
32018R0150[1]
A Bizottság (EU) 2018/150 végrehajtási rendelete (2018. január 30.) az (EU) 2016/1240 végrehajtási rendeletnek az állami intervencióra és magántárolási támogatásra jogosult tej és tejtermékek elemzési és minőségértékelési módszerei tekintetében történő módosításáról
A BIZOTTSÁG (EU) 2018/150 VÉGREHAJTÁSI RENDELETE
(2018. január 30.)
az (EU) 2016/1240 végrehajtási rendeletnek az állami intervencióra és magántárolási támogatásra jogosult tej és tejtermékek elemzési és minőségértékelési módszerei tekintetében történő módosításáról
AZ EURÓPAI BIZOTTSÁG,
tekintettel az Európai Unió működéséről szóló szerződésre,
tekintettel a közös agrárpolitika finanszírozásáról, irányításáról és monitoringjáról és a 352/78/EGK, a 165/94/EK, a 2799/98/EK, a 814/2000/EK, az 1290/2005/EK és a 485/2008/EK tanácsi rendelet hatályon kívül helyezéséről szóló, 2013. december 17-i 1306/2013/EU európai parlamenti és tanácsi rendeletre (1) és különösen annak 62. cikke (2) bekezdésének i) pontjára,
mivel:
(1) Az (EU) 2016/1238 felhatalmazáson alapuló bizottsági rendelet (2) és az (EU) 2016/1240 bizottsági végrehajtási rendelet (3) megállapítja az állami intervencióra és a magántárolási támogatásra vonatkozó szabályokat. A 273/2008/EK bizottsági rendelet (4) meghatározza azokat a módszereket, amelyeket annak értékelésekor kell alkalmazni, hogy a tej és a tejtermékek megfelelnek-e az állami intervenció és a magántárolási támogatás tekintetében a vonatkozó rendeletekben megállapított támogathatósági követelményeknek.
(2) A tej és a tejtermékek elemzési és minőségértékelési módszerei tekintetében végbement műszaki fejlődés fényében lényeges változtatásokra van szükség az ISO-szabványokra történő hivatkozások egyszerűsítése és korszerűsítése érdekében. Az egyértelműség és a hatékonyság érdekében, valamint tekintettel a 273/2008/EK rendeletben foglalt rendelkezések módosításainak mértékére és technikai jellegére, az említett rendelet vonatkozó rendelkezéseit be kell építeni az (EU) 2016/1240 végrehajtási rendeletbe.
(3) A tagállamokban az új szabványoknak és módszereknek való egységes megfelelés biztosítása érdekében a laboratóriumok számára elegendő időt kell biztosítani az eljárások felülvizsgálatára és a frissített módszerek alkalmazására.
(4) Ezért az (EU) 2016/1240 végrehajtási rendeletet ennek megfelelően módosítani kell.
(5) Az egyértelműség érdekében a 273/2008/EK rendeletet hatályon kívül kell helyezni.
(6) Az e rendeletben előírt intézkedések összhangban vannak a mezőgazdasági piacok közös szervezésével foglalkozó bizottság véleményével,
ELFOGADTA EZT A RENDELETET:
1. cikk
Az (EU) 2016/1240 végrehajtási rendelet a következőképpen módosul:
1) A 4. cikk a következőképpen módosul:
(a) az (1) bekezdés a következőképpen módosul:
i. a d) pont helyébe a következő szöveg lép:
"d) a vaj esetében: e rendelet IV. mellékletének I. és Ia. része;"
ii. az e) pont helyébe a következő szöveg lép:
"e) a sovány tejpor esetében: e rendelet V. mellékletének I. és Ia. része;"
(b) a (2) bekezdés helyébe a következő szöveg lép:
"(2) Az I., IV. és V. mellékletben említett, állami intervencióra jogosult gabonafélék, vaj és sovány tejpor minőségének meghatározásához alkalmazandó módszerek a vonatkozó európai vagy nemzetközi szabványoknak a legalább 6 hónappal az 1308/2013/EU rendelet 12. cikkében meghatározott állami intervenciós időszak első napját megelőzően hatályos, legújabb változatában megállapított módszerek."
2) A rendelet a következő 60a. cikkel egészül ki:
"60a. cikk
A tej és a tejtermékek állami intervenciójához és magántárolási támogatásához kapcsolódó ellenőrzésekre vonatkozó egyedi rendelkezés
(1) A vaj, a sovány tejpor és a sajt magántárolási támogatásra való jogosultságát a VI., VII., illetve VIII. mellékletben meghatározott módszereknek megfelelően kell megállapítani.
A szóban forgó módszereket a vonatkozó európai vagy nemzetközi szabványoknak a legalább 6 hónappal az 1308/2013/EU rendelet 12. cikkében meghatározott állami intervenciós időszak első napját megelőzően hatályos, legújabb változatára való hivatkozással kell megállapítani.
(2) Az e rendeletben meghatározott módszerek alkalmazásával végzett ellenőrzések eredményeit a IX. melléklettel összhangban kell értékelni."
(2) A mellékletek e rendelet mellékletének megfelelően módosulnak.
2. cikk
A 273/2008/EK rendeletet hatályát veszti.
3. cikk
Ez a rendelet az Európai Unió Hivatalos Lapjában való kihirdetését követő hetedik napon lép hatályba.
Ez a rendelet teljes egészében kötelező és közvetlenül alkalmazandó valamennyi tagállamban.
Kelt Brüsszelben, 2018. január 30-án.
a Bizottság részéről
az elnök
Jean-Claude JUNCKER
(1) HL L 347., 2013.12.20., 549. o.
(2) A Bizottság (EU) 2016/1238 felhatalmazáson alapuló rendelete (2016. május 18.) az 1308/2013/EU európai parlamenti és tanácsi rendeletnek az állami intervenció és a magántárolási támogatás tekintetében történő kiegészítéséről (HL L 206., 2016.7.30., 15. o.).
(3) A Bizottság (EU) 2016/1240 végrehajtási rendelete (2016. május 18.) az 1308/2013/EU európai parlamenti és tanácsi rendeletnek az állami intervenció és a magántárolási támogatás tekintetében történő alkalmazására vonatkozó szabályok megállapításáról (HL L 206., 2016.7.30., 71. o.).
(4) A Bizottság 273/2008/EK rendelete (2008. március 5.) a tej és tejtermékek elemzési és minőségértékelési módszerei tekintetében az 1255/1999/EK tanácsi rendelet alkalmazására vonatkozó részletes szabályok megállapításáról (HL L 88., 2008.3.29., 1. o.).
MELLÉKLET
Az (EU) 2016/1240 végrehajtási rendelet mellékletei a következőképpen módosulnak:
1. A IV. melléklet a következőképpen módosul:
a) az I. részben, a 2. pont második albekezdése helyébe a következő szöveg lép:
"Minden mintát külön kell elbírálni. Ismételt mintavétel és ismételt értékelés nem megengedett.";
b) a szöveg a következő Ia. résszel egészül ki:
"IA. RÉSZ
A sózatlan vaj állami intervenciója tekintetében alkalmazandó elemzési módszerek
| Paraméter | Módszer |
| Zsír (1) | ISO 17189 vagy ISO 3727, 3. rész |
| Víz | ISO 3727, 1. rész |
| Zsírmentes szárazanyag | ISO 3727, 2. rész |
| Zsírsavtartalom | ISO 1740 |
| Peroxidszám | ISO 3976 |
| Nem tejzsír | ISO 17678 |
| Érzékszervi tulajdonságok | ISO 22935, 2. és 3. rész, valamint az alábbi pontszámítási táblázat. |
Pontszámítási táblázat
| Külső | Állag | Szag és íz | |||
| Pontszám | Megjegyzések | Pontszám | Megjegyzések | Pontszám | Megjegyzések |
| 5 | Nagyon jó Ideális típus Legjobb minőség (egyenletesen száraz) | 5 | Nagyon jó Ideális típus Legjobb minőség (egyenletesen kenhető) | 5 | Nagyon jó Ideális típus Legjobb minőség (teljesen tiszta, finom illat) |
| 4 | Jó (nincs nyilvánvaló hiba) | 4 | Jó (nincs nyilvánvaló hiba) | 4 | Jó (nincs nyilvánvaló hiba) |
| 1, 2 vagy 3 | Bármilyen hiba | 1, 2 vagy 3 | Bármilyen hiba | 1, 2 vagy 3 | Bármilyen hiba” |
2. Az V. melléklet a következő Ia. résszel egészül ki:
"IA. RÉSZ
A sovány tejpor állami intervenciója tekintetében alkalmazandó elemzési módszerek
| Paraméter | Módszer |
| Fehérje | ISO 8968, 1. rész |
| Zsír | ISO 1736 |
| Víz | ISO 5537 |
| Savasság | ISO 6091 |
| Laktátok | ISO 8069 |
| Foszfatázvizsgálat | ISO 11816, 1. rész |
| Oldhatósági index | ISO 8156 |
| Égett szemcsék (2) | ADPI |
| Mikroorganizmusok | ISO 4833, 1. rész |
| Író | I. függelék |
| Oltós savó (3) | II. és III. függelék |
| Savanyú savó (4) | ISO 8069 vagy helyszíni ellenőrzések |
| Érzékszervi ellenőrzések (5) | ISO 22935, 2. és 3. rész |
I. függelék
SOVÁNY TEJPOR: A FOSZFATIDILSZERIN ÉS A FOSZFATIDIL-ETANOLAMIN MENNYISÉGI MEGHATÁROZÁSA
Módszer: fordított fázisú HPLC
1. CÉL ÉS ALKALMAZÁSI TERÜLET
A módszer a sovány tejporban (SMP) lévő foszfatidilszerin (PS) és a foszfatidil-etanolamin (PE) mennyiségi meghatározására szolgál, és alkalmas a sovány tejporban található írószárazanyag kimutatására.
2. FOGALOMMEGHATÁROZÁS
PS + PE tartalom: az itt meghatározott eljárás alapján meghatározott anyag tömegtörtje. Az eredményt 100 g porra megadott foszfatidil-etanolamin-dipalmitoil (PEDP) milligrammjában fejezzük ki.
3. A MÓDSZER ELVE
Aminofoszfolipidek kivonása tejporból készült tejből metil-alkohollal. A PS és a PE meghatározása fordított fázisú (RP) HPLC-vel és fluoreszcencia-észleléssel o-ftáldialdehid-(OPA-) származék formájában történik. A vizsgálati minta PS- és PE-tartalmát egy ismert mennyiségű PEDP-t tartalmazó standarddal való összehasonlítás alapján kapjuk meg.
4. VEGYSZEREK
Az összes vegyszernek analitikai tisztaságúnak kell lennie. A felhasznált víznek egyéb előírás hiányában desztillált víznek, vagy azzal egyenértékű tisztaságú víznek kell lennie.
4.1. Standardanyag: legalább 99 %-os tisztaságú PEDP
Megjegyzés: A standardanyagot - 18 °C-on kell tárolni.
4.2. A standard mintához és a vizsgálati minta előkészítéséhez használt vegyszerek
4.2.1. HPLC-minőségű metil-alkohol
4.2.2. HPLC-minőségű kloroform
4.2.3. Triptamin-monohidroklorid
4.3. Az o-ftáldialdehid-derivátum készítéséhez használt vegyszerek
4.3.1. NaOH, 12 M vizes oldat
4.3.2. Bórsav, 0,4 M vizes oldat NaOH-val 10,0 pH-ra állítva (4.3.1.)
4.3.3. 2-merkapto-etanol
4.3.4. o-ftáldialdehid (OPA)
4.4. Eluáló oldószerek HPLC-hez:
4.4.1. Az eluáló oldószereket HPLC-minőségű vegyszerek felhasználásával kell készíteni.
4.4.2. HPLC-minőségű víz
4.4.3. Tesztelt, fluorimetrikus tisztaságú metil-alkohol
4.4.4. Tetrahidrofurán
4.4.5. Nátrium-dihidrogén-foszfát
4.4.6. Nátrium-acetát
4.4.7. Ecetsav
5. ESZKÖZÖK
5.1. Analitikai mérleg, 1 mg-os pontosságú mérésre képes, 0,1 mg-os leolvashatósággal
5.2. Főzőpoharak, 25 és 100 ml űrtartalommal
5.3. 1 és 10 ml adagolására alkalmas pipetták
5.4. Mágneses keverő
5.5. 0,2, 0,5 és 5 ml-es adagolásra képes beosztásos pipetták
5.6. 10, 50 és 100 ml űrtartalmú mérőlombikok
5.7. 20 és 100 μl-es fecskendők
5.8. Ultrahangos fürdő
5.9. 27 000 × g-vel működő centrifuga
5.10. Mintegy 5 ml űrtartalmú üvegfiolák
5.11. 25 ml űrtartalmú beosztásos mérőhenger
5.12. 0,1 pH egységig pontos pH-mérő
5.13. HPLC-berendezés
5.13.1. Gradiens szivattyúrendszer, amely 200 baron 1,0 ml/min sebességgel tud működni
5.13.2. Automata mintavevő derivátumképző lehetőséggel
5.13.3. Oszlopfűtő egység, amely képes az oszlopot 30 °C ± 1 °C-on tartani
5.13.4. Fluoreszcencia-detektor, amely képes 330 nm gerjesztési hullámhosszal és 440 nm kibocsátási hullámhosszal működni
5.13.5. A görbe csúcsai alatti terület mérésére alkalmas integrátor vagy adatfeldolgozó szoftver
5.13.6. Egy LiChrospher® - 100 oszlop (250 × 4,6 mm) vagy azzal egyenértékű oszlop, 5 μm-es részecskenagyságú oktadecilszilánnal (C18) töltve
6. MINTAVÉTEL
A mintavételt a 707 ISO-szabvány szerint kell elvégezni.
7. ELJÁRÁS
7.1. Belső standard oldat készítése
7.1.1. Mérjünk ki egy 100 ml-es mérőlombikba (5.6.) 30,0 ± 0,1 mg triptamin-monokloridot (4.2.3.), és töltsük fel a jelig metil-alkohollal (4.2.1.).
7.1.2. Pipettázzunk 1 ml-t (5.3.) ebből az oldatból egy 10 ml-es mérőlombikba (5.6.), és töltsük fel a jelig metil-alkohollal (4.2.1.), hogy elérjük a 0,15 mM-triptaminkoncentrációt.
7.2. A vizsgálati minta oldatának elkészítése
7.2.1. Mérjünk ki egy 25 ml-es főzőpohárba (5.2.) 1,000 ± 0,001 g soványtejpor-mintát. Adjunk hozzá pipettával (5.3.) 10 ml 40 °C ± 1 °C-os desztillált vizet, és a mágneses keverőn (5.4.) keverjük 30 percig, hogy minden csomó feloldódjon.
7.2.2. Pipettázzunk 0,2 ml (5.5.) tejporból készült tejet egy 10 ml-es mérőlombikba (5.6.), majd a fecskendő használatával (5.7.) adjunk hozzá 100 μl 0,15 mM-os triptaminoldatot (7.1.), és töltsük fel a jelig metil-alkohollal (4.2.1.). Gondosan keverjük el felfordítással, és 15 percen át kezeljük ultrahanggal (5.8.).
7.2.3. Centrifugáljuk (5.9.) 27 000 × g erővel 10 percig, és a felülúszót egy üvegfiolába gyűjtsük össze (5.10.).
Megjegyzés: A vizsgálati mintából készült oldatot a HPLC-vizsgálat elvégzéséig 4 °C-os hőmérsékleten kell tárolni.
7.3. Külső standard oldat készítése
7.3.1. Mérjünk ki 55,4 mg PEDP-t (4.1.) egy 50 ml-es mérőlombikba (5.6.), és adjunk hozzá a beosztásos mérőhenger (5.11.) használatával 25 ml kloroformot (4.2.2.). Melegítsük a ledugaszolt lombikot 50 °C ± 1 °C-ra, és gondosan addig keverjük, míg a PEDP fel nem oldódik. Hűtsük vissza a lombikot 20 °C-ra, töltsük fel a jelig metil-alkohollal (4.2.1.), és felfordítással keverjük el.
7.3.2. Pipettázzunk (5.3.) ebből az oldatból 1 ml-t egy 100 ml-es mérőlombikba (5.6.), majd töltsük fel a jelig metil-alkohollal (4.2.1.). Pipettázzunk (5.3.) ebből az oldatból 1 ml-t egy 10 ml-es mérőlombikba (5.6.), adjunk hozzá 100 μl (5.7.) 0,15 mM-os triptaminoldatot (7.1.), majd töltsük fel a jelig metil-alkohollal (4.2.1.). Keverjük el felfordítással.
Megjegyzés: A referenciaoldatot a HPLC-elemzés elvégzéséig 4 °C-os hőmérsékleten kell tárolni.
7.4. A származékképző reagens elkészítése
Mérjünk ki 25,0 ± 0,1 mg OPA-t (4.3.4.) egy 10 ml-es mérőlombikba (5.6.), adjunk hozzá 0,5 ml (5.5.) metil-alkoholt (4.2.1.), és gondosan keverjük el, hogy az OPA feloldódjon. Töltsük fel a jelig bórsavoldattal (4.3.2.), és fecskendővel (5.7.) adjunk hozzá 20 μl 2-merkaptoetanolt (4.3.3.).
Megjegyzés: A származékképző reagenst barna üvegfiolában 4 °C-on kell tárolni, és az egy hétig stabil.
7.5. HPLC-meghatározás
7.5.1. Eluáló oldószerek (4.4.)
A. oldószer: 0,3 mM nátrium-dihidrogén-foszfát és 3 mM nátrium-acetát-oldat (6,5 ± 0,1-es pH-ra ecetsavval beállítva): metil-alkohol:tetrahidrofurán = 558:440:2 (v/v/v) arányban
B. oldószer: metil-alkohol
7.5.2. Javasolt eluáló gradiens:
| Idő (perc) | A. oldószer (%) | B. oldószer (%) | Áramlási sebesség (ml/min) |
| Kiindulás | 40 | 60 | 0 |
| 0,1 | 40 | 60 | 0,1 |
| 5,0 | 40 | 60 | 0,1 |
| 6,0 | 40 | 60 | 1,0 |
| 6,5 | 40 | 60 | 1,0 |
| 9,0 | 36 | 64 | 1,0 |
| 10,0 | 20 | 80 | 1,0 |
| 11,5 | 16 | 84 | 1,0 |
| 12,0 | 16 | 84 | 1,0 |
| 16,0 | 10 | 90 | 1,0 |
| 19,0 | 0 | 100 | 1,0 |
| 20,0 | 0 | 100 | 1,0 |
| 21,0 | 40 | 60 | 1,0 |
| 29,0 | 40 | 60 | 1,0 |
| 30,0 | 40 | 60 | 0 |
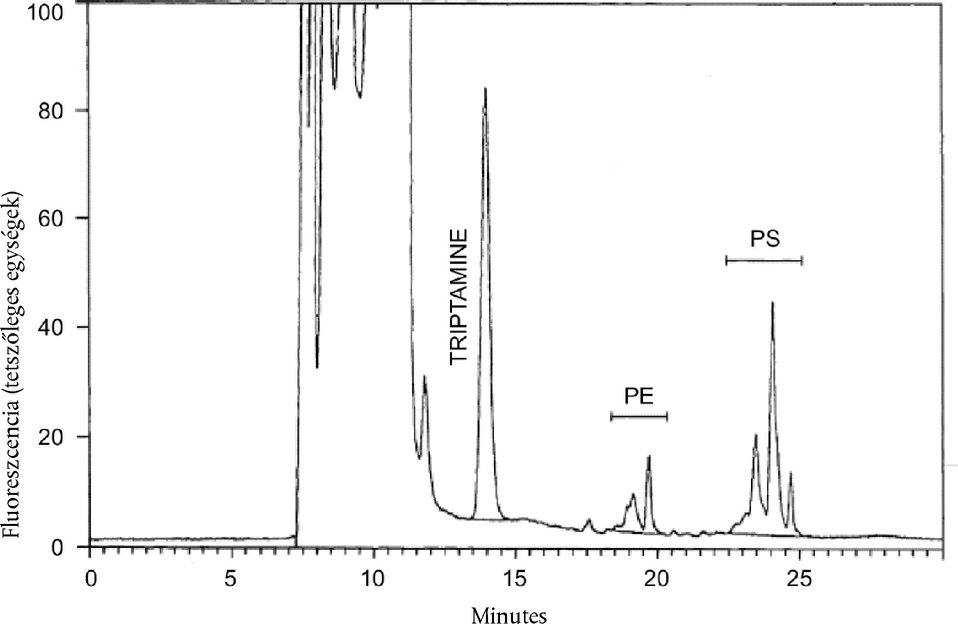
Megjegyzés: Annak érdekében, hogy az 1. ábrán bemutatott elválasztási képességet elérjük, az eluáló gradiens kismértékű megváltoztatására lehet szükség.
Oszlophőmérséklet: 30 °C.
7.5.3. Befecskendezett mennyiség: 50 μl származékképző reagens és 50 μl mintaoldat
7.5.4. Oszlop kalibrálása
A rendszer napi indításakor az oszlopot 100 %-os B. oldószerrel mossuk át egy negyed órán át, majd váltsunk át A:B = 40:60 arányú keverékre, és 1 ml/min áramlási sebességgel további negyed órán keresztül kalibráljuk. Végezzünk vak tesztet metil-alkohol (4.2.1.) injektálásával.
Megjegyzés: Hosszabb idejű tárolás előtt az oszlopot metil-alkohol:kloroform = 80:20 (v/v) arányú keverékével mossuk 30 percen át.
7.5.5. Határozzuk meg a PS + PE tartalmat a vizsgálati mintában
7.5.6. Végezzük el a kromatográfiás elemzés lépéseit a futtatástól-futtatásig terjedő időt állandó értéken tartva, hogy konstans retenciós időket kapjunk. A válaszjel számításához a külső standard oldatot (7.3.) minden 5-10 vizsgálati minta után fecskendezzük be.
Megjegyzés: Az oszlopot a 100 %-os B. oldószerrel (7.5.1.) minden 20-25 futtatás után legalább harminc percen át kell mosnunk.
7.6. Az integrálás módja
7.6.1. PEDP-csúcs
A PEDP egyetlen csúcsban eluálódik. A csúcs alatti területet völgytől-völgyig történő integrálással határozzuk meg.
7.6.2. Triptamincsúcs
A triptamin egyetlen csúcsban eluálódik (1. ábra). A csúcs alatti területet völgytől-völgyig történő integrálással határozzuk meg.
7.6.3. PS- és PE-csúcscsoportok
A leírt feltételek mellett (1. ábra), a PS két fő, részben egymástól el nem váló csúcsban eluálódik, amelyet egy kisebb csúcs előz meg. A PE három fő, részben egymástól el nem váló csúcsban eluálódik. Határozzuk meg minden csúcscsoport egész területét az alapvonalnak az 1. ábrán bemutatott illesztésével.
8. SZÁMÍTÁS ÉS AZ EREDMÉNYEK KIFEJEZÉSE
A vizsgálati minta PS- és PE-tartalmát a következők szerint lehet kiszámítani:
C = 55,36 × ((A2)/(A1)) × ((T1)/(T2))
ahol
C = a PS- vagy PE-tartalom a vizsgálati mintában (mg/100 g por)
A1 = a standardminta-oldat (7.3.) PEDP-csúcsa alatti terület
A2 = a PS- vagy PE-csúcs alatti terület a vizsgálati mintából készült oldatban (7.2.)
T1 = a standardminta-oldat (7.3.) triptamincsúcsa alatti terület
T2 = a triptamincsúcs alatti terület a vizsgálati mintából készült oldatban (7.2.).
9. A MÓDSZER PONTOSSÁGA
Megjegyzés: Az ismételhetőségre vonatkozó értékek kiszámítása az IDF nemzetközi szabványnak (*) megfelelően történt.
9.1. Ismételhetőség
Az ismételhetőség relatív szórása, amely az ugyanazon személy által, ugyanannak a készüléknek a használatával, ugyanolyan körülmények között, ugyanazon vizsgálati mintából, rövid időeltéréssel kapott független elemzési eredmények variabilitását fejezi ki, nem haladhatja meg a 2 %-os relatív értéket. Ha ilyen körülmények között két meghatározást végeznek el, akkor a két eredmény viszonylagos eltérése nem lehet nagyobb, mint az eredmények számtani középértékének 6 %-a.
9.2. Reprodukálhatóság
Ha két laboratóriumban más készülékeken, más személyek, más feltételek mellett végeznek el két meghatározást ugyanannak a vizsgálati mintának az elemzése céljából, akkor a két eredmény viszonylagos eltérése nem lehet nagyobb, mint az eredmények számtani középértékének 11 %-a.
10. HIVATKOZÁSOK
10.1. Resmini P., Pellegrino L., Hogenboom J.A., Sadini V., Rampilli M., "Detection of buttermilk solids in skimmilk powder by HPLC quantification of aminophospholipids". Sci. Tecn. Latt.-Cas., 39,395 (1988).
1. ábra
A foszfatidilszeril (PS) és foszfatidil-etanolamin (PE) OPA-származékainak HPLC-mintája sovány tejporból készült tej metil-alkoholos kivonatában. A jegyzőkönyvben szerepelnie kell a PS, PE és triptamin (belső standard) csúcsok integrálási módjának.
II. függelék
OLTÓS SAVÓ KIMUTATÁSA INTERVENCIÓS RAKTÁROZÁSRA SZÁNT SOVÁNY TEJPORBAN A KAZEINOMAKROPEPTIDEK NAGYTELJESÍTMÉNYŰ FOLYADÉKKROMATOGRÁFIÁS ELJÁRÁSSAL (HPLC) VALÓ MEGHATÁROZÁSA ÚTJÁN
1. TÁRGY ÉS ALKALMAZÁSI TERÜLET
Ez a módszer lehetővé teszi az oltós savó kimutatását intervenciós raktározásra szánt sovány tejporból a kazeinomakropeptidek meghatározása révén.
2. HIVATKOZÁS
ISO 707 nemzetközi szabvány: Tej és tejtermékek - Mintavételi útmutató
3. FOGALOMMEGHATÁROZÁS
Az oltóssavószárazanyag-tartalom a megadott eljárás alapján, kazeinomakropeptid-tartalom kimutatásával meghatározott, tömegszázalékban kifejezett mennyiség.
4. A MÓDSZER ELVE
- A sovány tejpor helyreállítása, zsír és fehérjék eltávolítása triklórecetsavas kicsapással és ezt követő centrifugálással vagy szűréssel;
- A kazeinomakropeptidek (CMP) mennyiségének meghatározása a felülúszóból nagyteljesítményű folyadékkromatográfiás eljárással (HPLC);
- A mintából kapott eredmények értékelése sovány tejporból és akár ismert mennyiségű savópor hozzáadásával, akár anélkül készült standard mintákkal való összehasonlítás révén.
5. VEGYSZEREK
Az összes vegyszernek analitikai tisztaságúnak kell lennie. A felhasznált víznek desztillált víznek vagy azzal legalább egyenértékű tisztaságú víznek kell lennie.
5.1. Triklór-ecetsav oldat
Oldjunk fel 240 g triklór-ecetsavat (CCl3COOH) vízben, és töltsük fel vízzel 1 000 ml-re. Az oldatnak tisztának és színtelennek kell lennie.
5.2. Eluáló oldat, 6,0 pH
Oldjunk fel 1,74 g dikálium-hidrogén-foszfátot (K2HPO4), 12,37 g kálium-dihidrogén-foszfátot (KH2PO4), valamint 21,41 g nátrium-szulfátot (Na2SO4) megközelítőleg 700 ml vízben. Szükség esetén foszforsav vagy kálium-hidroxid felhasználásával állítsuk be a pH-ját 6,0-ra.
Töltsük fel vízzel 1 000 ml-re, és homogenizáljuk.
Megjegyzés: Az eluens összetétele megváltoztatható, hogy megfeleljen a standardok tanúsítványának vagy az oszlop-töltőanyag gyártója ajánlásainak.
Használat előtt egy 0,45 μm pórusátmérőjű membránszűrőn szűrjük át az eluáló oldatot.
5.3. Öblítőoldat
Keverjünk el egy rész acetonitrilt (CH3CN) kilenc rész vízzel. A keveréket szűrjük át egy 0,45 μm pórusátmérőjű membránszűrőn.
Megjegyzés: Bármilyen más, csíraölő hatású öblítőoldat is alkalmazható, amely nem rontja az oszlopok elválasztóképességét.
5.4. Standard minták
5.4.1. Az e rendeletben foglalt követelményeknek megfelelő sovány tejpor (azaz [0]).
5.4.2. Ugyanez a sovány tejpor 5 % (m/m) szabványos összetételű oltós savóporral módosítva (azaz [5]).
6. ESZKÖZÖK
6.1. Analitikai mérleg
6.2. 2 200 g centrifugális erő elérésére képes opcionális centrifuga, körülbelül 50 ml-es, ledugózott vagy kupakkal ellátott centrifugacsövekkel felszerelve
6.3. Mechanikus rázógép
6.4. Mágneses keverő
6.5. Körülbelül hét centiméter átmérőjű üvegtölcsérek
6.6. Szűrőpapírok, közepes szűrőképességgel, körülbelül 12,5 cm átmérővel
6.7. Üvegszűrő-berendezés 0,45 μm pórusátmérőjű membránszűrővel
6.8. Beosztással ellátott pipetták, amelyek alkalmasak 10 ml adagolására (ISO 648, A osztály vagy ISO/R 835), vagy olyan adagolórendszer, amely két perc alatt képes 10,0 ml adagolására
6.9. 20,0 ml víz kb. 50 °C-on történő adagolására képes adagolórendszer.
6.10. Termosztáttal ellátott vízfürdő 25 ± 0,5 °C-ra állítva
6.11. HPLC-berendezés, részei:
6.11.1. Szivattyú
6.11.2. Injektor, kézi vagy automata, 15-30 μl űrtartalommal
6.11.3. Két sorosan kötött TSK 2 000-SW kromatográfiás oszlop (30 cm hosszú, belső átmérő 0,75 cm) vagy ezzel egyenértékű oszlopok (pl. egy TSK 2 000-SWxl, egy Agilent Technologies Zorbax GF 250), és egy I 125-el vagy azzal egyenértékű anyaggal töltött előtétoszlop (3 cm × 0,3 cm)
6.11.4. Termosztatikus oszlopkamra, 35 ± 1 °C-ra állítva
6.11.5. Változtatható hullámhosszúságú ultraibolya-sugárzás mérő, amely 205 nm-en 0,008 Å-érzékenységű méréseket tesz lehetővé.
6.11.6. Völgytől-völgyig történő integrálásra alkalmas integrátor
Megjegyzés: Szobahőmérsékleten tartott oszlopokkal is lehet dolgozni, de ekkor az elválasztóképességük valamivel kisebb. Ebben az esetben a hőmérséklet az elemzés egyik tartományában sem változhat ± 5 °C-nál többet.
7. MINTAVÉTEL
7.1. A mintákat az ISO 707 nemzetközi szabványban meghatározottak szerint kell venni. A tagállamok azonban alkalmazhatnak ettől eltérő mintavételi módszert is, feltéve hogy az teljesíti a fent említett szabványban foglalt alapelveket.
7.2. Tároljuk a mintát olyan körülmények között, amelyekkel kizárható, hogy az összetétele megváltozzon vagy lebomoljon.
8. ELJÁRÁS
8.1. A vizsgálati minta előkészítése
A tejport tegyük a tejpor terjedelménél kétszer nagyobb űrtartalmú edénybe, amely légmentesen zárható. Azonnal zárjuk le az edényt. Az edény ismételt felfordításával alaposan keverjük fel a benne lévő tejport.
8.2. Vizsgálati adag
Egy centrifugacsőbe (6.2.) vagy 50 ml-es ledugaszolt lombikba mérjünk ki a mintából 2000 ± 0,001 g-ot.
8.3. Zsír és fehérje eltávolítása
8.3.1. Adjunk 20,0 ml meleg vizet (50 °C) a vizsgálandó mennyiséghez. Oldjuk fel a port mechanikus rázógép segítségével ötperces rázatással (6.3.). Helyezzük a centrifugacsövet vízfürdőbe (6.10.), és engedjük 25 °C-ra hűlni.
8.3.2. Adjunk hozzá két perc alatt 10,0 ml kb. 25 °C-os triklór-ecetsav oldatot (5.1.), miközben a mágneses keverővel erőteljesen keverjük (6.4.). Helyezzük a csövet vízfürdőbe (6.10.), és hagyjuk ott 60 percig.
8.3.3. Centrifugáljuk (6.2.) 10 percig 2 200 g-n, vagy szűrjük át a szűrőpapíron (6.6.), kidobva az első 5 ml szűrletet.
8.4. Kromatográfiás meghatározás
8.4.1. Fecskendezzünk 15-30 μl pontosan kimért felülúszót vagy szűrletet (8.3.3.) a HPLC-berendezésbe (6.11.), amelynek percenként 1,0 ml eluáló oldat (5.2.) áramlási sebességgel kell működnie.
1. megjegyzés: A használt oszlopok belső átmérőjétől vagy az oszlop gyártójának utasításaitól függően más áramlási sebesség is használható.
2. megjegyzés: Minden megszakítás alkalmával öblítsük át vízzel az oszlopokat. Soha ne hagyjuk bennük az eluáló oldatot (5.2.).
Mielőtt 24 óránál hosszabb időre megszakítanánk a vizsgálatot, előbb öblítsük át az oszlopokat vízzel, majd mossuk legalább három órán át öblítőoldattal (5.3.) percenként 0,2 ml-es folyási sebesség mellett.
8.4.2. Az [E] vizsgálati minta kromatográfiás elemzésének eredményét egy kromatogram formájában kapjuk meg, ahol minden csúcsot a rá jellemző retenciós idő (RT) alapján tudunk azonosítani a következők szerint:
| II. csúcs: | a kromatogram második csúcsa, kb. 12,5 perces retenciós idővel |
| III. csúcs: | a kromatogram harmadik csúcsa, a CMP-nek megfelelően, kb. 15,5 perces retenciós idővel |
Az oszlopválasztás jelentősen befolyásolhatja az egyes csúcsok retenciós idejét.
Az integrátor (6.11.6.) minden csúcsra automatikusan kiszámolja az A területet:
| AII: | a II. csúcs alatti terület |
| AIII: | a III. csúcs alatti terület |
Lényeges, hogy még a mennyiségi értelmezés előtt minden kromatogram külalakját megvizsgáljuk, hogy felfedezzünk minden olyan rendellenességet, amely vagy a készülék vagy az oszlopok hibás működéséből ered, vagy a minta eredetére és jellegére vezethető vissza.
Kétség esetén az elemzést meg kell ismételni.
8.5. Kalibrálás
8.5.1. A standard mintára (5.4.) is alkalmazzuk pontosan ugyanazt az eljárást, mint amely a 8.2. és a 8.4.2. pont közötti leírásban szerepel.
Használjunk frissen készített oldatokat, mert a CMP 8 %-os triklór-ecetsavas környezetben hamar bomlik. A veszteség becsült értéke 30 °C-on óránként 0,2 %.
8.5.2. A minták kromatográfiás vizsgálatát megelőzően az oszlopokat az oldatban (8.5.1.) lévő standard minta (5.4.2.) ismételt befecskendezésével addig kondicionáljuk, amíg a CMP-nek megfelelő csúcs retenciós ideje konstanssá nem válik.
8.5.3. Ugyanannyi mennyiségű szűrletnek (8.5.1.) az injektálásával, mint amennyit a mintáknál használtunk, határozzuk meg az R-válaszjeleket.
9. AZ EREDMÉNYEK KIFEJEZÉSE
9.1. A számítás módszere és a képletek
9.1.1. Az R-válaszjelek kiszámítása:
| II. csúcs: | RII = 100/(AII[0]) |
ahol
RII = a II. csúcs válaszjelei,
AII [0] = a standard mintából [0] a 8.5.3. pont szerint nyert II. csúcsok alatti területek
| III. csúcs: | RIII = W/(AIII[5] – AIII[0]) |
ahol
RIII = a III. csúcs válaszjele,
AIII [0] és AIII [5] = a [0], illetve [5] standard minták esetén a 8.5.3. pontban nyert III. csúcs alatti területek
W = a savó mennyisége az [5] standard mintában, azaz 5.
9.1.2. A csúcsok alatti relatív terület az [E]mintában
SII[E] = RII × AII[E]
SIII[E] = RIII × AIII[E]
SIV[E] = RIV × AIV[E]
ahol
SII [E], SIII [E], SIV [E] = a II., III. és IV. csúcs alatti relatív területek az [E] mintában,
AII [E], AIII [E] = a 8.4.2. pontban nyert II. és III. csúcs alatti területek az [E] mintában,
RII, RIII = a 9.1.1. pontban számított válaszjelek.
9.1.3. Az [E] mintából kapott III. csúcs relatív retenciós idejének kiszámítása:
RRTIII[E] = (RTIII[E])/(RTIII[5])
ahol
RRTIII [E] = az [E] minta III. csúcsának relatív retenciós ideje,
RTIII [E] = az [E] mintából a 8.4.2. pont szerint nyert III. csúcs retenciós ideje,
RTIII [5] = a kontrollmintából [5] a 8.5.3. pont szerint nyert III. csúcs retenciós ideje.
9.1.4. A kísérletek azt mutatták ki, hogy a III. csúcs relatív retenciós ideje, vagyis az RRTIII [E] és a hozzáadott savópor mennyisége között 10 %-ig lineáris összefüggés van.
- Az RRTIII [E] < 1,000, ha a savótartalom > 5 %;
- az RRTIII [E] ≥ 1,000, ha a savótartalom ≤ 5 %.
Az RRTIII értékeire megengedhető bizonytalansági tényező ± 0,002.
Rendes esetben az RRTIII [0] értéke 1,034-től csak kis mértékben tér el. Az oszlopok állapotától függően az érték közeledhet az 1,000-hez, de annál mindig nagyobbnak kell lennie.
9.2. A mintában található oltós savópor százalékos mennyiségének kiszámítása:
W = SIII[E] - [1, 3 + (SIII[0] - 0, 9)]
ahol
W = az oltós savó tömegszázaléka az [E] mintában;
SIII [E] = az [E] vizsgálati minta 9.1.2. pont szerint nyert III. csúcsa alatti relatív terület;
1,3 = a III. csúcs alatti relatív átlagos terület a különféle származású nem módosított sovány tejporban meghatározott oltós savó gramm/100 g-ban kifejezve. Ez egy kísérletben nyert számadat;
SIII [0] = a III. csúcs alatti relatív terület, amely egyenlő RIII × AIII [0]. Ezek a 9.1.1. pontban és a 8.5.3. pontban kapott értékek;
(SIII [0] - 0,9) = az 1,3 relatív átlagos területre elvégzendő korrekció, ha SIII [0] nem egyenlő 0,9. Kísérleti úton a kontrollminta [0] III. csúcsa alatti relatív átlagos terület 0,9.
9.3. Az eljárás pontossága
9.3.1. Ismételhetőség
Az egy időben vagy rövid időeltéréssel egymás után, ugyanazon laboratóriumi személyzet által, ugyanazzal az eszközzel, azonos vizsgálati anyagon elvégzett két meghatározás különbsége nem haladhatja meg a 0,2 % m/m értéket.
9.3.2. Reprodukálhatóság
A két különböző laboratóriumban, azonos vizsgálati anyagon kapott két egyedi, egymástól független eredmény különbsége nem haladhatja meg a 0,4 % m/m értéket.
9.4. Értelmezés
9.4.1. Feltételezzük a savó hiányát a mintában, ha az SIII [E] III. csúcs alatti, a termék 100 grammjára jutó oltós savó grammban kifejezett relatív terület ≤ 2,0 + (SIII [0] - 0,9)
ahol
| 2,0 | a III. csúcs alatti relatív területre megengedett legnagyobb érték, figyelembe véve a III. csúcs alatti relatív átlagos területet, azaz 1,3-at, a sovány tejpor eltérő összetétele miatti bizonytalanságot és a módszer reprodukálhatóságát (9.3.2.), |
| (SIII [0] – 0,9) | elvégzendő korrekció, ha az SIII [0] terület különbözik 0,9-től (lásd a 9.2. pontot). |
9.4.2. Ha a III. csúcs, SIII [E] alatti relatív terület > 2,0 + (SIII [0] - 0,9) és a II. csúcs, SII [E] alatti relatív terület ≤ 160, az oltóssavó-tartalmat a 9.2. pontban jelzett módon határozzuk meg.
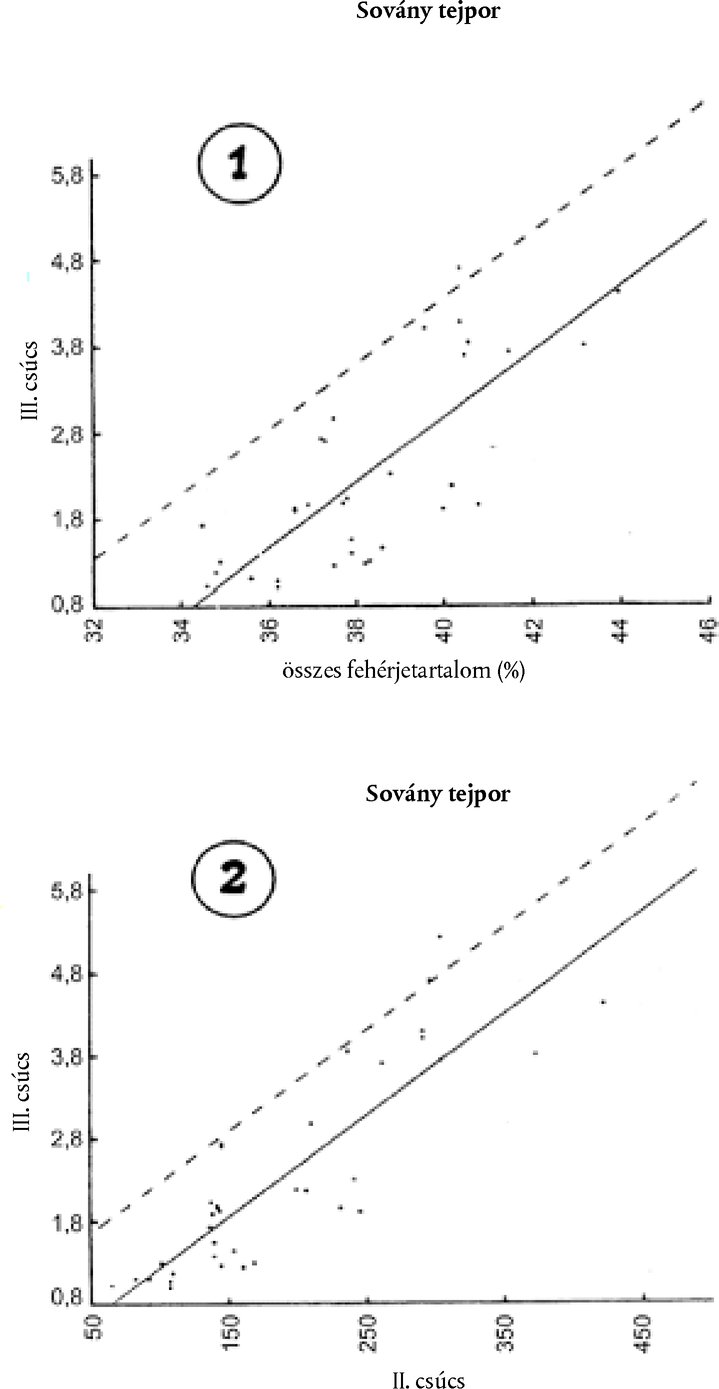
9.4.3. Ha a III. csúcs, SIII [E] alatti relatív terület > 2,0 + (SIII [0] - 0,9) és a II. csúcs, SII [E] alatti relatív terület ≤ 160, határozzuk meg az összes fehérjetartalmat (P %); majd vizsgáljuk meg az 1. és 2. grafikont.
9.4.3.1. A magas fehérjetartalommal rendelkező, módosítás nélküli sovány tejpor mintáinak elemzésével nyert adatok összeállítását az 1. és 2. grafikon mutatja.
A folyamatos vonal lineáris regressziót jelent, amelynek együtthatóit a legkisebb négyzetek módszerével számították ki.
A szaggatott egyenes vonal a III. csúcs alatti relatív terület felső határát az esetek 90 %-ában meg nem haladott valószínűséggel rögzíti.
Az 1. és 2. grafikon szaggatott egyenes vonalainak egyenletei a következők:
| SIII = 0,376 P % – 10,7 | (1. grafikon), |
| SIII = 0,0123 SII [E] + 0,93 | (2. grafikon), |
ahol:
SIII a III. csúcs alatti relatív terület, az összes fehérjetartalom vagy az SII [E] csúcs alatti relatív terület szerint számítva,
P % az összes fehérjetartalom tömegszázalékban kifejezve,
SII [E] a minta 9.1.2. pontban kiszámított relatív területe.
Ezek az egyenletek a 9.2. pontban említett 1,3-as számmal egyenértékűek.
A kapott SIII [E] relatív területe és az SIII relatív területe közötti különbség (T1 és T2) a következők szerint adódik: T1 = SIII[E] - [(0,376 P% - 10,7) + (SIII[0] - 0,9)]T2 = SIII[E] - [(0,0123 SII[E] + 0,93) + (SIII[0] - 0,9)]
| Ha T1 és/vagy T2 | nulla vagy kevesebb, nem határozható meg oltós savó jelenléte. |
| Ha T1 és T2 | nullánál több, oltós savó van jelen. |
Az oltóssavó-tartalmat a következő képlet alapján számíthatjuk ki: W = T2 + 0,91
ahol
0,91 a szaggatott és folyamatos vonal között a függőleges tengelyen mért távolság.
III. függelék
OLTÓSSAVÓ-SZÁRAZANYAG MEGHATÁROZÁSA SOVÁNY TEJPORBAN
1. CÉL: OLTÓSSAVÓ-SZÁRAZANYAG SOVÁNY TEJPORHOZ VALÓ HOZZÁADÁSÁNAK KIMUTATÁSA
2. HIVATKOZÁSOK: ISO 707 NEMZETKÖZI SZABVÁNY
3. FOGALOMMEGHATÁROZÁS
Az oltóssavószárazanyag-tartalom a megadott eljárás alapján, kazeinomakropeptid-tartalom kimutatásával meghatározott, tömegszázalékban kifejezett mennyiség.
4. A MÓDSZER ELVE
A mintákat fordított fázisú nagy teljesítményű folyadékkromatográfiás eljárással (HPLC) vizsgáljuk meg kazeinomakropeptid-A jelenlétére. A mintából kapott eredmények értékelése standard mintákkal való összehasonlítás révén történik, amelyek sovány tejporból, ismert mennyiségű savópor hozzáadásával, vagy anélkül készülnek. Az 1 %-nál (m/m) magasabb eredmények azt mutatják, hogy oltóssavó-szárazanyag van jelen.
5. VEGYSZEREK
Az összes vegyszernek analitikai tisztaságúnak kell lennie. A felhasznált víznek desztillált víznek vagy azzal legalább egyenértékű tisztaságú víznek kell lennie. Az acetonitrilnek spektroszkópiai minőségűnek vagy HPLC-minőségűnek kell lennie.
5.1. Triklór-ecetsav oldat
Oldjunk fel 240 g triklór-ecetsavat (CCl3COOH) vízben, és töltsük fel vízzel 1 000 ml-re. Az oldatnak tisztának és színtelennek kell lennie.
5.2. A. és B. eluens
A. eluens: Helyezzünk 150 ml acetonitrilt (CH3CN), 20 ml izopropanolt (CH3CHOHCH3), és 1,00 ml trifluor-ecetsavat (TFA, CF3COOH) egy 1 000 ml-es mérőlombikba. Töltsük fel vízzel 1 000 ml-re.
B. eluens: Helyezzünk 550 ml acetonitrilt, 20 ml izopropanolt és 1,00 ml TFA-t egy 1 000 ml-es mérőlombikba. Töltsük fel vízzel 1 000 ml-re. Használat előtt egy 0,45 μm pórusátmérőjű membránszűrőn szűrjük át az eluáló oldatot.
5.3. Az oszlop konzerválása
Az elemzéseket követően az oszlopot a B. eluenssel (egy gradiens útján) át kell mosni és ezt követően acetonitrillel elöblíteni (egy gradiens útján, 30 percig). Az oszlopot acetonitrilben kell tárolni.
5.4. Standard minták
5.4.1. Az intervenciós raktározás követelményeinek megfelelő sovány tejpor (azaz [0]).
5.4.2. Ugyanez a sovány tejpor 5 % (m/m) szabványos összetételű oltós savóporral módosítva (azaz [5]).
5.4.3. Ugyanez a sovány tejpor 50 % (m/m) szabványos összetételű oltós savóporral módosítva (azaz [50]).
6. ESZKÖZÖK
6.1. Analitikai mérleg
6.2. 2 200 g centrifugális erő elérésére képes opcionális centrifuga, körülbelül 50 ml-es, ledugózott vagy kupakkal ellátott centrifugacsövekkel felszerelve
6.3. Mechanikus rázógép
6.4. Mágneses keverő
6.5. Körülbelül hét centiméter átmérőjű üvegtölcsérek
6.6. Szűrőpapírok, közepes szűrőképességgel, körülbelül 12,5 cm átmérővel
6.7. Üvegszűrő-berendezés 0,45 μm pórusátmérőjű membránszűrővel
6.8. Beosztással ellátott pipetták, amelyek alkalmasak 10 ml adagolására (ISO 648, A osztály vagy ISO/R 835), vagy olyan adagolórendszer, amely két perc alatt képes 10,0 ml adagolására
6.9. 20,0 ml víz kb. 50 °C-on történő adagolására képes adagolórendszer.
6.10. Termosztáttal ellátott vízfürdő 25 ± 0,5 °C-ra állítva
6.11. HPLC-berendezés, részei:
6.11.1. Bináris gradiensű szivattyúrendszer
6.11.2. Injektor, kézi vagy automata, 100 μl kapacitással
6.11.3. Agilent Technologies Zorbax 300 SB-C3 kromatográfiás oszlop (25 cm hosszú, belső átmérő 0,46 cm) vagy ezzel egyenértékű, nagy belső átmérőjű, szilika alapú fordított fázisú oszlop
6.11.4. Termosztatikus oszlopkamra, 35 ± 1 °C-ra állítva
6.11.5. Változtatható hullámhosszúságú UV-detektor, amely 210 nm-en (szükség esetén magasabb hullámhosszon is, 220 nm-ig) 0,02 Å érzékenységű méréseket tesz lehetővé
6.11.6. A közös alapvonalra vagy völgytől-völgyig történő integrálásra alkalmas integrátor
Megjegyzés: Szobahőmérsékleten tartott oszlopokkal is lehet dolgozni, de ekkor a hőmérséklet az elemzés során sem változhat ± 1 °C-nál többet, ellenkező esetben túl nagy lesz a CMPA retenciós idejében fellépő variancia.
7. MINTAVÉTEL
7.1. A mintákat az ISO 707 nemzetközi szabványban meghatározottak szerint kell venni. A tagállamok azonban alkalmazhatnak ettől eltérő mintavételi módszert is, feltéve hogy az teljesíti a fent említett szabványban foglalt alapelveket.
7.2. Tároljuk a mintát olyan körülmények között, amely kizárja, hogy az összetétele megváltozzon vagy lebomoljon.
8. ELJÁRÁS
8.1. A vizsgálati minta előkészítése
A tejport tegyük a tejpor terjedelménél kétszer nagyobb űrtartalmú edénybe, amely légmentesen zárható. Azonnal zárjuk le az edényt. Az edény ismételt felfordításával alaposan keverjük fel a benne lévő tejport.
8.2. Vizsgálati adag
Egy centrifugacsőbe (6.2.) vagy egy alkalmas 50 ml-es ledugaszolt lombikba mérjünk ki a mintából 2,00 ± 0,001 g-ot.
Megjegyzés: Keverékek esetében a vizsgálati mintából olyan mennyiséget mérjünk ki, hogy a zsírtalanított mintaadag 2,00 g-nak feleljen meg.
8.3. Zsír és fehérje eltávolítása
8.3.1. Adjunk 20,0 ml meleg vizet (50 °C) a vizsgálandó mennyiséghez. Oldjuk fel a port mechanikus rázógép segítségével ötperces rázatással (6.3.). Helyezzük a centrifugacsövet vízfürdőbe (6.10.), és engedjük 25 °C-ra hűlni.
8.3.2. Adjunk hozzá 10,0 ml 25 °C hőmérsékletű triklór-ecetsav oldatot (5.1.) folyamatosan két percen át, miközben erőteljesen keverjük a mágneses keverő segítségével (6.4.). Helyezzük a csövet vízfürdőbe (6.10.), és hagyjuk ott 60 percig.
8.3.3. Centrifugáljuk (6.2.) 10 percig 2 200 g-n, vagy szűrjük át szűrőpapíron (6.6.). Öntsük ki az első 5 ml szűrletet.
8.4. Kromatográfiás meghatározás
8.4.1. A fordított fázisú HPLC-módszer kizárja a savanyú írópor jelenléte miatti hamis pozitív eredmények lehetőségét.
8.4.2. Mielőtt végrehajtanánk a fordított fázisú HPLC-elemzést, a gradienskörülményeket optimalizálni kell. A körülbelül 6 ml holttérfogattal (a térfogat az oldatok összefutási pontjától az injektorhurok térfogatáig bezárólag) rendelkező gradiensrendszerekhez 26 ± 2 perces CMPA-retenciós idő az ideális. Az ennél kisebb holttérfogattal rendelkező gradiensrendszerekhez (például 2 ml) optimális retenciós időnek 22 percet használjunk.
Vegyük az 50 % oltós savót tartalmazó, illetve nem tartalmazó standard mintákat (5.4.).
Injektáljunk 100 μl felülúszót vagy szűrletet (8.3.3.) a HPLC-berendezésbe, amely az 1. táblázat szerint megadott felderítő gradienskörülmények között működik.
1. táblázat
Felderítő gradienskörülmények a kromatográfiás vizsgálatok optimalizálásához
| Idő (perc) | Áramlási sebesség (ml/min) | % A | % B | Görbe |
| Kiindulás | 1,0 | 90 | 10 | * |
| 27 | 1,0 | 60 | 40 | lineáris |
| 32 | 1,0 | 10 | 90 | lineáris |
| 37 | 1,0 | 10 | 90 | lineáris |
| 42 | 1,0 | 90 | 10 | lineáris |
A két kromatogram összehasonlítása megmutatja a CMPA-csúcs helyét.
A következőkben meghatározott képlet alkalmazásával kiszámítható, hogy milyen kiindulási oldószer-összetételt kell alkalmazni a normál gradienshez (lásd 8.4.3.). % B = 10 - 2,5 + (13,5 + (RTcmpA - 26) / 6) × 30 / 27 % B = 7,5 + (13,5 + (RTcmpA - 26) / 6) × 1,11
ahol
RTcmpA : CMPA retenciós ideje a felderítő gradiensben
10 : a felderítő gradiens kiindulási % B-je
2,5 : a % B a középpontnál mínusz a % B a kiinduláskor a normál gradiensben
13,5 : a felderítő gradiens fél futamideje
26 : a CMPΑ szükséges retenciós ideje
6 : a felderítő és normál gradiens meredekségeinek aránya
30 : a % B kiinduláskor mínusz a % B 27 percnél a felderítő gradiensben
27 : a felderítő gradiens futamideje.
8.4.3. Vegyünk oldatot a vizsgálati mintákból
Fecskendezzünk 100 μl pontosan kimért felülúszót vagy szűrletet (8.3.3.) a percenként 1,0 ml eluáló oldat (5.2.) áramlási sebességgel üzemelő HPLC-berendezésbe.
Az elemzés kezdetén az eluens összetételét a 8.4.2. pont adja meg. Ez rendszerint közelíti az A:B = 76:24 arányt (5.2.). Közvetlenül az injektálás után egy lineáris gradiens indul, amely 27 perc elteltével a B-oldat 5 %-kal magasabb százalékos arányát eredményezi. Ezután elindul egy lineáris gradiens, amely az eluens összetételét öt perc múlva 90 % B arányra alakítja. Ez az összetétel öt percen keresztül fennmarad, ezt követően egy lineáris gradiens mentén öt perc alatt a kiindulási összetételre változik. A szivattyúrendszer belső térfogatától függően a következő injektálást az eredeti állapot visszaállása után 15 perccel lehet elvégezni.
1. megjegyzés: A CMPA retenciós idejének 26 ± 2 percnek kell lennie. Ezt az első gradiens kiindulási és végponti feltételeinek változtatásával lehet elérni. Mindazonáltal a % B különbségének a kiindulási és végponti körülmények között az első gradiensben 5 % B értékűnek kell maradnia.
2. megjegyzés: Az eluenseknek kellőképpen gázmenteseknek kell lenniük és így is kell maradniuk. Ez létfontosságú a gradiens-szivattyúrendszer megfelelő működéséhez. A CMPA-csúcs retenciós idejének szórása kisebb kell legyen 0,1 percnél (n = 10).
3. megjegyzés: Minden öt minta után ismét a referenciamintát [5] kell beinjektálni, és segítségével új R-válaszjelet kell meghatározni (9.1.1.).
8.4.4. A vizsgálati minta (E) kromatográfiás elemzésének eredményét egy kromatogram formájában kapjuk meg, ahol a CMPA-csúcsot a körülbelül 26 perces retenciós idő alapján lehet beazonosítani.
Az integrátor (6.11.6.) automatikusan kiszámolja a CMPA-csúcs H-csúcsmagasságát. Az alapvonal helyzetét minden kromatogramon ellenőrizni kell. Ha az alapvonal illesztése nem volt pontos, az elemzést vagy az integrálást ismételni kell.
Megjegyzés: Ha a CMPA-csúcs kellőképp elkülönül a többi csúcstól, völgytől-völgyig történő alapvonal-hozzárendelést kell alkalmazni, egyéb esetben merőlegeseket kell egy közös alapvonalra állítani, a kiindulási pont legyen közel a CMPA-csúcshoz (azaz nem t = 0 percnél!). Használjuk ugyanazt az integrálási módszert a standardnál és a mintáknál is, és közös alapvonal esetén ellenőrizzük a konzisztenciáját a minták és a standard esetében is.
Lényeges, hogy még a mennyiségi értelmezés előtt valamennyi kromatogram külalakját megvizsgáljuk, hogy felfedezzünk minden olyan rendellenességet, amely vagy a készülék vagy az oszlopok hibás működéséből ered, vagy a vizsgált minta eredetére és jellegére vezethető vissza. Kétség esetén az elemzést meg kell ismételni.
8.5. Kalibrálás
8.5.1. A standard mintákra (5.4.1. és 5.4.2.) is alkalmazzuk pontosan ugyanazt az eljárást, mint amely a 8.2.-8.4.4. pontig tartó leírásban szerepel. Használjunk frissen készített oldatokat, mert a CMP 8 %-os triklór-ecetsavas környezetben, szobahőmérsékleten hamar bomlik. Az oldat 4 °C-on 24 órán át stabil marad. Hosszú elemzési sorok esetében ajánlatos az automatikus injektorban hűtött mintatálcát használni.
Megjegyzés: A 8.4.2. pont kihagyható, ha a kiindulási feltételre korábbi elemzésekből ismert a % B értéke.
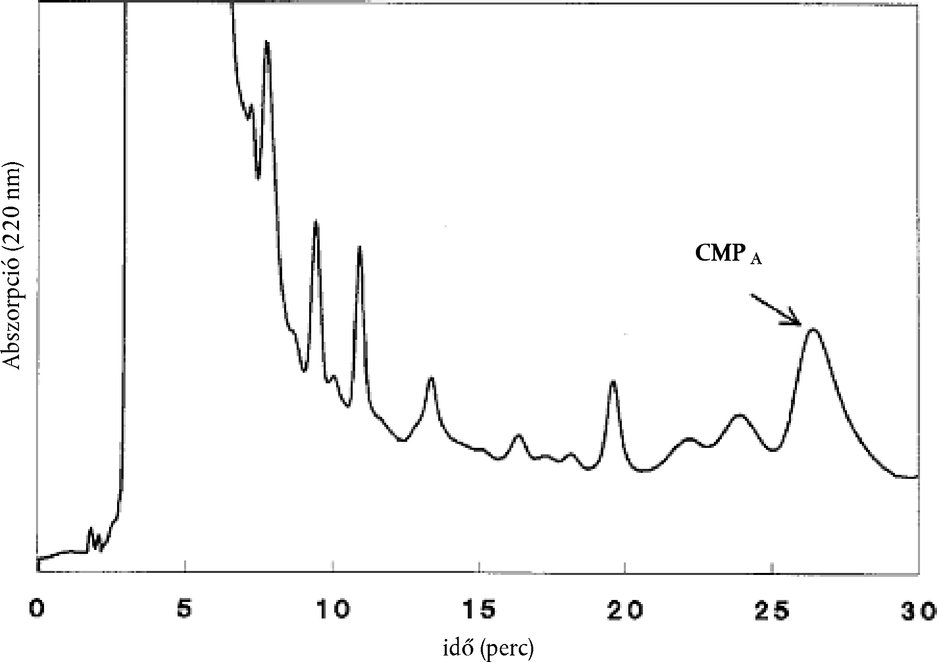
A referenciaminta [5] kromatogramjának az 1. ábrával analógnak kell lennie. Ezen a görbén a CMPA-csúcsot két kisebb csúcs előzni meg. Lényeges ugyanilyen elválasztást elérni.
8.5.2. A minták kromatográfiás meghatározása előtt injektáljunk 100 μl oltós savó nélküli standard mintát [0] (5.4.1.).
Ekkor a kromatogramon a CMPA-csúcs retenciós idejénél csúcs ne jelenjen meg.
8.5.3. Az R-válaszjelet úgy határozzuk meg, hogy a mintáknál használttal azonos mennyiségű szűrletet injektálunk (8.5.1.).
9. AZ EREDMÉNYEK KIFEJEZÉSE
9.1. A számítás módszere és a képletek
9.1.1. Az R-válaszjel kiszámítása:
CMPA-csúcs: R = W/H
ahol
R = a CMPA-csúcs válaszjele
H = a CMPA-csúcs magassága
W = a savó mennyisége a standard mintában [5].
9.2. A mintában található oltós savópor százalékos mennyiségének kiszámítása
W(E) = R × H(E)
ahol
W(E) = az oltós savó tömegszázaléka az (E) mintában
R = R a CMPA-csúcs válaszjele (9.1.1.)
H(E) = H(E) a CMPA-csúcs magassága az (E) mintában
Amennyiben W(E) nagyobb mint 1 %, és a retenciós ideje, valamint a standard minta [5] retenciós ideje közötti különbség kisebb mint 0,2 perc, akkor oltóssavó-szárazanyag van jelen.
9.3. Az eljárás pontossága
9.3.1. Ismételhetőség
Az egy időben vagy rövid időeltéréssel egymás után, ugyanazon laboratóriumi személyzet által, ugyanazzal az eszközzel, azonos vizsgálati anyagon elvégzett két meghatározás különbsége nem haladhatja meg a 0,2 % m/m értéket.
9.3.2. Reprodukálhatóság
Nincs meghatározva.
9.3.3. Linearitás
A 0-16 % oltóssavó-tartalom esetén lineáris összefüggést kell kapni, amelynek korrelációs együtthatója > 0,99.
9.4. Értelmezés
Az 1 %-os határ tartalmazza a reprodukálhatóság miatti bizonytalanságot.
1. ábra
Ni -4,6 standard
(*) IDF 135B/1991 nemzetközi szabvány. Tej és tejtermékek. Analitikai módszerek pontossági jellemzői. Vázlat a kollaboratív vizsgálati eljáráshoz."
3. A szöveg a következő mellékletekkel egészül ki:
"
VI. MELLÉKLET
A magántárolás keretében tárolt vaj esetében alkalmazandó elemzési módszerek
| Paraméter | Módszer |
| Zsír (6) | ISO 17189 vagy ISO 3727, 3. rész |
| Víz | ISO 3727, 1. rész |
| Zsírmentes szárazanyag (a só kivételével) | ISO 3727, 2. rész |
| Só | ISO 15648 |
VII. MELLÉKLET
A magántárolás keretében tárolt sovány tejpor esetében alkalmazandó elemzési módszerek
| Paraméter | Módszer |
| Zsír | ISO 1736 |
| Fehérje | ISO 8968, 1. rész |
| Víz | ISO 5537 |
VIII. MELLÉKLET
A magántárolás keretében tárolt sajtok esetében alkalmazandó elemzési módszerek
1. A függelékben meghatározott elemzési módszert kell használni annak biztosítására, hogy a csak és kizárólag juhtejből, kecsketejből, bivalytejből, illetve juhtej, kecsketej és bivalytej keverékéből készülő sajtok ne tartalmazzanak tehéntejkazeint.
A tehéntejkazein akkor minősül a mintában jelen lévőnek, ha az elemzett minta tehéntejkazein-tartalma egyenlő vagy nagyobb, mint a függelékben meghatározott, 1 % tehéntejet tartalmazó referenciaminta tehéntejkazein-tartalma.
2. A tehéntejkazeinnek az 1. pontban említett sajtokban történő kimutatására szolgáló módszerek alkalmazása engedélyezett, feltéve, hogy:
a) a kimutatási határ legfeljebb 0,5 %; és
b) nincsenek hamis pozitív eredmények; valamint
c) a tehéntejkazein megfelelő érzékenységgel még hosszabb érlelési időszakot követően is kimutatható, úgy ahogy a szokásos kereskedelmi körülmények mellett előfordulna.
Ha a fent említett követelmények bármelyike nem teljesül, a függelékben meghatározott módszereket kell alkalmazni.
Függelék
MÓDSZER A JUHTEJBŐL, KECSKETEJBŐL VAGY BIVALYTEJBŐL, ILLETVE JUH-, KECSKE- ÉS BIVALYTEJ ELEGYÉBŐL KÉSZÍTETT SAJTOKBAN TALÁLHATÓ TEHÉNTEJ ÉS KAZEINÁT KIMUTATÁSÁRA
1. TÁRGY
Juhtejből, kecsketejből vagy bivalytejből, illetve juh-, kecske- és bivalytej elegyéből készített sajtokban található tehéntej és kazeinátok kimutatása a γ-kazeinekre alkalmazott, plazmolízist követő izoelektromos fókuszálással.
2. ALKALMAZÁSI KÖR
Ez a módszer alkalmas a kezeletlen és hőkezelt tehéntej, valamint kazeinát nagy érzékenységű és specifikus kimutatására juhtejből, kecsketejből vagy bivalytejből, illetve juh-, kecske- és bivalytej elegyéből készített friss és érlelt sajtokban. Nem alkalmas azonban a tej és a sajt hőkezelt tehéntejsavófehérje-koncentrátumokkal történt hamisításának kimutatására.
3. A MÓDSZER ELVE
3.1. A kazeinek izolálása a sajtból és a referenciastandardból.
3.2. Az izolált kazeinek feloldása és alávetése a plazminos (EC.3.4.21.7.) bontásnak.
3.3. Izoelektromos fókuszálás alkalmazása a plazminnal kezelt kazeinekre karbamid jelenlétében, és a fehérjék megfestése.
3.4. A megfestett (a tehéntej jelenlétét bizonyító) γ3 és γ2-kazeinmintázatoknak az értékelése a vizsgálati mintából nyert mintázatok és az ugyanazon a gélen, a 0 %, illetve az 1 % tehéntejet tartalmazó referenciastandardokból kapott mintázatok összehasonlításával.
4. VEGYSZEREK
Ellenkező rendelkezés hiányában, analitikai tisztaságú vegyszereket kell alkalmazni. A víznek kétszeresen desztilláltnak vagy azzal egyenértékű tisztaságúnak kell lennie.
Megjegyzés: A következő részletek a laboratóriumban elkészített, karbamidtartalmú poliakrilamid gélekre vonatkoznak, amelyek méretei 265 × 125 × 0,25 mm. Ha más nagyságú vagy más típusú gélt használunk, a leválasztási körülményeket külön be kell állítani.
Izoelektromos fókuszálás
4.1. Vegyszerek a karbamidtartalmú poliakrilamid gél előállításához
4.1.1. Géltörzsoldat
Oldjuk fel a következőket vízben:
4,85 g akrilamid
0,15 g N, N′-metilén-bisz-akrilamid (BIS)
48,05 g karbamid
15,00 g glicerin (87 % m/m),
töltsük fel 100 ml-re, és hűtőszekrényben, barna üvegben tároljuk.
Megjegyzés: A fent említett fix tömegű, neurotoxikus akrilamidok helyett előnyösebb lehet a kereskedelemben kapható, előre bekevert akrilamid/BIS-oldat használata. Ha az ilyen oldat 30 % vegyesszázalék (m/v) akrilamidot és 0,8 % m/v BIS-t tartalmaz, 16,2 ml térfogatot kell belőle felhasználni a fix tömegek helyett. A törzsoldat eltarthatósági ideje legfeljebb 10 nap; ha vezetőképessége több mint 5 μS, 2 g Amberlite MB-3 vegyszerrel kell 30 percen keresztül történő keverés útján ionmentesíteni, majd egy 0,45 μm pórusátmérőjű membránszűrőn átszűrni.
4.1.2. Géloldat
Készítsük el a géloldatot úgy, hogy az adalékanyagokat és az amfolitokat (*) elkeverjük a géltörzsoldattal (lásd a 4.1.1. pontot).
9,0 ml törzsoldat
24 mg β-alanin
500 μl 3,5-9,5 pH közötti amfolit
250 μl 5-7 pH közötti amfolit
250 μl 6-8 pH közötti amfolit
Keverjük össze a géloldatot, és vákuumban vagy ultrahangos fürdőben gázmentesítsük két vagy három percen át.
Megjegyzés: A géloldatot közvetlenül a kiöntés előtt készítsük el (lásd a 6.2. pontot).
4.1.3. Katalizátoroldatok
4.1.3.1. N, N, N′ N′ - tetrametil-etilén-diamin (Temed).
4.1.3.2. 40 % m/v ammónium-perszulfát (PER):
Oldjunk fel vízben 800 mg PER-t, és töltsük fel vízzel 2 ml-re.
Megjegyzés: Mindig frissen készített PER-oldatot használjunk.
4.2. Kontaktfolyadék
Világítópetróleum vagy folyékony paraffin
4.3. Anódoldat
Oldjunk fel vízben 5,77 g foszforsavat (85 % m/m), és töltsük fel vízzel 100 ml-re.
4.4. Katódoldat
Oldjunk fel vízben 2,00 g nátrium-hidroxidot, és hígítsuk fel vízzel 100 ml-re.
A minta előkészítése
4.5. Regensek a fehérjék izolálására
4.5.1. Hígított ecetsav (25,0 ml jégecet vízzel 100 ml-re hígítva)
4.5.2. Diklórmetán
4.5.3. Aceton
4.6. Proteinoldó puffer
Oldjuk fel vízben a következőket:
5,75 g glicerin (87 % m/m)
24,03 g karbamid
250 mg ditio-treitol,
és töltsük fel 50 ml-re.
Megjegyzés: Hűtőszekrényben tárolva eltarthatósági ideje legfeljebb egy hét.
4.7. A kazeinek plazminos bontására használt vegyszerek
4.7.1. Ammónium-karbonát-puffer
Titráljunk 0,05 mol/l etilén-diamin-tetraecetsavat (EDTA, 1,46 g/100 ml) tartalmazó 0,2 mol/l ammónium-hidrogénkarbonát-oldatot (1,58 g/100 ml víz) 0,05 mol/l EDTA-t tartalmazó 0,2 mol/l ammónium-karbonát-oldattal (1,92 g/100 ml víz) 8 pH-ig.
4.7.2. Szarvasmarhaplazmin (EC.3.4.21.7.), aktivitása legalább 5 U/ml
4.7.3. ε-aminokapronsav-oldat enzimgátlásra
Oldjunk fel 2 624 g ε-aminokapronsavat (6-amino-n-hexánsav) 100 ml 40 térfogat-százalékos etil-alkoholban.
4.8. Standardok
4.8.1. Bizonylattal ellátott, 0 %, illetve 1 % tehéntejet tartalmazó, tejoltóenzimmel kezelt fölözött juhtej- és kecsketejkeverék referenciastandardot a Referenciaanyagok és Mérések Bizottsági Intézeténél (B-2440 Geel, Belgium) lehet beszerezni.
4.8.2. Tejoltóenzimmel kezelt bivalytejből előállított, 0 % és 1 % tehéntejet tartalmazó laboratóriumi belső standard készítése
A fölözött tejet nyers, ömlesztett bivalytejből, illetve szarvasmarhatejből 37 °C-on, 2 500 g-vel 20 percen át történő centrifugálással készítjük. A centrifugacsövet a tartalmával együtt hirtelen 6-8 °C-ra lehűtjük, majd a felső, zsíros réteget teljes egészében eltávolítjuk. Az 1 %-os standard elkészítéséhez 1 literes főzőpohárban adjunk 5,00 ml fölözött szarvasmarhatejet 495 ml fölözött bivalytejhez, pH-ját 10 vegyesszázalékos (m/v) hígított tejsav hozzáadásával állítsuk be 6,4-re. A hőmérsékletet állítsuk be 35 °C-ra és adjunk hozzá 100 μl borjúgyomorból nyert oltóenzimet (aktivitása 1:10 000, c. 3 000 U/ml), egy percen keresztül kavarjuk, majd a főzőpoharat alufóliával letakarva hagyjuk egy órán át 35 °C-on pihenni az alvadék kialakulásának elősegítésére. Miután az alvadék kialakult, a teljes beoltott tejet előzetes homogenizálás és a savó leszívása nélkül fagyasztva szárítjuk. A fagyasztva szárítást követően az anyagot finomra daráljuk, amíg homogén port nem kapunk. A 0 %-os standard elkészítéséhez ugyanezt a műveletsort a tiszta fölözött bivalytejjel kell elvégezni. A standardokat - 20 °C-on kell tárolni.
Megjegyzés: Ajánlatos a bivalytej tisztaságát a standardok elkészítése előtt a plazminos emésztéssel előkészített kazeinek izoelektromos fókuszálásával ellenőrizni.
A fehérje festéséhez használt vegyszerek
4.9. Fixálószer
Oldjunk fel vízben 150 g triklór-ecetsavat, és töltsük fel vízzel 1 000 ml-re.
4.10. Festékeltávolító oldat
Oldjunk fel 500 ml metil-alkoholt és 200 ml jégecetet 2 000 ml desztillált vízben.
Megjegyzés: A festékeltávolító oldatot minden nap frissen készítsük el; elkészíthető 50 % térfogat-százalékos metilalkohol- és 20 térfogat-százalékos jégecettörzsoldat egyenlő mennyiségeinek az összekeverésével.
4.11. Festékoldatok
4.11.1. Festékoldat (1. törzsoldat)
Mágneses keverő alkalmazásával oldjunk fel 3,0 g Coomassie G-250 brilliánskéket (C.I. 42655) 1 000 ml 90 térfogat-százalékos metil-alkoholban (megközelítőleg 45 percen át), és két, közepes sebességű, redős szűrőn szűrjük át.
4.11.2. Festékoldat (2. törzsoldat)
Oldjunk fel 5,0 g rézszulfát-pentahidrátot 1 000 ml 20 térfogat-százalékos jégecetben.
4.11.3. Festékoldat (munkaoldat)
Közvetlenül festés előtt öntsünk össze 125 ml-t mindkét törzsoldatból (4.11.1., 4.11.2.).
Megjegyzés: A festékoldatot a felhasználás napján kell elkészíteni.
5. ESZKÖZÖK
5.1. Üveglemezek (265 × 125 × 4 mm); gumihenger (15 cm széles); szintező asztal
5.2. Géltartó lemez (265 × 125 mm)
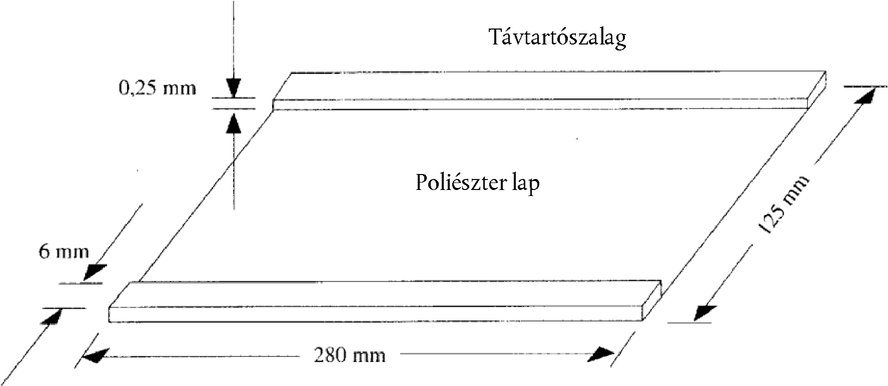
5.3. Fedőlemez (280 × 125 mm). Mindkét hosszanti végére ragasszunk fel egy-egy csíkban ragasztószalagot (280 × 6 × 0,25 mm) (lásd az 1. ábrát)
5.4. Elektrofókuszáló kamra hűtőlemezzel (pl. 265 × 125 mm) és megfelelő áramellátással (≥ 2,5 kV) vagy automata elektroforézis-készülék
5.5. Cirkulációs kriosztát, termosztátos vezérléssel 12 ± 0,5 °C-on
5.6. 3 000 g-ig állítható centrifuga
5.7. Elektródacsíkok (≥ 265 mm hosszban)
5.8. Műanyag csepegtetőpalackok az anód- és a katódoldat számára
5.9. Mintafelvivő eszköz (10 × 5 mm, viszkózus vagy alacsony fehérjeadszorpciós szűrőpapír)
5.10. Rozsdamentes acél vagy üvegfestő- és festékeltávolító-edények (például 280 × 150 mm műszertálca)
5.12. Szabályozható rúdhomogenizátor (10 mm tengelyátmérővel), percenkénti fordulatszám-tartomány 8 000-20 000 között
5.13. Mágneses keverő
5.14. Ultrahangos fürdő
5.15. Filmhegesztő gép
5.16. 25 μl-es mikropipetta
5.17. Vákuumbepárló vagy fagyasztva-szárító
5.18. Termosztatikus vezérlésű, 35 és 40 ± 1 °C-ra állítható vízfürdő rázógéppel
5.19. Denzitométer, λ = 634 nm-es leolvasóval felszerelve
6. ELJÁRÁS
6.1. A minta előkészítése
6.1.1. Kazeinek izolálása
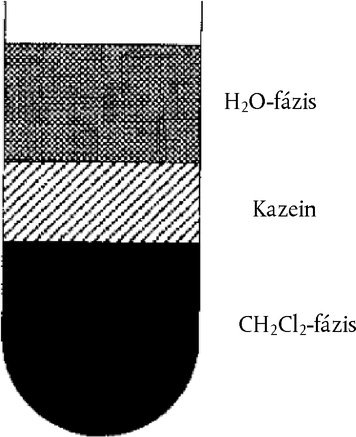
Egy 100 milliliteres centrifugacsőbe mérjünk ki 5 g szárazanyag-mennyiségnek megfelelő sajtot vagy a referenciastandardot, adjunk hozzá 60 ml desztillált vizet és homogenizáljuk a rúdhomogenizátorral (8 000-10 000-es percenkénti fordulatszám között). Állítsuk be hígított ecetsavval (4.5.1.) 4,6 pH-ra, és centrifugáljuk (5 perc, 3 000 g). A zsírt és a savót öntsük le, a maradékot pedig 20 000-es percenkénti fordulatszámon homogenizáljuk 40 ml desztillált vízben, amelynek a pH-ját előzetesen hígított ecetsavval (4.5.1.) 4,5 pH-ra állítottuk be, adjunk hozzá 20 ml diklórmetánt (4.5.2.), ismét homogenizáljuk és centrifugáljuk (5 perc, 3 000 g). Egy spatulával távolítsuk el a kazeinréteget, amely a vizes és a szerves fázis között található (lásd a 2. ábrát), és dekantáljuk mindkét fázist. A kazeint ismét homogenizáljuk 40 ml desztillált vízben (a fentiek szerint), és 20 ml diklórmetánnal (4.5.2.) centrifugáljuk. Addig ismételjük ezt a műveletet, amíg mindkét extraháló fázis színtelen nem lesz (két-három alkalommal). A fehérjemaradékot 50 ml acetonban (4.5.3.) homogenizáljuk, és egy közepes átfolyási sebességű, redős papírszűrőn szűrjük át. A szűrőn maradt maradékot két, külön 25 ml-es adagban acetonnal mossuk le, hagyjuk a levegőn megszáradni, vagy nitrogénnel szárítsuk, majd törjük finom porrá egy mozsárban.
Megjegyzés: száraz kazeinizolátumokat - 20 °C hőmérsékleten kell tartani.
6.1.2. A β-kazeinek plazminos bontása a γ-kazeinek kifejezettebbé tétele érdekében
Oszlassunk el 25 mg izolált kazeint (6.1.1.) 0,5 ml ammónium-karbonát-pufferben (4.7.1.) és 20 percig homogenizáljuk, például ultrahangos kezeléssel. Melegítsük fel 40 °C-ra, és adjunk hozzá 10 μl plazmint (4.7.2.), keverjük össze, majd folyamatos rázatás mellett inkubáljuk egy órán át 40 °C-on. Az enzim késleltetése érdekében adjunk hozzá 20 μl ε-aminokapronsav-oldatot (4.7.3.), majd 200 mg szilárd karbamidot és 2 mg ditio-treitolt.
Megjegyzés: A fókuszált kazeincsíkok nagyobb szimmetriájának elérése érdekében ajánlatos az oldatot a ε-aminokapronsav hozzáadását követően liofilizálni, majd a maradékot 0,5 ml proteinoldó-pufferben oldani (4.6.).
6.2. A karbamidtartalmú poliakrilamid gél elkészítése
Néhány csepp víz segítségével terítsük ki a géltartó lemezt (5.2.) egy üveglapra (5.1.) egy papírtörölközővel vagy itatóssal eltávolítva a felesleges vizet. A fedőlemezt (5.3.) egy másik üveglapra terítsük ki a távtartókkal (0,25 mm) együtt azonos módon. Fektessük a lapot vízszintesen egy szintező asztalra.
Adjunk hozzá az előkészített és légmentesített géloldathoz (4.1.2.) 10 μl Temed-oldatot (4.1.3.1.), keverjük össze és adjunk még hozzá 10 μl PER-oldatot (4.1.3.2.), ezt is alaposan keverjük össze, majd azonnal öntsük ki egyenletesen a fedőlemez közepére. A géltartó lemez egyik szélét arccal lefelé fektessük a fedőlemez mellé, és lassan engedjük rá úgy, hogy a két lemez között egy gélfilm képződjön, amely egyenletesen és buborékmentesen kitölti a helyet (3. ábra). Egy vékony spatula segítségével gondosan, egészen engedjük le a gélhordozó lemezt, és helyezzünk súlyként még három üveglemezt a tetejére. Miután a polimerizáció befejeződött (mintegy 60 perc elteltével), a polimerizálódott gélt az üveglemezek felfordításával emeljük át a hordozólemezről a fedőlemezre. A hordozólemez alsó felét gondosan tisztítsuk meg a maradványok és a karbamid eltávolítása érdekében. A gélszendvicset hegesszük bele egy filmcsőbe, és tároljuk hűtőszekrényben (legfeljebb hat hétig).
Megjegyzés: A fedőlemez a távtartókkal újra felhasználható. A poliakrilamid gél kisebb darabokra is felvágható, amely akkor ajánlott, ha kevesebb minta áll rendelkezésünkre, vagy egy automatikus elektroforézis készüléket használunk (két gél, 4,5 × 5 cm méretben).
6.3. Izoelektromos fókuszálás
A hűtőtermosztátot állítsuk be 12 °C-ra. A géltartó lemez hátulját petróleummal töröljük le, majd cseppentsünk néhány csepp petróleumot (4.2.) a hűtőblokk közepére. Ezután a gélszendvicset terítsük rá hordozó felülettel lefelé, óvatosan ügyelve arra, hogy ne képződjenek levegőbuborékok. Törüljünk le minden felesleges petróleumot, és távolítsuk el a fedőlemezt. Az elektródcsíkokat áztassuk bele az elektródos oldatokba (4.3., 4.4.), vágjuk le a gél hosszára, és fektessük le a megadott helyzetbe (az elektródok távolsága 9,5 cm).
Az izoelektromos fókuszálás feltételei:
6.3.1. Gélméret 265 × 125 × 0,25 mm
| SZÖVEG HIÁNYZIK |
Megjegyzés: Ha a gél vastagságát vagy szélességét megváltoztatjuk, az áramerősség és a teljesítmény értékeit megfelelő módon kell módosítani (például kettőzzük meg az áramerősség és teljesítmény értékeit, ha 265 × 125 × 0,5 mm-es gélt használunk).
6.3.2. A következőkben egy automatikus elektroforéziskészülék feszültségprogramja látható (2 db 5,0 × 4,5 cm gél), az elektródokat csíkok nélkül, közvetlenül a gélre helyezzük fel.
| SZÖVEG HIÁNYZIK |
A mintafelvivőt a 2. lépésben 0 Vh-nál helyezzük el.
A mintafelvivőt a 2. lépésben 30 Vh-nál távolítsuk el.
6.4. Fehérjefestés
6.4.1. Fehérjefixálás
Az áram kikapcsolása után azonnal távolítsuk el az elektródcsíkokat, és a gélt rögtön tegyük a 200 ml fixálószerrel (4.9.) töltött festő/festékeltávolító edénybe; folyamatos rázatás mellett hagyjuk benne 15 percig.
6.4.2. A géllemez mosása és festése
Alaposan öntsük le a fixálószert, és a géllemezt kétszer harminc másodpercig mossuk le alkalmanként 100 ml festékeltávolító szerben (4.10.). Öntsük le az eltávolító oldatot, és töltsük meg az edényt 250 ml festőoldattal (4.11.3.); 45 percig fessünk, miközben gyengén rázogatjuk az edényt.
6.4.3. Festék eltávolítása a géllemezről
Öntsük le a festőoldatot, kétszer mossuk le alkalmanként 100 ml festékeltávolító szerben (4.10.), majd rázzuk 15 percen át 200 ml festékeltávolító szerben, és ismételjük meg a festékeltávolítást legalább két-három alkalommal egészen addig, amíg a háttér tiszta és színtelen nem lesz. Ezt követően desztillált vízzel öblítsük le a géllemezt (2 × 2 perc), és levegőn (2-3 óra) vagy hajszárítóval (10-15 perc) szárítsuk meg.
1. megjegyzés: A fixálást, mosást, festést és festékeltávolítást 20 °C-on végezzük. Ne használjunk magas hőmérsékletet.
2. megjegyzés: Ha az érzékenyebb ezüstfestést (például a Pharmacia Biotech ezüstfestő fehérjekészlete, kódszáma 17-1150-01) választjuk, a plazminnal kezelt kazeinmintákat 5 mg/ml-re kell hígítani.
7. ÉRTÉKELÉS
Az értékelést az ismeretlen minta által adott fehérjemintázatnak a referenciastandardból ugyanazon a gélen kapott mintázattal történő összehasonlítása révén végezhetjük el. A juh-, kecske- és bivalytejből, illetve juh-, kecske- és bivalytej keverékéből készített sajtokban található tehéntej kimutatása a γ3- és γ2-kazeinek révén történik, amelyek izoelektromos fókuszálási pontjai a 6,5-ös pH és 7,5-ös pH közötti tartományba esnek (4a., 4b. és 5. ábra). A kimutatási határ kisebb mint 0,5 %.
7.1. Szemrevételezéssel történő értékelés
A szarvasmarha-eredetű tej mennyiségének szemrevételezéssel történő értékeléséhez ajánlatos a minták és a standardok koncentrációit úgy beszabályozni, hogy ugyanolyan intenzitást kapjunk a juh-, kecske- és/vagy bivalyeredetű γ2- és γ3-kazeinekre (lásd a "γ2 E,G,B" és "γ3 E,G,B" csíkokat a 4a., 4b. és 5. ábrán). Ezt követően az ismeretlen minta szarvasmarha-eredetű tejtartalma (1 %-nál kevesebb, egyenlő vagy több) közvetlenül megítélhető, ha a szarvasmarha-eredetű γ3- és γ2-kazeineket (lásd a "γ3 C" és "γ2 C" csíkot a 4 a., 4b. és az 5. ábrán) egybevetjük a 0 % és az 1 %-os referenciastandardokkal (juh és kecske esetében), valamint a laboratórium belső standardjával (bivalynál).
7.2. Denzitometriás értékelés
Ha rendelkezésre áll, használjunk denzitométert (5.19.) a szarvasmarha-eredetű és a juh-, kecske-, illetve bivalyeredetű γ2- és γ3-kazeinek adta csúcsok egymáshoz viszonyított arányának mérésére (lásd az 5. ábrát). Hasonlítsuk össze ezt az értéket az ugyanezen a gélen elemzett 1 %-os referenciastandard (juh, kecske), illetve a laboratóriumi belső standard (bivaly) γ2- és γ3-kazeincsúcsa által lefedett terület arányával.
Megjegyzés: A módszer akkor működik megfelelően, ha mindkét szarvasmarha-eredetű kazeinre, a γ2- és γ3-kazeinekre egyértelműen pozitív a jelzés az 1 %-os referenciastandardban, de nincs jel a 0 %-os standardban. Ellenkező esetben, az eljárást optimalizálni kell a módszer részletes leírását követve.
A mintát akkor fogjuk pozitívnak tekinteni, ha benne a szarvasmarha-eredetű γ2- és γ3-kazein, illetve az ezeknek megfelelő csúcsok alatti terület nagysága nagyobb vagy egyenlő az 1 %-os referenciastandardnál kapott szintnél.
8. HIVATKOZÁSOK
Addeo F., Moio L., Chianese L., Stingo C., Resmini P., Berner I, Krause I., Di Luccia A., Bocca A.: Use of plasmin to increase the sensitivity of the detection of bovine milk in ovine and/or caprine cheese by gel isoelectric focusing of γ2-caseins. Milchwissenschaft 45, 708-711 (1990).
Addeo F., Nicolai M.A., Chianese L., Moio L., Spagna Musso S., Bocca A., Del Giovine L.: A control method to detect bovine milk in ewe and water buffalo cheese using immunoblotting. Milchwissenschaft 50, 83-85 (1995).
Krause I., Berner I, Klostermeyer H.: Sensitive detection of cow milk in ewe and goat milk and cheese by carrier ampholyte - and carrier ampholyte/immobilized pH gradient - isoelectric focusing of γ-caseins using plasmin as signal amplifier. in: Electrophoresis-Forum 89 (B. J. Radola, ed.) pp 389-393, Bode-Verlag, München (1989).
Krause Ι., Belitz H.-D., Kaiser K.-P.: Nachweis von Kuhmilch in Schaf and Ziegenmilch bzw. -käse durch isoelektrische Fokussierung in harnstoffhaltigen Polyacrylamidgelen. Z. Lebensm. Unters. Forsch. 174, 195-199 (1982).
Radola B.J.: Ultrathin-layer isoelectric focusing in 50-100 μm polyacrylamide gels on silanised glass plates or polyester films. Electrophoresis 1, 43-56 (1980).
1. ábra
A fedőlemez sematikus rajza
2. ábra
A vizes és a szerves fázis között a centrifugálást követően kialakuló kazeinréteg
3. ábra
Ultravékony poliakrilamid gél öntésére alkalmas terítéses eljárás
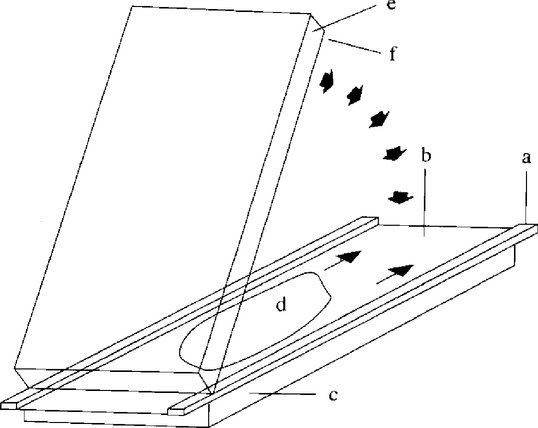
a = távtartószalag (0,25 mm); b = fedőlemez (5.3.); c, e = üveglapok (5.1.); d = géloldat (4.1.2.); f = géltartó lemez (5.2.).
4a. ábra
A juhsajtból és a kecskesajtból származó és különböző mennyiségű tehéntejet tartalmazó, plazminnal kezelt kazeinek izoelektromos fókuszálása
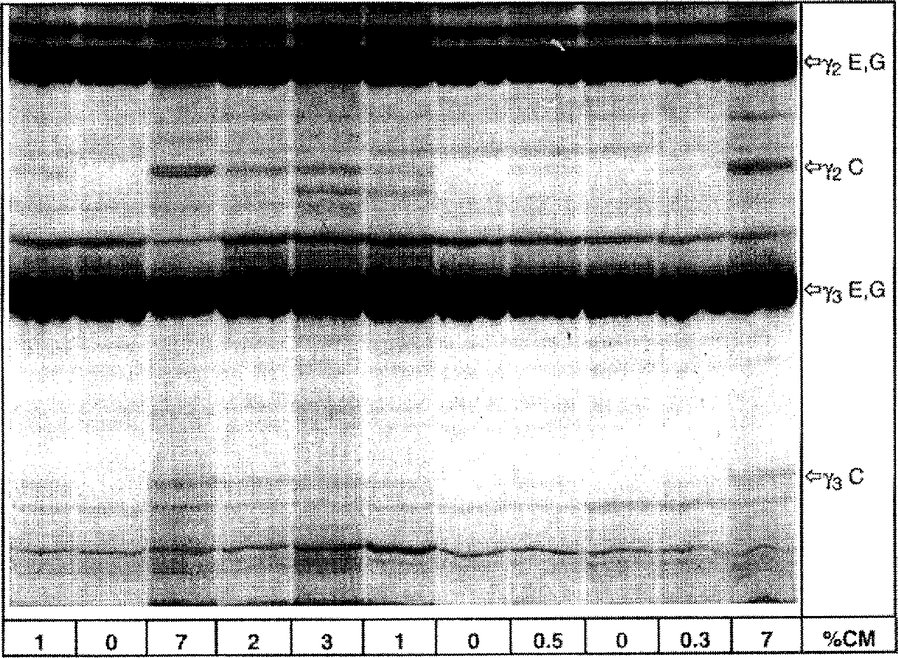
% CM = tehéntej százalékos aránya, C = tehén, E = juh, G = kecske
Az IEF-gél felső fele látható.
4b. ábra
A juhtej, kecsketej és bivalytej keverékéből készült sajtokból származó és különböző mennyiségű tehéntejet tartalmazó, plazminnal kezelt kazeinek izoelektromos fókuszálása
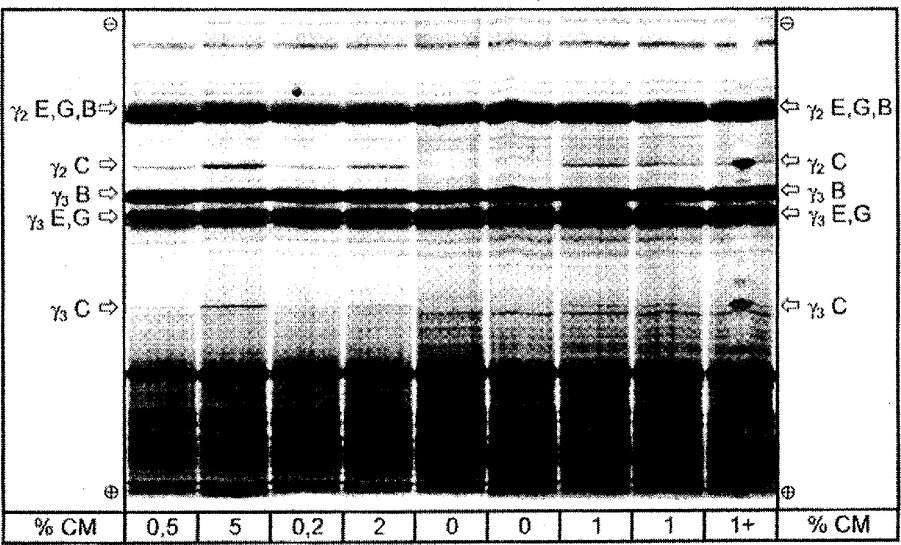
% CM = tehéntej százalékos aránya, 1 + = 1 % tehéntejet tartalmazó és a tiszta szarvasmarhakazein-csúccsal a fókuszált távolság felénél jelentkező minta C = tehén, E = juh, G = kecske, B = bivaly
Az IEF-gél teljes elválasztási távolsága látható.
5. ábra
A standardok (STD) és a juh-, illetve kecsketej keverékéből készült sajtokból származó minták denzitogrammjai izoelektromos fókuszálást követően
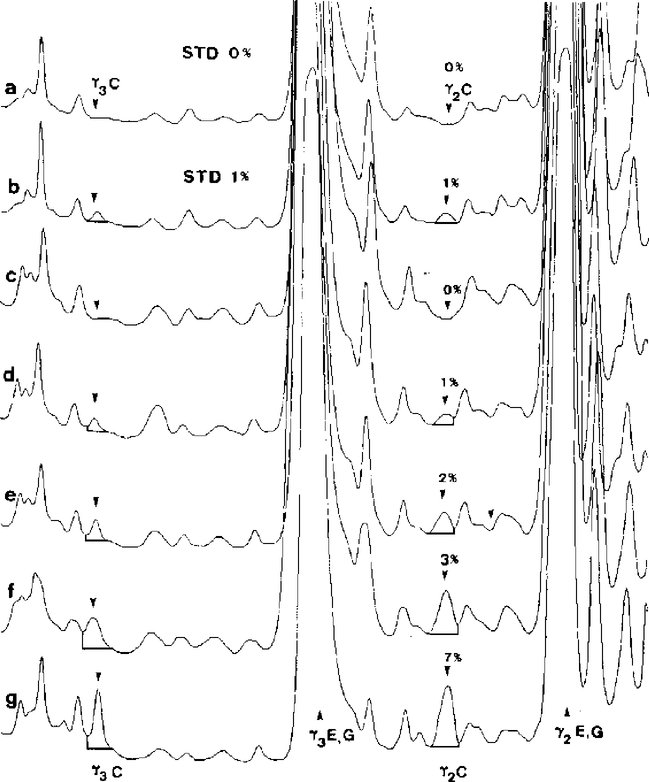
a, b = 0 és 1 % tehéntejet tartalmazó standardok; c-g = 0, 1, 2, 3 és 7 % tehéntejet tartalmazó sajtminták, C = tehén, E = juh, G = kecske.
Az IEF-gél felső felét a λ = 634 nm hullámhosszon mértük le.
IX. MELLÉKLET
Az elemzések értékelése
1. Minőségbiztosítás
Az elemzéseket a 882/2004/EK rendelet (**) 12. cikkének megfelelően kijelölt vagy a tagállam illetékes hatóságai által kijelölt laboratóriumokban kell végezni.
2. A mintavétel és az elemzési eredmények vitathatósága
1. A mintavételt az érintett termékre vonatkozó szabályozással összhangban kell végrehajtani. Amennyiben nem írnak elő kifejezetten a mintavételre vonatkozó rendelkezéseket, akkor az ISO 707 szabványban (Tej és tejtermékek - Mintavételi útmutató) megadott rendelkezések alkalmazandók.
2. Az elemzési eredmények laboratóriumi jegyzőkönyveinek elegendő információt kell tartalmazniuk ahhoz, hogy lehetőség nyíljon az eredményeknek a függelékkel összhangban történő értékeléséhez.
3. Az uniós szabályok szerinti kötelező elemzésekhez ellenmintát kell venni.
4. Amennyiben vita merül fel az eredményekkel kapcsolatban, a kifizető ügynökségnek a szóban forgó terméken újból el kell végeztetnie a szükséges elemzéseket, és a költséget a vesztes fél viseli.
A fent említett elemzést el kell végezni, feltéve hogy a termékből leplombált ellenminták állnak rendelkezésre, és az illetékes hatóság azokat az előírásoknak megfelelően tárolta. A gyártónak az első elemzés eredményeiről történt értesítést követő 7 munkanapon belül kérelmet kell benyújtania a kifizető ügynökséghez. Az elemzést a kifizető ügynökségnek a kérés beérkezését követően 21 munkanapon belül el kell végeztetnie.
5. A fellebbezés eredménye végleges.
6. Amennyiben a gyártó a mintavételtől számított öt munkanapon belül bizonyítani tudja, hogy a mintavételi eljárást nem végezték el helyesen, a mintavételt - ha lehetséges - meg kell ismételni. Amennyiben a mintavétel nem ismételhető meg, a szállítmányt el kell fogadni.
Függelék
Szállítmányok értékelése a jogszabály által előírt határértéknek való megfelelés szempontjából
1. Alapelv
Amennyiben az állami intervencióra és a magántárolási támogatásra vonatkozó jogszabályok részletes mintavételi eljárásokat állapítanak meg, ezeket az eljárásokat kell követni. Minden egyéb esetben az ellenőrzésnek alávetett szállítmányból véletlenszerűen vett legalább három mintaegységből álló mintát kell használni. Készíthető összetett minta. A kapott eredményeket össze kell hasonlítani a jogszabályban előírt határértékekkel 95 %-os megbízhatósági intervallum (kétszeres szórás) számításával, ahol a vonatkozó szórás attól függ, hogy (1) a módszert nemzetközi együttműködés során a σr és σR -re vonatkozó értékekkel validálták-e, vagy (2) házon belüli validálás esetén számítottak-e belső reprodukálhatóságot. Ez a megbízhatósági intervallum egyenlő lesz az eredmény mérési bizonytalanságával.
2. A módszert nemzetközi együttműködés során validálják
Ebben az esetben a σr ismételhetőségi szórást és a σR reprodukálhatósági szórást megállapították, és a laboratórium bizonyítani tudja a validált módszer teljesítményjellemzőinek való megfelelést.
Számítsuk ki az n ismételt mérések [Kép #1] számtani középértékét.
KÉP HIÁNYZIK
Kép #1
A következőképpen számítjuk ki [Kép #2] kiterjesztett bizonytalanságát (k = 2)
KÉP HIÁNYZIK
Kép #2
KÉP HIÁNYZIK
Ha az x mérési végeredményt az, x = y 1 + y 2, x = y 1 - y 2, x = y 1 · y 2 vagy x = y 1/y 2, formájú képletek alkalmazásával számítjuk ki, a szórások kombinálásának ilyen esetekben szokásos eljárását kell követni.
A szállítmány akkor tekintendő az UL előírt felső határértéknek nem megfelelőnek, ha
[Kép #3] ;
KÉP HIÁNYZIK
Kép #3
egyéb esetben az UL-nek megfelelőnek minősül.
A szállítmány akkor tekintendő az LL előírt alsó határértéknek nem megfelelőnek, ha
[Kép #4] ;
KÉP HIÁNYZIK
Kép #4
egyéb esetben az LL-nek megfelelőnek minősül.
3. Házon belüli validálás belső reprodukálhatósági szórás számításával
Azokban az esetekben, amikor ebben a rendeletben nem szereplő módszereket használnak és nem folytatnak pontossági méréseket, házon belüli validálást kell végrehajtani. Az U kiterjesztett bizonytalanság számításának képletében σr és σ R helyett a sir belső ismételhetőségi szórást és a si R belső reprodukálhatósági szórást kell használni.
A jogszabályban előírt határértéknek való megfelelés meghatározása során követendő szabályokat az 1. pont határozza meg. Amennyiben azonban a szállítmányt a jogszabályban előírt határértéknek nem megfelelőnek ítélik, a méréseket meg kell ismételni az e rendeletben meghatározott módszerrel, és az eredményt az 1. pont szerint kell értékelni.
(*) Az Ampholine® pH 3,5-9,5-es (Pharmacia) és a Resolyte® pH 5-7, valamint pH 6-8-as (BDH, Merck) termékek különösen alkalmasnak bizonyultak a γ -kazeinek kívánt szeparációjának elérésére.
(**) Az Európai Parlament és a Tanács 882/2004/EK rendelete (2004. április 29.) a takarmány- és élelmiszerjog, valamint az állat-egészségügyi és az állatok kíméletére vonatkozó szabályok követelményeinek történő megfelelés ellenőrzésének biztosítása céljából végrehajtott hatósági ellenőrzésekről (HL L 165, 2004.4.30., 1. o.).
"
(1) Az alkalmazandó módszert a kifizető ügynökséggel jóvá kell hagyatni.
(2) Az égett szemcsék elemzése rendszeresen végezhető. A szóban forgó elemzéseket azonban minden esetben el kell végezni, amennyiben nem végeznek érzékszervi ellenőrzéseket.
(3) Az alkalmazandó - egy vagy mindkét - módszert a kifizető ügynökséggel jóvá kell hagyatni.
(4) Az alkalmazandó módszert a kifizető ügynökséggel jóvá kell hagyatni.
(5) Az érzékszervi ellenőrzéseket a kifizető ügynökség által jóváhagyott kockázatalapú elemzést követően szükségesnek ítélt esetekben kell elvégezni.
(6) Az alkalmazandó módszert a kifizető ügynökséggel jóvá kell hagyatni.
(7) Mintaalkalmazás: Az előfókuszálást követően (1. lépés) pipettázzunk 18 μl mintát és standard oldatokat a mintafelvivőre (10 × 5 mm), azokat egymástól legalább 1 mm-es távolságra, az anódtól pedig hosszirányban legalább 5 mm-re helyezzük rá a gélre, és enyhén nyomjuk meg. A fenti körülmények mellett végezzük el a fókuszálást, és óvatosan távolítsuk el a mintafelvivő eszközt a minta 60 perces futtatását követően.
Lábjegyzetek:
[1] A dokumentum eredetije megtekinthető CELEX: 32018R0150 - https://eur-lex.europa.eu/legal-content/HU/ALL/?uri=CELEX:32018R0150&locale=hu