol/l sósav oldattal (4.2.) és gondosan keverjük össze. Ezek az oldatok rendre 0, 0,5, l, 2, 3, 4 és 5 μg/ml rezet tartalmaznak.
7.3. Meghatározás
Lásd az I/4. fejezetet (7.3.). Készítsük fel a spektrométert (5.) 324,8 nm-en történő mérésre.
8. AZ EREDMÉNYEK KIFEJEZÉSE
Lásd az I/4. fejezetet (8.).
A műtrágya tömegszázalékban kifejezett réztartalmát az alábbi egyenlet adja meg:
Cu% = [(xs-x")xVxD]/(Mx104
Ha az I/3. fejezetet használtuk:
Cu% = [(xs-xb)xVx2D]/(Mx104
ahol:
Cu = a műtrágya százalékban kifejezett réztartalma,
xs = a vizsgálati oldat koncentrációja (μg/ml) (6.2.),
xb = a vakoldat koncentrációja (μg/ml) (7.1.),
V = az I/1. vagy az I/2. fejezet szerint nyert kivonat térfogata (ml),
D = a 6.2. szerint végzett hígítás tényezője,
M = az I/1. vagy az I/2. fejezet szerint bemért minta tömege (g).
A D hígítási tényező kiszámítása: Ha az (a1), (a2), (a3), ... (ai) és (a) az alikvot részek, (v1), (v2), (v3), ... (vi) és (100) a hígításoknak rendre megfelelő térfogatok, akkor a D hígítási tényező:
D = (v1/a1) x (v2/a2) x (v3/a3) x.x.x.x. (vi/ai) x (100/a).
I/8. VAS MEGHATÁROZÁSA MŰTRÁGYAKIVONATOKBAN ATOMABSZORPCIÓS SPEKTROMETRIÁVAL
1. TÁRGY
A módszer a vas műtrágyakivonatokban való meghatározásának eljárását írja le.
2. ALKALMAZÁSI TERÜLET
Az eljárást azon műtrágyák az I/1. és I/2. fejezet szerint kivont mintáinak vizsgálatára lehet alkalmazni, amelyekre a 3. számú melléklet 1. függeléke az összes és/vagy a vízben oldható vastartalom megadását előírja.
3. A MÓDSZER ELVE
A kivonatok megfelelő kezelését és hígítását követően a vastartalmat atomabszorpciós spektrometriával határozzuk meg.
4. VEGYSZEREK
4.1. Sósav oldat, kb. 6 mol/l Lásd az I/4. fejezetet (4. L).
4.2. Sósav oldat, kb. 0,5 mol/l Lásd az I/4. módszert (4.2.).
4.3. Hidrogén-peroxid oldat (30% H2O2, d20 =1,11 g/ml), mikroelemektől mentes.
4.4. Lantánsó oldatok (10 g La literénként) Lásd az I/4. módszert (4.3.).
4.5. Vas kalibrációs oldatok
4.5.1. Vas törzsoldat (1000 μg/ml)
Mérjünk be 500 ml-es főzőpohárba 1 g tiszta vasdrótot 0,1 mg-os pontossággal, adjunk hozzá 200 ml 6 mol/l sósavat (4.1.) és 15 ml hidrogén-peroxid oldatot (4.3.). Melegítsük főzőlapon, amíg a vas teljesen fel nem oldódik. Amikor lehűlt, vigyük át veszteség nélkül egy 1000 ml-es mérőlombikba. Töltsük jelig vízzel és gondosan keverjük össze.
4.5.2. Vas munkaoldat (100 μg/ml)
Helyezzünk 20 ml törzsoldatot (4.5.1.) egy 200 ml-es mérőlombikba. Töltsük fel jelig 0,5 mol/l sósav oldattal (4.2.) és gondosan keverjük össze.
5. FELSZERELÉS
Atomabszorpciós spektrométer: lásd az I/4. módszert (5.). A műszer legyen felszerelve vasra jellemző sugarak (248,3 nm) kibocsátására alkalmas fényforrással.
6. A VIZSGÁLATI OLDAT ELŐKÉSZÍTÉSE
6.1. Kivont vas-oldat
Lásd az I/1. és/vagy az I/2., illetve értelemszerűen az I/3. fejezetet.
6.2. A mintaoldat előkészítése
Lásd az I/4. fejezetet (6.2.). A vizsgálati oldatnak 10 térfogat% lantánsó oldatot kell tartalmaznia.
7. ELJÁRÁS
7.1. Vakoldat készítése
Lásd az I/4. fejezetet (7.1.). A vizsgálati oldatnak 10 térfogat% a (6.2.) pontban használt lantánsó oldatot kell tartalmaznia.
7.2. A kalibrációs oldatok elkészítése Lásd az I/4. fejezetet (7.2.).
A vas számára optimális 0-10 (μg/ml meghatározási tartomány beállításához tegyünk rendre 0, 2, 4, 6, 8 és 10 ml munkaoldatot (4.5.2.) 100 ml-es mérőlombikokba. Ha szükséges, a sósav-koncentrációt állítsuk be úgy, hogy az a vizsgálati oldat sósav-koncentrációjához a lehető legközelebb essen. Tegyünk mindegyik mérőlombikba 10 ml-t a 6.2. pontban használt lantánsó oldatból. Töltsük fel 0,5 mol/l sósav oldattal (4.2.) és gondosan keverjük össze. Ezek az oldatok rendre 0, 2, 4, 6, 8 és 10 μg/ml vasat tartalmaznak.
7.3. Meghatározás
Lásd az I/4. fejezetet (7.3.). Készítsük fel a spektrofotométert (5.) 248,3 nm-en történő mérésre.
8. AZ EREDMÉNYEK KIFEJEZÉSE
Lásd az I/4. fejezetet (8.).
A műtrágya tömegszázalékban kifejezett vastartalmát az alábbi egyenlet adja meg:
Fe% = [(xs-xb)xVxD]/(Mx104
Ha az I/3. fejezetet használtuk:
ahol:
Fe = a műtrágya százalékban kifejezett vastartalma,
xs = a vizsgálati oldat koncentrációja (μg/ml) (6.2.),
xb = a vakoldat koncentrációja (μg/ml) (7. 1.),
V = az I/1. vagy az I/2. fejezet szerint nyert kivonat térfogata (ml),
D = a 6.2. pont szerint végzett hígítás tényezője,
M = az I/1. vagy a I/2. módszer szerint bemért minta tömege (g).
A D hígítási tényező kiszámítása: Ha az (a1), (a2), (a3) ... (ai) és (a) az alikvot részek, (v1), (v2), (v3), ... (vi) és (100) a hígításoknak rendre megfelelő térfogatok, akkor a D hígítási tényező:
D = (v1/a1)x (v2/a2 )x (v3/a3)x.x.x.x. (vi/ai) x (100/a).
I/9. MÓDSZER MANGÁN MEGHATÁROZÁSA MŰTRÁGYAKIVONATOKBAN ATOMABSZORPCIÓS SPEKTROMETRIÁVAL
1. TÁRGY
A módszer a mangán műtrágyakivonatokban való meghatározásának eljárását írja le.
2. ALKALMAZÁSI TERÜLET
Az eljárást azon műtrágyák az I/1. és az I/2. módszerrel kivont mintáinak vizsgálatára lehet alkalmazni, amelyekre a 3. számú melléklet 1. függeléke az összes és/vagy a vízben oldható mangántartalom megadását előírja.
3. A MÓDSZER ELVE
A kivonatok megfelelő kezelését és hígítását követően a mangántartalmat atomabszorpciós spektrometriával határozzuk meg.
4. VEGYSZEREK
4.1. Sósav oldat, kb. 6 mol/l Lásd az I/4. fejezetet (4.1.).
4.2. Sósav oldat, kb. 0,5 mol/l Lásd az I/4. fejezetet (4.2.).
4.3. Lantánsó oldatok (10 g La literenként) Lásd az I/4. fejezetet (4.3.).
4.4. Mangán kalibrációs oldatok
4.4.1. Mangán törzsoldat (1000 μg/ml)
Mérjünk be 250 ml-es főzőpohárba 1 g mangánt 0,1 mg-os pontossággal, adjunk hozzá 25 ml 6 mol/l sósav oldatot (4.3.). Melegítsük főzőlapon, amíg a mangán teljesen fel nem oldódik. Amikor lehűlt, vigyük át veszteség nélkül egy 1000 ml-es mérőlombikba. Töltsük jelig vízzel és gondosan keverjük össze.
4.4.2. Mangán munkaoldat (100 μg/ml)
Hígítsunk fel 20 ml törzsoldatot (4.4.1.) 0,5 M sósavban (4.2.) egy 200 ml-es mérőlombikban. Töltsük fel a 0,5 mol/l sósav oldattal (4.2.) és gondosan keverjük össze.
5. FELSZERELÉS
Atomabszorpciós spektrométer: lásd az I/4. módszert (5.). A műszer legyen felszerelve mangánnak megfelelő sugarak (279,6 nm) kibocsátására alkalmas fényforrással.
6. A VIZSGÁLATI OLDAT ELŐKÉSZÍTÉSE
6.1. Kivont mangán-oldat
Lásd az I/1. és/vagy az I/2. fejezetet, illetve értelemszerűen az I/3 fejezetet.
6.2. A vizsgálati oldat előkészítése
Lásd az I/4. fejezetet (6.2.). A vizsgálati oldat 10% (v/v) lantánsó oldatot tartalmazzon (4.3).
7. ELJÁRÁS
7.1. Vakoldat készítése
Lásd az I/4. fejezetet (7.1.). A vakoldat 10% (v/v), a 6.2.-ben alkalmazott lantánsót tartalmazzon.
7.2. A kalibrációs oldatok elkészítése Lásd az I/4. fejezetet (7.2.).
A mangán számára optimális 0-5 μg/ml meghatározási tartomány beállításához tegyünk rendre 0, 0,5, l, 2, 3, 4 és 5 ml munkaoldatot (4.4.2.) 100 ml-es mérőlombikokba. Ha szükséges, akkor a sósav koncentrációt állítsuk be úgy, hogy a vizsgálati oldatok sósav-koncentrációjához a lehető legközelebb essen. Adjunk mindegyik mérőlombikba 10 ml-t a 6.2. pontban használt lantánsó oldatból. Töltsük fel 100 ml-re a 0,5 M sósav oldattal (4.2.) és gondosan keverjük össze. Ezek az oldatok rendre 0, 0,5, 1, 2, 3, 4 és 5 μg/ml mangánt tartalmaznak.
7.3. Meghatározás
Lásd az I/4. fejezetet (7.3). Készítsük fel a spektrométert (5.) 279,6 nm-en történő mérésre.
8. AZ EREDMÉNYEK KIFEJEZÉSE
Lásd az I/4. fejezetet (8.).
A műtrágya tömegszázalékban kifejezett mangántartalmát az alábbi egyenlet adja meg:
Mn% = [(xs-xb)xVxD]/(Mx104
Ha az I/3. fejezetet használtuk:
Mn% = [(xs-x")xVx2D]/(Mx104
ahol:
Mn = a műtrágya százalékban kifejezett mangántartalma,
xs = a vizsgálati oldat koncentrációja (μg/ml) (6.2.),
xh = a vakoldat koncentrációja (μg/ml) (7.1.),
V = az I/1. vagy az I/2. fejezet szerint nyert kivonat térfogata (ml),
D = a 6.2. pont szerint végzett hígítás tényezője,
M = az I/1. vagy az I/2. fejezet szerint bemért minta tömege (g).
A D hígítási tényező kiszámítása: ha az (a1), (a2), (a3),... (ai) és (a) az alikvot részek, és (v1), (v2), (v3),... (vi) és (100) a hígításoknak rendre megfelelő térfogatok, akkor a D hígítási tényező:
D = (v1/a1) x (v2/a2) x (v3/a3) x.x.x.x. (vi/ai) X (100/a).
I/10. MOLIBDÉN MEGHATÁROZÁSA MŰTRÁGYAKIVONATOKBAN AMMÓNIUM-TIOCIANÁTTAL KÉPZETT KOMPLEXÉNEK SPEKTROFOTOMETRIÁS MÉRÉSÉVEL
1. TÁRGY
A módszer a molibdén műtrágyakivonatokban való meghatározásának eljárását írja le.
2. ALKALMAZÁSI TERÜLET
Az eljárást azon műtrágyák az I/1. és az I/2. módszerrel kivont mintáinak vizsgálatára lehet alkalmazni, amelyekre a 3. számú melléklet 1. függeléke az összes és/vagy a vízben oldható molibdén-tartalom megadását előírja.
3. A MÓDSZER ELVE
A molibdén (V) savas közegben SCN- ionokkal [MoO(SCN)5] - - komplexet képez.
A komplexet n-butil-acetáttal vonjuk ki. A zavaró ionok, így a vas-ionok a vizes fázisban maradnak. A sárga-narancs színt molekuláris abszorpciós spektrometriával 470 nm-en mérjük.
4. VEGYSZEREK
4.1. Hígított sósav (HCl) oldat, kb. 6 mol/l Lásd az I/4. fejezetet (4.1.).
4.2. Réz oldat (70 mg/l) 1 ,5 mol/l sósav oldatban
Mérjünk be 275 mg rézszulfátot (CuSO4.5H2O) 0,1 mg pontossággal és oldjuk fel 250 ml 6 mol/l sósav oldatban (4.1.) egy 1000 ml-es mérőlombikban. Töltsük jelig vízzel és gondosan rázzuk össze.
4.3. Aszkorbinsav oldat (50 g/l)
Oldjunk fel 50 g aszkorbinsavat (C6H8O6) vízben egy 1000 ml-es mérőlombikban. Töltsük jelig vízzel, rázzuk össze gondosan és tartsuk hűtőszekrényben.
4.4. n-butil-acetát
4.5. Ammónium-tiocianát oldat, 0,2 mol/l
Oldjunk fel 15,224 g NH4SCN-ot vízben egy 1000 ml-es mérőlombikban. Töltsük fel jelig vízzel, gondosan rázzuk össze és tartsuk sötét üvegben.
4.6. Ón-klorid oldat (50 g/l) 2 mol/l sósavban
Ennek az oldatnak tökéletesen tisztának kell lennie és közvetlenül a felhasználás előtt kell elkészíteni. Használjunk nagyon tiszta ón-kloridot, másképpen az oldat nem lesz tiszta.
100 ml oldat készítéséhez oldjunk fel 5 g SnCl2.2 H2O-ot 35 ml 6 mol/l sósavban (4.1.). Adjunk hozzá 10 ml-t a réz oldatból (4.2.). Vízzel töltsük jelig és gondosan keverjük össze.
4.7. Molibdén kalibrációs oldatok
4.7.1. Molibdén törzsoldat (500 μg/ml)
Tegyünk 0,1 mg pontossággal bemért 0,920 g ammónium-molibdenátot
[(NH4)6Mo7O24.4H2O] egy 1000 ml-es mérőlombikba és oldjuk fel 6 M sósavban (4.1.). Ugyanezzel az oldattal töltsük jelig, és gondosan keverjük össze.
4.7.2. Molibdén közbenső oldat (25 μg/ml).
A törzsoldat (4.7.1.) 25 ml-ét tegyük egy 500 ml-es mérőlombikba. Töltsük fel jelig 6 mol/l sósavval (4.1.) és gondosan keverjük össze.
4.7.3. Molibdén munkaoldat (2,5 μg/ml)
Vigyük át a közbenső oldat (4.7.2.) 10 ml-ét egy 100 ml-es mérőlombikba. Töltsük fel jelig 6 mol/l sósav oldattal és gondosan keverjük össze.
5. FELSZERELÉS
5.1. Molekuláris abszorpció mérésére alkalmas spektrométer 20 mm optikai úthosszú küvettákkal ellátva, 470 nm-en való mérésre beállítva.
5.2. 200 ml-es vagy 250 ml-es választótölcsérek.
6. A VIZSGÁLATI OLDAT ELŐKÉSZÍTÉSE
6.1. Kivont molibdén-oldat
Lásd az I/1. és/vagy az I/2., illetve értelemszerűen az I/3. fejezetet.
6.2. A vizsgálati oldat előkészítése
A kivonat (6.1.) alikvot részét hígítsuk meg 6 mol/l sósavoldattal (4.1.), hogy megfelelő molibdén koncentrációt kapjunk. Nevezzük a hígítási tényezőt D-nek.
Vegyünk ki egy alikvot részt (a) a kivonatból, ami 1-12 μg közötti mennyiségű molibdént tartalmaz és tegyük az választótölcsérbe (5.2.). Töltsük fel 50 ml-re a 6 mol/l sósavoldattal (4.1.).
7. ELJÁRÁS
7.1. A vakoldat készítése
Készítsünk vakoldatot úgy, hogy a kivonástól kezdve az egész eljárást megismételjük, a műtrágya-minta bemérése nélkül.
7.2. Kalibrációs sorozat készítése
Legalább hat, növekvő koncentrációjú kalibrációs oldatot készítsünk a spektrométer optimális érzékenységi tartományának megfelelő koncentráció tartományban.
A 0-12,5 μg molibdén tartományhoz tegyünk rendre 0, l, 2, 3, 4, és 5 ml munkaoldatot (4.7.3.) a választótölcsérekbe (5.2.). Egészítsük ki a térfogatukat 50 ml-re 6 M sósavval (4.1.). így a tölcsérek rendre 0, 2,5, 5, 7,5, 10 és 12,5 μg molibdént tartalmaznak.
7.3. A komplex kialakítása és elválasztása
Minden egyes választótölcsérbe (6.2., 7.1. és 7.2.) tegyünk az alábbi sorrendben:
- 10 ml réz oldatot (4.2.),
- 20 ml aszkorbinsav oldatot (4.3.).
Gondosan keverjük össze, várjunk két-három percet, azután adjunk hozzá:
- 10 ml n-butil-acetátot (4.4.), precíziós pipettát használva,
- 20 ml tiocianát oldatot (4.5.).
Rázzuk egy percig, hogy a komplex átmenjen a szerves fázisba; hagyjuk szétválni a fázisokat; ha a két fázis szétvált, az egész vizes fázist engedjük le és ontsuk el; majd a szerves fázist mossuk:
- 10 ml ón-klorid oldattal (4.6.).
Rázzuk egy percig. Hagyjuk szétválni és a teljes vizes fázist engedjük le. Gyűjtsük a szerves fázist kémcsőbe, ezáltal lehetővé válik, hogy a szuszpendált vízcseppeket is összegyűjtsük.
7.4. Meghatározás
Mérjük le a 7.3. szerint kapott oldatok abszorbanciáját 470 nm hullámhosszon, a 0 μg/ml molibdén kalibrációs oldattal (7.2.) szemben.
8. AZ EREDMÉNYEK KIFEJEZÉSE
Készítsük el a kalibrációs görbét úgy, hogy a kalibrációs oldatokban (7.2.) lévő molibdén μg-ban kifejezett mennyiségét az abszcisszán, a spektrométerről leolvasott megfelelő abszorbancia értékeket (7.4.) pedig az ordinátán ábrázoljuk.
A görbe segítségével határozzuk meg a molibdén tömegét a vizsgálati oldatban (6.2.) és a vakoldatban (7.1.). Ezeket a tömegeket nevezzük rendre xs-nek és Xb-nek.
A műtrágya tömegszázalékban kifejezett molibdén tartalma:
Mo% = [(xs-xb)xVxD]/(Mx104
Ha az I/3. fejezet szerint használtuk:
Mo% = [(xs-xb)xVx2D]/(Mx104
ahol:
Mo = a műtrágya százalékos molibdén tartalma,
a = az utolsó hígításból (6.2.) kivett alikvot térfogata, ml,
xs = a Mo tömege a vizsgálati oldatban (6.2.), mg,
xb = a Mo tömege a vakoldatban (7.1.), amelynek térfogata a vizsgálati oldat (6.2.) alikvot része térfogatának felel meg,
V = az I/1. vagy I/2. fejezet szerint nyert kivonat térfogata, ml,
D = 6.2. szerinti hígítás tényezője,
M = az I/1. vagy az I/2. fejezet szerint bemért minta tömege, g.
A D hígítási tényező számítása: ha (a1), (a2) egymást követő alikvot részek, és (v1), (v2) a megfelelő hígításaik térfogata, a hígítási tényező a következő:
D = (v1/a1)x(v2/a2).
I/11. CINK MEGHATÁROZÁSA MŰTRÁGYAKIVONATOKBAN ATOMABSZORPCIÓS SPEKTROMETRIÁVAL
1. TÁRGY
A módszer a cink műtrágyakivonatokban való meghatározásának eljárását írja le.
2. ALKALMAZÁSI TERÜLET
Az eljárást olyan műtrágyák az I/1. és az I/2. módszerrel kivont mintáinak vizsgálatára lehet alkalmazni, amelyekre a 3. számú melléklet 1. függeléke az összes és/vagy a vízben oldható cinktartalom megadását előírja.
3. A MÓDSZER ELVE
A kivonatok megfelelő kezelését és hígítását követően a cinktartalmat atomabszorpciós spektrometriával határozzuk meg.
4. VEGYSZEREK
4.1. Sósav oldat, kb. 6 mol/l Lásd az I/4. fejezetet (4.1.).
4.2. Sósav oldat, kb. 0,5 mol/l Lásd az I/4. fejezetet (4.2.).
4.3. Lantánsó oldatok (10 g La/liter) Lásd az I/4. fejezetet (4.3.).
4.4. Cink kalibrációs oldatok
4.4.1. Cink törzsoldat (l000 μg/ml)
1000 ml-es mérőlombikban oldjunk fel 1 g cinkport vagy -lemezkét 0,1 mg pontossággal bemérve, 25 ml 6 mol/l sósavban (4. L). Miután teljesen feloldódott, töltsük fel jelig vízzel és gondosan keverjük össze.
4.4.2. Cink munkaoldat (100 μg/ml)
Egy 200 ml-es mérőlombikban hígítsunk fel 20 ml törzsoldatot (4.4.1.) 0,5 mol/l sósav oldatban (4.2.). Töltsük fel jelig 0,5 mol/l sósav oldattal és gondosan keverjük össze.
5. FELSZERELÉS
Atomabszorpciós spektrométer: lásd az I/4. módszert (5.). A műszer legyen felszerelve cinkre jellemző sugarak (213,8 rím) kibocsátására alkalmas fényforrással. A spektrométernek alkalmasnak kell lennie háttérkorrekció elvégzésére.
6. A VIZSGÁLATI OLDAT ELŐKÉSZÍTÉSE
6.1. Kivont cink-oldat
Lásd az I/1. és/vagy az I/2., illetve értelemszerűen az I/3. fejezetet.
6.2. A vizsgálati oldat előkészítése
Lásd az I/4. fejezetet (6.2.). A vizsgálati oldat 10% (v/v) lantánsó oldatot tartalmazzon.
7. ELJÁRÁS
7.1. Vakoldat készítése
Lásd az I/4. fejezetet (7.1.). A vakoldat 10% (v/v), a 6.2. pontban alkalmazott lantánsó oldatot tartalmazzon.
7.2. A kalibrációs oldatok elkészítése Lásd az I/4. fejezetet (7.2.).
A cink számára optimális 0-5 μg/ml meghatározási tartomány beállításához tegyünk rendre 0, 0,5, l, 2, 3, 4 és 5 ml munkaoldatot (4.4.2.) 100 ml-es mérőlombikokba. Ha szükséges, a sósav-koncentrációt állítsuk úgy, hogy az a vizsgálati oldatok sósav-koncentrációjához a lehető legközelebb essen. Adjunk mindegyik mérőlombikba 10 ml-t a 6.2. pontban használt lantánsó oldatból. Töltsük fel 100 ml-re 0,5 mol/l sósav oldattal (4.2.) és gondosan keverjük Össze. Ezek az oldatok rendre 0, 0,5, l, 2, 3, 4 és 5 M-g/ml kobaltot tartalmaznak.
7.3. Meghatározás
Lásd az I/4. fejezetet (7.3.). Készítsük elő a spektrométert 213,8 nm-en történő mérésre.
8. AZ EREDMÉNYEK KIFEJEZÉSE
Lásd az I/4. fejezetet (8.).
A műtrágya tömegszázalékban kifejezett cinktartalmát az alábbi egyenlet adja meg:
Zn% = [(xs-xh)xVxD]/(Mx104
Ha az I/3. fejezetet használtuk:
Zn% = [(xs - xh)xVx2D]/(Mx104
ahol:
Zn a műtrágya százalékban kifejezett cinktartalma,
xs a (6.2.) vizsgálati oldat koncentrációja, μg/ml,
xb a (7.1.) vakoldat koncentrációja, μg/ml,
V az I/1. vagy I/2. fejezet szerint nyert kivonat térfogata, ml,
D a 6.2. pont szerint végzett hígítás tényezője,
M az I/1. vagy az I/2. fejezet szerint bemért minta tömege, g.
A D hígítási tényező kiszámítása: Ha az (a1), (a2), (a3),... (ai) és (a) az alikvot részek, és (v1), (v2), (v3),... (vi) és (100) a hígításoknak rendre megfelelő térfogatok, akkor a D hígítási tényező:
D = (v1/a1) x (v2/a2) x (v3/a3) x.x.x. (vi/ai) x (100/a).
J) 10%-NÁL NAGYOBB KONCENTRÁCIÓJÚ MIKROELEMEK
J/1. AZ ÖSSZES MIKROELEM KIVONÁSA
1. TÁRGY
Ez a módszer eljárást ír elő a következő mikroelemek kivonásához: összes bór, összes kobalt, összes réz, összes vas, összes mangán, összes molibdén és összes cink. A cél az, hogy lehetőleg minimális számú kivonást végezzünk, ahol lehetséges, ugyanazt a kivonatot használva a fent felsorolt mikroelemek összes mennyiségének meghatározására.
2. ALKALMAZÁSI TERÜLET
Az eljárás a 3. számú melléklet 1. függeléke hatálya alá eső, a következő mikroelemek közül egyet vagy többet tartalmazó műtrágyákra vonatkozik: bór, kobalt, réz, vas, mangán, molibdén és cink. Alkalmazandó minden olyan mikroelemre, amelynek bejelentett tartalma meghaladja a 10%-ot.
3. A MÓDSZER ELVE
Oldás forrásban lévő hígított sósavban.
Megjegyzés:
A kivonás empirikus és előfordulhat, hogy nem veszteségmentes, a terméktől vagy a műtrágya egyéb alkotórészeitől függően. Különösen bizonyos mangán-oxidok esetében előfordulhat, hogy a kivont mennyiség lényegesen kisebb, mint a termékben található mangán összes mennyisége. A műtrágyagyártó cég felelőssége annak biztosítása, hogy a bejelentett tartalom ténylegesen megfeleljen a módszer feltételei mellett kivont mennyiségnek.
4. VEGYSZEREK
Hígított sósav (HCl) oldat, kb. 6 mol/l.
Elegyítsünk 1 térfogatrész sósavat (d20 =1,18 g/ml) 1 térfogatrész vízzel.
Tömény ammónia oldat (NH4OH, d20 = 0,9 g/ml).
5. FELSZERELÉS
5.1. Hőfokszabályzós elektromos főzőlap.
5.2. pH-mérő műszer.
Megjegyzés:
Ha a kivonat bórtartalmának a meghatározása a cél, boroszilikát üvegedényt használni tilos! Mivel a módszer forralást ír elő, a teflon vagy a kvarcüveg használata előnyösebb. Amennyiben bórátokat tartalmazó mosogatószerrel mostuk el az üvegedényeket, alaposan öblítsük le azokat.
6. A MINTA ELKÉSZÍTÉSE Lásd B) fejezet.
7. ELJÁRÁS
7.1. Vizsgálati minta
Vegyünk 1 vagy 2 g tömegű műtrágyát, attól függően, hogy mennyi a termékben az elem deklarált részaránya. A következő táblázatot kell használni az olyan végső oldat elkészítéséhez, amely megfelelő hígítás után az egyes módszerekre előírt mérési tartományon belül lesz. A mintákat 1 mg pontossággal kell bemérni.
| A műtrágyában található mikroelem deklarált részaránya (%) | >10<25 | >25 |
| A vizsgálati minta tömege (g) | 2 | 1 |
| A mintában található elem tömege (mg) | > 200 < 500 | >250 |
| A kivonat térfogata, V (ml) | 500 | 500 |
| A kivonatban található elem koncentrációja (mg/l) | > 400 < 1000 | >500 |
Tegyük a mintát 250 ml-es főzőpohárba.
7.2. Az oldat elkészítése
Szükség esetén nedvesítsük meg a mintát egy kevés vízzel, óvatosan, kis részletekben adjunk hozzá műtrágya-grammonként 10 ml hígított sósavat (4.1.), majd öntsünk hozzá kb. 50 ml vizet. Fedjük le a főzőpoharat óraüveggel, és keverjük össze. Forraljuk fel a főzőlapon, és forraljuk 30 percig. Időnként megkeverve hagyjuk lehűlni. Veszteség nélkül ontsuk át 500 ml-es mérőlombikba. Töltsük jelig vízzel, és jól keverjük össze. Száraz szűrőn szűrjük át száraz tárolóedénybe. Az első részletet ontsuk el. A kivonatnak teljesen tisztának kell lennie.
Megjegyzés:
Amennyiben a tárolóedényt nem lehet bedugaszolni, ajánlatos a meghatározást a tiszta szűrlet alikvot részeivel azonnal elvégezni.
Olyan kivonat esetén, ahol a bórtartalmat kell meghatározni, tömény ammóniaoldattal állítsuk be a pH értéket 4 és 6 közé (4.2.).
8. MEGHATÁROZÁS
Az egyes mikroelemek meghatározását az azokra a mikroelemekre leírt módszerben megadott alikvot részből kell elvégezni.
A J/5., J/6., J/7., J/9. és a J/10. módszert nem lehet olyan elem meghatározására használni, amely kelát- vagy komplex kötésben van. Ilyenkor a meghatározás előtt a 10.3. módszert kell használni.
Atomabszorpciós spektrometriával (J/8. és J/11. módszer) végzett meghatározás esetén az ilyen kezelés nem feltétlenül szükséges.
J/2.VÍZOLDHATÓ MIKROELEMEK KIVONÁSA
1. TÁRGY
Ez a módszer eljárást ír elő a következő mikroelemek vízoldható formáinak a kivonására: bór, kobalt, réz, vas, mangán, molibdén és cink. A cél az, hogy - ahol csak lehet - minimális számú kivonást végezzünk, lehetőleg ugyanazt a kivonatot használva a fent felsorolt mikroelemek mennyiségének meghatározására.
2. ALKALMAZÁSI TERÜLET
Az eljárás olyan közösségi műtrágyákra vonatkozik, amelyek a 3. számú melléklet 1. függeléke hatálya alá esnek, és amelyek a következő mikroelemek közül egyet vagy többet tartalmaznak: bór, kobalt, réz, vas, mangán, molibdén és cink. Alkalmazandó minden olyan mikroelemre, amelynek bejelentett tartalma meghaladja a 10%-ot.
3. A MÓDSZER ELVE
A mikroelemeket a műtrágya 20 ± 2 °C hőmérsékletű vízzel való rázatásával extraháljuk.
Megjegyzés:
A kivonás empirikus és nem biztos, hogy veszteségmentes.
4. VEGYSZEREK
4.1. Hígított sósav (HCl) oldat, kb. 6 mol/l
Elegyítsünk 1 térfogatrész sósavat (d20 =1,18 g/ml) 1 térfogatrész vízzel.
5. FELSZERELÉS
Forgó keverő, kb. 35-40 l/min, fordulatszámra beállítva.
Megjegyzés:
Ha a kivonat bértartalmát kell meghatározni, ne használjunk boroszilikát üvegedényt. Teflon- vagy kvarcüveg-edény ajánlható ehhez a kivonáshoz. Amennyiben bórátokat tartalmazó mosogatószerrel mostuk el az üvegedényeket, alaposan öblítsük ki azokat.
6. A MINTA ELKÉSZÍTÉSE Lásd B) fejezet.
7. ELJÁRÁS
7.1. Vizsgálati minta
Mérjünk le a műtrágyából 1 vagy 2 g-ot, attól függően, hogy mennyi a termékben az elem bejelentett részaránya. A következő táblázatot kell használni a végső oldat elkészítéséhez, amely megfelelő hígítás után az egyes módszerek esetében a mérési tartományon belül fog esni. A mintákat 1 mg pontossággal kell bemérni.
| A műtrágya deklarált mikroelem tartalma (%) | >10<25 | >25 |
| A vizsgálati minta tömege (g) | 2 | 1 |
| Az elem tömege a mintában (mg) | > 200 < 500 | >250 |
| A kivonat térfogata, V (ml) | 500 | 500 |
| Az elem koncentrációja a kivonatban (mg/l) | > 400 < 1000 | >500 |
Tegyük a mintát 500 ml-es lombikba.
7.2. Az oldat elkészítése Öntsünk hozzá kb. 400 ml vizet.
Szorosan dugaszoljuk be a lombikot. Rázzuk jól össze kézzel, hogy diszpergáljuk a mintát, utána helyezzük rá a rázógépre, és rázassuk 30 percig.
Töltsük a lombikot jelig vízzel, és jól rázzuk össze.
7.3. A mintaoldat elkészítése
Szűrjük azonnal tiszta, száraz lombikba. Dugaszoljuk be a lombikot. A meghatározást közvetlenül a szűrés után végezzük.
Megjegyzés:
Ha a szűrlet fokozatosan zavarossá válik, készítsünk egy másik kivonatot a 7.1. és 7.2. pont szerint egy Ve térfogatú lombikban. Szűrjük le egy előzetesen kiszárított W térfogatú mérőlombikba, amelybe 5 ml hígított sósavat (4.1.) töltöttünk. A szűrést pontosan akkor fejezzük be, amikor a szűrlet a mérőlombik jelét elérte. Gondosan keverjük össze.
Ilyen körülmények között a V értéke az eredmények kifejezésekor a következő lesz:
V = VexW/(W-5)
Az eredmények kifejezésekor a hígítás ettől a V értéktől függ.
8. MEGHATÁROZÁS
A mikroelemek meghatározását az egyes mikroelemeknél leírt alikvot részekből kell elvégezni.
A J/5., J/6., J/7., J/9. és J/10. módszert nem lehet olyan elemek meghatározására használni, amelyek kelát- vagy komplex kötésben vannak. Ilyenkor a meghatározás előtt a J/3. módszert kell használni.
Atomabszorpciós spektrometriával (J/8. és J/11. módszer) végzett meghatározás esetén az ilyen kezelést el lehet hagyni.
J/3. SZERVES VEGYÜLETEK ELTÁVOLÍTÁSA MŰTRÁGYAKIVONATOKBÓL
1. TÁRGY
Ez a módszer eljárást ír elő a szerves vegyületeknek műtrágyakivonatból történő eltávolítására.
2. ALKALMAZÁSI TERÜLET
Az eljárást olyan, a J/1. és a J/2. módszerrel kivont műtrágyaminták elemzésére alkalmazandó, amelyekre a 3. számú melléklet 1. függeléke az összes elem, és/vagy a vízoldható elemek bejelentését írja elő.
Megjegyzés:
Ha szerves anyag kis mennyiségben van jelen, az rendszerint nem befolyásolja az atomabszorpciós spektrometriával végzett meghatározást.
3. A MÓDSZER ELVE
A kivonat alikvot részében a szerves anyagokat hidrogén-peroxiddal oxidáljuk.
4. VEGYSZEREK
Hígított sósav (HCl) oldat, körülbelül 0,5 mol/l.
Elegyítsünk 1 térfogatrész sósavat (d20 =1,18 g/ml) 20 térfogatrész vízzel.
Hidrogén-peroxid oldat (30% H2O2, d20 = 1,11 g/ml), mikroelemektől mentes.
5. FELSZERELÉS Hőfokszabályzós elektromos főzőlap.
6. ELJÁRÁS
Vegyünk a J/1. vagy a J/2. módszerrel nyert kivonatból 25 ml-t, és ontsuk 100 ml-es főzőpohárba. A J/2. módszer alkalmazása esetén öntsünk hozzá 5 ml hígított sósavat (4.1.). Ezután öntsünk hozzá 5 ml hidrogén-peroxid oldatot (4.2.). Fedjük le óraüveggel. Hagyjuk oxidálódni szobahőmérsékleten kb. 1 órán keresztül, majd lassan forraljuk fel, és hagyjuk fél óráig forrni. Ha az oldat lehűlt, szükség szerint adjunk hozzá még 5 ml hidrogén-peroxidot. Ezután forraljuk fel, hogy a felesleges hidrogén-peroxid eltávozzék. Hagyjuk lehűlni, és veszteség nélkül ontsuk át 50 ml-es mérőlombikba, majd töltsük jelig vízzel. Szükség esetén szűrjük le.
Amikor alikvot részt veszünk ki, és kiszámítjuk a termékben található mikroelem százalékos mennyiségét, ezt a hígítást figyelembe kell venni.
J/4. A MŰTRÁGYAKIVONATOKBAN LÉVŐ MIKROELEMEK MEGHATÁROZÁSA ATOMABSZORPCIÓS SPEKTROMETRIÁVAL (ÁLTALÁNOS ELJÁRÁS)
1. TÁRGY
Ez a fejezet általános eljárást ír elő a műtrágyakivonatokban található vas és cink mennyiségének a meghatározására atomabszorpciós spektrometriával.
2. ALKALMAZÁSI TERÜLET
Ez az eljárás a J/1. és a J/2. fejezet szerint kivont műtrágyaminták elemzésére szolgál, amelyekre a 3. számú mellékletének 1. függeléke az összes, és/vagy a vízoldható vas és cink bejelentését előírja. Az eljárásnak a különböző mikroelemekre való alkalmazása az egyes elemekre külön-külön előírt módszerekben van részletesen leírva.
Megjegyzés:
Kis mennyiségű szerves anyag jelenléte legtöbbször nem befolyásolja az atomabszorpciós spektrometriával végzett meghatározást.
3. A MÓDSZER ELVE
Azt követően, hogy szükség esetén kezeltük a kivonatot a zavaró kémiai anyagok csökkentése vagy kiküszöbölése végett, a kivonatot úgy hígítjuk, hogy a koncentrációja a meghatározandó mikroelem mérésére alkalmas hullámhosszon a spektrométer optimális tartományába essék.
4. VEGYSZEREK
4.1. Hígított sósav (HCl) oldat, kb. 6 mol/l
Elegyítsünk 1 térfogatrész sósavat (p = 1,18 g/ml) 1 térfogatrész vízzel.
4.2. Hígított sósav (HCl) oldat, kb. 0,5 mol/l
Elegyítsünk 1 térfogatrész sósavat (d20 =1,18 g/ml) 20 térfogatrész vízzel.
4.3. Lantánsó oldatok (10 g La/l)
Ez a reagens vas és cink meghatározására szolgál. Kétféle módon készíthető:
a) Sósavban oldott lantán-oxiddal (4.1.). Vigyünk 11,73 g lantán-oxidot (La2O3) 150 ml vízzel 1 literes mérőlombikba, és adjunk hozzá 120 ml 6 mol/l sósavat (4.1.). Hagyjuk feloldódni, majd egészítsük ki vízzel 1 literre, és gondosan keverjük össze. Ez az oldat kb. 0,5 mol/l-es lantánra.
b) Lantán-klorid, -szulfát vagy -nitrát oldattal. Oldjunk fel 26,7 g lantán-klorid-heptahidrátot (LaCl3.7H2O) vagy 31,2 g lantán-nitrát-hexahidrátot [La(NO3)3.6H2O] vagy 26,2 g lantán-szulfát-monahidrátot [La2(SO4)3.9H2O] 150 ml vízben, majd öntsünk hozzá 85 ml 6 M töménységű sósavat (4.1.). Hagyjuk feloldódni, majd egészítsük ki vízzel 1 literre. Jól keverjük össze. Ez az oldat kb. 0,5 mol/l lantánra.
4.4. Kalibráló oldatok
Ezek elkészítéséhez lásd az egyes mikroelemek meghatározási módszereit.
5. FELSZERELÉS
Atomabszorpciós spektrométer, a meghatározandó mikroelemekre jellemző sugárzást kibocsátó sugárforrással felszerelve. Az analitikus tartsa be a műszer használati utasítását, és legyen járatos a műszer használatában. A készülék tegye lehetővé háttérkorrekció alkalmazását, hogy szükség esetén használni lehessen (pl. Zn). Az alkalmazandó gázok: levegő és acetilén.
6. A VIZSGÁLATI OLDAT ELKÉSZÍTÉSE
6.1. A meghatározandó elemeket tartalmazó kivonat-oldatok elkészítése Lásd J/1., és/vagy J/2. fejezetet, valamint szükség szerint a J/3. fejezetet.
6.2. A vizsgálandó oldat kezelése
A J/1., a J/2. vagy a J/3. fejezet szerint nyert kivonat alikvot részét hígítsuk vízzel és/vagy sósavval (4.1.) vagy (4.2.) úgy, hogy a mérendő végső oldatban a meghatározandó elem olyan koncentrációjú legyen, amely megfelel az alkalmazott kalibrációs tartománynak (7.2.), a sósav-koncentráció pedig legalább 0,5 mol/l, legfeljebb pedig 2,5 M. Ez a művelet egy vagy több hígítási lépést igényelhet.
A végső oldatot úgy nyerjük, hogy egy alikvot rész hígított kivonatot 100 ml térfogatú mérőlombikba öntünk. Legyen ennek az alikvot résznek a térfogata (a) ml. Öntsünk hozzá 10 ml lantánsó oldatot (4.3.). Töltsük a jelig 0,5 mol/l töménységű sósavoldattal (4.2.), és jól keverjük össze. A hígítási tényezőt nevezzük D-nek.
7. ELJÁRÁS
7.1. Vakoldat készítése
Készítsünk vakoldatot oly módon, hogy a kivonási szakasztól kezdve végighaladunk az egész eljáráson, csak a műtrágya vizsgálati minta bevitelét hagyjuk el belőle.
7.2. Kalibráló oldatok készítése
Az egyes mikroelemekre megadott módszerrel készített kalibráló munkaoldatból készítsünk 100 ml-es mérőlombikokban legalább öt, növekvő koncentrációjú kalibráló oldatot, melyek koncentrációja a spektrométer optimális mérési tartományába esik. Szükség esetén állítsuk be a sósav koncentrációt úgy, hogy a lehető legjobban megközelítse a hígított mintaoldat koncentrációját (6.2). A vas vagy a cink meghatározásakor öntsünk hozzá 10 ml-t ugyanabból a lantánsó oldatból (4.3.), mint amilyent a (6.2.) pontban használtunk. Töltsük jelig 0,5 mol/l sósavoldattal (4.2.), és jól keverjük össze.
7.3. Meghatározás
Készítsük elő a spektrométert (5.) a meghatározáshoz, és állítsuk be az egyes mikroelemekre a módszerben megadott hullámhossz-értéket.
Poriasszuk be háromszor egymás után a kalibráló oldatokat (7.2.), a vizsgálandó oldatot (6.2) és a vakoldatot (7.1.). Közben jegyezzük fel az egyes eredményeket, és Öblítsük át a műszert desztillált vízzel az egyes porlasztások között.
Készítsük el a kalibrációs görbét oly módon, hogy az egyes kalibráló oldatokra (7.2.) a spektrométeren leolvasott értékek átlagát felvesszük az ordinátán, az elem μg/ml-ben kifejezett, megfelelő koncentrációját pedig az abszcisszán.
Ebből a görbéből határozzuk meg az Xs mintaoldatban (6.2.), és az Xb vakoldatban (7.1.) lévő mikroelem-koncentrációkat, μg/ml-ben kifejezve.
8. AZ EREDMÉNY MEGADÁSA
A műtrágyában lévő mikroelem (E) százalékarányát a következő képlettel adjuk meg:
E%=[(xs-xb)x VxD]/(Mx 104)
Ha a J/3. fejezetet használtuk:
E%=[(xs-xb)x Vx 2D]/(Mx 104)
ahol:
E = a meghatározott mikroelem mennyisége, a műtrágya százalékában kifejezve,
Xs = a mintaoldat (6.2.) koncentrációja, μg/ml-ben,
Xb = a vakoldat (7.1.) koncentrációja, μg/ml-ben,
V = a J/1. vagy J/2. fejezet szerint nyert kivonat mennyisége, ml-ben,
D = a 6.2. pontban végzett hígításnak megfelelő tényező,
M = a J/1. vagy J/2. fejezet szerint vett vizsgálati minta tömege, g-ban.
A D hígítási tényező számítása:
Ha (a1), (a2), (a3),..., (ai) és (a) az alikvot részek, és (v1), (v2), (v3),..., (vi) és (100) a megfelelő hígításokhoz tartozó ml-ben megadott mennyiségek, akkor a D hígítási tényező a következővel egyenlő:
D = (v1/a1) x (v2/a2) x (v3/a3) x...x (vi/ai) x (100/a).
J/5. MŰTRÁGYAKIVONATOKBAN LÉVŐ BÓR MEGHATÁROZÁSA ACIDIMETRIÁS TITRÁLÁSSAL
1. TÁRGY
Ez a fejezet eljárást ír elő a műtrágyakivonatok bórtartalmának a meghatározására.
2. ALKALMAZÁSI TERÜLET
Ezt az eljárást olyan, a J/1. vagy a J/2. módszerrel nyert műtrágyaminták kivonatának a vizsgálatára kell alkalmazni, amelyekre nézve a 3. számú melléklet 1. függeléke előírja az összes, és/vagy a vízoldható bórtartalom bejelentését.
3. A MÓDSZER ELVE
A borát és a mannit alábbi reakciója során mannit-bór komplex képződik:
C6H8(OH)6+H3BO3 -> C6H15O8B+H2O.
A komplexet nátrium-hidroxid oldattal 6,3 pH értékre titráljuk.
4. VEGYSZEREK
4.1. Metilvörös indikátoroldat
Oldjunk fel 0,1 g metilvöröst (C15H15N3O2) 50 ml etanolban (95%-os) 100 ml-es mérőlombikban. Töltsük jelig vízzel. Jól rázzuk össze.
4.2. Higított sósavoldat, kb. 0,5 mol/l
Elegyítsünk 1 térfogatrész sósavat (HCl) (d20 =1,18 g/ml) 20 térfogatrésznyi vízzel.
4.3. Nátrium-hidroxid oldat, kb. 0,5 mol/l
Szén-dioxid-mentesnek kell lennie. Oldjunk fel 20 g nátrium-hidroxid (NaOH) pelletet 1 literes, kb. 800 ml forralt vizet tartalmazó mérőlombikban. Amikor az oldat lehűlt, egészítsük ki 1000 ml-re forralt vízzel, és jól keverjük össze.
4.4. Nátrium-hidroxid mérőoldat, kb. 0,025 mol/l
Szén-dioxid-mentesnek kell lennie. A 0,5 mol/l nátrium-hidroxid oldatot (4.3.) forralt vízzel hígítsuk húszszorosára, és jól keverjük össze. Az oldat bórban (B) kifejezett értékét meg kell határozni (lásd 9. pont).
4.5. Bór kalibráló oldat (l00 μg/ml B)
1000 ml-es mérőlombikban oldjunk fel vízzel 0,5719 g, 0,1 mg pontossággal bemért bórsavat (H3BO3). Ontsuk fel vízzel, és jól keverjük össze. Ontsuk át műanyag palackba, és tároljuk hűtőszekrényben.
4.6. D-mannit (C6H14O6) por.
4.7. Nátrium-klorid (NaCl).
5. FELSZERELÉS
5.1. Üvegelektródos pH-mérő műszer
5.2. Mágneses keverő
5.3. 400 ml-es főzőpohár teflon keverőpálcával
6. A VIZSGÁLATI OLDAT ELKÉSZÍTÉSE
6.1. A bóroldat elkészítése
Lásd a J/1., a J/2. és, ahol szükséges, a J/3. fejezeteket.
7. ELJÁRÁS
Vizsgálat
Tegyünk egy 400 ml-es főzőpohárba (5.3.) alikvot résznyit (a) a kivonatból (6.1.), amely 2-4 mg B-t tartalmaz. Öntsünk hozzá 150 ml vizet.
Adjunk hozzá néhány csepp metilvörös indikátoroldatot (4.1.).
A J/2. fejezet szerint történő kivonás esetében 0,5 mol/l sósav (4.2.) hozzáadásával az indikátoroldat színátcsapási pontjáig savanyítsuk meg a kivonatot, azután adjunk hozzá további 0,5 ml 0,5 mol/l sósavat (4.2.).
3 g nátrium-klorid (4.7.) hozzáadása után forraljuk fel az oldatot a szén-dioxid kiűzése végett. Hagyjuk lehűlni. Helyezzük a főzőpoharat a mágneses keverőre (5.2.), és helyezzük bele az előzetesen kalibrált pH-mérő elektródokat (5.1.).
Állítsuk be a pH-értéket pontosan 6,3-ra, először a 0,5 mol/l nátrium-hidroxid oldattal (4.3.), utána a 0,025 mol/l NaOH oldattal (4.4.).
Adjunk hozzá 20 g D-mannitot (4.6.), oldjuk fel teljesen, és jól keverjük össze. Titráljuk a 0,025 mol/l nátrium-hidroxid oldattal (4.4.) 6,3 pH-értékre (legalább 1 percig stabil legyen). A szükséges térfogat legyen x1.
8. VAKOLDAT
Készítsünk vakoldatot oly módon, hogy megismételjük az egész eljárást az oldatkészítési szakasztól, csupán a műtrágyát hagyjuk ki. A szükséges térfogat legyen x0.
9. A NÁTRIUM-HIDROXID OLDAT (4.4.) BÓR (B) EGYENÉRTÉKE
Pipettázzunk 20 ml (2,0 mg B) kalibráló oldatot (4.5.) 400 ml-es főzőpohárba, és adjunk hozzá néhány csepp metilvörös indikátoroldatot (4.1.). Adjunk hozzá 3 g nátrium-kloridot (4.7.) és sósavoldatot (4.2.) az indikátoroldat (4.1.) színátcsapási pontjáig.
Egészítsük ki a térfogatot kb. 150 ml-re, és fokozatosan forraljuk fel a szén-dioxid kiűzése végett. Hagyjuk lehűlni. Helyezzük a főzőpoharat a mágneses keverőre (5.2.), és tegyük bele az előzetesen kalibrált pH-mérő elektródokat (5.1.). Állítsuk be a pH-értéket pontosan 6,3-ra, először a 0,5 mol/l nátrium-hidroxid oldattal (4.3.), majd a 0,025 mol/l NaOH oldattal (4.4.).
Adjunk hozzá 20 g D-mannitot (4.6.), oldjuk fel teljesen, és jól keverjük össze. Titráljuk a 0,025 mol/l nátrium-hidroxid oldattal (4.4.) 6,3 pH-értékre (legalább 1 percig stabil maradjon). A fogyott mérőoldat térfogata legyen V1.
Ugyanilyen módon készítsünk vakoldatot, a kalibráló oldatot 20 ml vízzel helyettesítve. A fogyott mérőoldat térfogata legyen V0.
A NaOH mérőoldat (4.4.) bór egyenértéke (F) mg/ml mértékegységben megadva a következő:
F (mg/ml-ben) = 2/(V1-V0).
1 ml pontosan 0,025 mol/l értékre beállított nátrium-hidroxid oldat 0,27025 mg bórnak felel meg.
10. AZ EREDMÉNY MEGADÁSA
A műtrágyában található bór százalékaránya a következő képlettel számítható:
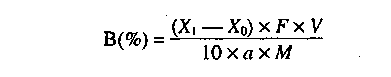
| ahol: |
| B (%) = a műtrágya százalékos bértartalma, |
| X1 = a vizsgálati mintára fogyott 0,025 mol/l nátrium-hidroxid oldat (4.4.) térfogata, ml-ben, |
| X0 = a vakoldatra fogyott 0,025 mol/l nátrium-hidroxid oldat M (4.4.) térfogata, ml-ben, |
| F = a 0,025 mol/l nátrium-hidroxid oldat M (4.4.) bor (B) értéke, mg/ml-ben, |
| V = a 10.1. vagy a 10.2. módszerrel nyert kivonat-oldat térfogata, ml-ben, |
| a = a kivonat-oldatból (6.1.) vett alikvot rész (7.1.) térfogata, ml-ben, |
| M = a 10.1. vagy a 10.2. módszerrel vett vizsgálati minta tömege, grammban. |
J/6. A MŰTRÁGYAKIVONATOKBAN TALÁLHATÓ KOBALT MEGHATÁROZÁSA GRAVIMETRIÁS MÓDSZERREL, 1-NITROZO-2-NAFTOLLAL
1. TÁRGY
A fejezet eljárást ír elő a műtrágyakivonatokban található kobalt meghatározására.
2. ALKALMAZÁSI TERÜLET
Az eljárást olyan műtrágyamintákból vett kivonatok vizsgálatára kell alkalmazni, amelyeket a J/1. vagy a J/2. eljárással nyertek, és amelyekre nézve a 3. számú melléklet 1. függeléke előírja a kobalttartalom bejelentését.
3. A MÓDSZER ELVE
A kobalt(III)-ion 1-nitrozo-2-naftollal vörös csapadékot, Co (C10H6ONO)3.2H2O-t ad. Miután a kivonatban lévő kobaltot kobalt(III)-ion formájára hoztuk, ecetsavas közegben 1-nitrozo-2-naftol oldattal lecsapatjuk. Szűrés után a csapadékot kimossuk, súlyállandóságig szárítjuk, és mint Co (C10H6ONO)3.2H2O-t mérjük.
4. VEGYSZEREK
4.1. Hidrogén-peroxid oldat (H2O2, ρ = 1,11 g/ml), 30%.
4.2. Nátrium-hidroxid oldat kb. 2 mol/l
Oldjunk fel 8 g nátrium-hidroxid pelletet 100 ml vízben.
4.3. Higított sósav oldat, kb. 6 mol/l
Elegyítsünk 1 térfogatrész sósavat (d20= 1,18 g/ml) 1 térfogatrész vízzel.
4.4. Ecetsav (99,7% CH3CO2H) (d20 = 1,05 g/ml).
4.5. 1:2 ecetsav oldat, kb. 6 mol/l
Elegyítsünk 1 térfogatrész ecetsavat (4.4.) 2 térfogatrész vízzel.
4.6. 1-nitrozo-2-naftol oldat 100 ml ecetsavban (4.4.). Adjunk hozzá 100 ml langyos vizet. Alaposan keverjük össze. Rögtön szűrjük le. Az így nyert oldatot azonnal fel kell használni.
5. FELSZERELÉS
5.1. Szűrőtégely P 16/ISO 4793-as, 4-es porozitású, 30 vagy 50 ml űrtartalmú.
5.2. Szárítószekrény, 130 ± 2 °C hőmérsékletre beállítva.
6. A VIZSGÁLATI OLDAT ELKÉSZÍTÉSE
6.1. A kobaltoldat elkészítése Lásd J/1. vagy J/2. fejezet.
6.2. A vizsgálandó oldat elkészítése
Tegyünk a kivonatból legfeljebb 20 mg kobaltot tartalmazó alikvot részt egy 400 ml-es főzőpohárba. Ha a kivonatot a 10.2. módszerrel készítettük, savanyítsuk meg 5 csepp sósavval (4.3.). Adjunk hozzá kb. 10 ml hidrogén-peroxid oldatot (4. L). Hagyjuk az oxidálószert 15 percig hidegen hatni, majd az oldat térfogatát egészítsük ki vízzel kb. 100 ml-re. Fedjük le a főzőpoharat óraüveggel. Forraljuk fel, és hagyjuk forrni kb. 10 percig. Hűtsük le. Cseppenként lúgosítsuk meg a nátrium-hidroxid oldattal (4.21.), amíg fekete kobalt-hidroxid kezd kicsapódni.
7. ELJÁRÁS
Adjunk az oldathoz 10 ml ecetsavat (4.4.), és vízzel egészítsük ki kb. 200 ml-re a térfogatát. Melegítsük forrásig. Folyamatos keverés közben bürettával cseppenként adjunk hozzá 20 ml 1-nitrozo-2-naftol oldatot (4.6.). Erőteljes keveréssel fejezzük be, hogy a csapadék tömörödjön (koaguláljon).
Szűrjük át az előre lemért szűrőtégelyen (5.1.), vigyázva, hogy a szűrő ne tömődjön. Ezt szem előtt tartva ügyeljünk arra, hogy a szűrés alatt a csapadék felületét mindig folyadékréteg borítsa.
Mossuk át a főzőpoharat hígított ecetsavval (4.5.), hogy az összes csapadékot eltávolítsuk, mossuk a Csapadékot a szűrőn hígított ecetsavval (4.5.), és azután mossuk át háromszor forró vízzel.
130 ± 2 °C hőmérsékletű szárítószekrényben (5.2.) szárítsuk súlyállandóságig.
8. AZ EREDMÉNY MEGADÁSA
1 mg Co(C10H6ONO)3.2H2O csapadék 0,096381 mg kobaltnak felel meg.
A műtrágyában található kobalt (Co) százalékarányát következő képlettel lehet kiszámítani:
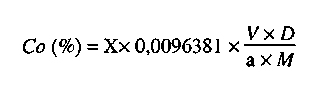
| ahol: |
| X = a csapadék tömege, mg, |
| V = a J/1. vagy J/2. fejezet szerint nyert kivonat-oldat mennyisége, ml, |
| a = az utolsó hígításból vett alikvot rész mennyisége, ml, |
| D = ennek az alikvot résznek a hígítási tényezője, |
| M = a vizsgálati minta tömege, g. |
J/7. MŰTRÁGYAKIVONATOKBAN LÉVŐ RÉZ MEGHATÁROZÁSA TITRIMETRIÁS MÓDSZERREL
1. TÁRGY
Ez a fejezet eljárást ír elő a műtrágyakivonatokban lévő réz meghatározására.
2. ALKALMAZÁSI TERÜLET
Ezt az eljárást olyan műtrágyamintákból vett kivonatokra kell alkalmazni, amelyeket a J/1. vagy a J/2. eljárással nyertek, és amelyekre nézve a 3. számú melléklet 1. függeléke a réztartalom bejelentését írja elő.
3. A MÓDSZER ELVE
A réz(II) ionokat savas közegben kálium-jodiddal redukáljuk:
2 Cu ++ + 4 I - -> 2 CuI + J2.
Az így felszabadult jódot nátrium-tioszulfát mérőoldattal, keményítő indikátor jelenlétében titráljuk a következő képlet szerint:
I2 + 2 Na2S2O3 -> 2 NaI + Na2S4O6.
4. VEGYSZEREK
4.1. Salétromsav (HNO3, d20 = 1,40 g/ml).
4.2. Karbamid [(NH2)2C= 0].
4.3. Ammónium-hidrogén-difluorid (NH4HF2) oldat, 10% (m/V) oldata. Az oldatot műanyag edényben kell tartani.
4.4. Ammónium-hidroxid oldat (1 + 1).
Elegyítsünk 1 térfogatrész ammóniát (NH4OH, ρ = 0,9 g/ml) 1 térfogatrész vízzel.
4.5. Nátrium-tioszulfát mérőoldat
Oldjunk fel 7,812 g nátrium-tioszulfát pentahidrátot (Na2S2O3.5H2O) vízzel, 1 literes mérőlombikban. Az oldat 1 ml-e 2 mg rézzel egyenértékű. Stabilizáljuk az oldatot néhány csepp kloroformmal. Az oldatot közvetlen fénytől védve, üvegedényben kell tartani.
4.6. Kálium-jodid (KI).
4.7. Kálium-tiocianát (KSCN) oldat 25% (m/V) Tartsuk az oldatot műanyag palackban.
4.8. Keményítő oldat (kb. 0,5%)
Tegyünk 2,5 g keményítőt egy 600 ml-es főzőpohárba. Öntsünk hozzá kb. 500 ml vizet. Keverés közben forraljuk fel. Hűtsük le szobahőmérsékletre. Az oldatot csak rövid ideig lehet eltartani. Az eltarthatóságot javíthatjuk, ha kb. 10 mg higany-jodidot adunk az oldathoz.
5. A VIZSGÁLATI OLDAT ELKÉSZÍTÉSE
A rézoldat elkészítése Lásd J/1. és J/2. fejezet.
6. ELJÁRÁS
6.1. Oldatkészítés titráláshoz
Tegyünk egy legalább 20-40 mg rezet tartalmazó alikvot részt az oldatból 500 ml-es Erlenmeyer-lombikba.
Rövid forralással űzzük el a jelen lévő oxigénfelesleget. A térfogatot egészítsük ki kb. 100 ml-re vízzel. Adjunk hozzá 5 ml salétromsavat (4.1.), forraljuk fel, és hagyjuk forrni kb. fél percig.
Vegyük le az Erlenmeyer-lombikot a fűtőkészülékről, adjunk hozzá kb. 3 g karbamidot (4.2.), és ismét forraljuk kb. fél percig.
Vegyük le a fűtőkészülékről, és adjunk hozzá 200 ml hideg vizet. Ha szükséges, hűtsük az Erlenmeyer-lombik tartalmát szobahőmérsékletre.
Fokozatosan adjunk hozzá ammónium-hidroxid oldatot (4.4.), amíg az oldat színe kékre változik, akkor adjunk még hozzá 1 ml-t feleslegben.
Adjunk hozzá 50 ml ammónium-hidrogén-difluorid oldatot (4.3.), és keverjük össze.
Adjunk hozzá 10 g kálium-jodidot (4.6.), és oldjuk fel.
6.2. Az oldat titrálása
Helyezzük az Erlenmeyer-lombikot mágneses keverőre. Tegyük a keverőrudat az Erlenmeyer-lombikba, és állítsuk be a keverőt a kívánt fordulatszámra.
Adjunk hozzá bürettával nátrium-tioszulfát mérőoldatot (4.5.) addig, amíg az oldatból felszabaduló jód barna színe halványabbá válik.
Adjunk hozzá 10 ml keményítő-oldatot (4.8.).
Folytassuk a nátrium-tioszulfát oldattal (4.5.) végzett titrálást, amíg a bíbor szín csaknem eltűnik.
Adjunk hozzá 20 ml kálium-tiocianát oldatot (4.7.), és folytassuk a titrálást addig, amíg az ibolyakék szín teljesen el nem tűnik.
Jegyezzük fel a felhasznált tioszulfát oldat mennyiségét.
7. AZ EREDMÉNY MEGADÁSA
1 ml nátrium-tioszulfát mérőoldat (4.5.) 2 mg réznek felel meg.
A műtrágyában lévő réz százalékarányát a következő képlettel lehet kiszámítani:
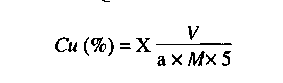
| ahol: |
| X = A felhasznált nátrium-tioszulfát oldat térfogata, ml-ben, |
| V = A J/1. és a J/2. fejezet szerinti kivonat-oldat térfogata, ml-ben, |
| A = Az alikvot rész térfogata, ml-ben, |
| M = A J/1. és a J/2. fejezet szerint kezelt vizsgálati minta tömege, g-ban. |
J/8. A MŰTRÁGYAKIVONATOKBAN LÉVŐ VAS MEGHATÁROZÁSA ATOMABSZORPCIÓS SPEKTROMETRIÁVAL
1. TÁRGY
Ez a fejezet eljárást ír elő a műtrágyakivonatokban lévő vas meghatározására.
2. ALKALMAZÁSI TERÜLET
Ezt az eljárást olyan, műtrágyamintákból vett kivonatokra kell alkalmazni, amelyeket a J/1. és a J/2. módszerrel nyertek, és amelyekre nézve a 3. számú melléklet 1. függeléke az összes, és/vagy a vízoldható vas bejelentését írja elő.
3. A MÓDSZER ELVE
A kivonat megfelelő kezelése és hígítása után a vastartalmat atomabszorpciós spektrometriával határozzuk meg.
4. VEGYSZEREK
4.1. Sósavoldat, kb. 6 mol/l Lásd J/4. fejezet (4.1.).
4.2. Sósavoldat, kb. 0,5 mol/l Lásd J/4. fejezet (4.2.).
4.3. Hidrogén-peroxid oldat (30% H2O2 d20 =1,11 g/ml), mikroelemektől mentes.
4.4. Lantánsó oldatok (10 g La/l) Lásd J/4. fejezet (4.3.).
4.5. Vas kalibráló oldat
4.5.1. Vas törzsoldat (l000 μg/ml)
Mérjünk 1 g tiszta vashuzalt 0,1 mg pontossággal 500 ml-es főzőpohárba, adjunk hozzá 200 ml 6 mol/l sósavat (4.1.), és 15 ml hidrogén-peroxid oldatot (4.3.). Melegítsük főzőlapon, amíg a vas teljesen feloldódik. Amikor lehűlt, vigyük át veszteség nélkül egy 1000 ml-es mérőlombikba. Töltsük fel jelig vízzel, és jól keverjük össze.
4.5.2. Vas munkaoldat (100 μg/ml)
Öntsünk 20 ml törzsoldatot (4.5.1.) 200 ml-es mérőlombikba. Töltsük jelig a 0,5 mol/l sósav oldattal (4.2.), és jól keverjük össze.
5. FELSZERELÉS
Atomabszorpciós spektrométer: lásd 10.4. módszer (5.). A műszer legyen ellátva vas mérésére alkalmas sugárforrással (248,3 nm).
6. A VIZSGÁLANDÓ OLDAT ELKÉSZÍTÉSE
6.1. Vas kivonat-oldat
Lásd: J/1., és/vagy J/2., és szükség szerint a J/3. fejezet.
6.2. A mintaoldat elkészítése
Lásd J/4. fejezet (6.2.). A mintaoldat 10% (v/v) lantánsó oldatot tartalmazzon.
7. ELJÁRÁS
7.1. Vakoldat készítése
Lásd J/4. fejezet (7.1.). A vakoldat 10% (v/v), a 6.2. pontban használt lantánsó oldatot tartalmazzon.
7.2. Kalibráló oldatok készítése Lásd J/4. fejezet (7.2.).
A vas 0-10 μ g/ml közötti optimális meghatározási tartományához öntsünk 0, 2, 4, 6, 8 és 10 ml munkaoldatot (4.5.2.) 100 ml-es mérőlombikokba. Ha szükséges, állítsuk be a sósav-koncentrációt, hogy a lehető legjobban megközelítse a mintaoldatét. Adjunk mindegyik mérőlombikba 10 ml, a 6.2. pontban használt lantánsó oldatot. Töltsük jelig a lombikot 0,5 M sósavoldattal (4.2.), és jól keverjük össze. Az oldatok rendre 0, 2, 4, 6, 8 és 10 μ g/ml vasat tartalmaznak.
7.3. Meghatározás
Lásd J/4. fejezet (7.3.). Készítsük elő a spektrométert (5.) a 248,3 nm-es hullámhosszon történő méréshez.
8. AZ EREDMÉNY MEGADÁSA
Lásd J/4. fejezet (8.).
A műtrágya százalékos vastartalmát a következő képlettel lehet kiszámítani:
Fe%=[xs- xb) x V xD]/(M x 104).
Ha a J/3. fejezet szerinteket használjuk:
Fe%=[xs-xb) x V x 2D]/(M x 104).
Ahol:
Fe = a vas mennyisége a műtrágya százalékában kifejezve,
Xs = a mintaoldat (6.2.) koncentrációja μg/ml-ben,
Xb = a vakoldat (7.1.) koncentrációja μg/ml-ben,
V = J/1. vagy J/2. fejezet szerint nyert kivonat mennyisége ml-ben,
D = a 6.2. pontban végzett hígítás tényezője,
M = a J/1. vagy J/2. fejezet szerint vett vizsgálati minta tömege grammban.
INNEN
A D hígítási tényező számítása: ha (a1), (a2), (a3)..., (ai) és (a) alikvot részek, és (v1), (v2), (v3),..., (vi) és (100) a hígításuknak megfelelő mennyiségek, ml-ben megadva, a D hígítási tényezőt a következő képlettel lehet számítani:
D=(v1/a1) x (v2/a2) x (v3/a3) x ...x (vi/ai) x (100/a).
J/9. MŰTRÁGYAKIVONATOKBAN LÉVŐ MANGÁN MEGHATÁROZÁSA TITRÁLÁSSAL
1. TÁRGY
Ez a módszer eljárást ír elő a műtrágyakivonatokban lévő mangán meghatározására.
2. ALKALMAZÁSI TERÜLET
Ezt az eljárást olyan műtrágyamintából nyert kivonatokra kell alkalmazni, amelyeket a J/1. és a J/2. fejezet alapján nyertek, és amelyekre a 3. számú melléklet 1. függeléke a mangán bejelentését írja elő.
3. A MÓDSZER ELVE
Ha a kivonatban kloridionok vannak jelen, akkor azokat a kivonat kénsavas forralásával el kell távolítani. A mangánt salétromsavas közegben nátrium-bizmutáttal oxidáljuk. A képződött kálium-permanganátot vas(II)-szulfát feleslegével redukáljuk. Ezt a felesleget kálium-permanganát oldattal titráljuk vissza.
4. VEGYSZEREK
4.1. Koncentrált kénsav (H2SO4, d20 = 1,84 g/ml).
4.2. Kénsav, kb. 9 mol/l
Gondosan elegyítsünk 1 térfogatrész koncentrált kénsavat (4.1.) 1 térfogatrész vízzel.
4.3. Salétromsav, 6 mol/l
Elegyítsünk 3 térfogatrész salétromsavat (HNO3, ρ =1,40 g/ml) 4 térfogatrész vízzel.
4.4. Salétromsav, 0,3 mol/l
Elegyítsünk 1 térfogatrész 6 M salétromsavat 19 térfogatrész vízzel.
4.5. Nátrium-bizmutát (NaBiO3) (85%).
4.6. Kovaföld.
4.7. Ortofoszforsav, 15 mol/l (H3PO4, ρ = 1,71 g/ml).
4.8. Vas-szulfát oldat, 0,15 mol/l
Oldjunk fel 41,6 g vas-szulfát.heptahidrátot (FeSO4.7 HaO) 1 literes mérőlombikban.
Adjunk hozzá 25 ml koncentrált kénsavat (4.1.), és 25 ml foszforsavat (4.7.). Töltsük fel 1000 ml-re. Keverjük össze.
4.9. Kálium-permanganát oldat, 0,020 mol/l
Mérjünk be 3,160 g kálium-permanganátot (KMnO4) 0,1 mg pontossággal. Oldjuk fel, és töltsük fel 1000 ml-re vízzel.
4.10. Ezüst-nitrát oldat, 0,1 mol/l
Oldjunk fel 1,7 g ezüst-nitrátot (AgNO3) vízben, és töltsük fel 100 ml-re.
5. FELSZERELÉS
5.1. Szűrőtégely, P16/ISO 4793 szerint, 4-es porozitású, 50 ml térfogatú, 500 ml-es szűrőlombikra szerelve.
5.2. Mágneses keverő.
6. A VIZSGÁLANDÓ OLDAT ELKÉSZÍTÉSE
6.1. Mangán kivonat-oldat
Lásd a J/1. és J/2. fejezetet. Ha nem ismeretes, hogy vannak-e jelen kloridionok, akkor egy csepp ezüst-nitrát oldattal (4.10.) végezzünk kísérletet ennek megállapítására.
6.2. Ha kloridionok nincsenek jelen, 10-20 mg mangánt tartalmazó alikvot részt vigyünk magas főzőpohárba. A térfogatot állítsuk be kb. 25 ml-re bepárlással vagy víz hozzáadásával. Adjunk hozzá 2 ml koncentrált kénsavat (4.1.).
6.3. Ha kloridionok vannak jelen, akkor azokat a következőképpen kell eltávolítani.
Vigyük a kivonatból 10-20 mg mangánt tartalmazó alikvot részt 400 ml-es magas főzőpohárba. Adjunk hozzá 5 ml 9 M kénsavat (4.2.). Vegyifülke alatt forraljuk főzőlapon mindaddig, amíg bőséges fehér füstfejlődést nem észlelünk. Addig folytassuk a forralást, amíg a térfogat kb. 2 ml-re nem csökken (a főzőpohár alján vékony, szirupszerű folyadékréteg látható). Hagyjuk lehűlni szobahőmérsékletre.
Óvatosan adjunk hozzá 25 ml vizet, és újra vizsgáljuk meg 1 csepp ezüst-nitrát oldat (4.10.) hozzáadásával, hogy vannak-e jelen kloridionok. Ha még mindig van klorid az oldatban, ismételjük meg a műveletet 5 ml 9 M kénsav (4.2.) hozzáadása után.
7. ELJÁRÁS
Adjunk a mintaoldatot tartalmazó 400 ml-es főzőpohárba 25 ml 6 M salétromsavat (4.3.), és 2,5 g nátrium-bizmutátot (4.5.). Keverjük erőteljesen 3 percig a mágneses keverőn (5.2.).
Adjunk hozzá 50 ml 0,3 M salétromsavat (4.4.), és keverjük újra. Szűrjük vákuumban, olyan szűrőtégelyen (5.1.) keresztül, amelynek alját kovafölddel (4.6.) borítottuk. Mossuk át a szűrőt néhányszor a 0,3 M salétromsavval (4.4.), amíg színtelen szűrletet nem kapunk.
Vigyük a szűrletet és a mosófolyadékot 500 ml-es főzőpohárba. Keverjük össze, és adjunk hozzá 25 ml 0,15 M vas-szulfát oldatot (4.8.). Ha vas-szulfát hozzáadásakor a szűrlet megsárgul, adjunk hozzá 3 ml 15 M ortofoszforsavat (4.7.)
Bürettából titráljuk a fölös vas-szulfátot 0,02 M kálium-permanganát oldattal (4.9.), amíg az elegy színe rózsaszínbe megy át, és ez a szín 1 percig állandó marad. Végezzünk vakpróbát azonos feltételek mellett, de a vizsgálati minta kihagyásával.
Megjegyzés:
Az oxidált oldatnak nem szabad gumival érintkeznie.
8. AZ EREDMÉNY MEGADÁSA
1 ml 0,02 kálium-permanganát oldat 1,099 mg mangánnak (Mn) felel meg.
A műtrágya százalékos mangántartalmát a következő képlettel lehet kiszámítani:
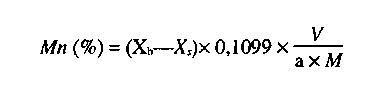
| ahol: |
| Xb = a vakpróbához felhasznált kálium-permanganát mennyisége ml-ben, |
| Xs = a vizsgálati mintához felhasznált kálium-permanganát mennyisége ml-ben, |
| V = a J/1. és J/2. fejezet szerint nyert kivonat-oldat mennyisége ml-ben, |
| a = a kivonatból vett alikvot rész mennyisége ml-ben, |
| M = a vizsgálati minta tömege g-ben. |
J/10. MŰTRÁGYAKIVONATOKBAN LÉVŐ MOLIBDÉN MEGHATÁROZÁSA GRAVIMETRIÁS MÓDSZERREL 8-HIDROXI-KINOLINNAL
1. TÁRGY
Ez a fejezet eljárást ír elő műtrágyakivonatokban lévő molibdén meghatározására.
2. ALKALMAZÁSI TERÜLET
Az eljárást olyan, műtrágyából vett minták kivonataira kell alkalmazni, amelyeket a J/1. és a J/2. módszerrel nyertek, és amelyekre a 3. számú melléklet 1. függeléke a molibdén bejelentését írja elő.
3. A MÓDSZER ELVE
A molibdént meghatározott körülmények között lecsapatott molibdenil-oxinát formájában határozzuk meg.
4. VEGYSZEREK
4.1. Kénsavoldat kb. 1 mol/l
Óvatosan töltsünk 55 ml kénsavat (H2SO4 ρ =1,84 g/ml) 1 literes, 800 ml vizet tartalmazó mérőlombikba. Keverjük össze. Lehűlés után egészítsük ki a térfogatát 1 literre. Keverjük össze.
4.2. Hígított ammónia oldat (1:3).
Elegyítsünk 1 térfogatrész koncentrált ammónia oldatot (NH4OH, d20=0,9 g/ml) 3 térfogatrész vízzel.
4.3. Hígított ecetsav oldat (1:3)
Elegyítsünk 1 térfogatrész tömény ecetsavat (99,7% CH3COOH, ρ = 1,049 g/ml) 3 térfogatrész vízzel.
4.4. Dinátrium-etilén-diamin-tetraacetát (EDTA)
Oldjunk fel 5 g Na2EDTA-t vízben, 100 ml-es mérőlombikban. Töltsük a jelig és keverjük össze.
4.5. Pufferoldat
100 ml-es mérőlombikban oldjunk fel 15 ml tömény ecetsavat és 30 g ammónium-acetátot vízben. Töltsük fel 100 ml-re.
4.6. Hidroxi-kinolin (oxin) oldat
100 ml-es mérőlombikban oldjunk fel 3 g 8-hidroxi-kinolint 5 ml koncentrált ecetsavban. Adjunk hozzá 80 ml vizet. Cseppenként adjunk hozzá ammónia oldatot (4.2.), amíg az oldat zavarossá nem válik, azután addig adjuk hozzá az ecetsavat (4.3.), amíg az oldat újra kitisztul. Töltsük fel vízzel 100 ml-re.
5. FELSZERELÉS
5.1. Szűrőtégely, P16/ISO 4793 szerint, 4-es porozitású, 30 ml térfogatú.
5.2. pH-mérő, üvegelektróddal.
5.3. Szárítószekrény, 130-135 °C közötti hőmérsékleten.
6. A VIZSGÁLATI OLDAT ELKÉSZÍTÉSE
6.1. A molibdén oldat elkészítése. Lásd a J/1. és a J/2. módszert.
7. ELJÁRÁS
7.1. A mintaoldat el készítése
Vigyünk 20-100 mg molibdént tartalmazó alikvot részt 250 ml-es főzőpohárba. Egészítsük ki a térfogatát vízzel 50 ml-re. Állítsuk be az oldat kémhatását kénsav oldat (4.1.) cseppenkénti adagolásával pH 5-re. Adjunk hozzá 15 ml EDTA oldatot (4.4.), azután 5 ml pufferoldatot (4.5.). Vízzel töltsük fel kb. 80 ml-re.
7.2. A csapadék kinyerése és tisztítása A csapadék kinyerése
Enyhén melegítsük az oldatot. Folyamatos keverés mellett adjuk hozzá az oxin oldatot (4.6.). Addig folytassuk a lecsapatást, amíg csapadék kiválása már nem észlelhető. Adjunk még hozzá annyi vegyszert, hogy a felülúszó oldat enyhén sárgára színeződjék. Általában 20 ml elegendő. Enyhén melegítsük a csapadékot még két-három percig.
Szűrés és mosás
Szűrjük szűrőtégelyen (5.1.). Öblítsük néhányszor 20 ml forró vízzel. Az öblítővíznek fokozatosan színtelenné kell válnia, jelezve azt, hogy oxin már nincs jelen.
7.3. A csapadék mérése
Szárítsuk a csapadékot súlyállandóságig (legalább 1 órán keresztül) 130-135 °C közötti hőmérsékleten. Hagyjuk exszikkátorban lehűlni, és mérjük le.
8. AZ EREDMÉNY MEGADÁSA
1 mg molibdenil-oxinát, MoC2(C9H6ON)2 0,2305 mg molibdénnek felel meg.
A műtrágya százalékos molibdén tartalmát a következő képlettel lehet kiszámítani:
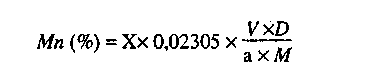
| ahol: |
| X = a molibdenil-oxinát csapadék tömege mg-ban, |
| V = a J/1. vagy J/2. fejezet szerinti kivonat-oldat mennyisége ml-ben, |
| a = az utolsó hígításból vett alikvot rész mennyisége ml-ben, |
| D = az alikvot rész hígítási tényezője, |
| M = a vizsgálati minta tömege g-ban. |
J/11. A MŰTRÁGYAKIVONATOKBAN LÉVŐ CINK MEGHATÁROZÁSA ATOMABSZORPCIÓS SPEKTROMETRIÁVAL
1. TÁRGYA
Ez a módszer eljárást ír elő a műtrágyakivonatokban lévő cink meghatározására.
2. ALKALMAZÁSI TERÜLET
Ezt az eljárást olyan, műtrágyamintákból vett kivonatokra kell alkalmazni, amelyeket a J/1. és a J/2. fejezettel nyertek, és amelyekre nézve a 3. számú melléklet 1. függeléke a cink bejelentését írja elő.
3. A MÓDSZER ELVE
A kivonat megfelelő kezelése és hígítása után a cinktartalmat atomabszorpciós spektrometriával határozzuk meg.
4. VEGYSZEREK
4.1. Sósav oldat, kb. 6 mol/l Lásd J/4. fejezet (4.1.).
4.2. Sósav oldat, kb. 0,5 mol/l Lásd J/4. fejezet (4.2.).
4.3. Lantánsó oldatok (10 g La/l)
Lásd J/4. fejezet, (4.3.).
4.4. Cink kalibráló oldatok
4.4.1. Cink törzsoldat (1000 μg/ml)
1000 ml-es mérőlombikban oldjunk fel 1 g 0,1 mg-os pontossággal bemért cinkport vagy -pelyhet 25 ml 6 mol/l töménységű sósavban (4.1.). Amikor teljesen feloldódott, töltsük fel vízzel, és jól keverjük össze.
4.4.2. Cink munkaoldat (100 μg/ml)
200 ml-es mérőlombikban hígítsunk a törzsoldatból (4.4.1.) 20 ml-t 0,5 mol/l sósav oldatban (4.2.). Töltsük jelig 0,5 M sósav oldattal és jól keverjük össze.
5. FELSZERELÉS
Atomabszorpciós spektrométer
Lásd a J/4. fejezetet (5.)- A készülék legyen ellátva cink mérésére alkalmas sugárforrással (213,8 nm). A spektrométer legyen alkalmas háttérkorrekció végzésére.
6. A VIZSGÁLATI OLDAT ELKÉSZÍTÉSE
6.1. Cink kivonat-oldat
Lásd a J/1. és/vagy J/2. fejezetet.
6.2. Mintaoldat készítése
Lásd a J/4. fejezetet (6.2.). A mintaoldat 10% (v/v) lantánsó oldatot (4.3.) tartalmazzon.
7. ELJÁRÁS
7.1. Vakoldat készítése
Lásd a J/4. (7.1.) fejezetet. A vakoldat 10% (v/v), a 6.2. pontban használt lantánsó oldatot tartalmazzon.
7.2. Kalibráló oldat készítése
Lásd a J/4. fejezetet (7.2.). A cink 0-5 m g közé eső optimális meghatározási tartományához vigyünk 0, 0,5, l, 2, 3, 4 és 5 ml munkaoldatot (4.4.2.) rendre 100 ml-es mérőlombikokba. Ha szükséges, állítsuk be a sósavoldat koncentrációját úgy, hogy a lehető legközelebb essék a mintaoldatéhoz. Adjunk mindegyik mérőlombikba 10 ml, a 6.2. pontban használt lantánsó oldatot. Töltsük jelig a lombikokat a 0,5 M sósavoldattal (4.2.), és jól keverjük össze.
Ezek az oldatok rendre 0, 0,5, l, 2, 3, 4 és 5 m g/ml cinket tartalmaznak.
7.3. Meghatározás
Lásd a J/4. fejezetet (7.3.). Készítsük elő a spektrométert (5.) 213,8 nm-en történő méréshez.
8. AZ EREDMÉNY MEGADÁS A
Lásd a J/4. fejezetet (8.).
A műtrágya százalékos cinktartalmát a következő képlettel lehet kiszámítani:
Zn%=[(Xs-Xb) x V x D]/(Mx 104).
Ha a J/3. fejezetet használtuk:
Zn%=[(Xs-Xb) x V x 2D]/(Mx 104).
ahol:
Zn = a cink mennyisége a műtrágya százalékában kifejezve,
Xs = a műtrágyaoldat koncentrációja μg/ml-ben,
Xb = a vakoldat koncentrációja μg/ml-ben,
V = a J/1. vagy J/2. fejezet szerint nyert kivonat-oldat mennyisége ml-ben,
D = a (6.2.) pontban végzett hígítás tényezője,
M = a J/1. vagy J/2. fejezet szerint vett vizsgálati minta tömege g-ban.
A D hígítási tényező számítása: ahol (a1), (a2), (a3),... (ai) és (a) az egymást követő alikvot részek, és (v1), (v2), (v3), ... (vi) és (100) a hígításuknak megfelelő mennyiségek, a D hígítási tényező:
D=(v1/a1) x (v2/a2) x (v3/a3)x ... x (vi/ai) x (100/a).
8. számú melléklet a 8/2001. (I. 26.) FVM rendelethez[82]
A magas nitrogéntartalmú egyszerű ammónium-nitrát műtrágyák jellemzőinek, robbanó képességének ellenőrzésére vonatkozó eljárások
A 2. számú melléklet 1. MŰTRÁGYÁK fejezetének 1.3. pontjában és a 3. számú melléklet 1. MŰTRÁGYÁK fejezetének 1.3. pontjában meghatározott tulajdonságok határértékeinek való megfelelés ellenőrzésére szolgáló módszerek.
I. A HŐKEZELÉSI CIKLUSOK ALKALMAZÁSÁNAK MÓDSZEREI
1. TÁRGY ÉS ALKALMAZÁSI TERÜLET
Ez a módszer meghatározza az olaj-visszatartási (porozitási) és robbanóképesség vizsgálat végrehajtása előtti hőkezelési ciklusok alkalmazására vonatkozó eljárásokat a magas nitrogéntartalmú egyszerű ammónium-nitrát műtrágyák esetében.
2. A 2. SZÁMÚ MELLÉKLET 1. MŰTRÁGYÁK FEJEZETÉNEK 1.3. PONTJÁBAN HIVATKOZOTT OLAJ-VISSZATARTÁSI VIZSGÁLATOT MEGELŐZŐ HŐKEZELÉSI CIKLUSOK
2.1. Alkalmazási terület
Ez az eljárás a műtrágya olaj visszatartásának meghatározása előtt alkalmazott hőkezelési ciklus eljárásra vonatkozik. Az alkalmazandó hőkezelési ciklusok száma kettő.
2.2. Alapelv és fogalom-meghatározás
A vizsgálati minta felmelegítése szoba hőmérsékletről 50 °C-os hőmérsékletre és ezen való tartása két órán keresztül (50 °C-os fázis). A minta lehűtése 25 °C-ra és ezen a hőmérsékleten való tartása két órán keresztül (25 °C-os fázis). A két egymást követő 50 °C-os és 25 °C-os fázis alkot egy hőkezelési ciklust. Az olajvisszatartás meghatározása céljából két hőkezelési ciklusnak kitett mintát 203 °C-os hőmérsékleten kell tartani.
2.3. Készülékek
Normál laboratóriumi felszerelés, különösen:
- 25 l, illetve 50 ± 1 °C-os termosztált vízfürdők,
- Erlenmeyer-lombikok, 150 ml-es.
2.4. Eljárás
Az egyes vizsgálati mintákból mérjünk 70 ± 5 grammot egy-egy Erlenmeyer-lombikba, és dugóval zárjuk le. Az egyes lombikokat helyezzük át minden két órában az 50 °C-os fürdőből a 25 °C-os fürdőbe és viszont. Az egyes fürdőkben a víz hőmérsékletét tartsuk állandó értéken, valamint a vizet tartsuk gyors keveréssel mozgásban annak érdekében, hogy a vízszint a minta szintje felett legyen. A dugót védjük meg a vízpára lecsapódástól habgumi kupakkal.
3. A 2. SZÁMÚ MELLÉKLET 1. MŰTRÁGYÁK FEJEZETÉNEK 1.3. PONTJÁBAN HIVATKOZOTT ROBBANÓKÉPESSÉG VIZSGÁLATOT MEGELŐZŐ HŐKEZELÉSI CIKLUSOK
3.1. Alkalmazási terület
Ez az eljárás a robbanó-képesség vizsgálat előtti hőkezelési ciklusos vizsgálatra vonatkozik. Az alkalmazandó hőkezelési ciklusok száma öt.
3.2. Alapelv és fogalom meghatározás
Vízálló tartályban a mintát a szoba hőmérsékletről melegítsük fel 50 °C-ra, és tartsuk ezen a hőmérsékleten egy órán keresztül (50 °C-os fázis). Ezután a mintát hűtsük le 25 °C-ra, és tartsuk ezen a hőmérsékleten egy órán keresztül (25 °C-os fázis). Az 50 °C-os és a 25 °C-os fázis egymás utáni kombinációja alkot egy hőkezelési ciklust. A megfelelő számú hőkezelési ciklus után a mintát tartsuk 20 ± 3 °C-os hőmérsékleten a robbanó-képesség vizsgálat végrehajtásáig.
3.3. Készülék
- 20 és 51 °C között termosztált vízfürdő, amelynek minimális melegedési és hűtési sebessége legalább 10 °C/h, vagy két vízfürdő, amelyek közül az egyik 20 °C-on termosztált, a másik 51 °C-on. A vízfürdőkben a vizet folyamatosan keverni kell; a fürdő méretének elég nagynak kell lennie ahhoz, hogy a víz megfelelő áramlását biztosítsa.
- Rozsdamentes acél tartály, amely mindenütt vízzáró, és a közepén termoelemmel van ellátva. A tartály külső szélessége 45 (2) mm, falvastagsága 1,5 mm (lásd az 1. ábrát). A tartály magassága és hosszúsága a vízfürdő méreteinek megfelelően választható meg, pl. a hosszúság 600 mm, a magasság 400 mm.
3.4. Eljárás
Helyezzünk egy robbantáshoz szükséges mennyiségű műtrágyát a tartályba, és zárjuk le a zárófedelet. Helyezzük a tartályt a vízfürdőbe. Melegítsük fel a vizet 51 °C-ra, és mérjük meg a hőmérsékletet a műtrágya közepén. Egy órával azután, hogy a hőmérséklet a középpontban elérte az 50 °C-t, a vizet hűtsük le. Egy órával azután, hogy a hőmérséklet a középpontban 25 °C-ra süllyedt, a vizet újra melegítsük fel a második ciklus megkezdéséhez.
1. ábra Az olajvisszatartás meghatározása
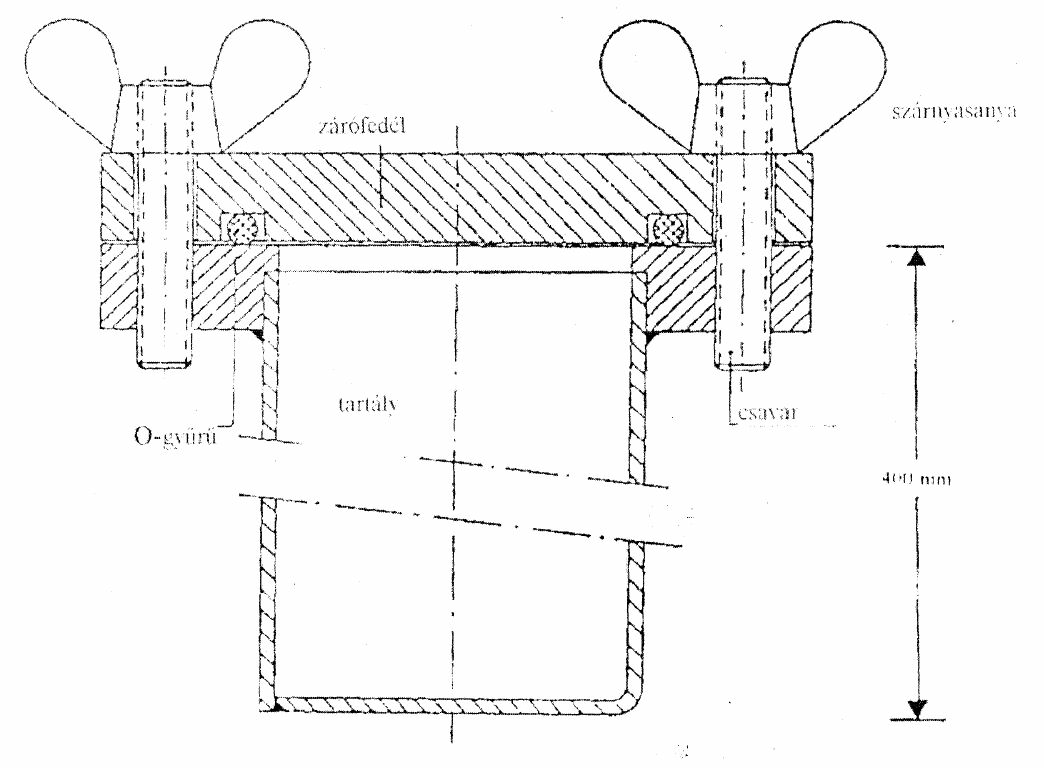
II. AZ OLAJVISSZATARTÁS MEGHATÁROZÁSA
1. TÁRGY ÉS ALKALMAZÁSI TERÜLET
Ez a módszer a magas nitrogéntartalmú egyszerű ammónium-nitrát műtrágyák olaj visszatartásának meghatározására vonatkozó eljárást határozza meg.
A módszer alkalmazható mind szemcsézett, mind granulált műtrágyáknál, amelyek nem tartalmaznak olajban oldódó anyagokat.
2. FOGALOMMEGHATÁROZÁS
Egy műtrágya olajvisszatartása: a műtrágya által visszatartott olaj tömegszázalékban kifejezett mennyisége a meghatározott vizsgálati feltételek között.
3. ALAPELV
A vizsgálati mennyiség meghatározott időre teljes bemerítésre kerül a gázolajba, amit a fölös olaj meghatározott feltételek mellett történő eltávolítása követ. Ezután a vizsgált anyagmennyiség tömegnövekedésének mérése történik.
4. REAGENS
Gázolaj
Maximális viszkozitás: 5 mP 40 °C-on. Sűrűség: 0,8 és 0,85 g/ml között 20 °C-on. Kéntartalom: < 1,0% (tömegszázalék) Hamu: < 0, 1 % (tömegszázalék)
5. KÉSZÜLÉKEK
Szokásos laboratóriumi felszerelés, és:
5.1. Legalább 0,01 gramm pontosságú mérleg
5.2. Főzőpoharak, 500 ml-es.
5.3. Műanyagból készült tölcsér, lehetőleg henger alakú fallal a felső végénél, megközelítőleg 200 mm átmérőjű.
5.4. A tölcsérbe (5.3.) illő vizsgáló szita, 0,5 mm lyukméretű.
Megjegyzés:
A tölcsér és a szita méretének olyannak kell lennie, hogy csupán néhány szemcse legyen egymáson és az olaj könnyen eltávozhasson.
5.5. Szűrőpapír, gyors szűrőképességű, krepp, puha, tömege 150 g/m2.
5.6. Abszorbens kendő (laboratóriumi minőség).
6. ELJÁRÁS
Két külön párhuzamos meghatározás történik gyors egymásutánban ugyanannak a vizsgálati mintának a különböző részletein.
6.1. A vizsgáló szita (5.4.) segítségével távolítsuk el a 0,5 mm-nél kisebb részecskéket. 0,01 gramm pontossággal mérjünk be 50 gramm mintát a főzőpohárba (5.2.). Adjunk elegendő mennyiségű gázolajat (4. rész) úgy, hogy teljesen befedje a szemcséket, és óvatosan keverjük, hogy az olaj az összes szemcse teljes felületét benedvesítse. Fedjük be a főzőpoharat óraüveggel, és hagyjuk állni egy órán keresztül 25 ± 2 °C-on.
6.2. Szűrjük le a főzőpohár teljes tartalmát a vizsgáló szitával (5.4.) ellátott tölcséren keresztül (5.3.). Hagyjuk a szűrőn a fennmaradt mennyiséget egy órán keresztül, hogy az olaj fölösleg legnagyobb, része lefolyhasson.
6.3. Egy sima felületen fektessünk két szűrőpapírt (5.5.) (500 x 500 mm) egymásra; hajtsuk mindkét szűrőpapír négy szélét felfelé körülbelül 40 mm szélességig, hogy ez megakadályozza a szemcsék leszóródását. Helyezzünk két réteg abszorbens kendőt a szűrőpapírok közepére. Ontsuk ki a szűrő (5.4.) teljes tartalmát az abszorbens kendőre, és puha, sima ecsettel terítsük el egyenletesen a szemcséket. Két perc elteltével emeljük fel az abszorbens kendők egyik oldalát, hogy a szemcsék átkerüljenek az alul lévő szűrőpapírokra, és terítsük el egyenletesen a szemcséket az ecsettel. Helyezzünk egy másik szűrőpapírt - hasonlóan felfelé hajtott szélekkel - a mintára, és körkörös mozdulatokkal, gyenge nyomást gyakorolva görgessük a szemcséket a két szűrőpapír között. Minden nyolcadik kör után tartsunk szünetet, és emeljük fel a szűrőpapírok szemben lévő oldalait, hogy a szélre került szemcsék visszakerüljenek a közepére. Alkalmazzuk a következő eljárást: végezzünk négy teljes körzést, először az óramutató járásával megegyezően, majd az óramutató járásával ellenkezően. Ezután görgessük vissza a szemcséket a fent leírt módon. Ezt az eljárást háromszor ismételjük meg (24 körzés, a szélek kétszer kerülnek felemelésre). Óvatosan helyezzünk új szűrőpapírt az alsó és a felette lévő lap közé, és a felső lap széleit felemelve engedjük a szemcséket legurulni az új lapra. Borítsuk be a szemcséket új szűrőpapírral, és ismételjük meg a fent leírt eljárást. Közvetlenül a görgetés után ontsuk a szemcséket kitárázott edénybe, és a visszatartott gázolaj tömegének meghatározásához újra mérjük meg 0,01 gramm pontossággal.
6.4. A görgetési eljárás megismétlése és a mérés megismétlése
Ha a visszatartott gázolaj mennyisége a vizsgálati anyagban meghaladja a 2,00 grammot, helyezzük a vizsgálati anyagot új szűrőpapírokra, és ismételjük meg a görgetési eljárást, a sarkokat a 6.3. résznek megfelelően felemelve (kétszer nyolc körzés, egy felemelés). Ezután mérjük meg újra a vizsgálati mennyiséget.
7. AZ EREDMÉNYEK KIFEJEZÉSE
7.1. A számítás módja és a képlet
Az olajvisszatartás mértékét az egyes meghatározások esetében a megszitált vizsgálati minta mennyiség tömegszázalékában kifejezve a következő egyenlet adja meg:
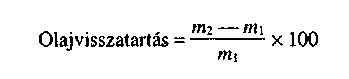
| ahol: |
| m1 = a szűrt vizsgálati mintamennyiség (6.1.) tömege grammban kifejezve; |
| m2 = a 6.3., illetve a 6.4. résznek megfelelő vizsgálati mintamennyiség tömege grammban kifejezve az utolsó |
| mérés alapján. Eredményként a két külön párhuzamos meghatározás számtani közepét vegyük. |
III. AZ ÉGHETŐ ALKOTÓRÉSZEK MEGHATÁROZÁSA
1. TÁRGY ÉS ALKALMAZÁSI TERÜLET
Ez a módszer a magas nitrogéntartalmú ammónium-nitrát műtrágyák éghetőanyag-tartalom meghatározását szolgáló eljárást írja le.
2. ALAPELV
A szervetlen töltőanyagokból keletkező szén-dioxidot előzetesen savval eltávolítjuk. A szerves alkotórészeket krómsav/kénsav keverék segítségével oxidáljuk. A képződött szén-dioxidot bárium-hidroxid oldatban elnyeletjük. A csapadékot sósavoldatban oldjuk fel, és nátrium-hidroxid oldattal történő visszatitrálással mérjük.
3. REAGENSEK
3.1. Analitikai minőségű króm-trioxid Cr2O3.
3.2. Kénsav, 60 v/v%:
Töltsünk 360 ml vizet egy egyliteres főzőpohárba, és óvatosan adjunk hozzá 640 ml kénsavat (sűrűség 20 °C-on = 1 ,83 g/ml).
3.3. Ezüst-nitrát: 0,1 mol/l-es oldat.
3.4. Bárium-hidroxid:
Mérjünk ki 15 gramm bárium-hidroxidot (Ba(OH)2.8 H2O), és teljesen oldjuk fel forró vízben. Hagyjuk kihűlni, és töltsük át egy egyliteres mérőlombikba. Töltsük fel a jelig, és keverjük össze. Szűrjük át redős szűrőpapíron.
3.5. Sósav: 0,1 M mérőoldat.
3.6. Nátrium-hidroxid: 0,1 mól/l mérőoldat.
3.7. Brómfenol-kék: 0,4 gramm/liter koncentrációjú vizes oldata.
3.8. Fenolftalein: 2 gramm/liter koncentrációjú 60% térfogat százalékos etanolos oldata;
3.9. Nátrium-karbonát: szemcseméret 1,0 és 1,5 mm között.
3.10. Ioncserélt víz, frissen forralt a szén-dioxid eltávolításáért.
4. KÉSZÜLÉK
4.1. Szokásos laboratóriumi felszerelés, különösen:
- 15 ml kapacitású szűrőtégely zsugorított üveggel; a korong átmérője: 20 mm; teljes magasság: 50 mm; porozitás 4 (pórusátmérő 5 és 15 mm között);
- 600 ml-es főzőpohár.
4.2. Sűrített nitrogén-adagoló.
4.3. A készülék a következő alkatrészekből áll, és lehetőség szerint csiszolt gömb csőcsatlakozással kerül összeszerelésre (lásd a 2. ábrát).
4.3.1. Körülbelül 200 mm hosszú és 30 mm átmérőjű abszorpciós cső (A), nátrium-karbonáttal (3.9.) töltött, amelyet üveggyapot dugó tart a helyén.
4.3.2. 500 ml-es B reakcióedény, kétnyakú gömblombik
4.3.3. Vigreux frakcionáló oszlop (C '), körülbelül 150 mm hosszú.
4.3.4. Dupla falú hűtő ©, 200 mm hosszú.
4.3.5. Drechsel-palack (D), amely az esetlegesen átdesztillálódó felesleges sav felfogására szolgál.
4.3.6. Jégfürdő (E) a Drechsel-palack hűtésére.
4.3.7. Két darab gázmosó (F1 és F2), 32 és 35 mm közötti átmérőjű, amelynek gázelosztója 10 mm-es alacsony porozitású zsugorított üveglemez.
4.3.8. Vákuumszivattyú és vákuumszabályozó készülék (G), amely T üvegcsövet tartalmaz az áramlási körbe helyezve, és a szabad vége egy Hoffman-szorítóval ellátott rövid gumicsövön keresztül egy vékony kapillárishoz csatlakozik.
Figyelem:
Forrásban lévő krómkénsav oldat használata csökkentett nyomás alatt levő készülékben veszélyes művelet, és megfelelő óvintézkedések megtételét igényli. :
5. ELJÁRÁS
5.1. Mintavétel elemzés céljára
Mérjünk ki körülbelül 10 gramm ammónium-nitrátot 0,001 gramm pontossággal.
5.2. A karbonátok eltávolítása
Helyezzük az elemzendő mintát a B reakcióedénybe. Adjunk hozzá 100 ml kénsavat (3.2.). Szobahőmérsékleten a szemcsék körülbelül 10 perc alatt feloldódnak. Állítsuk össze a készüléket a 2. ábrán látható módon. Csatlakoztassuk az abszorpciós cső (A) egyik végét a nitrogénforráshoz (4.2.) egy visszacsapó szelepen keresztül, amely 5-6 mm közötti higanyt tartalmaz, a másik végét pedig a reakcióedénybe vezető gázbevezető csőhöz csatlakoztassuk. Szereljük fel a lombikra (B) a Vigreux frakcionáló oszlopot (C') és a duplafalú hűtőt (C), és indítsuk el a hűtővizet. Állítsuk be a nitrogént úgy, hogy mérsékelten áramoljon az oldaton keresztül, forraljuk fel az oldatot, és tartsuk forrásban két percig. Ezután már nem szabad pezsegnie. Amennyiben mégis buborékok figyelhetők meg, folytassuk a forralást további harminc percig. Hagyjuk az oldatot legalább 20 percig hűlni, miközben a nitrogén tovább áramlik rajta keresztül.
Fejezzük be a készülék rajz szerinti összeszerelését úgy, hogy a hűtő csövét a Drechsel-palackhoz (D), a palackot pedig az F1 és F2 gázmosóhoz csatlakoztatjuk. Az összeszerelési művelet alatt a nitrogénnek folyamatosan áramolnia kell az oldaton keresztül. Gyorsan adjunk 50-50 ml bárium-hidroxid oldatot (3.4.) a gázmosók (F1 és F2) mindegyikébe.
Buborékoltassunk keresztül nitrogént körülbelül 10 percen át. Az oldatnak tisztának kell maradnia a gázmosókban. Amennyiben nem így lenne, a karbonát mentesítő eljárást meg kell ismételni.
5.3. Oxidáció és abszorpció
Miután kihúztuk a kétnyakú gömblombikból (4.3.2.) a nitrogéntápcsövet, gyorsan adagoljunk be 20 gramm króm-trioxidot (3.1.) és 6 ml ezüst-nitrát oldatot (3.3.) a reakcióedény (B) oldalsó nyakán keresztül. Csatlakoztassuk a készüléket a vákuumszivattyúhoz, és állítsuk be úgy a nitrogénáramlást, hogy az F1 és F2 zsugorított üveg gázmosón a buborékok állandó árama haladjon keresztül.
Melegítsük a reakcióedényt (B), amíg a folyadék forrni kezd, és tartsuk forrásban másfél órán keresztül. A nitrogénáramlás szabályozása miatt szükségesnek bizonyulhat a vákuumszabályozó szelep (G) utánállítása, mivel lehetséges, hogy a vizsgálat alatt kicsapódott bárium-karbonát eltömi a zsugorított üveg lemezeket. A művelet akkor kielégítő, ha az F2 gázmosóban lévő bárium-hidroxid oldat tiszta marad. Ellenkező esetben ismételjük meg a vizsgálatot. Hagyjuk abba a melegítést, és szedjük szét a készüléket. A bárium-hidroxid eltávolításához mossuk el mindegyik gázelosztót mind kívül, mind belül, és gyűjtsük össze a mosásterméket a megfelelő gázmosóban. Helyezzük a gázelosztókat, egyiket a másik után egy 600 ml-es főzőpohárba, amelyet később a meghatározás során fogunk használni.[83]
A zsugorított-üveg szűrőtégelyt használva gyorsan szűrjük meg vákuum alatt először az F2, majd az F1 gázmosó tartalmát. Gyűjtsük össze a csapadékot a gázmosók vízzel (3.10.) történő átmosásával, és mossuk át a tégelyt az ugyanebből a vízből vett 50 ml-es mennyiséggel. Helyezzük a szűrőtégelyt a 600 ml-es főzőpohárba, és adjunk hozzá körülbelül 100 ml vizet. Öntsünk körülbelül 50 ml kiforralt vizet mindegyik gázmosóba, és öt percen át buborékoltassunk nitrogént a gázelosztókon keresztül. Egyesítsük a vizet a főzőpohárból származóval. Ismételjük meg a műveletet még egyszer, hogy a gázelosztók átmosása alapos legyen.
5.4. A szerves anyagból származó karbonátok mérése
Adjunk öt csepp fenolftaleint (3.8.) a főzőpohár tartalmához. Az oldat színe pirosra változik. Titráljuk sósavval (3.5.), amíg a rózsaszín szín éppen eltűnik. Alaposan keverjük az oldatot, hogy biztosak lehessünk benne, a rózsaszín szín nem tér vissza. Adjunk hozzá öt csepp brómfenol-kéket, és titráljuk sósavval, amíg az oldat színe sárgává nem válik. Adjunk hozzá további 10 ml sósavat (3.5.).
Melegítsük az oldatot forráspontig, és folytassuk a forralását legfeljebb egy percig. Gondosan ellenőrizzük, hogy ne maradjon csapadék a folyadékban.
Hagyjuk kihűlni, és titráljuk vissza nátrium-hidroxid oldattal (3.6).
6. VAKPRÓBA
Hajtsunk végre vakpróbát ugyanezt az eljárást követve és az összes reagensből ugyanilyen mennyiséget használva.
7. AZ EREDMÉNYEK KIFEJEZÉSE
A minta tömegszázalékában, szénként kifejezett éghetőanyag-tartalmat (C) a következő képlet adja meg
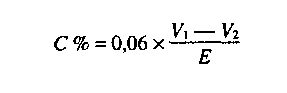
| ahol: |
| E = a vizsgálati anyagmennyiség tömege grammban kifejezve, |
| V1 = a fenolftalein színében bekövetkezett változás után hozzáadott 0,1 M sósav teljes mennyisége ml-ben kifejezve, |
| V2 = a visszatitrálásra használt 0,1 M nátrium-hidroxid mennyisége ml-ben kifejezve. |
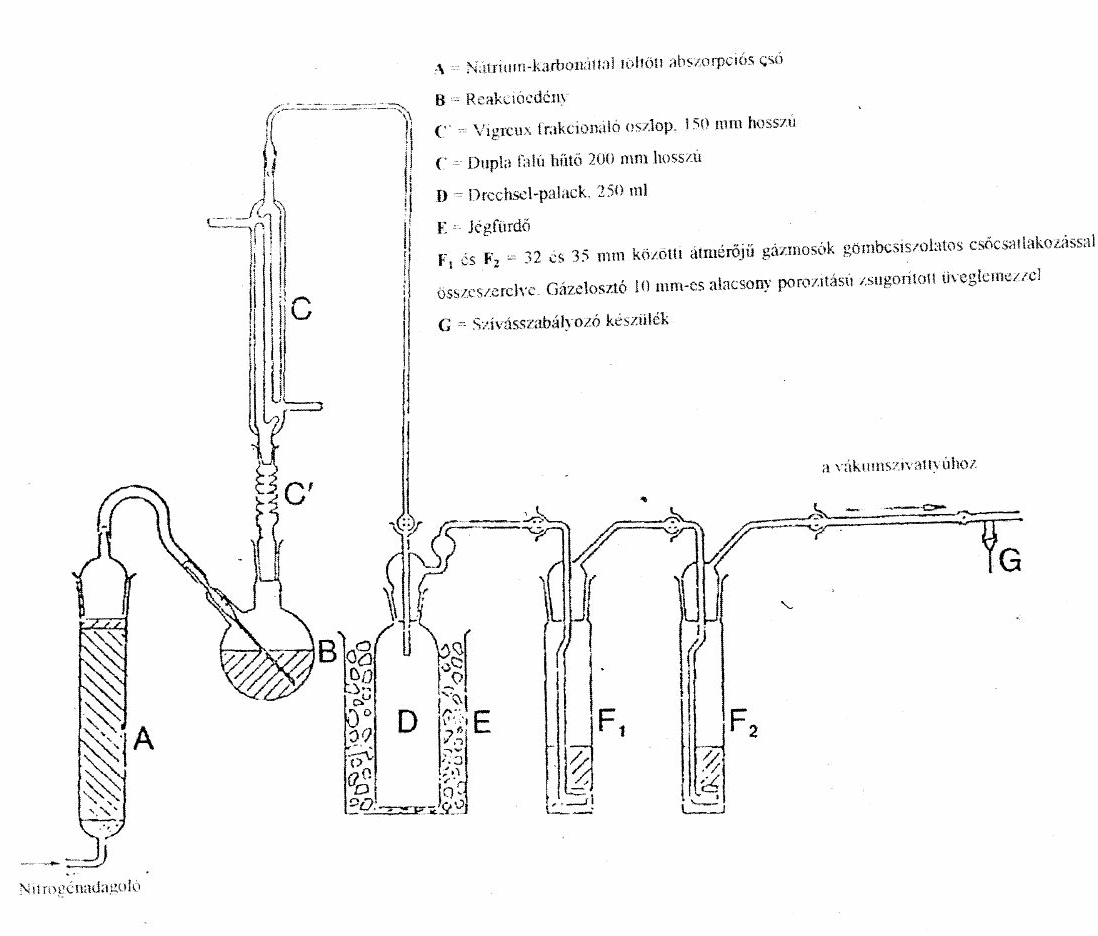
IV. A pH-ÉRTÉK MEGHATÁROZÁSA
1. TÁRGY ÉS ALKALMAZÁSI TERÜLET
Ez a módszer a magas nitrogéntartalmú egyszerű ammónium-nitrát műtrágyák oldatának pH-értékériek mérési eljárását határozza meg.
2. ALAPELV
Az ammónium-nitrát oldat pH-értékének mérése pH-mérő segítségével.
3. REAGENSEK
Desztillált vagy ioncserélt víz, szén-dioxid mentesített.
3.1. 20 °C-on 6,88 pH-értékű pufferoldat
Oldjunk fel 3,40 ± 0,01 gramm káliumdihidrogén-ortofoszfátot (KH2PO4) körülbelül 400 ml vízben. Azután oldjunk fel 3,55 ± 0,01 gramm dinátriumhidrogén-ortofoszfátot (Na2HPO4) körülbelül 400 ml vízben. Veszteség nélkül töltsük át a két oldatot egy 1000 ml-es mérőlombikba, töltsük fel jelig, és keverjük össze. Tartsuk ezt a folyadékot egy légmentesen záró edényben.
3.2. 20 °C-ón 4,00 pH-értékű pufferoldat
Oldjunk fel 10,2 ± 10,01 gramm káliumhidrogén-ftalátot (KHC3O4H4) vízben, töltsük át veszteség nélkül egy 1000 ml-es mérőlombikba, töltsük fel jelig, és keverjük össze.
Tartsuk ezt a folyadékot légmentesen záró edényben.
3.3. A kereskedelmi forgalomban beszerezhető pH-szabványoldatok használhatók.
4. KÉSZÜLÉK
Üveg és kálóméi vagy ezekkel egyenértékű elektródákkal felszerelt, 0,05 pH-egység érzékenységű pH-mérő.
5. ELJÁRÁS
5.1. A pH-mérő kalibrálása
Kalibráljuk a pH-mérőt (4.) 20 ± 1 °C-on a (3.1.), (3.2.) vagy (3.3.) pufferoldatok használatával. Vezessünk az oldat felületére lassú nitrogénáramot, és ezt az áramot tartsuk fenn az egész vizsgálat során.
5.2. Meghatározás
Töltsünk 100,0 ml vizet 10 (± 0,01) grammnyi mintára egy 250 ml-es főzőpohárban. Távolítsuk el az oldhatatlan anyagokat szűréssel, dekantálással vagy centrifugálással. Mérjük meg a tiszta oldat pH-értékét 20 ± 1 °C-on ugyanazzal az eljárással, mint a pH-mérő kalibrálásakor.
6. AZ EREDMÉNY KIFEJEZÉSE
Fejezzük ki az eredményeket pH-egységekben 0,1 egység pontossággal, és tüntessük fel az alkalmazott hőmérsékletet.
V. A SZEMCSEMÉRET MEGHATÁROZÁSA
1. TÁRGY ÉS ALKALMAZÁSI TERÜLET
Ez a módszer a magas nitrogéntartalmú ammónium-nitrát műtrágyák szitaelemzésének eljárását határozza meg.
2. ALAPELV
A vizsgálati minta szitálása három szitából álló szitasoron kézzel vagy mechanikus módon történik. Az egyes szitákon fennmaradt anyag tömege rögzítésre kerül, és kiszámítják a szükséges szitákon átjutott anyag százalékos arányát.
3. KÉSZÜLÉK
3.1. 200 mm átmérőjű drótszövet vizsgálati sziták egyenként 2,0 mm-es, 1,0 mm-es és 0,5 mm-es lyukmérettel a szabvány méreteknek megfelelően. Egy-egy fedél és egy-egy gyűjtőtartály mindegyik szitasorhoz.
3.2. 0,1 gramm mérési pontosságú mérleg.
3.3. Mechanikus szitarázó készülék (ha rendelkezésre áll), amely képes a vizsgálati minta mind függőleges, mind vízszintes irányú mozgatására.
4. ELJÁRÁS
4.1. A mintát körülbelül 100 grammos reprezentatív részekre osztjuk.
4.2. Mérjük meg az egyik ilyen részt 0,1 gramm pontossággal.
4.3. Rendezzük el a szitákból képzett szitasort emelkedő sorrendben: gyűjtőtartály, 0,5 mm-es, 1,0 mm-es, 2,0 mm-es, és helyezzük a lemért vizsgálati anyag mennyiséget a legfelső szitára. Illesszük a fedőt a szitasor tetejére.
4.4. Rázzuk kézzel vagy géppel, mind függőlegesen, mind vízszintesen mozgatva, és amennyiben a rázás kézzel történik, néha megütögetve. Folytassuk ezt az eljárást 10 percig, vagy addig, amíg az egyes szitákon egy perc alatt áthaladó anyag mennyisége már kevesebb, mint 0,1 gramm.
4.5. Távolítsuk el sorban a szitákat a szitasorból, és gyűjtsük össze az általuk felfogott anyagot, szükség esetén finom ecsettel seperjük le az anyagot a szita hátoldaláról.
4.6. Mérjük meg az egyes sziták által felfogott és a gyűjtőtartályban összegyűlt anyagot 0,1 grammos pontossággal.
5. AZ EREDMÉNYEK ÉRTÉKELÉSE
5.1. Számoljuk ki az egyes gyűjtött részek tömegértékét a résztömeg értékek összegének százalékos arányában (nem az eredeti mennyiséghez viszonyított százalékos arányra).
Számoljuk ki a százalékos arányt a gyűjtőtartályban (vagyis<0,5 mm): A%
Számoljuk ki a százalékos arányt a 0,5 mm-es szitán fennmaradt anyag vonatkozásában: B%
Számoljuk ki az 1,0 mm-es szitán áthaladt anyag mennyiségét, vagyis (A + B)%
A résztömegek összegének az eredeti tömeg 2%-án belül kell maradnia.
5.2. Legalább két külön elemzést kell elvégezni, és az A esetében az eredmények nem térhetnek el 1,0%-nál nagyobb mértékben, a B esetében pedig 1,5%-nál nagyobb mértékben. Egyéb esetben ismételjük meg a vizsgálatot.
6. AZ EREDMÉNYEK KIFEJEZÉSE
Rögzítsük a kétszeri méréssel kapott A és az A + B értékek középértékét.
VI. A KLÓRTARTALOM MEGHATÁROZÁSA (KLORIDIONKÉNT)
1. TÁRGY ÉS ALKALMAZÁSI TERÜLET
Ez a módszer a magas nitrogéntartalmú ammónium-nitrát műtrágyák klórtartalmának (kloridionként való) meghatározására vonatkozó eljárást határozza meg.
2. ALAPELV
A vízben oldott kloridionokat ezüst-nitráttal savas közegben potenciometrikus titrálással határozzuk meg.
3. REAGENSEK
Kloridmentes desztillált vagy ioncserélt víz.
3.1. Aceton.
3.2. Koncentrált salétromsav (sűrűség 20 °C-on = 1,40 g/ml).
3.3. Ezüst-nitrát 0,1 mól/l mérőoldat - ezt az oldatot barna üvegben tároljuk.
3.4. Ezüst-nitrát 0,004 mól/l mérőoldat - ezt az oldatot felhasználáskor készítsük el.
3.5. Kálium-klorid, 0,1 mól/l szabvány referenciaoldat. Mérjünk ki 0,1 gramm pontossággal 3,7276 gramm analitikai minőségű kálium-kloridot, amelyet megelőzően kemencében 130 °C-on egy órán keresztül szárítottunk, majd exszikkátorban hűtöttünk le a környezeti hőmérsékletre. Oldjuk fel kevés vízben, töltsük át az oldatot veszteség nélkül 500 ml-es mérőlombikba, hígítsuk fel a jelig, és keverjük össze.
3.6. Kálium-klorid, 0,004 mól/l szabvány referenciaoldat - ezt az oldatot felhasználáskor készítsük el.
4. KÉSZÜLÉK
4.1. Ezüstindikátor elektróddal és kálóméi referenciaelektróddal felszerelt potenciométer, amelynek érzékenysége 2 mV, és -500-tól + 500 mV-ig használatos.
4.2. Telített kálium-nitrát oldatot tartalmazó mérőhíd, amely a kalomel-elektródhoz (4.1.) csatlakozik, és amely a végein porózus dugaszokkal van lezárva.
Figyelem: A hídra nincs szükség, amennyiben ezüst- és higany1 szulfát elektródot használunk.
4.3 Mágneses keverő, teflonbevonatú rúddal.
4.4 Finom végű, 0,01 ml-es osztásokkal ellátott mikrobüretta.
5. ELJÁRÁS
5.1. Az ezüst-nitrát oldat titerének beállítása
Mérjünk 5,00 ml és 10,00 ml szabvány referencia kálium-klorid oldatot (3.6.), és töltsük két megfelelő méretű (például 250 ml-es), alacsony főzőpohárba. Végezzük el mindkét főzőpohár tartalmának titrálását az alábbiak szerint.
Adjunk 5 ml salétromsavat (3.2.), 120 ml acetont (3.1.) és annyi vizet, hogy a teljes mennyiség nagyjából 150 ml legyen. Helyezzük a mágneses keverő (4.3.) rúdját a főzőpohárba, és indítsuk be a keverőt. Merítsük be az ezüstelektródot (4.1.) és a híd (4.2) szabad végét az oldatba. Kössük össze az elektródokat a potenciométerrel (4.1.), és miután meggyőződtünk róla, hogy a készülék nullán áll, jegyezzük fel a kiinduló potenciál értékét.
Titráljunk a mikrobüretta (4.4.) használatával, először 4, illetve 9 ml ezüst-nitrát oldatot adagolva a használt szabvány referencia kálium-klorid oldatnak megfelelően. Folytassuk az adagolást 0,1 ml-es adagokban a 0,004 mol/l-es oldatok és 0,05 ml-es adagokban a 0,1 mol/l-es oldatok esetében. Az egyes hozzáadások után várjuk meg a potenciál stabilizálódását.
Egy táblázat első két oszlopába jegyezzük be a hozzáadott mennyiségeket és a megfelelő potenciálértékeket.
A táblázat harmadik oszlopában jegyezzük fel az E potenciál egymás utáni növekményeit ( D1E). A negyedik oszlopban jegyezzük fel a potenciálnövekmények ( D1E) közötti negatív vagy pozitív különbségeket (D2E). A titrálásnak akkor érünk a végére, amikor az ezüst-nitrát oldathoz hozzáadott 0,1 vagy 0,05 ml-es adag (V1) a D1E-nek maximális értéket biztosít.
A reakció végének megfelelő ezüst-nitrát oldat pontos mennyiségének (Veq) kiszámítására használjuk a következő képletet:
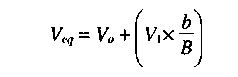
| ahol: |
| V0 = | az ezüst-nitrát oldat összes mennyisége ml-ben kifejezve, amely közvetlenül alacsonyabb annál a menynyiségnél, amely a Δ1E-nek maximális értéket biztosít, |
| V1 = | az ezüst-nitrát oldat utolsó hozzáadott adagjának (0,1 vagy 0,05 ml) mennyisége ml-ben kifejezve, |
| b = | a Δ2E utolsó pozitív értéke |
| B = | az utolsó pozitív és első negatív Δ2E abszolút értékének összege (lásd a példákat az 1. táblázatban). |
| 5.2. Vakpróba |
| Végezzünk vakpróbát, és vegyük figyelembe a végső eredmény kiszámolásánál. |
| A reagenseken végzett vakpróba V4 eredményét ml-ben a következő képlet adja meg: |
| ahol: |
| V2 = | a kálium-klorid szabvány referenciaoldat 10 ml-es mennyisége titrálására fogyott ezüst-nitrát oldat pontos mennyiségének (Veq) értéke ml-ben kifejezve, |
| V3 = | a kálium-klorid szabvány referenciaoldat 5 ml-es mennyisége titrálására fogyott ezüst-nitrát oldat pontos mennyiségének (Veq) értéke ml-ben kifejezve. |
5.3. Ellenőrző próba
A vakpróba egyúttal annak ellenőrzésére is szolgál, hogy a készülék megfelelően működik, és a vizsgálati eljárást helyesen hajtották végre.
5.4. Meghatározás
Vegyünk egy részt a mintából 10 és 20 gramm közötti mennyiségben, és mérjük meg 0,01 gramm pontossággal. Vigyük át veszteség nélkül egy 250 ml-es főzőpohárba. Adjunk hozzá 20 ml vizet, 5 ml salétromsavat (3.2.), 120 ml acetont (3.1.) és annyi vizet, hogy a teljes mennyiség körülbelül 150 ml legyen.
Helyezzük a mágneses keverő (4.3.) rúdját a főzőpohárba, helyezzük a főzőpoharat a keverőre, és hozzuk mozgásba a keverőt. Merítsük bele az ezüstelektródát (4.1.) és a híd (4.2.) szabad végét az oldatba, kössük össze az elektródákat a potenciométerrel (4.1.), majd miután meggyőződtünk róla, hogy a készülék nullán áll,jegyezzük fel a kiinduló potenciál értékét.
Mikrobürettából (4.4.) 0,1 ml-es részletekben történő adagolással titráljuk meg az ezüst-nitrát oldattal (3.3.). Az egyes hozzáadások után várjuk meg, amíg a potenciál stabilizálódik.
Folytassuk a titrálást az 5.1.-ben meghatározott módon, a harmadik bekezdéstől kezdve: "Egy táblázat első két oszlopába jegyezzük be a hozzáadott mennyiségeket és a megfelelő potenciálértékeket..."
6. AZ EREDMÉNYEK KIFEJEZÉSE
Az elemzés eredményét az elemzéshez kapott mintában található klór százalékos mennyiségeként fejezzük ki. A klór (Cl) százalékos mennyiségét a következő képlet alapján számoljuk ki:
ahol:
T = a használt ezüst-nitrát oldat molaritása,
V4 = a vakpróba (5.2.) eredménye ml-ben kifejezve,
V5= a Veq ml-ben kifejezett értéke a meghatározásnak (5.4.) megfelelően,
m = a vizsgálati anyag bemért adagjának tömege grammban kifejezve.
1. táblázat
Példa
| Az ezüst-nitrát oldat mennyisége V | Potenciál E | D1E | D2E |
| ml | mV | ||
| 4,80 | 176 | ||
| 4,90 | 211 | 35 | |
| 5,00 | 283 | 72 | + 37 |
| 5,10 | 306 | 23 | -49 |
| 5,20 | 319 | 13 | -10 |
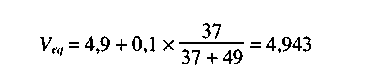
VII. A RÉZ MEGHATÁROZÁSA
1. TÁRGY ÉS ALKALMAZÁSI TERÜLET
Ez a módszer a magas nitrogéntartalmú egyszerű ammónium-nitrát műtrágyák réztartalmának meghatározására vonatkozó eljárást írja le.
2. ALAPELV
A mintát hígított sósavban oldjuk fel, és a réztartalmat atomabszorpciós spektrofotometriáyal határozzuk meg.
3. REAGENSEK
3.1. Sósav (sűrűség 20 °C-on =1,18 g/ml).
3.2. Sósav, 6 mol/l-es oldat.
3.3. Sósav, 0,5 mol/l-es oldat.
3.4. Ammónium-nitrát.
3.5. Hidrogén-peroxid, 30%-os
3.6. Rézoldat (törzsoldat): mérjünk ki 0,001 gramm pontossággal 1 gramm tiszta rezet, oldjuk fel 25 ml 6 mol/l-es sósav oldatban (3.2.), adjunk hozzá fokozatosan 5 ml hidrogén-peroxidot (3.5.), és hígítsuk fel 1 literre vízzel. Ebből az oldatból 1 ml 1000 μg rezet (Cu) tartalmaz.[84]
3.6.1. Rézoldat (hígított): hígítsunk fel 10 ml törzsoldatot (3.6.) 100 ml-re vízzel, majd a kapott oldatból 10 ml-t hígítsuk fel újra 100 ml-re vízzel, a legvégül kapott oldat 1 ml-e 10 μg rezet (Cu) fog tartalmazni. Ezt az oldatot közvetlenül a felhasználás előtt készítsük el.
4. KÉSZÜLÉK
Atomabszorpciós spektrofotométer réz lámpával (324,8 nm).
5. ELJÁRÁS
5.1. Az oldat előkészítése az elemzésre
Mérjünk ki 0,001 gramm pontossággal 25 grammot a mintából, helyezzük egy 400 ml-es főzőpohárba, óvatosan adjunk hozzá 20 ml sósavat (3.1.) (a szén-dioxid képződés miatt heves reakció jöhet létre). Szükség esetén adjunk hozzá még sósavat. Amikor a pezsgés abbamaradt, pároljuk szárazra, néha üvegpálcával kavargatva. Adjunk hozzá 15 ml 6 mol/l-es sósav oldatot (3.2.) és 120 ml vizet. Keverjük meg az üvegpálcával, amelyet hagyjunk a főzőpohárban, és fedjük be a főzőpoharat óraüveggel. Forraljuk óvatosan az oldatot, amíg a feloldódás teljes nem lesz, majd hűtsük le.
Töltsük át az oldatot egy 250 ml-es mérőlombikba, a főzőpoharat 5 ml 6 mol/l-es sósavval (3.2.) és kétszer 5 ml forró vízzel átmosva. Töltsük fel a jelig 0,5 mol/l-es sósavval (3.3.), és gondosan keverjük össze.
Szűrjük át rézmentes szűrőpapíron, az első 50 ml-t kiöntve.[85]
5.2. Vakoldat
Készítsünk vakoldatot, amelyből csupán a mintát hagytuk ki. Ezt vegyük figyelembe a végeredmény kiszámításánál.
5.3. Meghatározás
5.3.1. A minta és a vakpróba oldatok előkészítése
Hígítsuk fel a mintaoldatot (5.1.) és a vakpróbára szánt oldatot (5.2.) 0,5 mol/l-es sósav oldattal (3.3.), hogy a réz koncentrációja a spektrofotométer optimális mérési tartományán belül legyen. Rendes körülmények között hígításra nincsen szükség.
5.3.2. A kalibráló oldatok előkészítése
A réz standardoldat (3.6.) 0,5 mol/l-es sósav oldattal való hígításával (3.3.) készítsünk legalább öt, a spektrofotométer optimális mérési tartományának (0-tól 5,0 mg/l Cu) megfelelő standardoldatot. Mielőtt feltöltenénk jelig, adjunk mindegyik oldathoz annyi ammónium-nitrátot (3.4.), hogy a koncentráció 10 vegyes százalék legyen.
5.4. Mérés
Állítsuk be a spektrofotométert (4.) 324,8 nm hullámhosszra oxidáló levegő-acetilén lángot használva. Poriasszuk be egymás után háromszor az egyes oldatokat: a kalibráló oldatot (5.3.2.), a mintaoldatot és a vakoldatot (5.3.1.), és a készüléket a beporlasztások között minden alkalommal mossuk át desztillált vízzel. Szerkesszük meg az analitikai mérőgörbét az összes standardoldat átlagos abszorbanciáját az ordinátán ábrázolva, a hozzá tartozó μ g/ml-ben kifejezett rézkoncentrációkat pedig mint abszcisszán felvéve.
Az analitikai mérőgörbe segítségével határozzuk meg a rézkoncentrációt a végső minta oldatban és a vakoldatban.
6. Az eredmények kifejezése
A vizsgálati minta tömegének, az elemzés során végrehajtott hígításoknak és a vakpróba értékének figyelembevételével számoljuk ki a minta réztartalmát. Az eredményt mgCu/kg-ban fejezzük ki.
VIII. A ROBBANÓKÉPESSÉG MEGHATÁROZÁSA
1. TÁRGY ÉS ALKALMAZÁSI TERÜLET
Ez a módszer a magas nitrogéntartalmú egyszerű ammónium-nitrát műtrágyák robbanási ellenálló képességének meghatározására vonatkozó eljárást határozza meg.
2. ALAPELV
A vizsgálati mintát acélcsőbe zárjuk, és detonációs hatásnak vetjük alá robbanásindító töltet segítségével. A robbanás terjedését a csövet a vizsgálat alatt vízszintesen tartó ólomhengerek összenyomódásának mértékéből határozzuk meg.
3. ANYAGOK
3.1. Plasztikus robbanóanyag, amely 83-86% penthritet tartalmaz Sűrűség: 1500-1600 kg/m3
Detonációs sebesség: 7300-7700 m/s Súly: 500 ± 1 gramm
3.2. Hét hajlékony robbanózsinór darab nem fém hüvelyben Töltősúly: 11-13 g/m
Az egyes zsinórok hossza: 400 ± 2 mm
3.3. Másodlagos robbanóanyagból sajtolt pellet, mélyedéssel ellátva a detonátor befogadására.
Robbanóanyag: hexogén / viasz 95/5, tetril vagy más hasonló másodlagos robbanóanyag hozzáadott grafittal vagy anélkül.
Sűrűség: 1500-1600 kg/m3
Átmérő: 19-21 mm
Magasság: 19-23 mm
Középső mélyedés a detonátor befogadására: átmérő 7-7,3 mm, mélység 12 mm
3.4. ISO 65-1981-Heavy Series-ben meghatározott varrat nélküli acélcső,
névleges méretek DN 100 (4")
Külső átmérő: 113,1-115,0 mm
Falvastagság: 5,0-6,5 mm
Hossz: 1005 ( ± 2) mm
3.5. Fenéklemez
Anyag: jól hegeszthető minőségű acél
Méretek: 160 x 160 mm
Vastagság: 5-6 mm
3.6. Hat ólomhenger
Átmérő: 50 ( ± 1) mm
Magasság: 100-101 mm
Anyaguk: legalább 99,5%-os tisztaságú finomított ólom
3.7. Acéltömb
Hosszúság: legalább 1000 mm
Szélesség: legalább 150 mm
Magasság: legalább 150 mm
Tömeg: legalább 300 kg, ha nincs stabil alap az acéltömb számára
3.8. Műanyag vagy kartonpapír henger az indítótöltet számára
Falvastagság: 1,5-2,5 mm
Átmérő: 92-96 mm
Magasság: 64-67 mm
3.9. Detonátor (elektromos vagy nem elektromos) 8-tól 10-ig terjedő iniciációs erővel (Nobel sor szerinti erőssé)
3.10. Fakorong
Átmérő: 92-96 mm. Az átmérőnek illeszkednie kell az acélcső (3.4.) belső átmérőjéhez
Vastagság: 20 mm
3.11. A detonátorral (3.9.) egyező méretű farúd
3.12. Gombostű (maximális hosszúság: 20 mm), valamint tűzőgép
4. ELJÁRÁS
4.1. Az indítótöltet előkészítése az acélcsőbe való behelyezésre
A szükséges felszerelés meglététől függően két módszer közül választhatunk az indítótöltet robbanóanyagának beindításához.
4.1.1. Hétpontos egyidejű iniciálás
A használatra előkészített indítótöltetet a 3. ábra mutatja.
4.1.1.1. Fúrjunk lyukakat a fakorongba (3.10.) a korong tengelyével párhuzamosan a középponton és hat lyukon keresztül, amelyek egy 55 mm átmérőjű koncentrikus kör mentén szimmetrikusan helyezkednek el. A lyukak átmérője a használt gyújtózsinór (3.2.) átmérőjétől függően 6 és 7 mm között legyen (lásd a 3. ábra A-B részét).
4.1.1.2. Vágjunk le hét, egyenként 400 mm-es darabot a hajlékony gyújtózsinórból (3.2.). Tiszta vágásokkal és a végek azonnali leragasztásával kerüljük el a robbanóanyag bárminemű veszteségét a végeknél. Dugjuk át a hét zsinórdarabot a fakorong (3.10.) hét lyukán úgy, hogy a korong másik oldalán néhány cm kiálljon a végükből. 5-6 mm-re az egyes zsinórdarabok végétől szúrjunk gombostűt (3.12.) átlósan a zsinórdarabok textilhüvelyébe, és kenjük be ragasztóval a zsinórokat kívül a gombostűktől 2 cm-es sávban. Végül húzzuk meg a zsinórok hosszabbik végét, hogy a gombostűk érintkezzenek a fakoronggal.
4.1.1.3. Formáljuk a plasztik robbanóanyagot (3.1.) henger alakúra, amelynek átmérője a henger (3.8.) átmérőjétől függően 92 és 96 mm között van. Állítsuk ezt a hengert a talpára egy vízszintes felületen, és helyezzük bele a megformázott robbanóanyagot. Ezután helyezzük a hét gyújtózsinórt tartó fakorongot a henger tetejére, és nyomjuk le, bele a robbanóanyagba. Állítsuk be úgy a henger magasságát (64 és 67 mm között), hogy a legfelső pereme ne legyen feljebb a fa szintjénél. Végül kapcsok használatával rögzítsük a hengert a fakoronghoz, körben az egész kerületén.[86]
4.1.1.4. Csoportosítsuk úgy a hét gyújtózsinór szabad végeit a farúd (3.11.) körül, hogy a végeik mind egy, a rúdra merőleges síkba kerüljenek. Ragasztószalaggal rögzítsük őket egy kötegbe a farúd körül[87]
4.1.2. Központi iniciálás sajtolt pellettel
A használatra előkészített indítótöltetet a 2. ábra mutatja.
4.1.2.1. A sajtolt pellet előkészítése
A szükséges biztonsági intézkedések megtétele után helyezzünk 10 grammot a másodlagos robbanóanyagból (3.3.) egy 19 és 21 mm közötti átmérőjű formába, és sajtoljuk össze a megfelelő alakra és sűrűségre.
(Az átmérő-magasság aránya durván 1:1 legyen.)
A forma aljának közepén egy rögzítő-peceknek kell lennie, amely 12 mm magas, 7,0-7,3 mm átmérőjű (a használt detonátor átmérőjétől függően), és amely egy henger alakú mélyedést alakít ki a töltetben a detonátor későbbi behelyezéséhez.
4.1.2.2. Az indítótöltet előkészítése
Helyezzük a plasztik robbanóanyagot a hengerbe (3.8.), amely függőlegesen áll sík felületen, majd nyomjuk le fa sajtolószerszámmal, hogy a robbanóanyag henger alakú formát kapjon a közepén mélyedéssel. Helyezzük be az összesajtolt pelletet ebbe a mélyedésbe. Egy fakoronggal, amely középen 7,0-7,3 mm közötti méretű lyukat tartalmaz a detonátor behelyezésére, fedjük be a henger alakúra formált és az összesajtolt pelletet tartalmazó robbanóanyagot. Kereszt alakban ragasztószalaggal rögzítsük egymáshoz a fakorongot és a hengert. Győződjünk meg róla, hogy a korongba fúrt lyuk és az összesajtolt pelletben lévő mélyedés a farúd behelyezésével egy tengelyű lett.
4.2. Az acélcsövek előkészítése a robbanási vizsgálathoz
Az acélcső (3.4.) egyik végénél fúrjunk két átellenes helyen 4 mm átmérőjű lyukat merőlegesen az oldalfalon keresztül, 4 mm-re a végétől.
Rögzítsük hegesztéssel a fenéklapot (3.5.) a cső másik végéhez, teljesen kitöltve az alsó lap és a cső fala közötti derékszöget olvasztott fémmel, a cső teljes kerülete körül.
4.3. Az acélcső megtöltése és feltöltése (Lásd a 3., 4. és 5. ábrát)
4.3.1. A vizsgálati mintát, az acélcsövet és az indítótölíetet 20 ( ± 5) °C-ra kell kondicionálni. Két robbanási vizsgálathoz 16-18 kg vizsgálati anyag szükséges.
4.3.2. Állítsuk a csövet függőleges helyzetbe, hogy a négyzet alakú alsó fenéklemez szilárd, sima felületre, lehetőleg betonra támaszkodjon. Töltsük meg a csövet körülbelül magasságának egyharmadáig a vizsgálati mintával, és egymás után ötször ejtsük függőlegesen 10 cm-ről a földre, hogy a szemcséket vagy granulátumokat, amennyire csak lehetséges, tömörítsük a csőben. A tömörítés gyorsítása érdekében a ledobások között összesen 10-szer ütögessük meg az oldalfalat 750-1000 grammos kalapáccsal.
Ismételjük meg ezt a betöltési módszert a vizsgálati minta újabb adagjával. Végül további mennyiséget úgy adagoljunk, hogy az a cső 10-szer végzett felemelése és leejtése, valamint az eközben összesen 20 kalapácsütés által végzett tömörítés után a felső végétől számított 70 mm-es távolságig töltse meg a csövet.
Az acélcsőben a vizsgálati minta töltési magasságát úgy kell beállítani, hogy a később behelyezendő indítótöltet (4.1.1. vagy 4.1.2.) szoros érintkezésben legyen a mintával, annak teljes felületén.
4.3.3. Helyezzük be az indítótöltetet a csőbe úgy, hogy érintkezzen a mintával; a fakorong felső felületének 6 mm-rel a cső vége alatt kell lennie. Gondoskodjunk róla, hogy a robbanóanyag és a vizsgálati minta valóban szorosan érintkezzen, ha szükséges, a mintából kis mennyiségeket vegyünk el, vagy tegyünk hozzá. A 3. és 4. ábrán látható módon a cső nyitott végénél helyezzünk sasszegeket a lyukakon keresztül, hogy két végük a csőre támaszkodjon.
4.4. Az acélcső és az ólomhengerek elhelyezése
4.4.1. Számozzuk meg az ólomhengerek alját (3.6.) 1-től 6-ig. Csináljunk hat jelölést egymástól 150 mm-re a vízszintes alapon nyugvó acéltömb (3.7.) középvonalán úgy, hogy az első jelölés legalább 75 mm-re legyen a tömb szélétől. Helyezzünk függőlegesen minden egyes jelölésre egy ólomhengert úgy, hogy az egyes hengerek aljának közepét a jelölésre állítjuk.
4.4.2. A 4.3. szerint előkészített acélcsövet fektessük le vízszintesen az ólomhengerekre oly módon, hogy a cső tengelye párhuzamos legyen az acéltömb középvonalával, és a cső hegesztett vége 50 mm-rel nyúljon túl a 6. számú ólomhengeren. Hogy megakadályozzuk a cső legurulását, helyezzünk kisméretű faékeket az ólomhengerek és a cső fala közé (egyet minden oldalra), vagy helyezzünk kereszt alakban fadarabokat a cső és az acéltömb közé.
Megjegyzés: Győződjünk meg róla, hogy a cső érintkezik mind a hat ólomhengerrel; a cső felületének enyhe görbületét kiegyenlíthetjük a cső hosszanti tengelye körüli forgatásával; ha bármely ólomhenger túl hosszú lenne (100 mm), óvatosan ütögessük a kérdéses hengert kalapáccsal, ameddig el nem éri a szükséges magasságot.
4.5. Előkészület a robbantásra
4.5.1. Állítsuk össze a készüléket a 4.4. szerint egy bunkerben vagy más megfelelően előkészített föld alatti helyen (pl. bányában vagy alagútban). Ügyeljünk arra, hogy az acélcső hőmérséklete a robbantás előtt 20 ae 5 C-on maradjon.
Megjegyzés: Amennyiben a fenti robbantási helyek nem lennének biztosíthatóak, a műveletet el lehet végezni egy fagerendákkal betakart, betonbélelésű gödörben. A robbanás hatására acélszilánkok csapódhatnak szét nagy mozgási energiával, ezért a robbantást lakott területtől vagy közforgalmú utaktól megfelelő távolságra kell végezni.
4.5.2. Ha hétpontos iniciálással ellátott indítótöltetet használunk, ügyeljünk arra, hogy a gyújtózsinórokat a 4.1.1.4. ponthoz tartozó lábjegyzetben leírt módon feszítettük ki, és amennyire csak lehetséges, vízszintesen rendeztük el.
4.5.3. Végezetül távolítsuk el a farúdat, és tegyük be helyette a detonátort. A robbantást addig ne hajtsuk végre, ameddig a veszélyzónából nem evakuáltunk mindenkit, és a vizsgálatot végző személyzet nem vonult védett helyre.
4.5.4. Robbantsuk fel a robbanóanyagot.
4.6. Hagyjunk elég időt, hogy a füst (gáznemű és esetleg mérgező bomlástermékek, például nitrózus gázok) eloszoljanak, azután gyűjtsük be az ólomhengereket, és mérjük meg magasságukat nóniuszos tolómércével.
Jegyezzük fel az összes jelöléssel ellátott henger esetében az összenyomódás mértékét a 100 mm-es eredeti magasság százalékában kifejezve. Amennyiben a hengerek ferdén nyomódtak össze, mérjük meg a legkisebb és a legmagasabb értéket, és vegyük ezek középértékét.
4.7. Szükség esetén a robbanási sebesség folyamatos mérésére érzékelőt alkalmazhatunk; az érzékelőt a cső tengelyével egyező hosszanti irányban vagy az oldalfal mentén kell behelyezni.
4.8. Mintánként két robbantási vizsgálatot kell végrehajtani.
5. VIZSGÁLATI JELENTÉS
Az egyes robbantási vizsgálatok vizsgálati jelentéseiben a következő paraméterek értékeit kell feltüntetni
- az acélcső külső átmérőjének és falvastagságának ténylegesen mért értéke,
- az acélcső Brinell-keménysége,
- a cső és a minta hőmérséklete közvetlenül a robbantás előtt,
- az acélcsőben levő minta tényleges sűrűsége (kg/m3),
- az egyes ólomhengerek magassága a robbantás után, megadva a henger megfelelő sorszámát,
- az indítótöltet iniciálásának módja.
5.1. A vizsgálati eredmények értékelése
Amennyiben minden egyes robbantás esetén legalább egy ólomhenger összenyomódása nem haladja meg az 5%-os értéket, a vizsgálatot bizonyító erejűnek kell tekinteni.
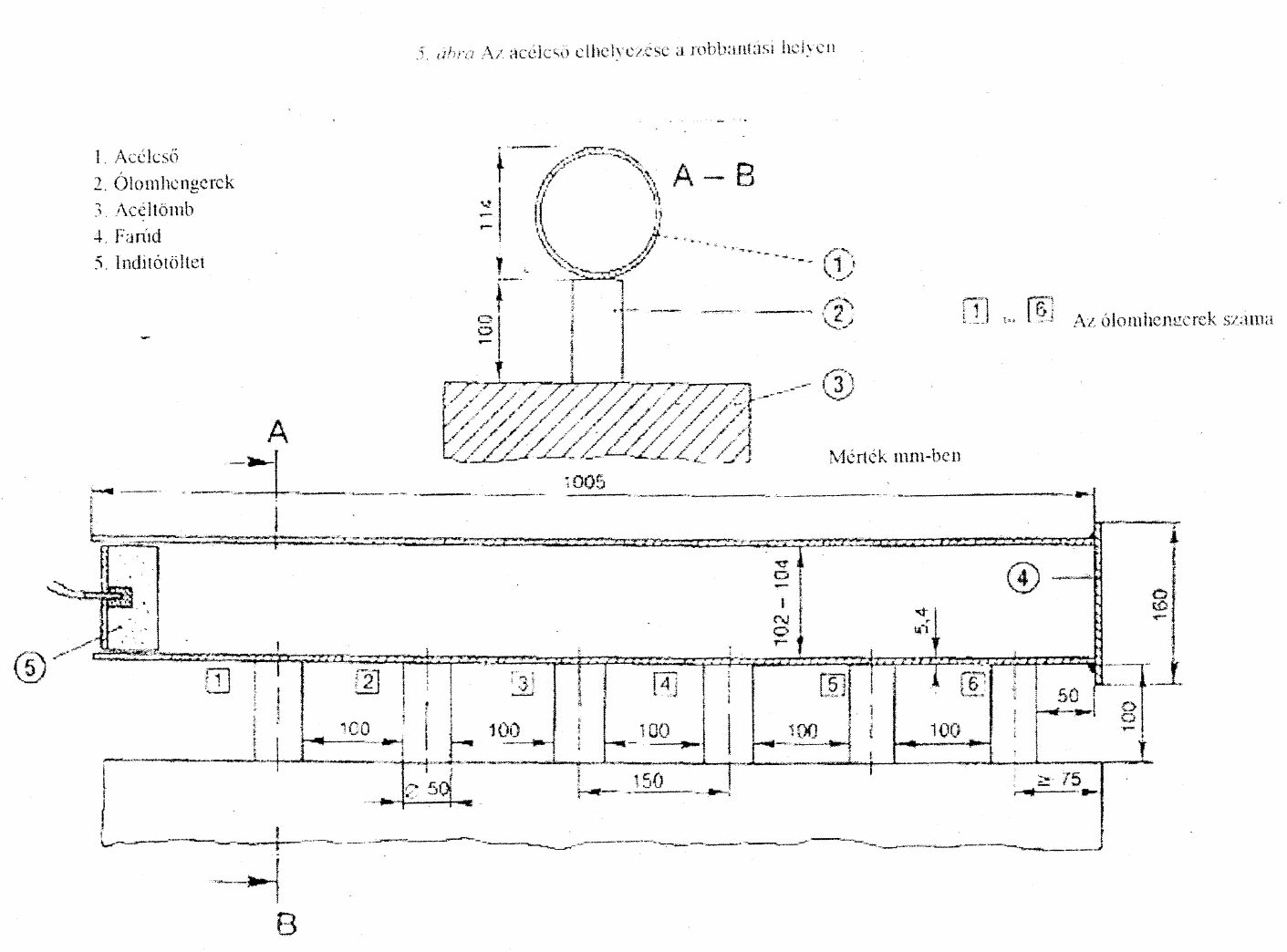
Lábjegyzetek:
[1] Megállapította az 50/2003. (V. 9.) FVM rendelet 1. § (1) bekezdése. Hatályos 2003.05.15.
[2] Megállapította az 50/2003. (V. 9.) FVM rendelet 1. § (2) bekezdése. Hatályos 2003.05.15.
[3] Beiktatta az 50/2003. (V. 9.) FVM rendelet 1. § (3) bekezdése. Hatályos 2003.05.15.
[4] Megállapította az 50/2003. (V. 9.) FVM rendelet 2. § -a. Hatályos 2003.05.15.
[5] Megállapította az 50/2003. (V. 9.) FVM rendelet 2. § -a. Hatályos 2003.05.15.
[6] Megállapította az 50/2003. (V. 9.) FVM rendelet 2. § -a. Hatályos 2003.05.15.
[7] Megállapította az 50/2003. (V. 9.) FVM rendelet 2. § -a. Hatályos 2003.05.15.
[8] Beiktatta az 50/2003. (V. 9.) FVM rendelet 2. § -a. Hatályos 2003.05.15.
[9] Beiktatta az 50/2003. (V. 9.) FVM rendelet 2. § -a. Hatályos 2003.05.15.
[10] Beiktatta az 50/2003. (V. 9.) FVM rendelet 2. § -a. Hatályos 2003.05.15.
[11] Beiktatta az 50/2003. (V. 9.) FVM rendelet 2. § -a. Hatályos 2003.05.15.
[12] Megállapította az 50/2003. (V. 9.) FVM rendelet 3. § (1) bekezdése. Hatályos 2003.05.15.
[13] Módosította a 97/2005. (X. 28.) FVM rendelet 11. § -a. Hatályos 2005.11.01.
[14] Megállapította az 50/2003. (V. 9.) FVM rendelet 4. § (1) bekezdése. Hatályos 2003.05.15.
[15] Megállapította az 50/2003. (V. 9.) FVM rendelet 4. § (2) bekezdése. Hatályos 2003.05.15.
[16] Megállapította az 50/2003. (V. 9.) FVM rendelet 5. § -a. Hatályos 2003.05.15.
[17] Megállapította az 50/2003. (V. 9.) FVM rendelet 6. § -a. Hatályos 2003.05.15.
[18] Megállapította az 50/2003. (V. 9.) FVM rendelet 6. § -a. Hatályos 2003.05.15.
[19] Megállapította az 50/2003. (V. 9.) FVM rendelet 7. § -a. Hatályos 2003.05.15.
[20] Megállapította az 50/2003. (V. 9.) FVM rendelet 7. § -a. Hatályos 2003.05.15.
[21] Módosította az 50/2003. (V. 9.) FVM rendelet 8. § (1) bekezdése. Hatályos 2003.05.15.
[22] Módosította az 50/2003. (V. 9.) FVM rendelet 8. § (1) bekezdése. Hatályos 2003.05.15.
[23] Módosította az 50/2003. (V. 9.) FVM rendelet 8. § (2) bekezdése. Hatályos 2003.05.15.
[24] Megállapította az 50/2003. (V. 9.) FVM rendelet 8. § (3) bekezdése. Hatályos 2003.05.15.
[25] Szerkezetét módosította az 50/2003. (V. 9.) FVM rendelet 9. § -a. Hatályos 2003.05.15.
[26] Beiktatta az 50/2003. (V. 9.) FVM rendelet 9. § -a. Hatályos 2003.05.15.
[27] Beiktatta az 50/2003. (V. 9.) FVM rendelet 9. § -a. Hatályos 2003.05.15.
[28] Beiktatta az 50/2003. (V. 9.) FVM rendelet 9. § -a. Hatályos 2003.05.15.
[29] Megállapította az 50/2003. (V. 9.) FVM rendelet 10. § -a. Hatályos 2003.05.15.
[30] Módosította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[31] Módosította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[32] Módosította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[33] Módosította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[34] Módosította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[35] Megállapította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[36] Módosította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[37] Megállapította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[38] Megállapította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[39] Módosította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[40] Megállapította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[41] Megállapította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[42] Megállapította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[43] Módosította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[44] Megállapította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[45] Megállapította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[46] Módosította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[47] Megállapította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[48] Megállapította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[49] Megállapította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[50] Megállapította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[51] Módosította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[52] Megállapította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[53] Megállapította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[54] Megállapította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[55] Megállapította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[56] Módosította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[57] Megállapította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[58] Megállapította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[59] Megállapította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[60] Megállapította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[61] Beiktatta az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[62] Módosította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[63] Megállapította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[64] Megállapította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[65] Módosította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[66] Módosította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[67] Módosította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[68] Módosította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[69] Megállapította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[70] Megállapította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[71] Megállapította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[72] Beiktatta az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[73] Megállapította az 50/2003. (V. 9.) FVM rendelet 11. § (1) bekezdése. Hatályos 2003.05.15.
[74] Beiktatta az 50/2003. (V. 9.) FVM rendelet 11. § (2) bekezdése. Hatályos 2003.05.15.
[75] Minden csomót szét kell oszlatni (szükség szerint úgy, hogy kivesszük, majd szétnyomva visszatesszük ezeket).
[76] A biuretet előzetesen 10%-os ammónia oldattal, majd acetonnal történő mosással és vákuumszárítással tisztíthatjuk.
[77] Lásd 9. függelék.
[78] Ha nem áll rendelkezésre mechanikus keverő, a lombikot 5 percenként kézzel is összerázhatjuk.
[79] Vegyük ki pipettával a műtrágya kivonat kb. 0,01 g
[80] 21 ml, ha a kicsapandó oldat több, mint 15 ml citrát oldatot tartalmaz (semleges citrát, Peterman vagy Joulie-féle lúgos citrát).
[81] 15 ml citrát (semleges Petermann vagy Joulie) oldatnál többet tartalmazó foszfát oldatoknál, ahol a savanyításhoz 21 ml tömény salétromsavat használtunk, a lecsapó reagensből 80 ml-t kell használni.
[82] Beiktatta az 50/2003. (V. 9.) FVM rendelet 11. § (2) bekezdése. Hatályos 2003.05.15.
[83] Ezüst-nitrát katalizátor jelenlétében a szerves anyagok nagyobb részének esetében a másfél órás reakcióidő elégségesnek bizonyul.
[84] A kereskedelemben kapható szabvány rézoldatot is használhatunk.
[85] Whatman 541 vagy ezzel egyenértékű.
[86] A korong átmérőjének minden esetben egyeznie kell a henger belső átmérőjével.
[87] NB: Amíg a hat szélső, a kerületen elhelyezkedő zsinórnak feszesnek kell lennie az összeállítás után, a középsőú zsinórnak némileg lazának kell maradnia.