
32008R0429[1]
A Bizottság 429/2008/EK rendelete ( 2008. április 25.) az 1831/2003/EK európai parlamenti és tanácsi rendeletnek a kérelmek elkészítése és megjelenési formája, valamint a takarmány-adalékanyagok értékelése és engedélyezése tekintetében történő végrehajtására vonatkozó részletes szabályokról
A Bizottság 429/2008/EK rendelete
(2008. április 25.)
az 1831/2003/EK európai parlamenti és tanácsi rendeletnek a kérelmek elkészítése és megjelenési formája, valamint a takarmány-adalékanyagok értékelése és engedélyezése tekintetében történő végrehajtására vonatkozó részletes szabályokról
(EGT-vonatkozású szöveg)
AZ EURÓPAI KÖZÖSSÉG BIZOTTSÁGA,
tekintettel az Európai Közösséget létrehozó szerződésre,
tekintettel a takarmányozási célra felhasznált adalékanyagokról szóló 1831/2003/EK európai parlamenti és tanácsi rendeletre (1) és különösen annak 7. cikke (4) és (5) bekezdésére,
az Európai Élelmiszer-biztonsági Hatósággal az 1831/2003/EK rendelet 7. cikkének (4) és (5) bekezdése alapján folytatott konzultációt követően,
mivel:
(1) A takarmány-adalékanyagok 1831/2003/EK rendelet alapján történő engedélyezésének eljárására vonatkozóan végrehajtási szabályok megállapítása szükséges, beleértve a kérelmek elkészítésére és megjelenési formájára vonatkozó szabályokat, valamint az ilyen adalékanyagok értékelésére és engedélyezésére vonatkozó szabályokat is. E szabályok hivatottak a takarmányokban lévő adalékanyagok értékelésére vonatkozó iránymutatások meghatározásáról szóló 87/153/EGK tanácsi irányelv (2) mellékletében megállapított rendelkezések helyébe lépni.
(2) E szabályokban rendelkezni kell arról, hogy a kérelemhez csatolt dokumentáció mely követelményeknek feleljen meg. Különösen az érintett adalékanyag azonosítása és jellemzése céljából benyújtandó tudományos adatokat, valamint a hatékonyságának és az emberek, az állatok és a környezet számára biztonságos jellegének bizonyítása érdekében benyújtandó vizsgálatokat kell meghatározni, az engedélykérelmeknek az Európai Élelmiszer-biztonsági Hatóság (a továbbiakban: a Hatóság) által történő megvizsgálása és értékelése céljából.
(3) Az adalékanyag jellegétől, illetve kérelmezett alkalmazási feltételeitől függően, a tulajdonságainak vagy hatásainak értékeléséhez szükséges vizsgálatok terjedelme változó lehet. Ezért a gazdasági szereplőknek bizonyos mértékű rugalmasságot kell engedni az érintett adalékanyag biztonságosságának és hatékonyságának bizonyítása céljából benyújtandó vizsgálatok és anyagok tekintetében. Azoknak a gazdasági szereplőknek, akik élnek ezzel a rugalmassággal, választásukat a dokumentációban indokolniuk kell.
(4) A Hatóság számára lehetővé kell tenni, hogy szükség esetén további tájékoztatást kérjen annak megállapítása céljából, hogy az adalékanyag megfelel-e az 1831/2003/EK rendelet 5. cikkében említett engedélyezési feltételeknek.
(5) A takarmányokban vagy vízben felhasználni kívánt adalékanyagok dokumentációjának összeállítása során elengedhetetlen a megfelelő minőségi szabványok alkalmazása annak biztosítása érdekében, hogy a laboratóriumi vizsgálatok eredményeit ne lehessen megkérdőjelezni.
(6) Ha szükséges, különleges követelményeket kell megállapítani az 1831/2003/EK rendelet 6. cikkének (1) bekezdésében említett egyes adalékanyag-kategóriák tekintetében.
(7) A kevésbé jelentős fajokra vonatkozó engedélyek beszerzésére irányuló erőfeszítéseknek - a megfelelő biztonsági szint megtartása mellett történő - ösztönzése érdekében különleges feltételeket kell megállapítani arra, hogy a főbb fajokon végzett vizsgálatok eredményeit figyelembe lehessen venni a kevésbé jelentős fajokra történő extrapolálás céljából.
(8) Az engedélykérelmekre vonatkozó végrehajtási szabályoknak különböző követelményekre kell tekintettel lenniük az élelmiszer-termelő állatok és az egyéb állatok vonatkozásában, amely utóbbiak esetében az ember mint fogyasztó szempontjából fontos biztonsági értékelés szempontjai nem jelentősek.
(9) A laboratóriumi állatok kísérleti vagy egyéb tudományos célból történő használatát magukban foglaló eljárások, valamint a kísérleti és egyéb tudományos célokra felhasznált állatok védelmére vonatkozó tagállami törvényi, rendeleti és közigazgatási rendelkezések közelítéséről szóló, 1986. november 24-i 86/609/EGK tanácsi irányelv (3) szerinti, állatokon végzett vizsgálatokat magukban foglaló eljárások alkalmazását a minimumra kell szorítani.
(10) A vizsgálatok szükségtelen ismétlését elkerülendő, egyszerűsített eljárásokról kell rendelkezni az élelmiszerben történő felhasználásra már engedélyezett adalékanyagok engedélyezése tekintetében.
(11) A 70/524/EGK tanácsi irányelv (4) szerint határozatlan időre már engedélyezett adalékanyagok tekintetében adott esetben a kérelmező számára biztosítani kell a lehetőséget arra, hogy a hatékonyságot, amennyiben vizsgálatok nem állnak rendelkezésre, bármely egyéb, a hatékonyság bizonyítására rendelkezésére álló dokumentummal bizonyítsa, különösen az érintett adalékanyag használatának hosszú történetére vonatkozó dokumentummal.
(12) Szabályokat kell megállapítani az engedélyek módosítására vonatkozó, az 1831/2003/EK rendelet 13. cikke (3) bekezdésének megfelelő kérelmek tekintetében.
(13) Úgyszintén szabályokat kell megállapítani az engedélyek megújítására vonatkozó, az 1831/2003/EK rendelet 14. cikke szerinti kérelmek tekintetében.
(14) A kérelem alátámasztása végett elvégzendő biztonsági és hatékonysági vizsgálatokra vonatkozó rendelkezéseket illetően átmeneti időszakot kell megjelölni, amelyben a jelenlegi szabályok továbbra is alkalmazandók. Az e rendelet hatálybalépése előtt benyújtott kérelmeket továbbra is a 87/153/EGK irányelv mellékletével összhangban kell kezelni. Ami a hatálybalépést követő bizonyos időszak folyamán benyújtott kérelmeket illeti, az egyes vizsgálatokhoz szükséges hosszú időtartamot figyelembe véve a kérelmezők számára lehetővé kell tenni, hogy választhassanak az e rendeletben előírt szabályok és a 87/153/EGK irányelv melléklete között. A végrehajtási szabályok kidolgozása a jelenlegi tudományos és szakmai ismeretek alapján történt, és ezeket szükség szerint az új fejleményekhez kell igazítani.
(15) Az e rendeletben előírt intézkedések összhangban vannak az Élelmiszerlánc- és Állat-egészségügyi Állandó Bizottság véleményével,
ELFOGADTA EZT A RENDELETET:
1. cikk
Fogalommeghatározások
E rendelet alkalmazásában a következő fogalommeghatározásokat kell alkalmazni:
1. "kedvtelésből tartott és egyéb, nem élelmiszer-termelő állatok": a szokásosan táplált, tenyésztett, illetve tartott, de emberek által nem fogyasztott fajokhoz tartozó állatok, kivéve a lovakat;
2. "kevésbé jelentős fajok": a szarvasmarhaféléktől (tej- és hústermelő állatok, beleértve a borjakat is), a juhféléktől (hústermelő állatok), a sertéstől, a csirkétől (beleértve a tojótyúkokat is), a pulykától és a lazacfélék (Salmonidae) közé tartozó halaktól eltérő, élelmiszer-termelő állatok.
2. cikk
Alkalmazás
(1) Az 1831/2003/EK rendelet 7. cikkében előírt, takarmány-adalékanyag engedélyezése iránti kérelem benyújtásához az I. mellékletben szereplő formanyomtatványt kell alkalmazni.
A kérelemhez csatolni kell a 3. cikkben előírt dokumentációt (a továbbiakban: dokumentáció), amely az 1831/2003/EK rendelet 7. cikkének (3) bekezdésében említett adatokat és dokumentumokat tartalmazza.
(2) Ha a kérelmező az 1831/2003/EK rendelet 18. cikkének megfelelően az (1) bekezdésben említett dokumentáció egyes részeinek bizalmas kezelését kéri, minden egyes dokumentum, illetve a dokumentum minden egyes része tekintetében ellenőrizhető módon igazolnia kell, hogy az adott információ felfedése versenyhelyzetét jelentősen sértheti. A bizalmas részeket a dokumentáció többi anyagától elkülönítve kell benyújtani, és nem lehet az 1831/2003/EK rendelet 7. cikke (3) bekezdésének h) pontjában említett összefoglalóban szerepeltetni. A kérelmező a Bizottság részére a csatolt igazolással együtt megküldi a dokumentáció azon részeinek egy példányát, amelyek bizalmas kezelését kéri.
3. cikk
Dokumentáció
(1) A dokumentációnak helytállóan és kielégítően bizonyítania kell, hogy a takarmány-adalékanyag megfelel az 1831/2003/EK rendelet 5. cikkében előírt engedélyezési feltételeknek.
(2) A dokumentáció elkészítésére és megjelenési formájára vonatkozó általános követelményeket a II. melléklet határozza meg.
A III. melléklet határozza meg azokat az egyedi követelményeket, amelyeket a dokumentációnak az adott esetben teljesítenie kell.
A hosszú távú vizsgálatok minimális időtartamát a IV. melléklet tartalmazza.
(3) A (2) bekezdéstől eltérve, a kérelmező olyan dokumentációt is benyújthat, amely nem felel meg a (2) bekezdés szerinti követelményeknek, feltéve hogy az e követelményeknek meg nem felelő valamennyi elemre vonatkozóan indokolást is csatol.
4. cikk
Átmeneti intézkedések
(1) Az e rendelet hatálybalépése előtt benyújtott engedélykérelmekre továbbra is a 87/153/EGK irányelv mellékletét kell alkalmazni.
(2) Azon engedélykérelmek tekintetében, amelyeket 2009. június 11-e előtt nyújtottak be, a kérelmezők dönthetnek úgy, hogy továbbra is a 87/153/EGK irányelv melléklete I. és II. részének III. és IV. szakaszát alkalmazzák a III. melléklet 1.3., 1.4., 2.1.3., 2.1.4., 2.2.3., 2.2.4., 3.3., 3.4., 4.1.3., 4.1.4., 4.2.3., 4.2.4., 5.3., 5.4., 6.3., 6.4., 7.3., 7.4., 8.3. és 8.4. pontjai helyett, valamint a IV. melléklet táblázatainak "Hosszú távú hatékonysági vizsgálatok minimális időtartama" című oszlopában megállapított rendelkezések helyett.
5. cikk
Hatálybalépés
Ez a rendelet az Európai Unió Hivatalos Lapjában való kihirdetését követő huszadik napon lép hatályba.
Ez a rendelet teljes egészében kötelező és közvetlenül alkalmazandó valamennyi tagállamban.
Kelt Brüsszelben, 2008. április 25-én.
a Bizottság részéről
Androulla VASSILIOU
a Bizottság tagja
(1) HL L 268., 2003.10.18, 29. o. A 378/2005/EK bizottsági rendelettel (HL L 59., 2005.3.5., 8. o.) módosított rendelet.
(2) HL L 64., 1987.3.7., 19. o. Hatályon kívül helyezte az 1831/2003/EK rendelet.
(3) HL L 358., 1986.12.18., 1. o. A 2003/65/EK európai parlamenti és tanácsi irányelvvel (HL L 230., 2003.9.16., 32. o.) módosított irányelv.
(4) HL L 270., 1970.12.14., 1. o. A legutóbb az 1800/2004/EK bizottsági rendelettel (HL L 317., 2004.10.16., 37. o.) módosított irányelv.
I. MELLÉKLET
A 2. CIKK (1) BEKEZDÉSÉBEN EMLÍTETT FORMANYOMTATVÁNY ÉS ADMINISZTRATÍV ADATOK
1. FORMANYOMTATVÁNY - EURÓPAI BIZOTTSÁG
EGÉSZSÉGÜGYI ÉS FOGYASZTÓVÉDELMI
FŐIGAZGATÓSÁG
(Cím)
Dátum: ...
Tárgy : Takarmány-adalékanyagnak az 1831/2003/EK rendelet szerinti engedélyezése iránti kérelem.
Takarmány-adalékanyag engedélyezése vagy takarmány-adalékanyag új alkalmazásának engedélyezése (az 1831/2003/EK rendelet 4. cikkének (1) bekezdése)
Már létező termék engedélyezése (az 1831/2003/EK rendelet 10. cikkének (2) vagy (7) bekezdése)
Meglévő engedély módosítása (az 1831/2003/EK rendelet 13. cikkének (3) bekezdése)
Takarmány-adalékanyag engedélyének megújítása (az 1831/2003/EK rendelet 14. cikke)
Sürgős engedélyezés (az 1831/2003/EK rendelet 15. cikke)
(Jelölje egyértelműen valamely kockába elhelyezett x-szel)
A kérelmező(k) és/vagy a Közösségen belüli képviselője(képviselői) (az 1831/2003/EK rendelet 4. cikkének (3) bekezdése), az 1831/2003/EK rendelet 7. cikke (3) bekezdésének a) pontjában előírt feltételek (név, cím....) alapján
...
...
benyújtja/ják ezt a kérelmet a következő termék takarmány-adalékanyagként történő engedélyezése iránt:
1.1. Az adalékanyag azonosítása és jellemzése
Adalékanyag neve (a II. melléklet 2.2.1.1. és 2.2.1.2. alpontjaiban meghatározottak szerinti hatóanyag(ok) vagy reagens(ek) leírása):
...
...
Kereskedelmi név (ha a jogosulthoz kötött engedélyekhez szükséges):
...
...
a következő adalékanyag-kategórián/kategóriákon és funkcionális csoporton/csoportokon (1) belül (felsorolás):
...
...
célfaj:
...
...
...
Az engedély jogosultjának neve: (az 1831/2003/EK rendelet 9. cikkének (6) bekezdése)
...
...
Ezt az adalékanyagot a takarmányokra vonatkozó szabályozásban a(z) ......./.../E(G)K irányelv, illetve a(z) .../.../EK rendelet a ... számon a következő adalékanyag-kategóriaként már engedélyezte:
...
Ezt az adalékanyagot az élelmiszerekre vonatkozó szabályozásban a(z) ......./.../E(G)K irányelv, illetve a(z) .../.../EK rendelet a ... számon a következőként már engedélyezte:
...
a következőben történő felhasználásra:
...
Ha a termék géntechnológiával módosított szervezetből (a továbbiakban: GMO) áll, ilyen szervezetet tartalmaz, vagy ilyenből állították elő, tüntesse fel a következő adatokat:
egyedi azonosító (65/2004/EK bizottsági rendelet (2)) (szükség szerint):
...
vagy az 1829/2003/EK európai parlamenti és tanácsi rendeletnek (3) megfelelően kiadott bármely engedély részletei:
...
vagy az 1829/2003/EK rendelet szerinti, még elbírálás alatt lévő engedélykérelem részletei:
...
1.2. Alkalmazási feltételek
1.2.1. Alkalmazás teljes értékű takarmányokban
Állatfaj vagy -kategória:
...
...
Maximális életkor és súly:
...
...
Minimális dózis (szükség szerint): mg vagy aktivitási egység (4) vagy telepképző egység (a továbbiakban: CFU) vagy ml/kg, 12 %-os nedvességtartalmú teljes értékű takarmányban
...
...
Maximális dózis (szükség szerint): mg vagy aktivitási egység vagy CFU vagy ml/kg, 12 %-os nedvességtartalmú teljes értékű takarmányban
...
...
A folyékony takarmányok esetében a minimális és a maximális dózis literben adható meg.
1.2.2. Alkalmazás vízben
Minimális dózis (szükség szerint): mg vagy aktivitási egység vagy CFU vagy ml/l, vízben
...
...
Maximális dózis (szükség szerint): mg vagy aktivitási egység vagy CFU vagy ml/l, vízben
...
...
1.2.3. Különleges alkalmazási feltételek (ha szükséges)
Állatfaj vagy -kategória:
...
...
Maximális életkor:
...
...
Minimális dózis (szükség szerint): mg vagy aktivitási egység vagy CFU/kg, 12 %-os nedvességtartalmú kiegészítő takarmányban
...
...
Maximális dózis (szükség szerint): mg vagy aktivitási egység vagy CFU/kg, 12 %-os nedvességtartalmú kiegészítő takarmányban
...
...
A folyékony takarmányok esetében a minimális és a maximális dózis literben adható meg.
Az alkalmazás feltételei, illetve korlátai (szükség szerint):
...
...
...
A kezelésre vonatkozó különös feltételek vagy korlátozások (szükség szerint):
...
...
...
...
Maximális maradékanyag-határérték (szükség szerint):
állatfaj vagy -kategória:
...
...
jelölő maradékanyag:
...
...
célszövetek vagy -termékek:
...
...
...
maximális maradékanyag szövetekben vagy termékekben (μg/kg):
...
...
...
a maradékanyagszintnek az MRL-értékek alá csökkenéséhez szükséges idő:
...
1.3. Referenciaminták
Közösségi referencialaboratóriumi (a továbbiakban: KRL) minta száma (ha alkalmazható):
...
Cikkszám/gyártási tétel kódja:
...
Gyártás időpontja:
...
Lejárat időpontja:
...
Koncentráció:
...
Tömeg:
...
Fizikai leírás:
...
Tárolóedény leírása:
...
Tárolási követelmények:
...
1.4. Kért módosítás (szükség szerint)
...
...
...
...
E kérelem másolata közvetlenül megküldésre került a dokumentációval együtt a Hatóság részére és a referenciamintákkal együtt a KRL részére.
Aláírás ...
1.5. Mellékletek:
teljes dokumentáció (csak a Hatóság részére);
a dokumentáció nyilvános összefoglalója;
a dokumentáció részletes összefoglalója;
a dokumentáció bizalmas kezelésre szánt részeinek jegyzéke és a dokumentáció érintett részeinek egy-egy példánya (csak a Bizottság és a Hatóság részére);
a kérelmező(k) adminisztratív adatainak másolata;
három minta a takarmány-adalékanyagból a KRL részére az 1831/2003/EK rendelet 7. cikke (3) bekezdésének f) pontja alapján (csak a KRL részére);
anyagbiztonsági adatlap (csak a KRL részére);
azonosítás és analízis bizonyítványa (csak a KRL részére); valamint
annak megerősítése, hogy a díj a KRL részére megfizetésre került (a 378/2005/EK rendelet (5) 4. cikke).
A nyomtatvány szükséges részeit töltse ki, a nem szükséges részeket törölje. Az eredeti kérelemnyomtatványt (más előírt mellékletekkel) közvetlenül az Európai Bizottság részére kell megküldeni.
2. A KÉRELMEZŐ(K) ADMINISZTRATÍV ADATAI - Valamely adalékanyag 1831/2003/EK rendelet szerinti engedélyezése iránti kérelem benyújtása esetében az elérhetőségi adatok
1. Kérelmező társaság vagy személy
a) A kérelmező személy vagy társaság neve
b) Cím (utca, házszám, irányítószám, város, ország)
c) Telefon
d) Telefax
e) E-mail (ha van)
2. Kapcsolattartó (a Bizottsággal, a Hatósággal és a KRL-lel folytatott levelezés teljes köre tekintetében)
a) A kapcsolattartó személy neve
b) Beosztása
c) Címe (utca, házszám, irányítószám, város és ország)
d) Telefon
e) Telefax
f) E-mail (ha van)
(1) Az állattenyésztésben alkalmazott adalékanyag kategóriáján belüli "egyéb, állattenyésztésben alkalmazott adalékanyag" funkcionális csoport esetében egyértelműen meg kell határozni, hogy az adalékanyagnak milyen funkciót szánnak.
(2) HL L 10., 2004.1.16., 5. o.
(3) HL L 268., 2003.10.18., 1. o. A legutóbb a 298/2008/EK rendelettel (HL L 97., 2008.4.9., 64. o.) módosított rendelet.
(4) Az "egység" fogalmát a kérelmezőnek kell meghatároznia.
(5) A Bizottság 378/2005/EK rendelete (2005. március 4.) a közösségi referencialaboratóriumnak a takarmány-adalékanyagok engedélyezési kérelmeivel kapcsolatos kötelezettségei és feladatai tekintetében az 1831/2003/EK európai parlamenti és tanácsi rendelet részletes végrehajtási szabályairól (HL L 59., 2005.3.5., 8. o.). A 850/2007/EK rendelettel (HL L 188., 2007.7.20., 3. o.) módosított rendelet.
II. MELLÉKLET
A 3. CIKK SZERINTI DOKUMENTÁCIÓVAL SZEMBEN TÁMASZTOTT ÁLTALÁNOS KÖVETELMÉNYEK
ÁLTALÁNOS SZEMPONTOK
Ez a melléklet meghatározza azokat a követelményeket, amelyekkel összhangban, az alábbiak céljából, az anyagokra, mikroorganizmusokra és készítményekre vonatkozó, az 1831/2003/EK rendelet 7. cikke alapján a dokumentációval benyújtandó vizsgálatok és adatok jegyzékét és jellemzőit meg kell állapítani:
- új takarmány-adalékanyagként történő engedélyezés,
- takarmány-adalékanyag új alkalmazásának engedélyezése,
- takarmány-adalékanyag meglévő engedélyének módosítása, vagy
- takarmány-adalékanyag engedélyének megújítása.
A dokumentációnak alkalmasnak kell lennie arra, hogy az alapján a tudomány jelenlegi állása szerint az adalékanyagok értékelését el lehessen végezni, és lehetővé kell tennie annak megítélését, hogy a szóban forgó adalékanyagok az 1831/2003/EK rendelet 5. cikkében megállapított engedélyezési alapelveknek megfelelnek-e.
A benyújtandó vizsgálatokat és azok terjedelmét az határozza meg, hogy milyen jellegű az adalékanyag, továbbá a kategória és a funkcionális csoport, az engedély típusa (nem jogosulthoz kötött, illetve jogosulthoz kötött), maga az anyag, a célállatok és az alkalmazási feltételek. A kérelmezőnek e melléklet és a III. melléklet alapján kell megítélnie, mely vizsgálatok és adatok nyújtandók be a kérelemmel.
A kérelmezőnek egyértelműen meg kell jelölnie azon okokat, amelyek miatt az e mellékletben, a III. mellékletben és a IV. mellékletben megkövetelt adatok bármelyikét a dokumentációból elhagyta, vagy azoktól eltért.
A dokumentáció részletes beszámolót tartalmaz az összes elvégzett vizsgálatról, amelyet az e mellékletben javasolt számozás szerint kell elkészíteni. A dokumentáció tartalmaz minden említésre került, közzétett tudományos adatra történő hivatkozást és ezen adatok másolatát, valamint minden olyan egyéb releváns vélemény másolatát, amelyet elismert tudományos testület adott ki korábban. Ha e vizsgálatokat egy európai tudományos testület a Közösségben hatályban lévő jogszabály alapján már értékelte, elegendő az értékelés eredményére hivatkozni. A korábban elvégzett és közzétett, vagy szakértői értékelés keretében végzett vizsgálatokból származó adatoknak egyértelműen ugyanarra az adalékanyagra kell vonatkozniuk, mint amely az engedélykérelem tárgya.
A vizsgálatokat, beleértve azokat, amelyeket korábban végeztek és tettek közzé, illetve amelyeket szakértői értékelés keretében végeztek, a helyes laboratóriumi gyakorlat alapelveinek alkalmazására és annak a vegyi anyagokkal végzett kísérleteknél történő alkalmazásának ellenőrzésére vonatkozó törvényi, rendeleti és közigazgatási rendelkezések közelítéséről szóló, 2004. február 11-i 2004/10/EK európai parlamenti és tanácsi irányelvvel (1) összhangban lévő, megfelelő minőségi szabványok (pl. a helyes laboratóriumi gyakorlat (GLP)) vagy a Nemzetközi Szabványügyi Szervezet. (ISO) megfelelő minőségi szabványai szerint kell megvalósítani és dokumentálni.
Ha in vivo vagy in vitro vizsgálatok Közösségen kívül történő elvégzésére kerül sor, a kérelmezőnek igazolnia kell, hogy a vizsgálatok helyszínei megfelelnek a Gazdasági Együttműködési és Fejlesztési Szervezet (OECD) helyes laboratóriumi gyakorlatra vonatkozó elveinek vagy az ISO-szabványoknak.
A fizikai-kémiai, toxikológiai és ökotoxikológiai jellemzők meghatározását a legutóbb a 2004/73/EK bizottsági irányelvvel (2) módosított, a veszélyes anyagok osztályozására, csomagolására és címkézésére vonatkozó törvényi, rendeleti és közigazgatási rendelkezések közelítéséről szóló, 1967. június 27-i 67/548/EGK tanácsi irányelv (3) által megállapított módszerek szerint, vagy pedig nemzetközi tudományos testületek által elismert, naprakésszé tett módszerek szerint kell végezni. Ezektől eltérő módszerek használatát indokolni kell.
Ösztönözni kell az in vitro módszerek alkalmazását, valamint olyan módszerek alkalmazását, amelyek finomítják vagy helyettesítik a laboratóriumi állatok segítségével végzett szokásos vizsgálatokat, illetve amelyek csökkentik az e vizsgálatok során használt állatok számát. Az ilyen módszereknek ugyanolyan minőségűeknek és ugyanolyan biztonsági szintűeknek kell lenniük, mint amilyen az általuk helyettesítendő módszer.
A takarmány, illetve víz tekintetében alkalmazott analitikai módszerek leírásának összhangban kell lennie a 2004/10/EK irányelvben és/vagy az EN ISO/IEC 17025. sz. szabványban megállapított GLP-szabályokkal. E módszereknek teljesíteniük kell a takarmány- és élelmiszerjog, valamint az állat-egészségügyi és az állatok kíméletére vonatkozó szabályok követelményeinek történő megfelelés ellenőrzésének biztosítása céljából végrehajtott hatósági ellenőrzésekről szóló, 2004. április 29-i 882/2004/EK európai parlamenti és tanácsi rendelet (4) 11. cikkében megállapított követelményeket.
Mindegyik dokumentációnak tartalmaznia kell egy nyilvános összefoglalót és egy részletes tudományos összefoglalót, hogy lehetővé tegye az érintett adalékanyag azonosítását és jellemzését.
Mindegyik dokumentációnak tartalmaznia kell - amennyiben az 1831/2003/EK rendelet 7. cikke (3) bekezdésének g) pontja megköveteli - egy forgalomba hozatalt követő felügyeleti tervet, valamint egy, az 1831/2003/EK rendelet 7. cikke (3) bekezdésének e) pontjában említett, címkézésre vonatkozó javaslatot.
Biztonsági értékelés
Ez az értékelés olyan vizsgálatokon alapul, amelyek célja az adalékanyag alkalmazása biztonságosságának bizonyítása a következők tekintetében:
a) a célfajok tekintetében, a takarmányba vagy vízbe keverés legnagyobb javasolt mértéke esetében, valamint egy biztonsági küszöb megállapítása érdekében e mérték többszöröse esetében;
b) azon fogyasztók tekintetében, akik olyan állatokból készült élelmiszertermékeket fogyasztanak, amelyek az adalékanyagot, annak maradékanyagait vagy anyagcseretermékeit fogyasztották. Ilyen esetben a biztonságról maximális maradékanyag-határértékek (a továbbiakban: MRL-ek) megállapítása révén, valamint a megengedhető napi bevitelre (a továbbiakban: ADI) vagy a biztonságos bevitel felső határára (a továbbiakban: UL) alapozott, a maradékanyagszintnek az MRL-értékek alá csökkenéséhez szükséges idő megállapítása révén kell gondoskodni;
c) azon személyek tekintetében, akik belélegzés, nyálkahártyával érintkezés, szembe kerülés vagy bőrrel való érintkezés által az adalékanyagnak várhatóan ki lesznek téve, miközben az adalékanyagot kezelik, vagy előkeverékekhez, teljes értékű takarmányhoz vagy vízhez keverik hozzá, vagy pedig az érintett adalékanyagot tartalmazó takarmányt vagy vizet használnak;
d) állatok és emberek tekintetében, az antimikrobiális rezisztenciát hordozó gének kiválasztása és terjesztése szempontjából; valamint
e) a környezet tekintetében, magának az adalékanyagnak az eredményeként, vagy az adalékanyagból - közvetlenül és/vagy az állatok anyagcseréje révén - származó termékek eredményeként.
Ha egy adalékanyag több összetevőből áll, mindegyiket külön lehet értékelni a fogyasztók biztonsága szempontjából, majd mérlegelni lehet az együttes hatást (amennyiben kimutatható, hogy az összetevők között nincs kölcsönhatás). Vagy pedig a teljes keveréket kell értékelni.
Hatékonysági értékelés
Ez az értékelés olyan vizsgálatokon alapul, amelyek célja valamely adalékanyag hatékonyságának bizonyítása az 1831/2003/EK rendelet 6. cikkének (1) bekezdésében és I. mellékletében meghatározottak szerinti funkciójából adódó célok szempontjából.
1. I. SZAKASZ: A DOKUMENTÁCIÓ ÖSSZEFOGLALÁSA
1.1. Az 1831/2003/EK rendelet 7. cikke (3) bekezdésének h) pontja szerinti nyilvános összefoglaló
A kérelmező az érintett adalékanyag fő tulajdonságait tartalmazó összefoglalót nyújt be. Az összefoglaló semmilyen bizalmas adatot nem tartalmazhat, felépítése pedig a következő:
1.1.1. Tartalom
a) a kérelmező(k) neve;
b) az adalékanyag azonosítása;
c) az előállítás módja és az analitikai módszer;
d) az adalékanyag biztonságosságára és hatékonyságára vonatkozó vizsgálatok;
e) az alkalmazás javasolt feltételei; valamint
f) a forgalomba hozatalt követő felügyeleti terv.
1.1.2. Leírás
a) a kérelmező(k) neve és címe
Ezt az adatot minden esetben meg kell adni, függetlenül a takarmány-adalékanyag engedélyének típusától (engedély jogosultjához kötött vagy a jogosulthoz nem kötött). Ha egy dokumentációt kérelmezők csoportja nyújt be, mindegyikük nevét fel kell tüntetni.
b) az adalékanyag azonosítása
Az adalékanyag azonosítása a II., illetve a III. melléklet szerint megkövetelt információk összefoglalóját tartalmazza, a takarmány-adalékanyag engedélyének típusától függően. Különösen: az adalékanyag nevét, a kategória és funkcionális csoport szerinti, javasolt besorolást, a célfajokat/állatkategóriákat és a dózisokat.
c) az előállítás módja és az analitikai módszer
Ismertetni kell a gyártási folyamatot.
Ismertetni kell az adalékanyagnak mint olyannak hatósági ellenőrzés céljából előkeverékekben és takarmányokban történő analízise során alkalmazandó analitikai módszerek általános eljárásait, az e mellékletben és a III. mellékletben előírtak szerint. Szükség szerint, az e melléklet és a III. melléklet alapján benyújtott adatok alapján, ki kell térni az adalékanyagoknak, illetve azok anyagcseretermékeinek hatósági ellenőrzés céljából állati eredetű élelmiszerben történő analízise során alkalmazandó módszer(ek) eljárására.
d) az adalékanyag biztonságosságára és hatékonyságára vonatkozó vizsgálatok
Közölni kell az adalékanyag biztonságosságára és hatékonyságára vonatkozó, az elvégzett különféle vizsgálatokra alapozott következtetést. A vizsgálatok eredményeit - a kérelmező(k) következtetésének alátámasztása végett - táblázatos formában is fel lehet tüntetni. Az összefoglalóban csak a III. mellékletben előírt vizsgálatokat kell szerepeltetni.
e) az alkalmazás javasolt feltételei
A kérelmező(k)nek meg kell jelölnie/jelölniük az alkalmazás általa/uk javasolt feltételeit. Különösen, a kérelmezőnek ismertetnie kell a vízben, illetve takarmányban való alkalmazás mértékét, és emellett a kiegészítő takarmányban való alkalmazás részletes feltételeit. Arról is tájékoztatást kell adni, ha a takarmányban vagy vízben való alkalmazás, illetve elkeverés más módszerét használják. Ismertetni kell minden esetleges egyedi alkalmazási feltételt (pl. összeférhetetlenséget), egyedi címkézési követelményt, valamint azt az állatfajt, amelynek az adalékanyagot szánják.
f) javaslat a forgalomba hozatalt követő felügyeleti tervre
Ez a rész csak azokra az adalékanyagokra alkalmazandó, amelyek az 1831/2003/EK rendelet 7. cikke (3) bekezdésének g) pontja szerint, nem tartoznak az ugyanazon rendelet 6. cikke (1) bekezdésében említett a) vagy b) kategóriába, valamint amelyek a géntechnológiával módosított szervezetekből (GMO-k) álló, azokat tartalmazó vagy azokból előállított termékek forgalmazására vonatkozó közösségi jogszabályok hatálya alá tartozó adalékanyagok.
1.2. A dokumentáció tudományos összefoglalója
Be kell nyújtani egy tudományos összefoglalót, amely az e mellékletben és a III. mellékletben foglaltak szerint, a kérelem alátámasztására benyújtott dokumentumok mindegyik részének részleteit tartalmazza. Ebben az összefoglalóban a kérelmező(k) következtetéseire is ki kell térni.
Az összefoglalónak az e mellékletben szereplő sorrendet kell követnie, és a különböző részek mindegyikét a dokumentáció megfelelő oldalszámára utalva kell tárgyalnia.
1.3. A dokumentumok és más adatok listája
A kérelmezőnek a kérelem alátámasztására benyújtott dokumentáció köteteit számozással és címmel kell ellátnia. Csatolnia kell a kötetek és oldalszámok részletes mutatóját.
1.4. Szükség szerint, a dokumentáció bizalmas kezelésre szánt részeinek listája
Ez a lista a dokumentáció megfelelő köteteit és oldalszámait tartalmazza.
2. II. SZAKASZ: AZ ADALÉKANYAG AZONOSÍTÁSA, JELLEMZÉSE ÉS ALKALMAZÁSÁNAK FELTÉTELEI; ANALITIKAI MÓDSZEREK
Az adalékanyagot teljeskörűen azonosítani és jellemezni kell.
2.1. Az adalékanyag megjelölése
2.1.1. Az adalékanyag neve
Ha szükséges, az engedélyjogosulthoz kötött adalékanyagok esetében javaslatot kell tenni a kereskedelmi névre.
2.1.2. Besorolásra vonatkozó javaslat
Javaslatot kell tenni az adalékanyagnak fő funkciói szerint, az 1831/2003/EK rendelet 6. cikke és I. melléklete alapján, egy vagy több kategóriába és funkcionális csoportba történő besorolására.
Ha az azonos hatóanyagok vagy reagensek más ismert (pl. élelmiszerben, humán-orvostudományban vagy állatorvos-tudományban, mezőgazdaságban és iparban történő) alkalmazására vonatkozóan bármilyen adat áll rendelkezésre, azt is meg kell adni. Meg kell jelölni a hatóanyag bármely más, takarmány- vagy élelmiszer-adalékanyagként, vagy állatgyógyászati készítményként történő használatára vonatkozó, illetve bármely más típusú engedélyt.
2.1.3. Minőségi és mennyiségi összetétel (hatóanyag/reagens, egyéb összetevők, szennyezőanyagok, tételenkénti eltérés)
Az adalékanyag hatóanyaga(i)/reagense(i) és valamennyi egyéb összetevője jegyzékét el kell készíteni, megadva azok végterméken belüli tömegszázalékát. Meg kell határozni a hatóanyag(ok)/reagens(ek) tételenkénti, minőségi és mennyiségi eltérését.
A mikroorganizmusok esetében: grammonkénti telepképző egységben (CFU/gr) kifejezve meg kell határozni az életképes sejtek és spórák számát.
Az enzimek esetében: minden egyes feltüntetett (fő) aktivitást ismertetni kell, és meg kell adni a végterméken belüli minden egyes aktivitás egységeinek számát. A releváns mellékaktivitásokra is ki kell térni. Definiálni kell az aktivitási egységeket, mégpedig lehetőleg a szubsztrátból percenként kibocsátott, μmol-ban kifejezett termékmennyiségként, feltüntetve a pH-t és a hőmérsékletet is.
Ha az adalékanyag hatóanyaga hatóanyagok vagy reagensek keveréke, amelyek mindegyike egyértelműen (minőségileg és mennyiségileg) meghatározható, a hatóanyag-/reagens-összetevőket külön-külön kell leírni, és a keveréken belüli arányukat meg kell adni.
Azokat az egyéb keverékeket, amelyekben az alkotóelemeket nem lehet egyetlen kémiai képlettel leírni és/vagy amelyekben nem minden alkotóelem azonosítható, az aktivitásukhoz hozzájáruló alkotóelem(ek)re és/vagy a tipikus fő alkotóelem(ek)re utalással kell jellemezni.
A Hatóság esetleges, az 1831/2003/EK rendelet 8. cikkének (2) bekezdése szerinti kiegészítő tájékoztatás iránti kérelmének sérelme nélkül, a kérelmező elhagyhatja az állattenyésztésben alkalmazott adalékanyagok, kokcidiosztatikumok és hisztomonosztatikumok kategóriájába nem tartozó, és az 1829/2003/EK rendelet hatálya alá nem tartozó adalékanyagokba szánt hatóanyagoktól, illetve reagensektől eltérő, biztonsági kockázatot nem jelentő egyéb összetevők leírását. A dokumentációban szereplő valamennyi vizsgálatnak minden esetben a ténylegesen a kérelem tárgyát képező adalékanyagon kell alapulnia, miközben tartalmazhat információt az egyéb lehetséges, különféle készítményekről, amelyeket elő lehetne állítani. Megengedhető a harmadik felek dokumentumaiba beépített, belső azonosító, és nyilatkozatot kell csatolni, amely felsorolja az azonosítókat és megerősíti, hogy az azonosító(k) arra/azokra a kiszerelés(ek)re vonatkozik/vonatkoznak, amely(ek) a kérelem tárgya(i).
2.1.4. Tisztaság
A kérelmező azonosítja és számszerűsíti a kémiai és mikrobiális szennyeződéseket, az olyan mérgező, illetve egyéb nem kívánatos tulajdonságokkal rendelkező anyagokat, amelyeket nem szándékosan adtak az adalékanyaghoz, és amelyek nem járulnak hozzá az adalékanyag aktivitásához. Továbbá, a fermentációs termékek esetében, a kérelmező köteles megerősíteni, hogy az adalékanyagban nincsenek termelő szervezetek. Ismertetni kell a termelési tételek szennyezőanyagok és szennyeződések szempontjából történő rutin átvizsgálása során alkalmazott protokollt.
Minden rendelkezésre bocsátott adatnak az adalékanyag specifikációjára vonatkozó javaslatot kell alátámasztania.
Az előállítási folyamattól függő egyedi követelmények felsorolása, amelyek megfelelnek a jelenlegi közösségi jogszabályoknak, alább szerepel.
2.1.4.1. Olyan adalékanyagok, amelyek engedélyezése engedélyjogosulthoz kötött
Az olyan adalékanyagok esetében, amelyek engedélyezése engedélyjogosulthoz kötött, meg kell adni a gyártó által alkalmazott konkrét folyamatra vonatkozó, az egyéb kapcsolódó célokra használt meglévő szabványokon alapuló releváns adatokat. Használhatók az élelmiszer-adalékanyagokkal foglalkozó közös FAO-WHO szakértői bizottság (JECFA) specifikációi, illetve az Európai Közösségből származó élelmiszer-adalékanyag engedélyekben használt specifikációk.
2.1.4.2. Olyan adalékanyagok, amelyek engedélyezése nem engedélyjogosulthoz kötött
Az olyan takarmány-adalékanyagok esetében, amelyek engedélyezése engedélyjogosulthoz nem kötött, használhatók az egyéb kapcsolódó célra alkalmazott meglévő szabványok, illetve az olyan szabványok, amelyek az Európai Közösségben engedélyezett vagy a JECFA-tól származó, élelmiszer-adalékanyagokra vonatkozó specifikációkat tartalmaznak. Ha ilyen szabványok nem állnak rendelkezésre, vagy a gyártási folyamat miatt indokolt, legalább a következő részletes adatokat kell ismertetni, és ezek tekintetében a koncentrációt megadni:
- mikroorganizmusok esetében: mikrobiológiai szennyezés, mikotoxinok, nehézfémek;
- fermentációs (mikroorganizmusokat hatóanyagként nem tartalmazó) termékek esetében: ugyanazok a követelmények, mint a mikroorganizmus termékek esetében (lásd fent). Továbbá fel kell tüntetni azt is, hogy a végtermékbe milyen mértékben kevertek elhasznált tápoldatot.
- növényekből nyert anyagok esetében: mikrobiológiai és botanikai szennyezés (pl. különösen ricinusmag, gyomnövénymagok, anyarozs), mikotoxinok, növényvédőszer-szennyezés, oldószerek maximális értéke, valamint, adott esetben, olyan, toxikológiai kockázatot jelentő anyagok, amelyekről tudott, hogy az eredeti növényben előfordulnak;
- állatokból nyert anyagok esetében: mikrobiológiai szennyezés, nehézfémek és adott esetben oldószerek maximális értékei;
- ásványi anyagok esetében: nehézfémek, dioxin és PCB-k (poliklórozott bifenilek);
- kémiai szintézis és folyamatok útján előállított termékek esetében: azonosítani kell és meg kell adni a koncentrációját minden, a szintézis folyamataiban használt vegyi anyagnak és bármely, a végtermékben megmaradó köztes terméknek.
Ha szükséges, a mikotoxinok analízisre történő kiválasztását a különböző mátrixok szerint kell végezni.
2.1.5. A termék egyes formáinak fizikai állapota
A szilárd halmazállapotú készítmények esetében adatokat kell szolgáltatni a szemcseméret-eloszlásról, a szemcse alakjáról, a sűrűségről, a térfogatsűrűségről, a porképző képességről, valamint a fizikai tulajdonságokat befolyásoló folyamatok alkalmazásáról. A folyékony halmazállapotú készítmények esetében, a viszkozitásra és a felületi feszültségre vonatkozó adatokat kell megadni. Ha az adalékanyagot vízben való felhasználásra szánják, be kell mutatni az oldhatóságot, illetve a diszperzió mértékét.
2.2. A hatóanyag(ok)/reagens(ek) jellemzése
2.2.1. Leírás
Meg kell adni a hatóanyag vagy reagens minőségi leírását. Ebben ki kell térni a hatóanyag vagy reagens tisztaságára és származására, továbbá minden más releváns jellemzőre.
2.2.1.1. Vegyi anyagok
A kémiailag jól meghatározott anyagokat a szokásos elnevezésükre, az IUPAC (Elméleti és Alkalmazott Kémia Nemzetközi Uniója) nómenklatúra szerinti kémiai nevükre, egyéb nemzetközi szokásos elnevezésekre és rövidítésekre és/vagy a Chemical Abstract Service Number (CAS)-számra utalással kell leírni. Meg kell adni a szerkezeti és a molekuláris képletet, valamint a molekulatömeget.
Az aromaanyagként használt, kémiailag meghatározott vegyületek esetében, meg kell adni az érintett kémiai csoportra vonatkozó FLAVIS-számot. A növényi kivonatok esetében meg kell adni a fitokémiai jelölőket.
Azokat a keverékeket, amelyekben az alkotóelemek nem írhatók le egyetlen kémiai képlettel és/vagy amelyekben nem mindegyik alkotóelem azonosítható, a keverék aktivitásához hozzájáruló alkotóelem(ek)re és/vagy a tipikus fő alkotóelem(ek)re utalással kell jellemezni. A stabilitás értékelésének és a nyomonkövethetőségnek a lehetővé tétele érdekében azonosítani kell a jelölő vegyületet.
Az enzimek és enzimkészítmények esetében, minden egyes bejelentett aktivitás tekintetében azt a számot és rendszertani megnevezést kell megadni, amelyet a Nemzetközi Biokémiai Unió (IUB) az "Enzim-nómenklatúra" legfrissebb kiadásában javasolt. Ami a nómenklatúrában még nem szereplő aktivitásokat illeti, az IUB nómenklatúraszabályaival összhangban lévő rendszertani megnevezést kell használni. Köznyelvi nevek is elfogadhatók azzal a feltétellel, hogy egyértelműek, és használatuk a dokumentáció egészében következetes, valamint első említésükkor egyértelműen kapcsolhatók a rendszertani megnevezéshez és az IUB-számhoz. Meg kell adni minden egyes enzimaktivitás biológiai eredetét.
Szintén ismertetni kell a fermentációs vegyi anyagok mikrobiális eredetét (lásd a 2.2.1.2. Mikroorganizmusok című pontot).
2.2.1.2. Mikroorganizmusok
Az eredetet minden mikroorganizmus esetében meg kell jelölni, függetlenül attól, hogy azt termékként vagy előállításhoz használt törzsként alkalmazzák.
A termékként vagy előállításhoz használt törzsként alkalmazott mikroorganizmusok esetében, minden módosítási lépést fel kell tüntetni. Minden egyes mikroorganizmus nevét és taxonómiai besorolását a nemzetközi nevezéktani szabályzatokban legutóbb közzétett adatok szerint kell megadni. A mikrobiális törzseket egy nemzetközileg elismert törzsgyűjteményben (lehetőleg az Európai Unióban) kell letétbe helyezni, és azokat az adalékanyag engedélyezett élettartamáig a törzsgyűjteménynek kell fenntartania. Be kell nyújtani a gyűjtemény által kibocsátott letétbehelyezési tanúsítványt, amelyben meg kell jelölni azt a felvételi számot, amelyen a törzset nyilvántartják. Ezenkívül, ismertetni kell minden, a törzs egyedi azonosításához szükséges, releváns morfológiai, fiziológiai és molekuláris jellemzőt, továbbá a törzs genetikai stabilitása igazolásának módját. A GMO-k esetében ismertetni kell a genetikai módosításokat. Minden egyes GMO tekintetében meg kell adni a géntechnológiával módosított szervezetek egyedi azonosítóinak kialakítására és hozzárendelésére szolgáló rendszer létrehozásáról szóló, 2004. január 14-i 65/2004/EK bizottsági rendeletben említettek szerinti egyedi azonosítót.
2.2.2. Releváns tulajdonságok
2.2.2.1. Vegyi anyagok
Ismertetni kell a fizikai és kémiai tulajdonságokat. Szükség szerint meg kell adni a disszociációs állandót, a pKa-t, az elektrosztatikus tulajdonságokat, az olvadáspontot, a forráspontot, a sűrűséget, a gőznyomást, az oldhatóságot vízben és szerves oldószerekben, a Kow-t és a Kd/Koc-t, a tömegspektrometriás és az abszorpciós spektrumokat, az NMR-adatokat, a lehetséges izomereket és minden egyéb megfelelő fizikai tulajdonságot.
A fermentáció útján előállított anyagok nem rendelkezhetnek olyan antimikrobiális aktivitással, amely a humán- és az állategészségügyben alkalmazott antibiotikumok szempontjából jelentős.
2.2.2.2. Mikroorganizmusok
- Toxinok és virulencia faktorok
Ki kell mutatni, hogy toxinok, illetve virulencia faktorok nincsenek jelen, vagy nem jelentenek kockázatot. Azon baktériumtörzsek tekintetében, amelyek olyan taxonómiai csoporthoz tartoznak, amelynek egyes tagjairól ismert, hogy képesek toxinok vagy más virulencia faktorok termelésére, megfelelő teszteket kell végezni annak molekuláris és - szükség szerint - sejtszinten történő bizonyítására, hogy nem jelentenek semmilyen kockázatot.
Azon mikroorganizmus-törzsek esetében, amelyek nyilvánvalóan biztonságos alkalmazása nincs dokumentálva, illetve amelyek biológiája egyelőre kevéssé feltárt, teljes körű toxikológiai vizsgálat elvégzése szükséges.
- Antibiotikum-termelés és antibiotikum-rezisztencia
Az adalékanyagként vagy termelő törzsként használt mikroorganizmusok nem rendelkezhetnek antibiotikus aktivitással, illetve nem lehetnek alkalmasak olyan antibiotikus anyagok termelésére, amelyek a humán- és az állategészségügyben használt antibiotikumok lehetnek.
Az adalékanyagként való alkalmazásra szánt mikroorganizmus-törzsek nem járulhatnak hozzá az antibiotikumokkal szemben rezisztens, az állatok bélflórájában és a környezetben már jelen lévő gének tárának további gyarapodásához. Következésképpen, minden baktériumtörzset tesztelni kell a humán- és az állatgyógyászatban használt antibiotikumokkal szembeni rezisztencia szempontjából. Rezisztencia kimutatása esetén meg kell állapítani a rezisztencia genetikai alapját, valamint annak valószínűségét, hogy a rezisztenciát a bélflórában található más organizmusnak is átadják.
A szerzett antimikrobiális rezisztenciát hordozó mikroorganizmus-törzsek nem használhatók takarmány-adalékanyagként, kivéve ha bizonyítható, hogy a rezisztencia kromoszómamutáció(k) eredménye, és nem örökíthető át.
2.3. Gyártási folyamat, beleértve az esetleges különleges feldolgozási eljárásokat
Ismertetni kell a gyártási folyamatot annak érdekében, hogy meg lehessen határozni a folyamat azon kritikus pontjait, amelyek a hatóanyag(ok)/reagens(ek), illetve adalékanyag tisztaságát befolyásolhatják. A gyártási folyamatban alkalmazott vegyi anyagokról anyagbiztonsági adatlapot kell benyújtani.
2.3.1. Hatóanyag(ok)/reagens(ek)
Be kell nyújtani az adalékanyag hatóanyaga(i)/reagens(ei) előállítása során alkalmazott gyártási folyamat (pl. kémiai szintézis, erjesztés, tenyésztés, szerves anyagból történő kivonás vagy desztillálás) leírását, szükség szerint folyamatábrán szemléltetve. Meg kell adni a fermentációs/tenyésztési közegek összetételét. Részletesen le kell írni a tisztítási módszereket.
Az adalékanyagok forrásaként használt és zárt rendszerben tenyésztett, genetikailag módosított mikroorganizmusok (a továbbiakban: GMM-ek) esetében a 90/219/EK tanácsi irányelvet (5) kell alkalmazni. Csatolni kell az erjesztési folyamatok (tápközeg, erjesztési körülmények és a fermentációs termékek továbbfeldolgozása) leírását is.
2.3.2. Adalékanyag
Az adalékanyag gyártási folyamatának részletes leírását kell benyújtani. Ki kell térni az adalékanyag készítésének kulcsfontosságú szakaszaira, beleértve a hatóanyag(ok)/reagens(ek) és más összetevők hozzáadását, valamint bármely olyan, ezt követő feldolgozási lépést, amely az adalékanyag-készítést befolyásolja, szükség szerint folyamatábrán szemléltetve.
2.4. Az adalékanyag fizikai-kémiai és technológiai tulajdonságai
2.4.1. Stabilitás
A stabilitás mérése általában a hatóanyag(ok)/reagens(ek) vagy azok aktivitásának/életképességének analitikai nyomon követése által történik. Az enzimek esetében a stabilitás a katalitikus hatás elvesztésére tekintettel határozható meg; a mikroorganizmusok esetében az életképesség elvesztésére tekintettel; az aromaanyagok esetében az íz elvesztésére tekintettel. Egyéb kémiai keverékek/kivonatok esetében a stabilitás egy vagy több megfelelő jelölőanyag koncentrációjának nyomon követése segítségével mérhető fel.
Az adalékanyag stabilitása
Vizsgálni kell az adalékanyag minden egyes kiszerelésének a különböző környezeti körülményeknek (fény, hőmérséklet, pH, nedvesség, oxigén és csomagolóanyag) való kitettség esetén jellemző stabilitását. A forgalomba hozott adalékanyag várható szavatossági idejét legalább kettő, az alkalmazási feltételek valószínű körére (pl., 25 oC, 60 % relatív páratartalom (HR) és 40 oC, 75 % HR) kiterjedő modellhelyzetre kell alapozni.
Az előkeverékekben és takarmányokban használt adalékanyag stabilitása
Az előkeverékekben és takarmányokban használt adalékanyagok esetében, kivéve az aromavegyületeket, az adalékanyag minden egyes kiszerelésének stabilitását az előkeverékek és takarmányok közös gyártási és tárolási körülményei között kell vizsgálni. Az előkeverékekben végzett stabilitási vizsgálatoknak legalább féléves terjedelműnek kell lenniük. A stabilitást lehetőleg nyomelemeket tartalmazó előkeverékekkel kell tesztelni; különben az adalékanyagot "nyomelemekkel nem keverendő" címkével kell ellátni.
A takarmányokban végzett stabilitási vizsgálatoknak legalább három hónapig kell tartaniuk. A kérelem által érintett fő állatfajok tekintetében a stabilitást általában pépes és pellet (beleértve a pelletkészítés és más kezelési formák hatását) takarmányban kell ellenőrizni.
A vízben való használatra szánt adalékanyagok esetében, az adalékanyag minden egyes kiszerelésének stabilitását vízben, a gyakorlati alkalmazást szimuláló körülmények között kell vizsgálni.
Stabilitás elvesztése esetén, továbbá amikor szükséges, jellemezni kell a potenciális bomlástermékeket.
Olyan analízisekről kell adatokat benyújtani, amelyek legalább a tárolási időszak kezdetén egy megfigyelést és ezen időszak végén egy megfigyelést foglalnak magukban.
Ha szükséges, a vizsgálatoknak tartalmazniuk kell a tesztekhez használt előkeverékek, illetve takarmányok részletes mennyiségi és minőségi összetételét.
2.4.2. Homogenitás
Ki kell mutatni az adalékanyag (kivéve az aromavegyületeket) előkeverékekben, takarmányokban, illetve vízben történő homogén eloszlásra való képességét.
2.4.3. Egyéb jellemzők
Ismertetni kell egyéb jellemzőket, úgymint porképző képesség, elektrosztatikus tulajdonságok vagy folyadékokban való diszperziós képesség.
2.4.4. Fizikai-kémiai összeegyeztethetetlenségek és kölcsönhatások
Be kell mutatni, hogy takarmánnyal, hordozóanyagokkal, más jóváhagyott adalékanyagokkal vagy gyógyszerkészítményekkel milyen fizikai-kémiai összeegyeztethetetlenségek és kölcsönhatások várhatók.
2.5. Az adalékanyag alkalmazásának feltételei
2.5.1. Az állatok takarmányozása során történő alkalmazás javasolt módja
Az e rendelet IV. mellékletében felsorolt kategóriákkal összhangban fel kell tüntetni az állatfajokat és -kategóriákat, az állatok korcsoportját és termelési fázisait. Meg kell említeni a lehetséges ellenjavallatokat. Meg kell határozni a takarmányban, illetve vízben javasolt használatot.
Az előkeverékek, takarmányok, illetve ivóvíz esetében az alkalmazás javasolt módszerének és az alkalmazás mértékének részleteit meg kell adni. Emellett szükség szerint ki kell térni a teljes értékű takarmányban javasolt dózisra, az alkalmazás javasolt időtartamára és a javasolt, a maradékanyagszintnek az MRL-értékek alá csökkenéséhez szükséges időre. Indokolni kell, ha valamely adalékanyag kiegészítő takarmányban való különleges használatát javasolják.
2.5.2. A felhasználók/dolgozók biztonságával kapcsolatos adatok
2.5.2.1. Vegyi anyagok
A 88/379/EGK irányelv 10. cikke végrehajtásának megfelelően, a veszélyes készítményekre vonatkozó különös információk rendszerével kapcsolatos részletes rendelkezések meghatározásáról és megállapításáról szóló, 1991. március 5-i 91/155/EGK bizottsági irányelv (6) követelményeinek megfelelő formában készített anyagbiztonsági adatlapot kell benyújtani. Ha szükséges, a foglalkozási kockázatokat megelőző intézkedéseket, valamint a gyártás, a kezelés, a használat és az ártalmatlanítás során alkalmazandó védekezései eszközöket kell javasolni.
2.5.2.2. Mikroorganizmusok
A munkájuk során biológiai anyagokkal kapcsolatos kockázatoknak kitett munkavállalók védelméről (hetedik egyedi irányelv a 89/391/EGK irányelv 16. cikkének (1) bekezdése értelmében) szóló, 2000. szeptember 18-i 2000/54/EK európai parlamenti és tanácsi irányelv (7) szerinti osztályozást kell benyújtani. Azon mikroorganizmusok tekintetében, amelyek nem az említett irányelvben szereplő 1. csoportba tartoznak, tájékoztatást kell adni a vevőknek ahhoz, hogy megtehessék a munkavállalóik tekintetében az említett irányelv 3. cikkének (2) bekezdése szerinti megfelelő védintézkedéseket.
2.5.2.3. Címkézési követelmények
Az 1831/2003/EK rendelet 16. cikkében megállapított címkézési és csomagolási rendelkezések megsértése nélkül, fel kell tüntetni minden egyedi címkézési követelményt és - adott esetben - egyedi alkalmazási és kezelési feltételt (beleértve az ismert összeférhetetlenségeket és ellenjavallatokat), valamint a helyes használatra vonatkozó utasításokat.
2.6. Analitikai módszerek és referenciaminták
Az analitikai módszereket az ISO (vagyis az ISO 78-2) által ajánlott szabványos megjelenési formában kell benyújtani.
Az 1831/2003/EK rendelet és a 378/2005/EK rendelet szerint, az e szakaszban szereplő analitikai módszereket a KRL értékeli. A KRL a Hatóság részére értékelési jelentést nyújt be, amelyben közli, hogy ezek a módszerek alkalmasak-e a kérelem tárgyát képező takarmány-adalékanyag hatósági ellenőrzése során történő alkalmazásra. A KRL értékelésében a 2.6.1. és 2.6.2. szakaszokban megjelölt módszerekre összpontosít.
Ha az állati eredetű élelmiszerekben található állatgyógyászati készítmények maximális maradékanyag-határértékeinek megállapítására szolgáló közösségi eljárás kialakításáról szóló, 1990. június 26-i 2377/90/EGK tanácsi rendelet (8) MRL-t állapított meg a kérelem tárgyát képező anyagra vonatkozóan, a 2.6.2. szakaszt a KRL-nek nem kell értékelnie. A kérelmező a 2.6.2. szakaszt úgy állítja össze, hogy ugyanazt a módszert, információkat és adatokat (beleértve a vonatkozó frissítéseket) adja meg, amelyeket az Európai Gyógyszerértékelő Ügynökség (EMEA) részére a 2377/90/EGK rendelet V. mellékletével összhangban, valamint "A gyógyszertermékekre irányadó szabályok az Európai Unióban" sorozat 8. kötetével, a "Hirdetmény kérelmezők számára és iránymutatás" című dokumentummal összhangban nyújt be.
Ha a KRL, a Hatóság vagy a Bizottság szükségesnek tartja, a 2.6.3. szakaszban leírt analitikai módszerekre szintén kiterjedhet az értékelés.
A 378/2005/EK rendelettel összhangban, a kérelmező a technikai dokumentáció értékelése előtt a referenciamintákat, a lejárat időpontja előtt pedig a helyettesítő mintákat közvetlenül a KRL rendelkezésére bocsátja.
A kérelmezőknek a KRL által a 378/2005/EK rendelet 12. cikkével összhangban létrehozott részletes iránymutatásra kell hivatkozniuk.
2.6.1. A hatóanyagra vonatkozó analitikai módszerek
Részletesen jellemezni kell az(oka)t a minőségi és - szükség szerint - mennyiségi analitikai módszer(eke)t, amellyel/amelyekkel a hatóanyag(ok)nak/reagens(ek)nek az adalékanyagban, az előkeverékekben, a takarmányokban és - adott esetben - a vízben javasolt maximális és minimális mértékének való megfelelés megállapítható.
2.6.1.1. E módszereknek ugyanazon követelményeket kell teljesíteniük, amelyeket a hatósági ellenőrzés során alkalmazott analitikai módszereknek a 882/2004/EK rendelet 11. cikkében foglaltak szerint teljesíteniük kell. Különösen, a következő követelmények közül legalább egyet kell teljesíteniük:
- megfelelnek a vonatkozó közösségi szabályoknak (pl. közösségi analitikai módszereknek), amennyiben ilyenek léteznek,
- megfelelnek nemzetközileg elismert szabályoknak vagy protokolloknak, pl. azoknak, amelyeket az Európai Szabványügyi Bizottság (CEN) fogadott el, vagy amelyeket nemzeti jogszabályban fogadtak el (pl. CEN szabványmódszerek),
- alkalmasak a rendeltetésük betöltésére, tudományos eljárások szerint történik a kidolgozásuk, továbbá hitelesítésük a körvizsgálatokra vonatkozó, nemzetközileg elismert protokoll (pl. ISO 5725 vagy IUPAC) szerint végzett gyűrűpróba keretében történik, vagy
- a 2.6.1.2. szakaszban említett, jellemzésre szolgáló paraméterek tekintetében hitelesítésük belső szinten történik, az analitikai módszerek belső szinten történő hitelesítésére vonatkozó nemzetközi harmonizált iránymutatások (9) szerint.
2.6.1.2. A módszer(ek) részletes jellemzésében a 882/2004/EK rendelet III. mellékletében meghatározott, megfelelő tulajdonságokra kell kitérni.
2.6.1.3. A belső szinten hitelesített módszerek teljesítményjellemzőit a módszernek egy második, akkreditált és független laboratóriumban történő tesztelésével kell ellenőrizni. Az ilyen tesztek eredményeit be kell mutatni, bármely olyan más információval együtt, amely alátámasztja a módszer hatósági ellenőrző laboratórium részére történő átadhatóságát. A függetlenség miatt, és a kérelmező által benyújtott dokumentáció értékelésében való részvétel miatt, amennyiben a második laboratórium a nemzeti referencialaboratóriumok azon konzorciumának tagja, amely a közösségi referencialaboratóriumot a 378/2005/EK rendeletben foglaltak értelmében segíti, a laboratórium - amint a közösségi referencialaboratórium átveszi a kérelmet - érdekeltségi nyilatkozatot köteles küldeni a közösségi referencialaboratóriumnak, leírva, hogy milyen munkát végez a kérelemmel kapcsolatban, és a kérelem értékelésében nem vehet részt.
2.6.1.4. A közösségi referencialaboratórium a Hatóságnak küldött értékelési jelentésében a 882/2004/EK rendelet III. mellékletében említett megfelelő jellemzőket választhat ki.
2.6.1.5. Az anyagok különös csoportjaira (pl. enzimek) vonatkozó módszerek tekintetében teljesítményjellemzők állapíthatók meg a közösségi referencialaboratórium (KRL) által a 378/2005/EK rendelet 12. cikkével összhangban készített részletes iránymutatásban.
2.6.2. Analitikai módszerek az adalékanyag, illetve annak anyagcseretermékei élelmiszerben található maradékanyagainak meghatározására
Az adalékanyag jelölő maradékanyagai és/vagy anyagcseretermékei célszövetekben és állati termékekben való kimutatására vonatkozó minőségi és mennyiségi analitikai módszerek részletes jellemzését kell bemutatni.
2.6.2.1. E módszereknek ugyanazon követelményeket kell teljesíteniük, amelyeket a hatósági ellenőrzés során alkalmazott analitikai módszereknek a 882/2004/EK rendelet 11. cikkében foglaltak szerint teljesíteniük kell. Különösen, a módszereknek a 2.6.1.1. alpontban említett követelmények közül legalább egynek meg kell felelniük.
2.6.2.2. A módszer(ek) részletes jellemzésének a 882/2004/EK rendelet III. mellékletében foglalt megfelelő jellemzőket kell tartalmaznia, továbbá e jellemzés során figyelembe kell venni a 2002/657/EK bizottsági határozatban (10) foglalt követelményeket. Szükség szerint tekintettel kell lenni a 96/23/EK tanácsi irányelv szerint bizonyos anyagok és azok maradékanyagai élő állatból származó termékekben történő kimutatására alkalmazandó analitikai módszerek megállapításáról szóló bizottsági határozatokban megállapított ugyanazon teljesítménykritériumokra.
A mennyiségi meghatározási határ (LOQ) egyik módszer esetében sem haladhatja meg a megfelelő MRL felét, és azt egy legalább az MRL felétől kétszereséig terjedő skálán kell validálni.
2.6.2.3. A belső szinten hitelesített módszerek teljesítményjellemzőit a módszernek egy második, akkreditált és független laboratóriumban történő tesztelésével kell ellenőrizni. Az ilyen tesztek eredményeit be kell mutatni. A függetlenség miatt, és a kérelmező által benyújtott dokumentáció értékelésében való részvétel miatt, amennyiben a második laboratórium a nemzeti referencialaboratóriumok (NRL-ek) azon konzorciumának tagja, amely a közösségi referencialaboratóriumot a 378/2005/EK rendeletben foglaltak értelmében segíti, a laboratórium - amint a közösségi referencialaboratórium átveszi a kérelmet - érdekeltségi nyilatkozatot köteles küldeni a közösségi referencialaboratóriumnak, leírva, hogy milyen munkát végez a kérelemmel kapcsolatban, és a kérelem értékelésében nem vehet részt.
2.6.2.4. A közösségi referencialaboratórium a Hatóságnak küldött értékelési jelentésében a 2.6.2.2. alpontban említettek közül megfelelő jellemzőket választhat ki.
2.6.2.5. Az anyagok különös csoportjaira (pl. enzimek) vonatkozó módszerek tekintetében teljesítményjellemzők állapíthatók meg a közösségi referencialaboratórium által a 378/2005/EK rendelet 12. cikkével összhangban készített részletes iránymutatásban.
2.6.3. Az adalékanyag azonosítására és jellemzésére vonatkozó analitikai módszerek
A kérelmezőnek ismertetnie kell a 2.1.3., 2.1.4., 2.1.5., 2.2.2., 2.4.1., 2.4.2., 2.4.3. és 2.4.4. pontban felsorolt jellemzők meghatározására alkalmazott módszereket.
A 378/2005/EK rendelettel módosított 1831/2003/EK rendelet II. melléklete értelmében, az e szakasz alapján benyújtott módszerek értékelését is el lehet végezni, ha a Hatóság vagy a Bizottság a kérelem értékelése szempontjából azt fontosnak tartja.
Ajánlott, hogy az e szakasz alapján ismertetett módszerek nemzetközileg elismertek legyenek. A nemzetközi elismertséggel nem rendelkező módszereket teljeskörűen ismertetni kell. Ilyen esetben a vizsgálatokat akkreditált és független laboratóriumoknak kell végezniük, és azokat megfelelő minőségi szabványok (pl. GLP a 2004/10/EK irányelv vagy ISO-szabványok) szerint kell dokumentálni.
Az adalékanyag azonosítására és jellemzésére szolgáló módszereknek ugyanazon követelményeket kell teljesíteniük, amelyeket a hatósági ellenőrzés során alkalmazott analitikai módszereknek a 882/2004/EK rendelet 11. cikkében foglaltak szerint teljesíteniük kell, különösen ha jogi követelményeket is megállapítottak (pl. szennyeződések, nem kívánatos anyagok tekintetében).
3. III. SZAKASZ: AZ ADALÉKANYAG BIZTONSÁGOSSÁGÁRA VONATKOZÓ VIZSGÁLATOK
Az e szakaszban és a külön mellékletekben szereplő vizsgálatok arra irányulnak, hogy lehetővé tegyék a következők értékelését:
- az adalékanyag célfajokban történő használatának biztonságossága,
- az antimikrobiális rezisztencia kiválasztásával és/vagy átadásával, valamint az enteropatogének fokozott jelenlétével és ürítésével összefüggő bármilyen kockázat,
- az érintett adalékanyagot tartalmazó vagy azzal kezelt takarmánnyal táplált állatokból származó élelmiszerrel összefüggően a fogyasztót érintő kockázatok, továbbá olyan kockázatok, amelyek az adalékanyag maradékanyagait vagy anyagcseretermékeit tartalmazó élelmiszer fogyasztásából adódhatnának,
- kockázatok azok számára, akik várhatóan az adalékanyagot önmagában vagy előkeverékekbe, illetve takarmányokba keverve kezelik, és azzal belélegzés útján, vagy más nyálkahártya-szöveten keresztül, szembe kerülés vagy bőrrel való érintkezés útján kerülnek kapcsolatba, és
- magának az adalékanyagnak, illetve az adalékanyagból nyert termékeknek - vagy közvetlenül és/vagy az állatok anyagcseréje útján - a környezetre gyakorolt káros hatásaival összefüggő kockázatok.
3.1. Az adalékanyag biztonságos használatára vonatkozó vizsgálatok a célállatok tekintetében
Az ebben a szakaszban szereplő vizsgálatok a következők értékelésére irányulnak:
- az adalékanyag magukban a célfajokban történő használatának biztonságossága, és
- az antimikrobiális rezisztencia kiválasztásával és/vagy átadásával, valamint az enteropatogének fokozott jelenlétével és ürítésével összefüggő bármilyen kockázat.
3.1.1. A célfajokra vonatkozó toleranciavizsgálatok
A toleranciavizsgálat célja annak a korlátozott felmérése, hogy az adalékanyag rövid távon mennyire mérgező a célállatok számára. E vizsgálat célja továbbá egy biztonsági küszöb megállapítása arra az esetre, ha az adalékanyagot az ajánlottnál nagyobb dózisban fogyasztják. Az ilyen toleranciavizsgálatot úgy kell végezni, hogy bizonyítékot szolgáltasson a biztonságosságra mindegyik olyan célfaj/célállat-kategória tekintetében, amelyre a kérelem kiterjed. Bizonyos esetekben elfogadható, hogy a hatékonysági tesztek egyikébe a toleranciavizsgálat néhány elemét beépítik, feltéve hogy az e vizsgálatokra vonatkozóan alább megadott követelmények teljesülnek. Az e szakaszban ismertetett valamennyi vizsgálatnak a II. szakaszban ismertetett adalékanyagra kell vonatkoznia.
3.1.1.1. A toleranciavizsgálat a felépítése szerint legalább három csoportra terjed ki:
- az adalékanyagot nem fogyasztó csoport,
- a legnagyobb ajánlott dózist fogyasztó csoport, és
- a legnagyobb ajánlott dózis többszörösét fogyasztó kísérleti csoport.
A kísérleti csoportban adott adalékanyagnak általában a legnagyobb ajánlott dózis tízszeresének kell lennie. A tesztállatoknál rendszeresen figyelemmel kell kísérni a klinikai hatások látható megjelenését, a teljesítményjellemzőket, adott esetben a termékminőséget, a hematológiai és rutin vérkémiai értékek vizsgálatának eredményét, továbbá egyéb olyan paramétereket, amelyek valószínűsíthetően összefüggnek az adalékanyag biológiai tulajdonságaival. Figyelembe kell venni a laboratóriumi állatokon végzett toxikológiai vizsgálatokból ismert kritikus végpontokat. Szintén be kell számolni ebben a szakaszban minden olyan esetleges káros hatásról, amelyet a hatékonysági tesztek során tapasztaltak. A toleranciavizsgálat során bekövetkező indokolatlan haláleseteket boncolásos vizsgálat, és szükség szerint szövettani vizsgálat útján ki kell vizsgálni.
Ha bizonyítható, hogy a legnagyobb ajánlott dózis 100-szorosa elviselhető, nincs szükség hematológiai vagy rutin vérkémiai értékek vizsgálatára. Ha a termék csak a legnagyobb ajánlott dózis tízszeresénél kisebb mértékig viselhető el, a vizsgálatot úgy kell kialakítani, hogy ki lehessen számítani az adalékanyagra vonatkozó biztonsági küszöböt, valamint további végpontokat (boncolásos vizsgálat, szükség szerint szövettani vizsgálat és más megfelelő kritérium útján) kell megadni.
Néhány adalékanyag esetében, toxikusságuktól és lebomlásuktól, illetve alkalmazásuktól függően lehetséges, hogy nincs szükség toleranciavizsgálatok elvégzésére.
Az alkalmazott kísérleti felépítés keretében a megfelelő statisztikai erőt is figyelembe kell venni.
3.1.1.2. A toleranciavizsgálatok időtartama
1. táblázat
A toleranciavizsgálatok időtartama: Sertés
| Célállatok | A vizsgálatok időtartama | A célállatok jellemzője |
| Szopós malac | 14 nap | Lehetőleg 14 naptól az elválasztásig |
| Elválasztott malac | 42 nap | Az elválasztás után 42 napig |
| Hízósertés | 42 nap | A vizsgálat kezdetén a testtömeg ≤ 35 kg |
| Tenyészkoca | 1 ciklus | A megtermékenyítéstől az elválasztási időszak végéig |
Ha a kérelem szopós malacokra és elválasztott malacokra vonatkozik, elegendőnek tekintendő a kombinált vizsgálat (14 nap a szopós malacok és 28 nap az elválasztott malacok esetében). Ha bizonyítást nyert, hogy az elválasztott malacok tűrik az adalékanyagot, a hízósertések tekintetében már nincs szükség külön vizsgálatra.
2. táblázat
A toleranciavizsgálatok időtartama: Baromfi
| Célállatok | A vizsgálatok időtartama | A célállatok jellemzője |
| Hízócsirke/tojástermelés céljából nevelt csirke | 35 nap | A kikeléstől |
| Tojótyúk | 56 nap | Lehetőleg a tojásrakási időszak első harmadában |
| Hízópulyka | 42 nap | A kikeléstől |
A hízócsirkére, illetőleg a hízópulykára vonatkozó tűrési adatok felhasználhatók a tojónak/tenyészállatnak nevelt csirke, illetőleg pulyka tekintetében a tűrés bizonyítására.
3. táblázat
A toleranciavizsgálatok időtartama: Szarvasmarha
| Célállatok | A vizsgálatok időtartama | A célállatok jellemzője |
| Hízóborjú | 28 nap | Kezdeti testtömeg ≤ 70kg |
| Tenyészborjú; hízó- vagy tenyészmarha | 42 nap | |
| Tejelő tehén | 56 nap |
Ha a kérelem tenyészborjakra és hízómarhákra vonatkozik, elegendőnek tekintendő a kombinált vizsgálat (mindegyik időszakban 28 nap).
4. táblázat
A toleranciavizsgálatok időtartama: Juh
| Célállatok | A vizsgálatok időtartama | A célállatok jellemzője |
| Tenyészbárány és húsbárány | 28 nap |
5. táblázat
A toleranciavizsgálatok időtartama: Lazacfélék és más halak
| Célállatok | A vizsgálatok időtartama | A célállatok jellemzője |
| Lazac és pisztráng | 90 nap |
A 90 napos időtartam helyett vizsgálat végezhető, ha a halaknak a teszt kezdetén mért kezdeti testtömege legalább kétszeresére gyarapszik.
Ha az adalékanyagot kizárólag a tenyészállományban kívánják használni, a toleranciavizsgálatokat az ívási időszakhoz lehető legközelebb eső időszakban kell végezni. A toleranciavizsgálatoknak 90 napig kell tartaniuk, és figyelmet kell szentelni az ikrák minőségének és életben maradásának.
6. táblázat
A toleranciavizsgálatok időtartama: Kedvtelésből tartott és egyéb nem élelmiszer-termelő állatok
| Célállatok | A vizsgálatok időtartama | A célállatok jellemzője |
| Kutyák és macskák | 28 nap |
7. táblázat
A tolerancia vizsgálatok időtartama: Nyúl
| Célállatok | A vizsgálatok időtartama | A célállatok jellemzője |
| Hízónyúl | 28 nap | |
| Nőstény tenyésznyúl | 1 ciklus | A megtermékenyítéstől az elválasztási időszak végéig |
Ha a kérelem szopósnyulakra és elválasztott nyulakra vonatkozik, elegendőnek tekintendő egy 49 napos (a születés után egy héttel kezdődő) időtartam, amelynek a nőstény nyulakra az elválasztásig kell kiterjednie.
Ha az adalékanyagot különleges és az adott állatkategóriára vonatkozó fogalommeghatározásban szereplőnél rövidebb időszakban alkalmazzák, azt a javasolt alkalmazási feltételek szerint kell az állatnak adni. A megfigyelési időszak azonban nem lehet 28 napnál rövidebb, és magában kell foglalnia a releváns végpontot (pl. a tenyészkocák esetében az adott gesztációs időszakot illetően az élve született malacok száma, vagy a laktációs időszakot illetően az elválasztott malacok száma és tömege).
3.1.1.3. Vizsgálati körülmények
A vizsgálatokról egyenként kell beszámolni, valamennyi kísérleti csoport részletes adatait megadva. A vizsgálat leírását az általános leíró adatok tekintetében gondosan ki kell dolgozni. Különösen, fel kell tüntetni a következőket:
1. állomány: helye és mérete; takarmányozási és tartási feltételek, takarmányozási módszer; vízben élő fajok, a mezőgazdasági üzemben használt tartályok és karámok mérete és száma, fényviszonyok és vízminőség, beleértve a víz hőmérsékletét és sótartalmát;
2. állatok: fajok (az emberi fogyasztásra szánt, vízben élő fajok esetében az azonosításhoz a köznyelvi nevüket kell megadni, majd azt követően zárójelben a két tagból álló latin nevüket), fajta, életkor (vízben élő fajok esetében méret), nem, azonosítási eljárás, fiziológiás állapot és általános egészségi állapot;
3. a tesztelés időpontja és pontos időtartama: az elvégzett vizsgálatok időpontja és jellege;
4. étrendek: az étrend(ek) gyártásának és mennyiségi összetételének leírása a felhasznált összetevők, a releváns tápanyagok (analizált értékek) és az energia szempontjából. Takarmánybeviteli adatok;
5. a hatóanyag(ok) vagy reagensek (és adott esetben az összehasonlítási céllal alkalmazott anyagok) takarmányokon belüli koncentrációját ellenőrző analízissel kell megállapítani, a megfelelő, elismert módszereket használva: a gyártási tételek referenciaszáma(i);
6. a tesztcsoportok és kontrollcsoportok száma, az állatok száma az egyes csoportokban: a tesztekben részt vevő állatok számának lehetővé kell tennie a statisztikai elemzést. Meg kell nevezni az alkalmazott statisztikai értékelési módszereket. A jelentésnek a tesztekben részt vevő valamennyi állatra és/vagy kísérleti egységre ki kell terjednie. Be kell jelenteni az adatok hiánya vagy elveszése miatt nem értékelhető eseteket, és azoknak az állatcsoportokon belüli eloszlását osztályozni kell;
7. jelenteni kell a kezelés egyedekben illetve csoportokban tapasztalt bármely nemkívánatos következményének időbeli megjelenését és gyakoriságát (megadva a vizsgálat során alkalmazott megfigyelési program részleteit); valamint
8. a gyógykezelések/megelőző kezelések, ha szükségesek, nem kerülhetnek kölcsönhatásba az adalékanyag javasolt hatásmechanizmusával, és ezeket külön kell nyilvántartani.
3.1.2. Mikrobiális vizsgálatok
Vizsgálatokat kell végezni annak megállapítása céljából, hogy képes-e az adalékanyag a humán-, illetve állatgyógyászatban használt antibiotikumokkal szembeni keresztrezisztenciát előidézni, a célfajokban terepfeltételek között rezisztens baktériumtörzseket kiválasztani, az emésztőcsatornában jelen lévő opportunista kórokozókra gyakorolt hatásokat előidézni, valamint ürítést, illetve zoonózist okozó mikroorganizmusok exkrécióját okozni.
Ha a hatóanyag(ok) a takarmánykoncentráció szintjén antimikrobiális aktivitással rendelkeznek, szabványosított eljárások szerint meg kell határozni a releváns baktériumfajokra vonatkozóan a minimális inhibitor koncentrációt (MIC). Amennyiben lényeges antimikrobiális aktivitás mutatható ki, meg kell állapítani, hogy képes-e az adalékanyag in vitro és a célfajokban rezisztens baktériumtörzseket kiválasztani, valamint az érintett antibiotikumokkal szemben keresztrezisztenciát előidézni (11).
Minden mikrobiális adalékanyag esetében, és olyan egyéb adalékanyagok esetében, amelyeknél várható a bélflórára gyakorolt hatás, az alkalmazás ajánlott mértéke szerinti vizsgálatokat kell végezni. E vizsgálatokban bizonyítani kell, hogy az adalékanyag használata nem teremt olyan feltételeket, amelyek a potenciálisan kórokozó mikroorganizmusok túlzott elszaporodását és ürítését okozzák.
A nyomon követendő mikroorganizmusok kiválasztása a célfajoktól függ, de annak ki kell terjednie az érintett, zoonózist okozó fajokra, függetlenül attól, hogy azok a célállatokban előidéznek-e tüneteket vagy sem.
3.2. Az adalékanyag biztonságos használatára vonatkozó vizsgálatok a fogyasztók tekintetében
A vizsgálatok célja az adalékanyag biztonságos használatának értékelése a fogyasztók tekintetében és az adalékanyagból vagy anyagcseretermékeiből származó, esetleges maradékanyagok azonosítása az adalékanyagot tartalmazó vagy az adalékanyaggal kezelt takarmányt, illetve vizet fogyasztó állatokból származó élelmiszerekben.
3.2.1. Az anyagcserével és a maradékanyaggal kapcsolatos vizsgálatok
Az adalékanyag anyagcsereútjának meghatározása a célfajban döntő lépés a maradékanyagok azonosításában és mennyiségi meghatározásában az adalékanyagot tartalmazó takarmányt vagy vizet fogyasztó állatokból származó, élelmezési célra alkalmas szövetekben és termékekben. Vizsgálatokat kell benyújtani az anyag (és ennek anyagcseretermékei) felszívódására, eloszlására, metabolizmusára és kiürülésére vonatkozóan.
A vizsgálatokat nemzetközileg validált tesztmódszerek szerint és a hatályos európai jogszabályokkal vagy az OECD módszertani részletekre vonatkozó irányelveivel összhangban, és a helyes laboratóriumi gyakorlat elveinek megfelelően kell elvégezni. A vizsgálatok során tiszteletben kell tartani az európai közösségi jogszabályok által meghatározott, állatjóllétre vonatkozó jogszabályokat, és a vizsgálatokat, amennyiben nem szükséges, nem szabad megismételni.
A célállat(ok)on az anyagcserével és a maradékanyaggal kapcsolatos vizsgálatokat a takarmányba kevert hatóanyaggal (nem pedig tömés útján, kivéve ha kellően indokolt) kell végezni.
Az élelmezési célra alkalmas szövetekben és termékekben levő összmaradékanyag több mint 10 %-át, valamint a trágyában levő összmaradékanyag 20 %-át kitevő anyagcseretermékek kémiai szerkezetét meg kell határozni. Ha a hatóanyag anyagcsereútjának vizsgálata során bármilyen toxikológiai vonatkozású veszély merül fel, a fent említett küszöbértékek alatti anyagcseretermékeket meg kell határozni.
A maradékanyagok kinetikai vizsgálata alapján fogják meghatározni a fogyasztók expozícióját, és ha szükséges, a maradékanyagszintnek az MRL-értékek alá csökkenéséhez szükséges időt, valamint a maximális maradékanyag-határértékeket (az MRL-eket). Javasolni kell egy jelölő maradékanyagot.
Néhány maradékanyag esetében, az anyag természetétől illetve felhasználásától függően, az anyagcsere- és a maradékanyag-vizsgálat nem mindig feltétlenül szükséges.
3.2.1.1. Anyagcsere-vizsgálatok
Az anyagcsere-vizsgálatok célja az adalékanyag felszívódásának, eloszlásának, biológiai úton történő átalakulásának és kiválasztódásának becslése a célfajban.
A szükséges vizsgálatok a következők:
1. a hatóanyag egyszeri dózisú alkalmazását követő anyagcsere-egyensúlyi vizsgálat a javasolt alkalmazási (a teljes mennyiség megfelel a napi bevitelnek) és esetleg többszörös dózisnál (amennyiben szükséges) a felszívódás, valamint az eloszlás (plazma/vér) és a kiválasztódás (vizelet, epe, széklet, tej vagy tojás, kilélegzett levegő, kopoltyún keresztül történő kiválasztódás) hozzávetőleges sebességének és mértékének megállapítása céljából hím és nőstény állatokban, adott esetben; és
2. a metabolittartalom komplex elemzése, az anyagcseretermék(ek) meghatározása a kiválasztott anyagokban és a szövetekben, és az eloszlást a kiválasztott anyagokban és a szövetekben a megjelölt anyag ismételt dózisának adagolását követően kell megállapítani a vérplazma-analízis alapján meghatározott, stabil állapotban (metabolikus egyensúlyban) levő állatoknál. Az alkalmazott dózis a használatra javasolt legmagasabb adagnak kell, hogy megfeleljen, és azt a takarmányhoz kell keverni.
3.2.1.2. Maradékanyagok vizsgálata
Figyelembe kell venni az élelmezési célra alkalmas szövetekből és termékekből nem eltávolítható maradékanyagok mennyiségét és jellegét.
A maradékanyagok vizsgálatát minden olyan anyagon el kell végezni, amely anyagcsere-vizsgálatot igényel.
Ha az anyag a testnedvek vagy szövetek természetes alkotóeleme, vagy természetes körülmények között nagy mennyiségben van jelen az élelmiszerben vagy a takarmányban, a maradékanyagok vizsgálata a nem kezelt csoport és a kérelem tárgyát képező legnagyobb adalékanyag-dózissal kezelt csoport szövet/termék összehasonlítására korlátozódik.
A főbb fajok esetében a vizsgálatok során egyidejűleg kell értékelni a toxikológiai jelentőségű összmaradékanyagot és azonosítani a hatóanyag jelölő maradékanyagát az élelmezési célra alkalmas szövetekben (máj, vese, izom, bőr, bőr és zsírszövet), valamint termékekben (tej, tojás és méz). A jelölő maradékanyag az a vizsgálatra kiválasztott anyag, amelynek koncentrációja ismert összefüggésben van a szövetekben található, toxikológiai jelentőségű teljes maradékanyag-mennyiséggel. A vizsgálatok során ki kell mutatni a maradékanyagok szövetekben vagy termékekben tapasztalt maradandóságát is, a maradékanyagszintnek az MRL-értékek alá csökkenéséhez szükséges idő megállapítása érdekében.
A maradékanyagszintnek az MRL-értékek alá csökkenéséhez szükséges idő megállapítása érdekében a mintavételi állatok és/vagy termékek javasolt legkisebb száma minden időpontban a következő:
- élelmezési célra alkalmas szövetek:
- szarvasmarha, kecske, sertés és kevésbé jelentős fajok 4,
- baromfifajok 6,
- lazacfélék és egyéb halfajok 10.
- termékek:
- tej 8 minta időpontonként,
- tojás 10 tojás időpontonként,
- méz 8 minta időpontonként.
A nemek megfelelő eloszlását figyelembe kell venni.
A maradékanyagokat a nulla értékű, a maradékanyagszintnek az MRL-értékek alá csökkenéséhez szükséges időnél (egyensúlyi állapot) és legalább három másik időpontban kell mérni.
Javasolni kell egy jelölő maradékanyagot.
A felszívódási, eloszlási és kiürülési vizsgálatokat, beleértve a főbb anyagcseretermékek meghatározását is, a legalacsonyabb NOAEL értékű laboratóriumi állatfajon, vagy alapértelmezésben (mindkét nembeli) patkányon kell elvégezni. Kiegészítő vizsgálatra lehet szükség egyedi anyagcseretermékek esetében, amennyiben ezek a metabolitok a célfajban termelődnek és a laboratóriumi fajban nem képződik jelentős mennyiség belőlük.
3.2.1.3. Anyagcsere- és eloszlásvizsgálat
Az anyagcseremérleget, a metabolittartalom-elemzést és a főbb anyagcseretermékek meghatározását magában foglaló vizsgálatot kell elvégezni a vizeletből és a székletből. A vizsgálatot további adatokkal kell kiegészíteni, ha egy másik laboratóriumi faj fogékonysága szembetűnően különbözik a patkányétól.
3.2.1.4. A maradékanyagok biológiai hasznosulása
Az állati termékekben kötött állapotban jelenlevő maradékanyagok fogyasztók szempontjából lehetséges kockázatainak felmérése során tekintetbe lehet venni egyéb biztonságossági tényezőket is a biológiai hasznosulásuk meghatározása során, megfelelő laboratóriumi állatok és elismert módszerek alkalmazásával.
3.2.2. Toxikológiai vizsgálatok
Az adalékanyag biztonságossága laboratóriumi állatokon in vitro és in vivo elvégzett toxikológiai vizsgálatokból állapítható meg. A toxikológiai vizsgálatok általában a következő méréseket foglalják magukban:
1. akut toxicitás;
2. genotoxicitás (mutagenitás, klasztogenitás);
3. szubkrónikus orális toxicitás;
4. krónikus orális toxicitás/karcinogenitás;
5. reprodukciós toxicitás, beleértve a teratogenitást; és
6. egyéb vizsgálatok.
Indokolt esetben, a hatóanyag és maradékanyagai biztonságának értékelésében hasznos kiegészítő információkat nyújtó további vizsgálatokat kell végezni.
E vizsgálatok eredményei alapján meg kell állapítani a toxikológiai NOAEL-t.
Kiegészítő vizsgálatra lehet szükség bizonyos különleges anyagcseretermékek esetében, amennyiben ezek a metabolitok a célfajban termelődnek és a laboratóriumi fajban nem képződik jelentős mennyiség belőlük. Ha embereken végzett anyagcsere-vizsgálatok állnak rendelkezésre, az adatokat figyelembe kell venni az esetleges kiegészítő vizsgálatok típusának eldöntése során.
A toxikológiai vizsgálatot a hatóanyagon kell elvégezni. Ha a hatóanyag fermentációs termékben található, tesztelni kell a fermentációs terméket. A tesztelt fermentációs terméknek azonosnak kell lennie azzal, amelyet a kereskedelemben forgalmazott adalékanyagban fognak alkalmazni.
A vizsgálatokat nemzetközileg validált tesztmódszerek szerint és a hatályos európai jogszabályokkal vagy az OECD módszertani részletekre vonatkozó irányelveivel összhangban, és a helyes laboratóriumi gyakorlat elveinek megfelelően kell elvégezni. A laboratóriumi állatokat felhasználó vizsgálatok során tiszteletben kell tartani az európai közösségi jogszabályok által meghatározott, állatjóllétre vonatkozó jogszabályokat, és a vizsgálatokat, amennyiben nem szükséges, nem szabad megismételni.
3.2.2.1. Akut toxicitás
Az akut toxicitási vizsgálat a vegyület toxicitásának osztályozására és korlátozott jellemzésére szolgál.
Az akut toxicitási vizsgálatot legalább két emlősfajon kell elvégezni. Adott esetben az egyik laboratóriumi faj helyettesíthető egy célfajjal.
Nem szükséges a pontos LD50-értéket meghatározni; a minimális letális dózis hozzávetőleges meghatározása elegendő. A maximális dózis nem haladhatja meg a 2 000 mg/testtömeg kg-ot.
A vizsgálatba bevont állatok számának csökkentése és szenvedéseik mérséklése céljából az akut toxicitás vizsgálatára folyamatosan új módszereket fejlesztenek ki. Az ilyen új módszerekkel végzett vizsgálatok a megfelelő validálást követően elfogadhatók.
A vizsgálatok során a 402. (akut dermális toxicitás), a 420. (rögzített dózis módszere), a 423. (akut toxicitási osztályozási módszer) és a 425. (dóziscsökkentési és -növelési eljárás) OECD-irányelvet kell szem előtt tartani.
3.2.2.2. Genotoxicitás-vizsgálatok, beleértve a mutagenitást is
A mutagén és genotoxikus tulajdonságokkal rendelkező hatóanyagok, adott esetben anyagcseretermékeik és bomlástermékeik azonosítása céljából, különböző genotoxicitás-vizsgálatból álló sorozatot kell végezni, tetszés szerinti kombinációban. A vizsgálatokat emlősök metabolikus aktiválása mellett és anélkül is el kell végezni, ha szükséges, és figyelembe kell venni a tesztelt anyag kompatibilitását a tesztrendszerrel.
Az alapvető tesztsorozat a következőket tartalmazza:
1. génmutációk indukálása baktériumokban és/vagy emlőssejtekben (lehetőleg egér limphoma tk vizsgálattal);
2. kromoszóma-rendellenességek indukálása emlőssejtekben; és
3. in vivo tesztek emlősfajokban.
A fent említett vizsgálatok eredményétől függően kiegészítő vizsgálatokra lehet szükség, az anyag teljes toxicitási profiljának és tervezett felhasználásának figyelembevételével.
Az eljárások összhangban kell legyenek a 471. (Salmonella typhimurium - reverz mutáció vizsgálat), a 472. (Escherichia coli - reverz mutáció vizsgálat), a 473. (in vitro emlős kromoszóma-rendellenességi vizsgálat), a 474. (emlős vörösvértest mikronukleusz vizsgálat), a 475. (emlőscsontvelőkromoszóma-rendellenességi vizsgálat), a 476. (in vitro emlőssejt gének mutáció vizsgálata) vagy a 482. (in vitro mentesítő DNS szintézis emlőssejtekben) OECD-irányelvvel, valamint egyéb, az in vitro és az in vivo vizsgálatokra vonatkozó OECD-irányelvekkel.
3.2.2.3. Szubkrónikus ismételt dózisú orális toxicitásvizsgálatok
A hatóanyag szubkrónikus toxikus potenciáljának vizsgálata céljából egy rágcsálófajon legalább egy 90 napos vizsgálatot szükséges elvégezni. Amennyiben szükséges, második vizsgálatot lehet végezni egy nem rágcsálófajon. A hatóanyagot szájon át, legalább további három szinten egy kontrollcsoportnak is be kell adni a dózisválasz kimutatása érdekében. A maximális dózis eredményeként általában várható a káros hatások megjelenése. A legalacsonyabb dózis nem válthat ki semmilyen, toxicitásra utaló jelet.
E vizsgálatok eljárásainak összhangban kell lenniük a 408. (rágcsálók) és a 409. (nem rágcsálók) OECD-irányelvvel.
3.2.2.4. Krónikus orális toxicitási vizsgálatok (beleértve a karcinogenitási vizsgálatokat)
A hatóanyag krónikus toxikus és karcinogén potenciáljának vizsgálata céljából egy rágcsálófajon egy legalább 12 hónapig tartó, szájon keresztüli krónikus toxicitási vizsgálatot kell végezni. A kiválasztott fajoknak a megjelent tudományos adatok alapján, beleértve a 90 napos vizsgálatok eredményeit is, a célra legalkalmasabbaknak kell lenniük. A patkány az alapértelmezett faj. Ha második vizsgálatra van szükség, egy rágcsáló és egy nem rágcsálófajt kell alkalmazni erre. A hatóanyagot szájon át, legalább további három szinten egy kontrollcsoportnak is be kell adni a dózisválasz kimutatása érdekében.
Ha a krónikus toxicitási vizsgálatot karcinogenitási vizsgálat kíséri, a vizsgálati időt 18 hónapra kell kiterjeszteni egerek és hörcsögök, valamint 24 hónapra patkányok esetében.
A karcinogenitási vizsgálatok nem feltétlenül szükségesek, ha a hatóanyag és a metabolitjai:
1. állandó negatív eredményt adnak a genotoxicitási vizsgálatok során;
2. szerkezetük nem hasonlít az ismert karcinogénekére; és
3. a krónikus toxicitási vizsgálatok során semmilyen, potenciális (pre)neopláziára utaló hatást nem mutatnak.
Az eljárásoknak összhangban kell lenniük a 452. (krónikus toxicitási vizsgálatok) és a 453. (kombinált krónikus toxicitási/karcinogenitási vizsgálatok) OECD-irányelvvel.
3.2.2.5. Reprodukciós toxicitási vizsgálatok (beleértve a prenatalis fejlődési toxicitást)
A hatóanyag adagolását követően a hím vagy a nőstény szaporodási funkciók esetleges gyengülésének, vagy az utódokra kifejtett káros hatás meghatározásának érdekében vizsgálni kell a szaporodási funkciót:
1. kétgenerációs reprodukciós toxicitási vizsgálatok során; és
2. prenatalis fejlődési toxicitási vizsgálatok (teratogenitási vizsgálatok) során.
Új vizsgálatok esetében az állatok kísérletes alkalmazását csökkentő, validált alternatív módszereket lehet alkalmazni.
3.2.2.5.1. Kétgenerációs reprodukciós toxicitási vizsgálatok
A reprodukciós funkciókat vizsgálni kell, legalább két utódnemzedékre (F1, F2) kiterjedően, legalább egy, általában rágcsáló, fajra vonatkozóan, és e vizsgálatok teratogenitási vizsgálatokkal kombinálhatók. A vizsgált anyagot megfelelő időben a párzás előtt, szájon keresztül kell beadni a hím és nőstény állatoknak. Az adagolást az F2 utódnemzedék elválasztásáig kell folytatni.
A termékenységet, a vemhességet, az ellést, az anyai viselkedést, a szoptatást, valamint az F1 utódok növekedését és fejlődését a termékenyüléstől az ivarérésig, és az F2 utódok fejlődését az elválasztásig gondosan meg kell figyelni és dokumentálni kell. A reprodukciós toxicitási vizsgálatokra vonatkozó eljárásoknak összhangban kell lenniük a 416. OECD-irányelvvel.
3.2.2.5.2. Prenatalis fejlődési toxicitásvizsgálatok (teratogenitási vizsgálatok)
Céljuk az adalékanyagnak kitett vemhes nőstényekre és az embrió, illetve a magzat fejlődésére gyakorolt bármilyen kóros hatás kimutatása, a beágyazódástól a teljes vemhességi idő leteltéig. E hatások magukban foglalják a megnövekedett toxicitást a vemhes nőstényekben, az embrió- és a magzathalált, a módosult magzati fejlődést és a kóros szerkezeti rendellenességet, valamint a magzatfejlődési rendellenességet.
Az első vizsgálatban felhasznált faj általában a patkány. Amennyiben negatív vagy bizonytalan a teratogenitási vizsgálat eredménye, másik fejlődési toxicitásvizsgálatot kell elvégezni egy második állatfajban, lehetőleg a nyúlban. Ha a patkányon végzett vizsgálat pozitív a teratogenitást illetően, nincs szükség a második fajon végzett vizsgálatra, kivéve ha az alapvető vizsgálatok összegzése azt jelzi, hogy az ADI (a megengedhető napi bevitel) a patkányokon végzett teratogenitási vizsgálatokra épülne. Ekkor az e végponthoz rendelt legérzékenyebb faj meghatározása céljából szükség van a második fajon végzett vizsgálatra is. Az eljárásoknak összhangban kell lenniük a 414. OECD-irányelvvel.
3.2.2.6. Egyéb specifikus toxikológiai és farmakológiai vizsgálatok
Indokolt esetben, a hatóanyag és maradékanyagai biztonságosságának értékelésében hasznos kiegészítő információkat nyújtó, további vizsgálatokat kell végezni. E vizsgálatok tartalmazhatják a farmakológiai hatás vizsgálatát, a juvenil (serdülés előtti) állatokban tapasztalható hatást, az immunotoxicitást és a neurotoxicitást.
3.2.2.7. A még nem észlelhető káros hatásszint (NOAEL) meghatározása
A NOAEL általában toxikológiai hatások vizsgálatára támaszkodik, de a farmakológiai hatások adott esetben jobban megfelelhetnek a célnak.
A legalacsonyabb NOAEL-értéket kell kiválasztani. A mg/testtömeg-kilogramm/nap formában kifejezett legalacsonyabb NOAEL-érték meghatározásánál tekintetbe kell venni, adott esetben, a nagymértékben hasonló szerkezetű vegyi anyagra vonatkozó, az előző szakaszokban feltüntetett minden előző eredményt, az összes vonatkozó megjelent adattal (beleértve bármilyen vonatkozó információt a hatóanyagnak az emberre gyakorolt hatásáról) és információval együtt.
3.2.3. A fogyasztói biztonság felmérése
A fogyasztói biztonságot a megállapított ADI (megengedhető napi bevitel) és a hatóanyagnak vagy anyagcseretermékeinek az élelmiszerből történő, elméletileg kiszámított bevitele közötti összehasonlítás révén kell felmérni. A vitaminok és a nyomelemek esetében az ADI helyett az UL (a biztonságos bevitel felső határa) alkalmazható.
3.2.3.1. Javaslat a hatóanyag(ok) megengedhető napi bevitelére (ADI)
A megengedhető napi bevitel (ADI) (az adalékanyag, vagy az adalékanyaghoz hasonló anyag mg-ban kifejezett értéke személyenként és naponta) kiszámításához a legalacsonyabb NOAEL-értéket (mg/testtömeg-kilogramm) el kell osztani a megfelelő biztonsági faktorral és meg kell szorozni a 60 kg átlagos emberi testsúllyal.
Adott esetben javaslatot kell tenni az ADI-re. Az ADI lehet "nem meghatározott" értékű is, az állatkísérletek alacsony toxicitása miatt. Nem lehet javaslatot tenni az ADI-re, ha a hatóanyag az emberre nézve genotoxikus vagy karcinogén tulajdonságokat mutat.
Az ADI beállításához általában az szükséges, hogy a hatóanyag a célfajokban és a laboratóriumi fajokban hasonló módon metabolizálódjon (ld. 3.2.1.4 A maradékanyagok biológiai hasznosulása), ami biztosítja azt, hogy a fogyasztók ugyanannak a maradékanyagnak legyenek kitéve, mint a toxikológiai vizsgálatokban alkalmazott állatok. Amennyiben nincs hasonlóság, az ADI beállítható egy második laboratóriumi fajon is, vagy a célfajra jellemző metabolitokkal végzett kiegészítő vizsgálattal.
Az adott hatóanyagra vonatkozó ADI meghatározására szolgáló biztonsági tényezőnek figyelembe kell vennie a NOAEL meghatározására szolgáló biológiai hatásokat és felhasznált adatok minőségét, e hatások fontosságát az emberre vonatkozóan és a hatások reverzibilitását, valamint bármilyen, a maradékanyagnak emberre vonatkozó közvetlen hatás(ai)val kapcsolatos ismeretet.
Az ADI meghatározásánál legalább 100-as értékű biztonsági tényezőt kell alkalmazni (ha teljes toxikológiai adatcsomag áll rendelkezésre). Elfogadható az alacsonyabb biztonsági faktor abban az esetben, ha a hatóanyagnak az emberre vonatkozó adatai ismertek. Magasabb biztonsági faktorok alkalmazhatók, ha az adatokban további bizonytalansági tényezők merülnek fel, vagy abban az esetben, ha a NOAEL-t valamely egyedi kritikus végpont, így például a teratogenitás alapján, állapítják meg.
3.2.3.2. A biztonságos bevitel felső határa (UL)
Néhány adalékanyag esetében megfelelőbb a biztonsági felmérést az UL-re alapozni, amely egy adott tápanyag teljes, krónikus, maximális napi bevitelét jelenti (minden forrásból származóan) és amely (nemzeti vagy nemzetközi tudományos testületek szerint) valószínűleg nem fejt ki a fogyasztók egészségére vagy a fogyasztók egy specifikus csoportjának egészségére káros hatásokat.
A dokumentációnak tartalmaznia kell olyan adatokat, amelyek azt bizonyítják, hogy az adalékanyag alkalmazása nem eredményezne olyan helyzetet, amelyben - a tápanyag minden lehetséges forrását tekintetbe véve - az UL túllépésére kerülne sor.
Világosan jelezni kell, ha a táplálék-adalékanyagból vagy ennek anyagcseretermékeiből származó maradékanyagok mennyisége az állati termékekben meghaladja az e termékekben normálisnak tekintett vagy várt értéket.
3.2.3.3. A fogyasztók expozíciója
Az összes forrásból származó adalékanyagnak és/vagy anyagcseretermékeinek teljes bevitele a fogyasztók vonatkozásában nem haladhatja meg az ADI vagy az UL értéket.
Az állati eredetű élelmiszerből származó elméleti bevitel kiszámításához figyelembe kell venni az adalékanyag alkalmazása végeztével az állati szövetekben és termékekben mért koncentrációt (az összmaradékanyag számtani középarányosa és a legmagasabb egyszeres érték). Ezen túlmenően, ha szükséges, meg kell határozni, a maradékanyagszintnek az MRL-értékek alá csökkenéséhez szükséges, különböző időpontokban a legrosszabb esetre vonatkozó emberi napi táplálékbeviteli értékeket.
A több fajnál alkalmazott adalékanyagok esetén a szövetek terhelését külön kell meghatározni az emlősök, a madarak és a halak vonatkozásában, és a legmagasabb értékeket kell alapul venni. A szövetek expozíciós adatait, adott esetben, a tejre és a tojásra vonatkozó expozícióval is ki kell egészíteni. Például, ha az adalékanyagot tejelő emlősök és tojástermelő szárnyasok vonatkozásában alkalmazzák, az egyes, élelmezési célra alkalmas szövetekben mért legmagasabb értékeket a tej- és a tojásfogyasztás esetében tapasztalt értékekhez kell hozzáadni. Ha az adalékanyagot halak, tojástermelő szárnyasok és tejelő emlősök vonatkozásában alkalmazzák, az egyes, élelmezési célra alkalmas szövetekben mért legmagasabb értékeket a tojás- és a tejfogyasztás esetében tapasztaltértékekhez kell hozzáadni. Egyéb kombinációkat kell előirányozni ugyanilyen módon.
Bizonyos esetekben (pl. egyes tápértékkel rendelkező vagy érzékszervi tulajdonságokat javító adalékanyagok esetén vagy kevésbé jelentős fajoknak szánt adalékanyagok esetén) az ember tekintetében később indokolt lehet az expozíció pontosítása reálisabb fogyasztói adatok felhasználásával, megőrizve azonban a leghagyományosabb megközelítést. Ahol lehetséges, a közösségi adatokra kell alapozni a vizsgálatot.
1. táblázat
Elméleti fogyasztói adatok (szövetek vagy termékek grammban)
| Emlősök | Szárnyasok | Halak | Egyéb | |
| Izom | 300 | 300 | 300 (12) | |
| Máj | 100 | 100 | — | |
| Vese | 50 | 10 | — | |
| Zsír | 50 (13) | 90 (14) | — | |
| +Tej | 1 500 | — | — | |
| +Tojás | — | 100 | — | |
| +Méz | 20 |
3.2.3.4. Javaslat a maximális maradékanyag-határértékekre (MRL-ek)
A maximális maradékanyag-határérték azt a maximális (a fogyasztható nedves szövet vagy termék kilogrammjában μg-ban megadott jelölő maradékanyagként kifejezett) maradékanyag-koncentrációt jelenti, amelyet a Közösség az élelmiszerben törvényesen megengedettként vagy elismertként fogadhat el. Ez azon a maradékanyag-típuson és -mennyiségen alapul, amelyet - az ADI-val kifejezve - úgy kell tekinteni, hogy az emberi egészségre semminemű toxikológiai veszélyt nem jelent. Az MRL-t nem lehet az ADI nélkül megállapítani.
A takarmány-adalékanyagok MRL-jeinek megállapításakor figyelembe kell venni az egyéb forrásokból származó maradékanyagokat (pl. a növényi eredetű táplálékot). Továbbá az MRL csökkenthető, annak érdekében, hogy megfeleljen a takarmány-adalékanyagok alkalmazási körülményeinek, és addig a mértékig, ameddig gyakorlati analitikai módszerek állnak rendelkezésre.
Adott esetben egyedi (a természetes szövet vagy termék kilogrammjában mg-ban megadott jelölő maradékanyagként kifejezett) MRL-eket kell meghatározni a cél állatfaj különböző szöveteire vagy termékeire. A különböző szöveteknél és termékeknél megállapított, egyedi MRL-értékeknél figyelembe kell venni a felhasználásra szánt állatfajok szöveteiben/termékeiben lévő maradékanyagok kiürülési kinetikáját. A variabilitást szokásos körülmények között a középérték 95 %-os konfidencia intervallumának alapján kell kiszámolni. Ha a konfidencia-határértéket nem lehet kiszámolni a minták csökkent száma miatt, a variabilitást a legmagasabb egyéni értékkel kell kifejezni.
A kokcidiosztatikumok és a hisztomonosztatikumok maximális maradékanyag-határértékeinek vizsgálatát az állatorvosi készítményekre vonatkozó hatályos szabályok alapján kell elvégezni (8. kötet "A gyógyszerkészítményekre irányadó szabályok az Európai Unióban - Hirdetmény a kérelmezők számára és iránymutatások. Állatgyógyászati termékek. Állatgyógyászati készítmények maximális maradékanyag-határértékeinek (MRL-jeinek) megállapítása az állati eredetű élelmiszerekben". 2005. október).
A kokcidiosztatikumoktól és a histomonosztatikumoktól eltérő kategóriájú adalékanyagokra vonatkozó maximális maradékanyag-határértékek meghatározását célzó vizsgálatokról szükség esetén e melléklet rendelkezik.
A (3.2.3.3. pont alapján kiszámolt) összmaradékanyagnak való fogyasztói expozíció meghatározásakor a különböző szövetekre és termékekre javasolt MRL-ek megállapításához figyelembe kell venni a jelölő maradékanyagnak az összmaradékanyaghoz viszonyított arányát (2. táblázat).
2. táblázat
Az MRL kiszámításánál alkalmazott meghatározások
| i-j | Az egyes szövetek/termékek (máj, vese, izom, bőr+zsír, tej, tojás, méz) különböző időpontokban |
| MRLi-j | Maximális maradékanyag-határérték a szövetekben/termékekben (mg jelölő anyag kg-1) |
| Qti-j | Az 1. táblázatban megállapított napi emberi fogyasztás az egyes szövetek/termékek vonatkozásában (kg), vagy ennek pontosítása |
| TRCi-j | Összmaradékanyag-koncentráció az egyes szövetekben/termékekben (mg kg-1) |
| MRCi-j | Jelölő maradékanyag-koncentráció az egyes szövetekben/termékekben (mg kg-1) |
| RMTRi-j | Az egyes szövetek/termékek MRCi-j-nek a TRCi-j-re vonatkoztatott aránya |
| DITRi-j | Az összmaradékanyagból (mg) kiszámított napi étrendi bevitel az egyes szövetek/termékek vonatkozásában DITRi-j = Qti-j × TRCi-j |
| DITRMRLi-j | Az MRL-értékekből (mg) kiszámított napi étrendi bevitel az egyes szövetek/termékek vonatkozásában DITRMRLi-j = Qti-j × MRLi-j × RMTRi-j -1 |
A mért TRC- és MRC-értéket a 3. táblázatban megadott modellbe kell megfelelően beilleszteni, és a többi értéket ki kell számolni. Amennyiben nem áll rendelkezésre a teljes adatsorozat azért, mert az értékek a kimutatási határ (LOD) alatt vannak, elfogadható az RMTR extrapolálása.
Az MRL-érték kiszámolása csak abban az estben végezhető el, ha az egyéni DITR-értékek összege az ADI-érték alatt van. Ha meghaladja az ADI-értéket, hosszabb kiürülési idejű, vagy alacsonyabb dózisú adatokkal elvégzett másik megoldás alkalmazható. Az MRL-re vonatkozó első javaslatot az MRC-értéket útmutatóul használva és az analitikai módszer LOQ-ját tekintetbe véve lehet megtenni. A javasolt MRL-értékek alapján kapott DITRMRL összegének nem szabad meghaladnia az ADI-értéket és az egyéni DITR-értékek összegéhez közeli értékűnek kell lennie. Ha az érték meghaladja az ADI-t, alacsonyabb MRL-et kell javasolni és meg kell ismételni az összehasonlítást.
Bizonyos adalékanyagok esetén a maradékanyagok mennyisége az MRL-érték alatt lehet a tejben, a tojásban vagy a húsban, amely azonban csökkentheti az élelmiszer minőségét bizonyos élelmiszer-feldolgozási eljárások során. Az ilyen adalékanyagok esetén indokolt ezenkívül az "(élelmiszer-)feldolgozási eljárással kompatibilis maradékanyag-maximum" (MPCR) alkalmazása az MRL-értékek meghatározása mellett.
3. táblázat
Az MRL-javaslat kiszámítására szolgáló modell
| Máj | Vese | Izom | bőr+zsír | Tej | Tojás | Méz | Összeg | |
| TRC (15) (mg kg-1) | — | |||||||
| MRC (16) (mg kg-1) | — | |||||||
| RMTR (16) | — | |||||||
| DITR (17) (mg) | ||||||||
| javasolt MRL (mg kg-1) | — | |||||||
| DITRMRL(mg) |
3.2.3.5. A maradékanyagszintnek az MRL-értékek alá csökkenéséhez szükséges időre vonatkozó javaslat
A maradékanyagszintnek az MRL-értékek alá csökkenéséhez szükséges időn azt az időtartamot értjük, amely az adalékanyag adagolásának a megszüntetését követően ahhoz szükséges, hogy a maradékanyagszintek az MRL-értékek alá csökkenjenek.
3.3. A felhasználókat/dolgozókat érintő, az adalékanyag biztonságos használatára vonatkozó vizsgálatok
A dolgozókat elsősorban a belégzés útján történő, illetve a topikális expozíció veszélyezteti az adalékanyaggal szemben, annak gyártása, kezelése és használata során. Például a mezőgazdasági dolgozók potenciális expozíciója az adalékanyaggal szemben, annak kezelése és bekeverése közben. Az anyagok kezelési módjáról kiegészítő információkat kell adni.
Mellékelni kell a dolgozókra vonatkozó kockázatfelmérést. Az előállító üzemben szerzett tapasztalatok gyakran fontos információforrásként szolgálnak a dolgozókat érintő, a levegőben vagy topikális úton történő, az adalékanyaggal szembeni expozíció tekintetében. Különös jelentőséggel bírnak azok az adalékanyagok/adalékanyaggal kezelt takarmányok és/vagy állati kiválasztott anyagok, amelyek száraz por alakúak vagy azokká válhatnak, és azok a takarmány-adalékanyagok, amelyek allergénpotenciállal rendelkezhetnek.
3.3.1. A felhasználókat/dolgozók biztonságát érintő toxikológiai kockázatfelmérés
A dolgozókat érintő kockázatokat fel kell mérni az adalékanyag azon formáján végzett vizsgálatsorozatban, amelyre a kérelem vonatkozik. Akut inhalációs toxicitás-vizsgálatokat kell végezni, kivéve ha a termék valószínűsíthetően nem képez belélegezhető port vagy permetet. Bőrirritációra vonatkozó vizsgálatokat kell végezni, és ha ezek negatív eredményeket mutatnak, akkor a nyálkahártya-irritációt (pl. szem) fel kell mérni. Az allergizáló és a bőrszenzibilizáló hatásokat is fel kell mérni. A fogyasztók biztonságát célzó toxicitási adatokat (ld. 3.2.2.) kell alkalmazni az adalékanyag potenciális toxicitásának a meghatározására az emberi szervezetre nézve. Mindezt közvetlen méréssel és specifikus vizsgálattal kell megállapítani, amennyiben szükséges.
3.3.1.1. A légutakra gyakorolt hatás
Bizonyítani kell, hogy a levegőben lévő por mennyisége nem veszélyes a dolgozók egészségére. Szükség esetén a vonatkozó adatoknak ki kell terjednie:
- a laboratóriumi állatokon végrehajtott inhalációs vizsgálatokra,
- az irodalomban megjelent epidemiológiai adatokra és/vagy a kérelmező üzeméből származó saját adatokra és/vagy az irritációra, és
- a légúti szenzitizációs vizsgálatokra.
Akut toxicitási vizsgálatra akkor van szükség, ha 50 μm-nél kisebb átmérőjű részecskék vagy cseppek a termék súlyának több mint 1 százalékát teszik ki.
Az eljárásoknak összhangban kell lenniük a 403. OECD-irányelvvel. Ha szubkrónikus toxicitási vizsgálatokra van szükség, azoknak a 412. OECD-irányelvet (ismételt adagolású, inhalációs toxicitás: 28 vagy 14 napos vizsgálat) vagy a 413. irányelvet (szubkrónikus inhalációs toxicitás: 90 napos vizsgálat) kell követniük.
3.3.1.2. A szemre és bőrre gyakorolt hatások
Amennyiben rendelkezésre áll, közvetlenül kell bizonyítani azt, hogy az ismert érintkezési helyzetekben nem lép fel az emberre veszélyes irritáció és/vagy szenzitizációs hatás. Ezt ki kell egészíteni a megfelelő adalékanyag alkalmazásával állatokon végrehajtott, validált bőr- és szemirritációs vizsgálatokból, valamint szenzitizációs potenciálvizsgálatokból nyert adatokkal. Az allergizáló és a bőrszenzibilizáló hatásokat is meg kell határozni. Az eljárásoknak összhangban kell lenniük a 404. (bőrirritáció/korrózió), a 405. (szemirritáció/korrózió), a 406. (bőrszenzitizálás) és a 429. (bőrszenzitizálás - helyi nyirokcsomó-vizsgálat) OECD-irányelvvel.
Ha akár az irodalomban közölt adatok, akár a specifikus in vitro tesztek alapján korróziót okozó tulajdonságot állapítottak meg, nem végezhetők további in vivo tesztek.
A dermális toxicitást szem előtt kell tartani, ha az adalékanyag belélegezve toxikus. Az eljárásoknak összhangban kell lenniük a 402. OECD-irányelvvel (akut dermális toxicitás).
3.3.1.3. Szervezeti toxicitás
A fogyasztók biztonságával kapcsolatos előírások, valamint egyéb követelmények betartása érdekében összegyűjtött, a toxicitásra vonatkozó adatokat (beleértve az ismételt dózistoxicitás-, mutagenitás-, karcinogenitás- és reprodukciós vizsgálatokat és az anyagcsereutat) a szervezeti toxicitás felmérésére kell felhasználni.
3.3.1.4. Expozíciós felmérés
Információt kell szolgáltatni arról, hogy az adalékanyag használata milyen módon eredményezhet expozíciós hatást (akár belégzés útján, akár bőrön keresztül vagy táplálékfelvétel útján). Ennek az információnak lehetőség szerint tartalmaznia kell egy mennyiségi felmérést, amely például a levegőben lévő jellemző koncentrációra, a bőrszennyeződésre vagy a táplálékfelvételre vonatkozik. Amennyiben mennyiségi információk nem állnak rendelkezésre, elegendő információval kell szolgálni a megfelelő expozíciós felméréshez.
3.3.2. Az expozíció ellenőrzésére szolgáló intézkedések
A toxikológiai és expozíciós felmérésekből származó információk felhasználásával következtetést kell levonni a felhasználók/munkavállalók egészségét érintő kockázatokra vonatkozóan (belélegzés, irritáció, szenzitizáció és szervezeti toxicitás). Az expozíció csökkentésére vagy megszüntetésére irányuló óvintézkedéseket lehet javasolni. Bármely maradékanyag-kockázattal szembeni védekezés során azonban a személyi védőfelszerelések alkalmazását csak végső megoldásként célszerű elrendelni, amennyiben az ellenőrző intézkedések már életbe léptek. Célszerűbb például a termék kiszerelésének a megváltoztatását fontolóra venni.
3.4. Az adalékanyag környezeti szempontú alkalmazási biztonságára vonatkozó vizsgálatok
Fontos szem előtt tartani a takarmány-adalékanyagok környezetre gyakorolt hatását, mivel a takarmány-adalékanyagokat rendszerint hosszú ideig adagolják, ez gyakorta az állatok nagy csoportjait érinti, és a hatóanyag(ok) jelentős mértékben anyavegyületként vagy annak metabolitja(i)ként ürül(nek) ki.
Az adalékanyagok környezeti hatásának meghatározásához progresszív eljárást kell alkalmazni. A további vizsgálatot nem igénylő adalékanyagok azonosítása céljából az I. szakasz során minden adalékanyagot azonosítani kell. A többi adalékanyag esetében a vizsgálatok második szakaszának (II. szakasz) elvégzésére van szükség, amely során kiegészítő információ nyerhető arra vonatkozóan, hogy milyen további vizsgálatok elvégzésére lehet szükség. A vizsgálatot a 67/548/EGK irányelv szerint kell végrehajtani.
3.4.1. A kockázatbecslés I. szakasza
A kockázatbecslés I. szakaszának célja annak megállapítása, hogy az adalékanyagnak vagy anyagcseretermékeinek van-e jelentős környezeti hatása, valamint annak eldöntése, hogy szükség van-e a vizsgálatok II. szakaszára (ld. a döntéshozatali folyamatábrát).
Hacsak nincs tudományosan megalapozott ok az ellenkezőjére, a kockázatbecslés II. szakaszát nem kell elvégezni a következő két feltétel egyikének teljesülése esetén:
a) az adalékanyag kémiai jellege és biológiai hatása, valamint felhasználási feltételei alapján megállapítható, hogy a hatás mértéke elhanyagolható, azaz amennyiben az adalékanyag:
- fiziológiai vagy természetes anyag, amely nem növeli meg jelentős mértékben a természetes koncentrációját, vagy
- ha az adalékanyagot nem élelmiszer-termelő állatoknak szánják;
b) a legrosszabb esetre vonatkozó becsült környezeti koncentráció (PEC) túl alacsony ahhoz, hogy aggodalomra adjon okot. A PEC értékét minden érintett környezeti elemre meg kell határozni (ld. alább), feltételezve hogy a táplálékkal bekerült adag 100 %-ban anyavegyületként ürül ki.
Ha a kérelmező nem tudja bizonyítani, hogy az adalékanyag teljesíti e mentességi feltételek valamelyikét, akkor el kell végezni a hatásvizsgálat II. szakaszát.
3.4.1.1. A szárazföldi állatoknak szánt adalékanyagok
Amikor az állattenyésztésből származó trágya a termőföldre kerül, a takarmány-adalékanyagok alkalmazása a talaj, a talajvíz és a felszíni vizek szennyeződéséhez vezethet (a szennyvíz és a vízelvezetés révén).
A talaj tekintetében megállapított legrosszabb esetre vonatkozó PEC (PECtalaj) valószínűleg a maradékanyag fő alkotóelemeinek maximális kiválasztása során keletkező trágyának a mezőgazdasági területre történő kiszórása következtében alakul ki. Ha a PECtalaj (alapértelmezés: 5 cm-es mélység) kevesebb mint 10 μg/kg, nincs szükség további vizsgálatra.
Ha a PEC a talajvíz szennyeződése esetében (PECgw) megbízható felmérés alapján 0,1 μg/liternél alacsonyabbra tehető, akkor az adalékanyag talajvízre vonatkozó környezeti hatásvizsgálatának II. szakaszát nem szükséges elvégezni.
3.4.1.2. A vízi állatoknak szánt adalékanyagok
A takarmány-adalékanyagok használata az akvakultúrákban az üledék és a víz szennyeződéséhez vezethet. A ketrecekben tenyésztett halak esetében a környezeti kockázatbecslésnél az érintett környezeti elem az üledék. A szárazföldi rendszerekben tenyésztett halak esetében a felszíni vizekbe jutó szennyvizet tekintik a legnagyobb veszély okának a környezet vonatkozásában.
Az üledék tekintetében megállapított legrosszabb esetre vonatkozó PEC (PECüledék) valószínűleg a maradékanyag fő alkotóelemeinek maximális kiválasztása során keletkező trágyának az üledékben való lerakódása következtében alakul ki. Ha a PECüledék (alapértelmezés: 20 cm-es mélység) kevesebb mint 10 μg/kg, nincs szükség további vizsgálatra.
Ha a felszíni vizek esetében a PEC (PECfszv) kevesebb mint 0,1 μg/l, nincs szükség további vizsgálatra.
I. szakasz Döntéshozatali folyamatábra
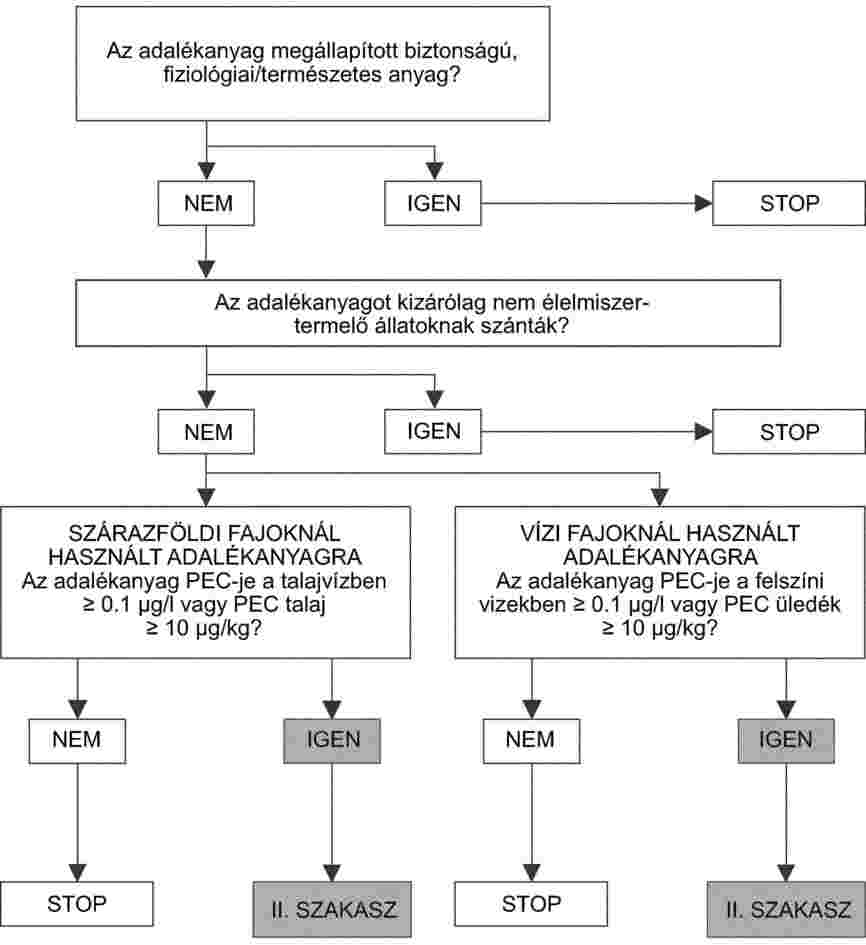
3.4.2. A kockázatbecslés II. szakasza
A II. szakasz célja az, hogy megállapítsa, lehet-e az adalékanyagnak hatása a környezetben a nem célfajokra, beleértve a vízi és a szárazföldi fajokat egyaránt, illetve bejuthat-e az adalékanyag a talajvíz olyan rétegeibe, ami már nem elfogadható. Az adalékanyagnak a célfajba való adagolását követően gyakorlati szempontból nem megfelelő az adalékanyag hatását minden olyan természeti fajra nézve megvizsgálni, amely esetlegesen ki van téve e hatásnak. A megvizsgált rendszertani szintek helyettesítőként vagy jelzőként szolgálnak a természeti fajok tekintetében.
A kockázatbecslés II. szakasza a kockázati hányados megközelítésén alapszik, amelyben a kapott PEC és a becsült hatásmentes koncentráció (PNEC) értékét hasonlítják össze az érintett környezeti elemek vonatkozásában. A PNEC-et kísérletileg megállapított végpontokban meghatározzák és elosztják a megfelelő kockázatbecslési tényezővel. A PNEC-értéket minden érintett környezeti elemre meg kell határozni.
A kockázatbecslés II. szakasza a PEC pontosításával kezdődik és kétlépcsős környezeti kockázatbecslési eljárást alkalmaz.
Az első, II.A. szakasz során az adalékanyag korlátozott számú metabolikus útjának és hatásának vizsgálatát alkalmazva az érintett környezeti elemek expozícióján és az ezekben észlelt hatásokon alapuló, konzervatív kockázatvizsgálat történik. Ha a PEC-nek a PNEC-hez viszonyított aránya egynél (1) kisebb, nincs szükség további kockázatbecslési vizsgálatra, kivéve ha bioakkumuláció várható.
Ha a PEC/PNEC arány nem elfogadható kockázatot jósol (az arány > 1), a környezeti kockázatbecslés pontosítására a kérelmezőnek el kell végeznie a II.B. vizsgálatot.
3.4.2.1. II.A. szakasz
Az I. szakasz során figyelembe vett környezeti elemeken kívül a felszíni vizekre vonatkozó PEC meghatározásakor figyelembe kell venni a lefolyó víztöbbletet és a lecsapolást.
Az egyes érintett környezeti elemek vonatkozásában a PEC pontosítható azokkal az adatokkal, amelyeket nem vettek figyelembe az I. szakasz során. A pontosított PEC ellenőrzésekor a következőkre kell figyelmet fordítani:
a) a hatóanyag(ok)nak és anyagcseretermékei(k)nek a trágyában/halürülékben lévő koncentrációja, azt követően, hogy az adalékanyagot a javasolt dózisszinten beadják az állatoknak. A számításnál figyelembe kell venni a kiválasztott anyagok mennyiségét és a dózisok nagyságát;
b) a hatóanyag(ok)/anyagcseretermékei(k) a trágya rendes felhasználása és tárolása mellett végbemenő potenciális hígulása, a trágyázás előtt, valamint a termőföldeken való alkalmazását megelőző tárolás során;
c) a hatóanyag(ok)nak/anyagcseretermékeinek a talajban történő adszorpciója/deszorpciója, akvakultúra esetében az üledékképződés, lehetőleg a talaj-/üledékvizsgálattal meghatározva (OECD 106);
d) a talaj- vagy vízi üledékrendszerekben történő lebomlás, (OECD 307, illetve 308); és
e) egyéb olyan tényezők, mint például a fotolízis, a hidrolízis, a párolgás, a szántás során végbemenő hígulás.
Az összes érintett környezeti elem esetében elvégzett ilyen számítások során kapott legmagasabb PEC-értékét kell elfogadni és figyelembe venni a kockázatbecslés II. szakaszának céljaira.
Ha a talajban magas szintű tartós megmaradásra (az összetevő eredeti koncentrációjának 90 %-os lebomlása: DT90 > 1 év) lehet számítani, akkor figyelembe kell venni a bioakkumuláció lehetőségét.
Ezt követően, az érintett környezeti elemek különböző táplálkozási szintjeit érintő, súlyos káros hatásokat okozó adalékanyag- (vagy metabolit-) szinteket kell meghatározni. Ezek többnyire akut vizsgálatok, amelyek során az OECD irányelveit vagy más, azokkal egyenértékűnek elismert irányelveket kell követni. A szárazföldi környezet tekintetében előírt vizsgálatok a következők: toxicitás földigilisztára, háromfajta szárazföldi növényre, a talajban lévő mikroorganizmusokra gyakorolt hatások (pl. a nitrogénkötésre gyakorolt hatások esetében). A vízi környezet esetében előírt vizsgálatok a következők: toxicitási vizsgálat halak; Daphnia magna, algák és egy üledéklakó szervezet esetében. Tengeri ketrecek esetében az üledéklakó szervezetek különböző rendszertani kategóriába tartozó három faját kell megvizsgálni.
A PNEC-értéket minden érintett környezeti elemre meg kell határozni. A PNEC rendszerint úgy számítható ki, hogy a fent említett ökotoxicitás-vizsgálatok során megállapított káros hatásra vonatkozó legalacsonyabb értéket, az alkalmazott végponttól és a vizsgálati fajok számától függően, egy legalább 100-as biztonsági tényezővel kell elosztani.
A bioakkumulációs potenciál az n-oktanol/víz megoszlási együtthatója, a Log Kow alapján határozható meg. Ha az érték ≥ 3, ez arra utal, hogy az anyag felhalmozódhat a környezetben. A másodlagos mérgezés veszélyének felbecslésére a II.B. szakaszban esetleges biokoncentrációs faktor (BKF) vizsgálatot kell megfontolni.
3.4.2.2. II. B. szakasz (részletesebb ökotoxikológiai vizsgálatok)
Azon adalékanyagok esetében, amelyeknél a kockázatbecslés II.A. szakaszát követően, azok környezeti veszélyeire vonatkozóan továbbra is kétségek merülnek fel, a biológiai fajokra gyakorolt hatásokra vonatkozó részletesebb vizsgálatok elvégzése szükséges azon környezeti elem(ek) tekintetében, amelyek a II.A. szakaszba tartozó vizsgálatok során lehetséges érintettnek mutatkoznak. Ebben az esetben további vizsgálatokat kell végezni a megfelelő állat-, növény- és mikrobafajokra gyakorolt krónikus és specifikusabb hatások meghatározására. Ez a kiegészítő információ alacsonyabb biztonsági faktor alkalmazását teszi majd lehetővé.
Számos kiadvány, mint pl. az OECD-irányelvek, közzétették a megfelelő kiegészítő ökotoxicitás-vizsgálatokat. E vizsgálatokat körültekintően kell kiválasztani annak érdekében, hogy azok megfeleljenek azon körülményeknek, amelyek mellett az adalékanyag és/vagy annak anyagcseretermékei kibocsátásra kerülhetnek és szétszóródhatnak a környezetben. A szárazföldi elemre gyakorolt hatásvizsgálat (PNECtalaj) a következő krónikus hatásvizsgálatokat tartalmazhatja: a földigilisztákra gyakorolt hatás vizsgálata, további vizsgálatok a talaj mikroflórára gyakorolt hatásra vonatkozóan, több gazdaságilag fontos növényfajon végrehajtott vizsgálat, a mezei gerincteleneken (beleértve a rovarokat), valamint vadon élő madarakon végrehajtott vizsgálatok.
A pontosabb hatásbecslés a víz/üledék vonatkozásában a II.A. szakaszban meghatározott, legérzékenyebb vízi/üledéklakó szervezeteken végzett krónikus toxicitásvizsgálatokra épülhet.
A bioakkumulációs vizsgálati eljárásoknak, amennyiben ezekre szükség van, összhangban kell lenniük a 305. OECD-irányelvvel.
4. IV. SZAKASZ: AZ ADALÉKANYAG HATÉKONYSÁGÁRA VONATKOZÓ VIZSGÁLATOK
A vizsgálatoknak minden javasolt alkalmazás hatékonyságát bizonyítaniuk kell és az 1831/2003/EK rendelet 5. cikke (3) bekezdésében felsorolt jellemzők közül legalább eggyel kell rendelkezniük, az említett rendelet 6. cikke és I. melléklete által meghatározott/előírt adalékanyag-kategóriáknak és -csoportoknak megfelelően. Az ilyen típusú vizsgálatoknak ezen felül lehetővé kell tenniük az adalékanyagnak az EU bevett gazdálkodási gyakorlata szerinti hatékonyságára vonatkozó értékelést is.
Az alkalmazott kísérleti tervet az adalékanyag felhasználására vonatkozóan, valamint az állatfaj és -kategória szerint kell megindokolni. Állatokon végzett vizsgálatok esetében a vizsgálatokat úgy kell elvégezni, hogy az állatok egészségi állapota és a tenyésztési feltételek ne befolyásolják hátrányosan az eredmények értelmezését. Minden kísérlet esetén ismertetni kell a pozitív és a negatív hatást, a technológiait és a biológiait egyaránt. Az állati termékek megkülönböztető jellemzőit hátrányosan befolyásoló hatások hiányát szintén bizonyítani kell. A vizsgálatok ideális esetben meg kell feleljenek egy elismert, külső szerv által auditált minőségbiztosítási rendszer által megállapított kritériumoknak. Minőségbiztosítási rendszer hiányában bizonyítani kell, hogy a vizsgálatot szakképzett személyzet végezte, a megfelelő felszerelés és berendezés alkalmazásával, valamint kinevezett vizsgálatvezető irányításával.
A vizsgálati protokoll gondos összeállítását a vizsgálatvezető végzi, szem előtt tartva az általános leíró adatokat, mint például az alkalmazott módszerek, műszerek és anyagok leírásával, az állat fajának, fajtájának és tenyészetének részletes ismertetésével, számuk megadásával és tartási, takarmányozási körülményeik részletezésével. Minden, állaton végzett vizsgálat esetén a kísérleti körülményeket a 3.1.1.3. pont szerint kell ismertetni. A végső jelentést, az alapadatokat, a vizsgálati terveket, valamint a jól körülírt és jellemzett tesztelőanyagokat referenciaként meg kell őrizni a jövőre vonatkozóan.
Az adalékanyagra vonatkozó legalacsonyabb ajánlott adagolás hatékonyságát igazoló vizsgálatokat az érzékeny paramétereket alkalmazva negatív és tetszés szerinti pozitív kontrollcsoporttal összehasonlítva kell megtervezni. E vizsgálatoknak tartalmazniuk kell a legnagyobb ajánlott adagot is, abban az esetben, amikor ez ajánlott. Nem javasolt egyetlen vizsgálati terv, a vizsgálatok megtervezését és kivitelezését a rugalmas tudományos megítélés alapján kell végezni.
Figyelmet kell fordítani az adalékanyag, egyéb adalékanyagok és/vagy állatgyógyászati készítmények és/vagy az étrend összetevői közötti ismert vagy potenciális, biológiai vagy vegyi kölcsönhatásokra, abban az esetben, amikor ez lényeges a szóban forgó hatóanyag hatékonyságának szempontjából (pl. a mikrobiális eredetű adalékanyag kompatibilitása a kokcidioszatikumokkal és a hisztomonosztatikumokkal, vagy a szerves savakkal).
4.1. In vitro vizsgálatok
Laboratóriumi vizsgálatokkal bizonyítani kell a hatékonyságot a takarmány tulajdonságait befolyásoló minden technológiai és néhány érzékszervi tulajdonságot javító adalékanyag vonatkozásában. A vizsgálatot olyan jellemző anyagok skálájára kell tervezni, amelynél az adalékanyagot alkalmazni fogják. Az eredményt lehetőség szerint paraméter nélküli teszttel kell értékelni és a várható változást P ≤ 0,05 valószínűséggel kell kimutatni.
A hatékonysági eredmények alátámasztására, különösen a gyomor és bélrendszer aspektusait in vitro szimuláló vizsgálatokat egyéb adalékanyagokra is alkalmazni lehet. E vizsgálatoknak statisztikai szempontból értékelhetőeknek kell lenniük.
4.2. Állatokon végzett rövid távú hatékonysági vizsgálatok
Biológiai hasznosulási vizsgálatok révén meghatározható, hogy az új kiszerelésű vagy eredetű tápanyag vagy színezék milyen mértékig képes egy már jóváhagyott vagy elfogadott, vele egyenértékű adalékanyagot helyettesíteni.
Emésztési és anyagcsere-egyensúlyi vizsgálatok végezhetők az állatkísérletek alátámasztására, a hatásmechanizmus bizonyítására. Egyes esetekben, különösen a környezeti előnyök tekintetében, megfelelőbb az egyensúlyi vizsgálatokkal bizonyítani a hatékonyságot, amelyek előnyben részesíthetők a hosszú távú hatékonysági vizsgálatokkal szemben. E kísérletek során a javasolt felhasználási körülményeknek megfelelő számú, valamint fajú és kategóriájú állatot kell alkalmazni.
Indokolt esetben egyéb, állatokon végzett rövid távú hatékonysági vizsgálat javasolható, és ezek helyettesíthetik az állatokon végzett hosszú távú hatékonysági vizsgálatokat, amennyiben ez teljes mértékben indokolt.
4.3. Állatokon végzett hosszú távú hatékonysági vizsgálatok
A vizsgálatokat legalább két különböző helyen kell végezni.
Az alkalmazott kísérleti tervnek figyelembe kell vennie a megfelelő statisztikai erőt, valamint az 1-es és 2-es típusú kockázatot. Az eljárásnak megfelelőképpen érzékenynek kell lennie ahhoz, hogy az adalékanyag bármilyen hatását kimutassa a legalacsonyabb ajánlott dózis alkalmazásánál (1 α típusú kockázat, általánosságban véve P ≤ 0,05, és rágcsálók, kevésbé jelentős fajok, kedvtelésből tartott állatok, valamint nem élelmiszer-termelő állatok esetében P ≤ 0,1) és statisztikai ereje elégséges kell, hogy legyen annak érdekében, hogy a kísérleti eljárás a vizsgálati céloknak megfeleljen. A 2 β típusú kockázat általában alacsonyabb vagy egyenlő 20 %-kal, és 25 % a kérődzők, kevésbé jelentős fajok, kedvtelésből tartott állatok és nem élelmiszer-termelő fajok esetében, ennélfogva az erő (1-β) nagyobb vagy egyenlő 80 %-kal (75 % a kérődzők, kevésbé jelentős fajok, kedvtelésből tartott állatok és nem élelmiszer-termelő fajok esetében).
Elismert tény, hogy egyes adalékanyagok természete miatt nehéz meghatározni az optimális eredményt lehetővé tevő kísérleti körülményt. Ennélfogva a meta-analízis alkalmazásának lehetőségét meg kell fontolni abban az esetben, ha a vizsgálatok száma meghaladja a hármat. Ezért hasonló eljárási tervet kell alkalmazni minden vizsgálatra úgy, hogy az adatok homogenitása alkalomadtán tesztelhető legyen, valamint (amennyiben a tesztek ezt szükségessé teszik) P ≤ 0,05 szintű statisztikai értékelésre összegyűjthető legyen.
4.4. Célállatokon végzett hosszú távú hatékonysági vizsgálatok időtartama
Általában a hatékonysági vizsgálatoknak igazodniuk kell a kérelmezett alkalmazási időszakhoz.
A hatékonysági vizsgálatot az Európai Unió gazdálkodási gyakorlatának megfelelően kell végezni és annak a IV. mellékletben meghatározott minimális időtartamúnak kell lennie.
Ha az adalékanyagot egyedi és rövidebb ideig alkalmazzák, mint amelyet az adott állatkategóriára meghatároztak, ennek a javasolt felhasználási feltételek szerint kell történnie. A megfigyelési időszak ennek ellenére nem lehet 28 napnál rövidebb és a megfelelő végpontokat kell tartalmaznia (pl. a tenyésztési anyakocák esetében az élve született malacok száma a vemhességi időszakban, vagy a malacok száma és súlya az elválasztást követően a szoptatási periódusban).
Egyéb állatfajok vagy -kategóriák esetében, amelyekre a minimális vizsgálati időszakot nem határozták meg a IV. mellékletben, az adagolás időszakát kell figyelembe venni, a javasolt felhasználási körülményeknek megfelelően.
4.5. A takarmány-adalékanyagok kategóriáira és funkcionális csoportjaira vonatkozó hatékonysági követelmények
In vivo vizsgálatnak kell alávetni minden olyan adalékanyagot, amelynek hatása lehet az állatokra.
Az állattenyésztésben alkalmazott adalékanyagok minden kategóriája, valamint a kokcidiosztatikumok és hisztomonosztatikumok esetében a hatékonyságot legalább három hosszú távú hatékonysági vizsgálattal kell bizonyítani. Egyes, állattenyésztésben alkalmazott és egyéb, az állatokra ható adalékanyag esetében azonban elfogadható a rövid távú hatékonysági vizsgálat, ha a hatékonyság egyértelműen bizonyított.
Az állatokra közvetlenül nem ható, egyéb adalékanyag-kategóriák esetében legalább egy, in vitro hatékonysági vizsgálatot kell végezni.
4.6. A nem várt hatású állati termékekre vonatkozó vizsgálatok
Annak bizonyítására, hogy az adalékanyagnak nincs negatív vagy egyéb nemkívánatos hatása az adalékanyaggal táplált állatokból származó élelemnek az érzékszervekre ható vagy táplálkozási (egészségügyi és indokolt esetben, technológiai) jellemzőire, megfelelő mintavételre van szükség a hatékonysági vizsgálatok egyike során. Két csoportot kell megvizsgálni: egy olyat, amelyik nem kapott adalékanyagot; és egy maximális ajánlott dózisú adalékanyaggal kezelt csoportot. Az így kapott adatok alapján végezhető el a statisztikai felmérés. E vizsgálatok elmulasztását megfelelő módon kell igazolni.
5. V. SZAKASZ: FORGALOMBA HOZATALT KÖVETŐ FELÜGYELETI TERV
Egyes adalékanyag-kategóriákra vonatkozóan, az adalékanyagok használatából eredő, bármilyen közvetlen vagy közvetett, azonnali, késleltetett vagy előre nem megjósolható, az ember vagy állatok egészégére, vagy a környezetre gyakorolt hatások kimutatása és azonosítása érdekében, a forgalomba hozatalt követő felügyeletre vonatkozó javaslatot kell benyújtani a 1831/2003/EK rendelet 7. cikke (3) bekezdésének g) pontja értelmében.
A felügyeleti tervet eseti alapon kell részletezni, a felügyeleti tervben megkövetelt különböző feladatokat ellátó személyek (pl. kérelmező, felhasználók) feltüntetésével, akik a felügyeleti terv megfelelő beállításáért és kivitelezéséért felelnek, valamint meg kell határozni, hogy milyen eljárás szerint tájékoztassák az illetékes hatóságokat az adalékanyag használatának biztonságosságával kapcsolatos minden új információról. A Bizottságot és a Hatóságot értesíteni kell minden észlelt negatív hatásról, az 1831/2003/EK rendelet 12. cikkében meghatározott felügyeleti rendelkezések sérelme nélkül.
Abban az esetben, ha a hatóanyag egyben elismert antibiotikum és kimutatták, hogy takarmány-adalékanyagként való alkalmazása során rezisztens baktériumtörzsek képződnek, a forgalomba hozatalt követő felügyeleti terv részeként üzemi vizsgálatokat kell végezni az adalékanyaggal szembeni bakteriális rezisztencia nyomon követésére.
A kokcidiosztatikumok és hisztomonosztatikumok esetében üzemi kísérlet keretében kell vizsgálni az Eimeria spp., illetőleg a Histomonas meleagridis rezisztenciáját, lehetőleg az engedélyezési szakasznak a második részében.
(1) HL L 50., 2004.2.20., 44. o.
(2) HL L 152., 2004.4.30., 1. o.
(3) HL L 196., 1967.8.16., 1. o. Legutóbb a 2006/121/EK európai parlamenti és tanácsi irányelvvel (HL L 396., 2006.12.30., 852. o.) módosított irányelv; helyesbítve: HL L 136., 2007.5.29., 281. o.
(4) HL L 165., 2004.4.30.
(5) HL L 117., 1990.5.8., 1. o. A legutóbb a 2005/174/EK bizottsági határozattal (HL L 59., 2005.3.5., 20. o.) módosított irányelv.
(6) HL L 76., 1991.3.22., 35. o. A legutóbb a 2001/58/EK irányelvvel (HL L 212., 2001.8.7., 24. o.) módosított irányelv.
(7) HL L 262., 2000.10.17., 21. o.
(8) HL L 224., 1990.8.18., 1. o. A legutóbb a 203/2008/EK bizottsági rendelettel (HL L 60., 2008.3.5., 18. o.) módosított rendelet.
(9) M. Thompson et al.: Az analitikai módszerek egységes laboratóriumi hitelesítésére vonatkozó harmonizált iránymutatások (IUPAC Technikai Jelentés) Pure Appl. Chem. (Elméleti és Alkalmazott Kémia), 74. kötet, 5. szám, 835-855. o., 2002.
(10) HL L 221., 2002.8.17., 8. o. A legutóbb a 2004/25/EK határozattal (HL L 6., 2004.1.10., 38. o.) módosított határozat.
(11) A következő címen egy nem teljes lista áll rendelkezésre: www.efsa.europa.eu/en/science/feedap/feedap_opinion/993.html
(12) Izom és bőr természetes arányban.
(13) A disznó esetében 50g zsír és bőr természetes arányban.
(14) Zsír és bőr természetes arányban.
(15) Szem előtt tartva a javasolt időt, amely a maradékanyagszintnek az MRL-értékek alá csökkenéséhez szükséges.
(16) Ideális esetben a TRC-vel azonos időpontban meghatározott.
(17) A TRC-értékek alapján kiszámolt.
III. MELLÉKLET
A 3. CIKK SZERINTI DOKUMENTÁCIÓ ÁLTAL AZ 1831/2003/EK RENDELET 7. CIKKÉNEK (5) BEKEZDÉSÉBEN FOGLALT BIZONYOS ADALÉKANYAG-KATEGÓRIÁK, ILLETVE BIZONYOS KÜLÖNLEGES HELYZETEK TEKINTETÉBEN TELJESÍTENDŐ, KÜLÖNLEGES KÖVETELMÉNYEK
Az 1831/2003/EK rendelet előirányozza az adalékanyagok engedélyezésére vonatkozó dokumentáció elkészítésének szükség szerinti kiegészítő támogatását, az adalékanyagok valamennyi kategóriája vagy egyéb egyedi célok vonatkozásában, az 1831/2003/EK rendelet 7. cikkének (5) bekezdése szerint.
Az alábbiakra vonatkozó dokumentáció elkészítéséhez szükséges különleges követelmények felsorolása:
1. Technológiai adalékanyagok
2. Érzékszervi tulajdonságokat javító adalékanyagok
3. Tápértékkel rendelkező adalékanyagok
4. Az állattenyésztésben alkalmazott adalékanyagok
5. Kokcidiosztatikumok és hisztomonosztatikumok
6. A főbb fajokon végzett vizsgálatok eredményeinek extrapolálása kevésbé jelentős fajokra
7. Kedvtelésből tartott állatok és nem élelmiszer-termelő állatok
8. Élelmiszerekben korábban engedélyezett takarmány-adalékanyagok
9. Engedélyek módosítása
10. Engedélyek megújítása
11. Az 70/524/EGK irányelvben korábban engedélyezett egyes adalékanyagok újraértékelése
A kérelmeket a fent felsoroltak közül egynél több különleges követelménynek megfelelően lehet benyújtani.
Általános feltételek
Az e szakaszokban előírt bármely adatnak a dokumentációból történő elhagyását meg kell indokolni.
1. TECHNOLÓGIAI ADALÉKANYAGOK
1.1. I. szakasz: a dokumentáció összefoglalása
A II. melléklet I. szakaszát teljes mértékben alkalmazni kell.
1.2. II. szakasz: az adalékanyag azonossága, jellemzői és felhasználási feltételei; elemzési módszerek
A II. melléklet II. szakaszát a következőképpen kell alkalmazni:
- az engedélyjogosulthoz nem kötött adalékanyagokra a 2.1.2., 2.1.3., 2.1.4., 2.1.4.2., 2.2., 2.3.1., 2.3.2., 2.4.1., 2.4.2., 2.4.4., 2.5., 2.6. szakaszt kell alkalmazni,
- az engedélyjogosulthoz kötött egyéb adalékanyagokra a teljes II. szakaszt kell alkalmazni.
1.3. III. szakasz: az adalékanyag biztonságosságára vonatkozó vizsgálatok
A II. melléklet 3.1., 3.2. és 3.4. alpontját nem kell alkalmazni a szilázs-adalékanyagokra, ha kimutatható, hogy:
- a végleges takarmányban semmilyen hatóanyag(ok) vagy a vonatkozó anyagcseretermékek vagy aktív reagens(ek) nem található(k), vagy
- a hatóanyagok vagy reagensek a szilázs természetes összetevőiként fordulnak elő, és az adalékanyag alkalmazása nem növeli számottevően a koncentrációjukat az adalékanyag felhasználása nélkül készült szilázshoz viszonyítva (vagyis abban az esetben, amikor nincs számottevő expozícióbeli változás).
Egyéb esetekben a II. melléklet 3. szakaszát teljes mértékben alkalmazni kell.
1.3.1. Az adalékanyag biztonságos használatára vonatkozó vizsgálatok a célállatokra vonatkozóan
A xenobiotikumokra (1) a II. melléklet teljes 3.1 alpontját alkalmazni kell
1.3.1.1. A célfajokra vonatkozó toleranciavizsgálatok
A szilázs-adalékanyagok tekintetében:
- a terméket az alapétrendhez kell adagolni és az eredményeket az ugyanazon étrendű negatív kontrollhoz kell viszonyítani. Az alapétrend az adalékanyag felhasználása nélkül készült egyetlen szilázsforrást tartalmazhat.
- a toleranciavizsgálathoz kiválasztott dózis a silózott anyagban megszokott körülmények között jelen levő koncentráció többszöröse kell, hogy legyen, amennyiben ez kétséget kizáróan megállapítható. Különleges figyelmet kell fordítani az élő mikroorganizmusokat tartalmazó termékekre, valamint a mikroorganizmusok túlélési és szaporodási képességére a silózás folyamán.
A toleranciavizsgálatokat többnyire a kérődzőfajokra, általában a tejelő tehenekre lehet korlátozni. Egyéb fajokon végzett vizsgálatokra csak abban az esetben van szükség, ha a silózott takarmány jellege jobban megfelel a nem kérődzőknél való használatra.
Egyéb anyagok:
Az egyéb olyan anyagok tekintetében, amelyeknek technológiai adalékanyagként való használata engedélyhez kötött és amelyeket takarmányban való alkalmazásra még nem engedélyeztek, bizonyítani kell azt, hogy ezek a legmagasabb javasolt dózisszint mellett nem ártalmasak az állatokra nézve. E bizonyítás korlátozódhat egyetlen, a legérzékenyebb fajok egyikén, vagy egyetlen laboratóriumi állatfajon elvégzett kísérletre.
1.3.1.2. Mikrobiális vizsgálatok
A II. melléklet 3.1.2. alpontját teljes mértékben alkalmazni kell.
1.3.2. Az adalékanyag biztonságos használatára vonatkozó vizsgálatok a fogyasztók tekintetében
1.3.2.1. Az anyagcserével és a maradékanyaggal kapcsolatos vizsgálatok
Nincs szükség az anyagcserével és a maradékanyaggal kapcsolatos vizsgálatokra, ha:
1. a hatóanyag vagy ennek anyagcseretermékei nincsenek jelen a takarmányban a takarmányozás időpontjában; vagy
2. a kiválasztott anyag változatlanul jelenik meg, vagy az anyagcseretermékei bizonyítható módon alapvetően nem szívódnak fel; vagy
3. a hatóanyag fiziológiás összetevők formájában szívódik fel; vagy
4. az adalékanyag aktív összetevő(i) kizárólag mikroorganizmusok vagy enzimek).
Akkor sincs szükség anyagcsere-vizsgálatra, ha a hatóanyag természetes körülmények között nagy mennyiségben van jelen az élelmiszerben vagy a takarmányban, vagy a testnedvek és szövetek természetes alkotóeleme. Ezekben az esetekben azonban maradékanyag-vizsgálatot kell végezni, amely a nem kezelt csoport és a legmagasabb javasolt dózisszint mellett kezelt csoport szöveteiben és termékeiben levő maradékanyagszint összehasonlítására korlátozódhat.
1.3.2.2. Toxikológiai vizsgálatok
Nincs szükség toxikológiai vizsgálatokra, ha:
1. a hatóanyag vagy ennek anyagcseretermékei nincsenek jelen a takarmányban a takarmányozás időpontjában; vagy
2. a hatóanyag fiziológiás összetevők formájában szívódik fel; vagy
3. a termék a silózott takarmányban általában megtalálható vagy az élelmiszer előállítása során már alkalmazott mikroorganizmusokból áll; vagy
4. a termék mikroorganizmusokból származó, nagy tisztasági fokú enzimekből áll, amelyek biztonságos felhasználhatósági dokumentációval rendelkeznek.
A fentebb nem kizárt mikroorganizmusokon és enzimeken genotoxicitási (beleértve a mutagenitási) és szubkrónikus orális toxicitási vizsgálatra van szükség. A genotoxicitás-vizsgálatokat nem szabad élő sejtek jelenlétében végezni.
A fentebb nem kizárt xenobiotikumok tekintetében a II. melléklet 3.2.2. alpontját teljes mértékben alkalmazni kell.
Egyéb anyagok esetében az esetenkénti megközelítést kell alkalmazni, szem előtt tartva az expozíció fokát és módját.
1.3.2.3. A fogyasztói biztonság felmérése
A II. melléklet 3.2.3. alpontját teljes mértékben alkalmazni kell az élelmiszer-termelő állatok tekintetében.
1.3.3. A felhasználókat/dolgozókat érintő, az adalékanyag biztonságos használatára vonatkozó vizsgálatok
A II. melléklet 3.3. alpontját teljes mértékben alkalmazni kell. Az enzimet és mikroorganizmust tartalmazó adalékanyag feltételezetten légúti szenzitizáló hatású, kivéve ha meggyőző bizonyíték van az ellenkezőjére.
1.3.4. Az adalékanyag használatának biztonságára vonatkozó vizsgálatok a környezet tekintetében
A II. melléklet 3.4. alpontját teljes mértékben alkalmazni kell. A szilázs-adalékanyagok tekintetében figyelembe kell venni az adalékanyag hatását a silózás során a kazalsilóból vagy silóból származó szennyvíz termelődésére.
1.4. IV. szakasz: az adalékanyag hatékonyságára vonatkozó vizsgálatok
A technológiai adalékanyag a takarmányok jellemzőinek javítására vagy stabilizálására szolgál, de általában nincs közvetlen biológiai hatása az állattenyésztésre. Az elfogadhatónak minősített módszerekben leírt megfelelő kritériumok segítségével, a tervezett felhasználási körülmények között és megfelelő kontrolltakarmányokkal összehasonlítva kell bizonyítani az adalékanyag hatékonyságát.
A hatékonyságot, kivéve a radionuklidokkal való szennyeződést korlátozó anyagokét, in vitro vizsgálatokkal kell megállapítani. Az egyes funkcionális csoportok megfelelő végpontjait a következő táblázat tartalmazza.
Az egyes technológiai adalékanyagokra vonatkozó végpontok
| SZÖVEG HIÁNYZIK |
Szilázs-adalékanyagok
A silózási folyamat során kívánt hatás kimutatása céljából (2) független vizsgálatokat kell végezni. A vizsgálatokat az alábbi kategóriák (amelyekben valamennyi takarmány, vagy pontosabban meg nem határozott takarmányok szerepelnek) mindegyikének egy irányadó esetén kell elvégezni:
- könnyen silózható takarmány: > 3 % oldható szénhidrát a friss alapanyagban (pl. teljes őrlésű kukorica, perje, rozsnok vagy cukorrépapép),
- mérsékelten nehezen silózható takarmány: 1,5 - 3,0 % oldható szénhidrát a friss alapanyagban (pl. réti perje, nád vagy fonnyasztott lucerna),
- nehezen silózható takarmány: < 1,5 % oldható szénhidrát a friss alapanyagban (pl. csomós ebír, vagy hüvelyes növények).
Ahol a szárazanyagra (a továbbiakban: SzA) vonatkoztatott takarmány-alkategóriákra korlátozódik az elvárás, a szárazanyag-választékot világosan meg kell jelölni. A vizsgálatokat a takarmány választékát képviselő alapanyagon kell végezni, és ahol lehetséges, különböző botanikai mintákat kell használni.
A különleges takarmányok vizsgálatához specifikus tesztekre van szükség.
A vizsgálat időtartamának általában 90 naposnak vagy hosszabbnak kell lennie, állandó hőmérsékleten mérve (ajánlott hőmérsékleti intervallum: 15 - 25 oC). A rövidebb vizsgálati periódust indokolni kell.
Rendszerint a következő, negatív kontrollal összehasonlított paraméterekre vonatkozó mérési adatokat kell megadni:
- szárazanyag és kiszámolt szárazanyag-veszteség (illékony anyagokhoz igazítva),
- pH-csökkenés,
- az illékony zsírsavak koncentrációja (pl. ecetsav, vajsav és propánsav), valamint a tejsav,
- az alkoholok koncentrációja (etanol),
- ammóniakoncentráció (g/kg össznitrogén), és
- vízben oldódó szénhidrátok tartalma.
Indokolt esetben, kiegészítésként egyéb, a specifikus kérelmet igazoló mikrobiológiai és kémiai paramétereket kell figyelembe venni (pl. a laktózt asszimiláló élesztők, a Clostridiumok, a Listeriak és a biogén aminok száma).
Az elérni kívánt hatást a szennyvíz csökkentésének vonatkozásában a teljes vizsgálati időszakban termelődött szennyvíz térfogatához viszonyítva kell megítélni, szem előtt tartva a környezetre gyakorolt valószínű hatását (pl. a szennyvízre vonatkozó ökotoxicitás, vagy a biológiai oxigén igénye). A szennyvíz termelődésének a csökkenését közvetlen módon kell bizonyítani. A siló kapacitásának elegendően nagynak kell lennie ahhoz, hogy a szennyvíz nyomás hatására távozzon belőle. A vizsgálat időtartama rendszerint 50 nap kell legyen. Amennyiben ettől eltérő ideig tartó vizsgálatot alkalmaznak, ennek okát igazolni kell.
A növelt aerob stabilitást negatív kontroll ellenében kell kimutatni. A levegő hatásának kitett takarmány tekintetében a stabilitási vizsgálatoknak legalább hétnaposaknak kell lenniük és az adalékanyag stabilitása legalább két nappal hosszabb kell legyen a nem kezelt kontrollhoz viszonyítva. Ajánlott, hogy a vizsgálatot 20 oC-os környező hőmérsékleten végezzék, és a háttérhőmérsékletnél 3 oC-kal vagy annál nagyobb mértékben tapasztal hőmérsékletemelkedést instabilitási mutatónak tekintsék. A hőmérsékletmérés a CO2-termelődés mérésével helyettesíthető.
1.5. V. SZAKASZ: forgalomba hozatalt követő felügyeleti terv
E szakaszt az 1831/2003/EK rendelet 7. cikke (3) bekezdése g) pontjának előírása szerint kell alkalmazni. Azaz forgalomba hozatalt követő felügyeleti tervre csak géntechnológiával módosított szervezeteknek minősülő, vagy géntechnológiával módosított szervezetekből előállított adalékanyagok esetén van szükség.
2. ÉRZÉKSZERVI TULAJDONSÁGOKAT JAVÍTÓ ADALÉKANYAGOK
2.1. Színezékek
2.1.1. I. szakasz: a dokumentáció összefoglalása
A II. melléklet I. szakaszát teljes mértékben alkalmazni kell.
2.1.2. II. szakasz: az adalékanyag azonossága, jellemzői és felhasználási feltételei; elemzési módszerek
A II. melléklet II. szakaszát a következőképpen kell alkalmazni:
- az engedélyjogosulthoz nem kötött adalékanyagokra a 2.1.2., 2.1.3., 2.1.4., 2.1.4.2., 2.2., 2.3.1., 2.3.2., 2.4.1., 2.4.2., 2.4.4., 2.5. és 2.6. bekezdést kell alkalmazni,
- az engedélyjogosulthoz kötött egyéb adalékanyagokra a teljes II. szakaszt kell alkalmazni.
2.1.3. III. szakasz: az adalékanyag használatának biztonságára vonatkozó vizsgálatok
A II. melléklet 3.3. alpontját teljes mértékben alkalmazni kell valamennyi adalékanyag tekintetében.
1) Azon anyagok esetében, amelyekkel etetve az állatokat, az állati eredetű élelmiszer színesebb lesz, a II. melléklet III. szakaszának 3.1., 3.2. és 3.4. alpontja teljes mértékben alkalmazandó.
2) Olyan anyagok esetében, amelyek a takarmány színének javítására vagy helyreállítására szolgálnak, a III. szakasz 3.1. alpontjában leírt vizsgálatokat kell elvégezni olyan állatokon, amelyek az ajánlott dózisban kapták az adalékanyagot. A bizonyítás a meglévő tudományos irodalomra való hivatkozás révén is elvégezhető. A II. melléklet III. szakaszának 3.2. és 3.4. alpontját kell alkalmazni.
3) A díszhalak és -madarak színét kedvezően befolyásoló anyagok esetében, a III. szakasz 3.1. alpontjában leírt vizsgálatokat kell elvégezni olyan állatokon, amelyeknél az ajánlott dózisban alkalmazták az adalékanyagot. A bizonyítás a meglévő tudományos irodalomra való hivatkozás révén is elvégezhető. A 3.2. és a 3.4. alpontot azonban nem kell alkalmazni.
2.1.4. IV. szakasz: az adalékanyag hatékonyságára vonatkozó vizsgálatok
A II. melléklet IV. szakaszát teljes mértékben alkalmazni kell.
a) Az állati eredetű élelmiszert megszínező anyagok esetében:
az adalékanyaggal a javasolt alkalmazási feltételek mellett kezelt állatokból nyert termékek színváltozásának mérése a megfelelő módszertan alkalmazásával történik. Ki kell mutatni, hogy az adalékanyag alkalmazása nem befolyásolja kedvezőtlenül a termék stabilitását vagy az élelmiszer érzékszervi és élelmezési minőségét. Elvileg, ha az adott anyagnak az állati termékek összetételére és jellemzőire gyakorolt hatása jól dokumentált, akkor a hatékonyság egyéb vizsgálatokkal (pl. biológiai hasznosulási vizsgálatokkal) megfelelőképpen bizonyítható.
b) A takarmányt megszínező vagy annak színét helyreállító anyagok esetében:
a hatékonyságot a célzott alkalmazási körülményeket tükröző, kontroll takarmánnyal való összehasonlítást alkalmazó, megfelelő laboratóriumi vizsgálatokkal kell bizonyítani.
c) A díszhalak és -madarak színét kedvezően befolyásoló anyagok esetében:
a hatás(okat) bizonyító vizsgálatokat olyan állatokon kell végezni, amelyek az adalékanyagot az ajánlott adagban kapták. A színváltozást a megfelelő módszertani eljárással kell mérni. A hatékonyság egyéb kísérletes vizsgálat (pl. biológiai hasznosulás) révén, vagy a tudományos irodalomra való hivatkozás révén is bizonyítható.
2.1.5. V. szakasz: forgalomba hozatalt követő felügyeleti terv
E szakasz az 1831/2003/EK rendelet 7. cikke (3) bekezdése g) pontjának az előírása szerint alkalmazandó. Azaz, forgalomba hozatalt követő felügyeleti tervre csak géntechnológiával módosított szervezetnek minősülő, vagy géntechnológiával módosított szervezetekből előállított adalékanyagok esetén van szükség.
2.2. Aromaanyagok
2.2.1. I. szakasz: a dokumentáció összefoglalása
A II. melléklet I. szakaszát teljes mértékben alkalmazni kell.
2.2.2. II. szakasz: az adalékanyag azonossága, jellemzői és felhasználási feltételei; elemzési módszerek
Általában a "természetes termékek" csoportja esetében, a teljes növények, állatok és egyéb szervezetek, ezek részei vagy az ezekből nagyon kis mértékű feldolgozással, így zúzással, őrléssel vagy szárítással nyert termékek (pl. számos gyógynövény és fűszer), nem minősülnek az érzékszervi tulajdonságokat javító adalékanyag-kategória aromák nevet viselő funkcionális csoportjába tartozónak.
Az e termékek engedélyezése iránti kérelem értékelése céljából az aromaanyagokat a következőképpen osztályozzák:
1. Természetes termékek:
1.1. Növénytani szempontból meghatározott természetes termékek.
1.2. Nem növényi eredetű természetes termékek.
2. Természetes vagy azzal megegyező, szintetikus, vegyi szempontból meghatározott aromaanyagok
3. Mesterséges anyagok.
A kérelem tárgyát képező termék vonatkozó csoportját meg kell jelölni. Abban az esetben, ha a termék nem illeszkedik egyik említett csoportba sem, ezt meg kell említeni és indokolni kell.
2.2.2.1. Hatóanyag(ok)/reagens(ek) jellemzése
A II. melléklet 2.2. alpontját teljes mértékben alkalmazni kell.
Továbbá:
Minden aromaanyag-csoport esetében, a kifejezetten a takarmányban és az élelmiszerben levő aromaanyag azonosítására szolgáló, vonatkozó azonosítószám(okat) [mint például FLAVIS (3), Európa Tanács (4), JECFA (élelmiszer-adalékanyagokkal foglalkozó közös FAO-WHO szakértői bizottság), CAS (5) vagy bármilyen nemzetközileg elfogadott számozási rendszer], ha rendelkezésre állnak, mindig meg kell adni.
1. Növénytani szempontból meghatározott természetes termékek.
A növénytani szempontból meghatározott, természetes termékek jellemzésének tartalmaznia kell az eredeti növény tudományos nevét, növénytani osztályozását (család, nemzetség, faj, adott esetben az alfaj és a változat nevét), valamint a közönséges neveket és a rokon értelmű szavakat a lehetséges legtöbb európai nyelven, vagy egyéb nyelv(eken) (mint pl. a termesztési vagy az eredethelyen alkalmazott nyelv(ek)), ahol ez(ek) rendelkezésre állnak. A növény felhasznált részei (levél, virág, magvak, gyümölcs, gumó) és a kevésbé ismert növényeknél a termőhely, a növény-meghatározási kritériumok és e növények egyéb vonatkozó jellemzői. Meg kell határozni a kivonat fő összetevőit és azok mennyiségét és az értéktartományát vagy variabilitását. Különleges figyelmet kell szentelni a szennyeződéseknek, a II. szakasz 2.1.4. alpontjában leírtaknak megfelelően. Jelenteni kell az emberre vagy az állatokra nézve azon toxikológiai jelentőségű anyagoknak a koncentrációját (6), amelyek a kivonat alapjául szolgáló növényben előfordulhatnak.
Az aromaanyag forrásául szolgáló növény, ennek részei vagy az ebből származó termékek farmakológiai vagy vonatkozó jellemzőit teljes körű vizsgálatnak kell alávetni, és jelenteni kell.
2. Nem növényi eredetű természetes termékek.
A fentiekkel megegyező megközelítés alkalmazható.
3. Természetes vagy azzal megegyező, vegyileg meghatározott aromaanyagok
A II. melléklet 2.2.1.1. alpontjának általános követelményein kívül meg kell jelölni az aromaanyag eredetét.
2.2.2.2. Termelési és gyártási módszerek
A II. melléklet 2.3. alpontját teljes mértékben alkalmazni kell.
A vegyileg nem jól meghatározott természetes termékek, általában kivonással nyert, nagyszámú alkotórészből álló összetett keverékek esetén mellékelni kell a kivonási eljárás részletes leírását. A leírásban ajánlott a vonatkozó terminológia használata, mint pl. illóolaj, abszolút, tinktúra, kivonat és hasonló kifejezések (7), amelyeket széleskörűen alkalmaznak a növénytanilag meghatározott aromaanyagok extrahálási eljárásának a leírására. Az extrahálás során használt oldószereket meg kell nevezni, valamint le kell írni azokat az óvintézkedéseket, amellyel elkerülték, hogy az aromaanyagban oldószer maradjon a kivonás után és meg kell adni a toxikológiai szempontból fontos maradékanyagszinteket, ha a maradékanyag jelenléte nem elkerülhető. A kivonás jellemzésére alkalmazott kifejezések tartalmazhatják a kivonási módszerre való hivatkozást.
2.2.2.3. Elemzési módszerek
1. A természetes termékek esetén (akár növénytanilag meghatározott vagy nem növényi eredetű), amelyek nem tartalmaznak az emberre vagy az állatokra toxikológiai szempontból veszélyes anyagokat, a II. melléklet 2.6. alpontjának szabványelőírásai a termék főbb vagy tipikus összetevőinek elemzésére alkalmas, egyszerűbb minőségi analitikai módszerekkel helyettesíthetők.
2. A természetes vagy az ezeknek megfelelő szintetikus, kémiailag meghatározott aromák esetén, amelyek az emberre vagy az állatokra nézve nem minősülnek toxikológiai jelentőségű anyagnak, a II. melléklet 2.6. alpontjának szabványelőírásai egyszerűbb minőségi analitikai módszerekkel helyettesíthetők.
A II. melléklet teljes 2.6. alpontját alkalmazni kell minden egyéb aromaanyagra, mint például azok a természetes kivonatok, amelyek toxikológiai jelentőségű anyagokat tartalmaznak, továbbá az olyan, természetes vagy ezeknek megfelelő szintetikus, kémiailag meghatározott aromák, amelyek maguk is toxikológiai jelentőségű anyagok, valamint a mesterséges aromaanyagok.
2.2.3. III. szakasz: az adalékanyag biztonságosságára vonatkozó vizsgálatok
Minden aromaanyag esetén meg kell adni az állatok expozícióját és a felvett anyag mennyiségét, a természetes expozícióból adódóan és az aromaanyagnak a takarmányhoz való adagolását követően egyaránt.
A mesterséges anyagok csoportjába tartozó aromaanyagok esetében a II. melléklet III. alpontját teljes mértékben alkalmazni kell.
2.2.3.1. Az adalékanyag biztonságos használatára vonatkozó vizsgálatok a célállatok tekintetében
1. Természetes termékek (növénytanilag meghatározott vagy nem növényi eredetűek)
E termékek biztonságossága megállapítható a termék főbb és jellemző összetevői alapján, valamint toxikológiai szempontból potenciális veszélyt jelentő anyagokat szem előtt tartva. Ha a főbb és jellemző összetevőket vegyileg meghatározott aromaanyagként vagy adalékanyagként még nem engedélyezték, ellenőrizni kell, hogy toxikológiai szempontból jelentenek-e potenciális veszélyt az emberekre vagy az állatokra nézve, és az összetevők toxikológiai tulajdonságait a II. melléklet 3.1. alpontjával összhangban kell benyújtani.
2. Természetes vagy azzal megegyező, vegyileg meghatározott aromaanyagok
Ha ezek a vegyületek emberi alkalmazásra engedélyezett aromaanyagok, a célfajok biztonságossága meghatározható a kérelmező által javasolt takarmánynak a célfajba történő beviteli szintje és az emberek által az élelmiszerből származó aromaanyagok beviteli szintjének összehasonlításával. Az emberek tekintetében a biztonságosság meghatározására alkalmazott anyagcsere- és toxikológiai adatokat be kell nyújtani.
Minden egyéb esetben, amely különbözik attól az esettől, amikor mindkét beviteli szint megegyező, mint például, amikor a kérelmező által a célállat tekintetében javasolt beviteli szint jóval magasabb az emberi táplálékkal bevittnél vagy abban az esetben, amikor az anyag jelenléte nem engedélyezett az élelmiszerben, a célállatok szempontjából a biztonságosság a következő adatok figyelembevételével határozható meg: toxikológiai küszöbérték alapelve (8), a rokon összetevők rendelkezésre álló toxikológiai és anyagcsere-adatai, és a hasonló vegyi szerkezetű anyagok esetében riadó megfontolása (a 2232/96/EK európai parlamenti és tanácsi rendelet (9) értelmében egy értékelési program elfogadásához szükséges intézkedések megállapításáról szóló, 2000. július 18-i 1565/2000/EK bizottsági rendelet analógiájára).
A toleranciavizsgálatokra csak küszöbértéket meghaladó esetben van szükség, vagy abban az esetben, ha ez nem határozható meg.
2.2.3.2. Az adalékanyag biztonságos használatára vonatkozó vizsgálatok a fogyasztók tekintetében
Bizonyítani kell, hogy az aromaanyag anyagcseretermékei nem halmozódnak fel olyan állati eredetű termékben, amely potenciális toxikológiai veszélyt jelenthet az emberekre nézve. Abban az esetben, ha a kérelem tárgyát képező aromaanyag használata a takarmányhoz való adagolása eredményeként az állati eredetű élelmiszerben maradékanyag képződésében nyilvánul meg, a fogyasztókra vonatkozó részletes expozíciós számítást kell benyújtani.
a) Az anyagcserével és a maradékanyaggal kapcsolatos vizsgálatok
1. Természetes termékek (növénytanilag meghatározott vagy nem növényi eredetűek)
E termékeknek a takarmányban aromaanyagként való alkalmazásából eredő biztonságossága az emberekre nézve az aromaanyag metabolizmusát illetően, a (célállatban végbemenő) metabolizmusnak és az aromaanyag főbb és jellemző alkotórészei maradékanyagainak, valamint a potenciális toxikológiai veszélyt jelentő anyagoknak a kivonatbeli hiányán alapszik.
Ha a fő és a jellemző összetevőket vegyileg meghatározott aromaanyagként még nem engedélyezték, vagy az aromaanyag beviteli szintje a célállat tekintetében jóval magasabb az emberi táplálékkal bevittnél, a II. melléklet 3.2.1. alpontját teljes mértékben alkalmazni kell.
2. Természetes vagy azzal megegyező, vegyileg meghatározott aromaanyagok
Ha e termékeket aromaanyagként nem engedélyezték az emberek vonatkozásában, vagy ha az aromaanyag beviteli szintje a célállat tekintetében jóval magasabb az emberi táplálékkal bevittnél, be kell nyújtani a metabolikus útra vonatkozó, rendelkezésre álló adatokat, amelyek alapján az élelmezési célra alkalmas szövetekben vagy termékekben való potenciális felhalmozódás felbecsülhető a II. melléklet 3.2.1 alpontjának megfelelően.
b) Toxikológiai vizsgálatok
1. Természetes termékek (növénytanilag meghatározott vagy nem növényi eredetűek)
E termékeknek a takarmányban aroma-adalékanyagként való alkalmazásából eredő biztonságossága az emberek vonatkozásában a fő és a jellemző alkotórészek toxikológiai vizsgálati adatain, valamint a potenciális toxikológiai veszélyt jelentő anyagoknak a kivonatbeli hiányán alapszik.
Teljes körű toxikológiai vizsgálat szükséges abban az esetben, ha a fő vagy jellemző összetevőkön végzett anyagcsere-vizsgálatok kimutatják, hogy az aromaanyag felhalmozódott az állati szövetekben vagy termékekben és a potenciális toxikológiai veszélyt jelentő érték a célállatok vonatkozásában a küszöbértéket meghaladja. A teljes körű toxikológiai vizsgálat magában foglalja a genotoxicitási, beleértve a mutagenitási és szubkrónikus orális toxicitásvizsgálatot, a II. melléklet 3.2.2. alpontjának megfelelően.
2. Természetes vagy azzal megegyező, vegyileg meghatározott aromaanyagok
A II. melléklet 3.2.2. alpontjának megfelelő genotoxicitási, beleértve a mutagenitási és a szubkrónikus orális toxicitásvizsgálatot magában foglaló, teljes körű toxikológiai vizsgálat szükséges abban az esetben, ha az ezeken az anyagokon végzett anyagcsere-vizsgálatok kimutatják, hogy anyagfelhalmozódás történt az állati szövetekben vagy termékekben és a potenciális toxikológiai veszélyt jelentő érték a célállatok vonatkozásában a küszöbértéket meghaladja.
2.2.3.3. A felhasználókat és dolgozókat érintő, az adalékanyag biztonságos használatára vonatkozó vizsgálatok
A II. melléklet 3.3. alpontját teljes mértékben alkalmazni kell.
2.2.3.4. Az adalékanyag használatának biztonságára vonatkozó vizsgálatok a környezet tekintetében
A II. melléklet 3.4. alpontját teljes mértékben alkalmazni kell.
2.2.4. IV. szakasz: az adalékanyag hatékonyságára vonatkozó vizsgálatok
A bizonyítékot az ízesítő tulajdonságokra vonatkozóan, általában a közzétett irodalom alapján kell benyújtani. Ez gyakorlati tapasztalattal is bizonyítható, amikor rendelkezésre áll, ellenkező esetben megkövetelhető az állatokon végzett vizsgálat.
Teljeskörűen meg kell vizsgálni és jelenteni kell, ha a kérelem tárgyát képező termék egyéb szerepet is betölt a takarmányban, az állatban vagy az állati eredetű élelmiszerben, mint amely az 1831/2003/EK rendelet I. mellékletében szereplő aromaanyag-összetevők meghatározásában szerepel.
2.2.5. V. szakasz: forgalomba hozatalt követő felügyeleti terv
E szakasz az 1831/2003/EK rendelet 7. cikke (3) bekezdése g) pontjában előírtaknak megfelelően alkalmazandó. Azaz forgalomba hozatalt követő felügyeleti tervre csak olyan adalékanyagok esetén van szükség, amelyek géntechnológiával módosított szervezetek, vagy géntechnológiával módosított szervezetekből származnak.
3. TÁPÉRTÉKKEL RENDELKEZŐ ADALÉKANYAGOK
3.1. I. szakasz: a dokumentáció összefoglalása
A II. melléklet I. szakaszát teljes mértékben alkalmazni kell.
3.2. II. szakasz: az adalékanyag azonossága, jellemzői és felhasználási feltételei; elemzési módszerek
A II. melléklet II. szakaszát a következőképpen kell alkalmazni:
- a konkrét engedélyjogosulthoz nem kötött adalékanyagokra a 2.1.2, 2.1.3, 2.1.4, 2.1.4.2, 2.2, 2.3.1, 2.3.2, 2.4.1, 2.4.2, 2.4.4, 2.5, 2.6 bekezdést kell alkalmazni,
- a konkrét engedélyjogosulthoz kötött egyéb adalékanyagokra a teljes II. szakaszt kell alkalmazni.
3.3. III. szakasz: az adalékanyagok biztonságosságára vonatkozó vizsgálatok
3.3.1. Az adalékanyag használatának biztonságára vonatkozó vizsgálatok a célfajok tekintetében
3.3.1.1. A célfajokra vonatkozó toleranciavizsgálatok
1. Nem szükséges a vizsgálat a 82/471/EGK irányelvben engedélyezett karbamid, az aminosavak és azok sói, valamint ezek analóg vegyületeinek tekintetében, valamint nyomelemek és vitaminok összetevői, provitaminok és hasonló hatású, vegyileg jól meghatározott anyagok esetében, amelyek nem rendelkeznek akkumulatív képességgel és amelyeket a 70/524/EGK irányelv takarmány-adalékanyagként már jóváhagyott.
2. Azon adalékanyagok esetében, amelyek a "vitaminok, provitaminok és hasonló hatású, kémiailag egyértelműen azonosítható anyagok" csoportba tartoznak és akkumulálódhatnak, a toleranciát csak azon összetevők vonatkozásában kell kimutatni, amelyek hatékonyak lehetnek vagy bizonyítottan különböznek az egyértelműen azonosított vitamin(ok)tól. Egyes esetekben a tolerenciavizsgálat lépései (tervezés vagy kritérium) a hatékonysági vizsgálatok egyikével kombinálhatók.
3. A toleranciát bizonyítani kell a karbamid-származékok, az aminosav-analógok és előzetesen nem engedélyezett nyomelem-összetevők vonatkozásában. A fermentációs termékek esetében a toleranciát bizonyítani kell, kivéve ha a hatóanyag a nyers fermentációs terméktől elkülönített és nagy tisztasági fokú, vagy a termelő szervezet hosszú időn át tartó biztonságos használatát bizonyították, valamint ismert a biológiai háttere, kizárva a toxikus anyagcseretermékek esetleges képződését.
4. Ahol a kérelem minden állatfajt és kategóriát érint, elegendő egyetlen, a legérzékenyebb fajon (vagy akár a megfelelő laboratóriumi állaton) és a legújabb ismeretek alapján végzett toleranciavizsgálat.
3.3.1.2. Mikrobiális vizsgálatok
A II. melléklet 3.1.2. alpontját teljes mértékben alkalmazni kell.
3.3.2. Az adalékanyag biztonságos használatára vonatkozó vizsgálatok a fogyasztók tekintetében
3.3.2.1. Az anyagcserével és a maradékanyaggal kapcsolatos vizsgálatok
Rendszerint nincs szükség anyagcsere-vizsgálatokra. A karbamid-származékok esetében a hatékonysági vizsgálatok során a kérődzők anyagcseréjét kell vizsgálni.
Maradékanyag- vagy lerakódási vizsgálatokat csak a "vitaminok, provitaminok és hasonló hatású, kémiailag egyértelműen azonosítható anyagok" csoportjába tartozó adalékanyagokon kell végezni, amelyek akkumulálódhatnak a szervezetben, valamint a nyomelem-összetevők funkcionális csoportjai esetében, amelyeknél megnőtt a biológiai hasznosulás. Ebben az esetben a II. melléklet 3.2.1. alpontjában leírt eljárást nem kell alkalmazni. A követelmény a kérelmezett anyag legnagyobb adagjával kezelt csoportnak és egy pozitív kontrollnak a szövetekben vagy a termékben meghatározott szintjére korlátozódik.
3.3.2.2. Toxikológiai vizsgálatok
E vizsgálatok a fermentációs termékekre és a még nem engedélyezett adalékanyagokra vonatkoznak. A fermentációs termékek esetében genotoxicitási és szubkrónikus toxicitásvizsgálatokat kell benyújtani, kivéve ha:
1. a hatóanyagot elkülönítik a nyers fermentációs terméktől és nagymértékben megtisztítják; vagy
2. a termelő szervezet hosszú időn át tartó biztonságos használatát bizonyították, valamint elegendően ismert a biológiai háttere, a toxikus anyagcseretermékek esetleges képződésének kizárására.
Ahol a termelő szervezet olyan csoport tagja, amelyben egyes törzsek toxintermelő képességükről ismertek, azokat kifejezetten ki kell zárni.
3.3.2.3. A fogyasztói biztonság felmérése
A II. melléklet 3.2.3. alpontját teljes mértékben alkalmazni kell.
3.3.3. A felhasználókat és dolgozókat érintő, az adalékanyag biztonságos használatára vonatkozó vizsgálatok
A II. melléklet 3.3. alpontját teljes mértékben alkalmazni kell.
3.3.4. Az adalékanyag használatának biztonságára vonatkozó vizsgálatok a környezet tekintetében
A II. melléklet 3.4. alpontját teljes mértékben alkalmazni kell a nyomelemek összetevői közé tartozó új hatóanyagok tekintetében.
3.4. IV. szakasz: az adalékanyag hatékonyságára vonatkozó vizsgálatok
Nincs szükség hatékonysági vizsgálatra a karbamid, az aminosavak sói és ezek takarmány-adalékanyagként jóváhagyott analóg vegyületei esetében, valamint a takarmány-adalékanyagként jóváhagyott nyomelem-összetevők, és a vitaminok, a provitaminok és ezekkel hasonló hatású, takarmány-adalékanyagként jóváhagyott, vegyileg jól meghatározott anyagok esetében.
A hatékonyság bizonyítása céljából rövid távú hatékonysági vizsgálatra van szükség a karbamid, az aminosavak sói és ezek takarmány-adalékanyagként még nem jóváhagyott analóg vegyületeinek tekintetében, valamint a takarmány-adalékanyagként még nem jóváhagyott nyomelem-összetevők, valamint a vitaminok, a provitaminok és ezekhez hasonló hatású, takarmány-adalékanyagként még nem jóváhagyott, vegyileg jól meghatározott anyagok esetében.
Egyéb anyagoknál, amelyek esetében a tápanyagra gyakorolt hatásra van szükség, legalább egy hosszú távú hatékonysági vizsgálatot kell alkalmazni a II. melléklet 4. szakasza értelmében.
Amennyiben szükséges, vizsgálatokkal kell igazolni, hogy az adalékanyag eleget tesz az állat tápanyagigényének. A vizsgálatoknak magukban kell foglalniuk az állat igényei alatt levő koncentrációjú táplálékot tartalmazó étrenddel táplált tesztcsoportot. Mindazonáltal kerülni kell a súlyos deficienciás kontrollcsoportot alkalmazó vizsgálatok alkalmazását. Általában elegendő a hatékonyságot egyetlen állatfajon vagy kategórián, beleértve a laboratóriumi állatokat, bemutatni.
3.5. V. szakasz: forgalomba hozatalt követő felügyeleti terv
E szakaszt az 1831/2003/EK rendelet 7. cikke (3) bekezdésének g) pontja alapján kell alkalmazni.
4. AZ ÁLLATTENYÉSZTÉSBEN ALKALMAZOTT ADALÉKANYAGOK
4.1. Enzimektől és mikroorganizmusoktól eltérő, állattenyésztésben alkalmazott adalékanyagok
4.1.1. I. szakasz: a dokumentáció összefoglalása.
A II. melléklet I. szakaszát teljes mértékben alkalmazni kell.
4.1.2. II. szakasz: az adalékanyag azonossága, jellemzői és felhasználási feltételei; elemzési módszerek
A II. melléklet II. szakaszát teljes mértékben alkalmazni kell.
4.1.3. III. szakasz: az adalékanyagok biztonságosságára vonatkozó vizsgálatok
4.1.3.1. Az adalékanyag biztonságos használatára vonatkozó vizsgálatok a célállatok tekintetében
A II. melléklet 3.1. alpontját teljes mértékben alkalmazni kell.
4.1.3.2. Az adalékanyag biztonságos használatára vonatkozó vizsgálatok a fogyasztók tekintetében
1. Az anyagcserével és a maradékanyaggal kapcsolatos vizsgálatok
Nincs szükség e vizsgálatokra, ha:
- az anyag vagy anyagcseretermékei bizonyíthatóan változatlan alakban választódnak ki, vagy bizonyíthatóan eleve nem szívódnak fel, vagy
- az anyag fiziológiás összetevő(k) formájában és az összetevő(k) fiziológiás szintjén szívódik fel.
Nincs szükség anyagcsere-vizsgálatra, ha az anyag természetes körülmények között nagy mennyiségben van jelen az élelmiszerben vagy a takarmányban, vagy ha a testnedvek és szövetek természetes alkotóeleme. Ezekben az esetekben azonban maradékanyag-vizsgálatot kell végezni, amely a nem kezelt csoport és a legmagasabb ajánlott dózissal kezelt csoport szöveteiben vagy termékeiben található maradékanyagszintek összehasonlítására korlátozódhat.
Egyéb esetekben a II. melléklet 3.2.1. szakaszát teljes mértékben alkalmazni kell.
2. Toxikológiai vizsgálatok
Nincs szükség toxikológiai vizsgálatra, ha a hatóanyag fiziológiás összetevő(k) alakjában szívódik fel.
A xenobiotikumok esetében a II. melléklet 3.2.2. alpontját teljes mértékben alkalmazni kell.
Egyéb anyagok esetében az esetenkénti megközelítést kell alkalmazni, szem előtt tartva az expozíció mértékét és módját és az e szakaszokban előírt bármely adatnak a dokumentációból való elhagyását meg kell indokolni.
3. A fogyasztói biztonság felmérése
A II. melléklet 3.2.3. alpontját teljes mértékben alkalmazni kell az élelmiszer-termelő állatok tekintetében.
4.1.3.3. A felhasználókat és dolgozókat érintő, az adalékanyag biztonságos használatára vonatkozó vizsgálatok
A II. melléklet 3.3. alpontját teljes mértékben alkalmazni kell.
4.1.3.4. Az adalékanyag használatának biztonságára vonatkozó vizsgálatok a környezet tekintetében
A II. melléklet 3.4. alpontját teljes mértékben alkalmazni kell.
4.1.4. IV. szakasz: az adalékanyag hatékonyságára vonatkozó vizsgálatok
A II. melléklet IV. szakaszát teljes mértékben alkalmazni kell.
1. Az állatok tenyésztését, teljesítményét vagy jólétét kedvezően befolyásoló adalékanyagok és az "egyéb állattenyésztési adalékanyagok" funkcionális csoport esetében.
A hatást a célfajonkénti vagy kategóriánkénti összefüggésben lehet csak kimutatni. Az adalékanyag jellemzőitől függően az eredményes intézkedések a teljesítményjellemzők (pl. a takarmány hatékonysága, a napi átlagos testsúlygyarapodás, a fokozott mértékű állattenyésztés), a hasított test összetétele, a csorda teljesítménye, a szaporodási paraméterek vagy az állatjóllét. A hatás módját rövid távú hatékonysági vizsgálatokkal vagy a megfelelő végpontot mérő laboratóriumi vizsgálatokkal bizonyítani kell.
2. Az állattenyésztés környezeti következményeit kedvezően befolyásoló adalékanyagok
E környezetet kedvezően befolyásoló adalékanyagok tekintetében (pl. csökkent nitrogén vagy foszforkiürülés vagy csökkent metánképződés, ízetlenség), a célfajok vonatkozásában három, az állatokra nézve kiemelkedően hasznos hatású, rövid távú vizsgálattal bizonyítható a hatékonyság. A vizsgálatok során figyelembe kell venni az adalékanyagra adott esetleges adaptív választ.
4.1.5. V. szakasz: forgalomba hozatalt követő felügyeleti terv
E szakaszt az 1831/2003/EK rendelet 7. cikke (3) bekezdésének g) pontja alapján kell alkalmazni.
4.2. Az állattenyésztésben alkalmazott adalékanyagok: enzimek és mikroorganizmusok
4.2.1. I. szakasz: a dokumentáció összefoglalása
A II. melléklet I. szakaszát teljes mértékben alkalmazni kell.
4.2.2. II. szakasz: az adalékanyag azonossága, jellemzői és felhasználási feltételei; elemzési módszerek
A II. melléklet II. szakaszát teljes mértékben alkalmazni kell.
4.2.3. III. szakasz: az adalékanyagok biztonságosságára vonatkozó vizsgálatok
4.2.3.1. Az adalékanyag biztonságos használatára vonatkozó vizsgálatok a célállatok tekintetében
A II. melléklet 3.1.1. alpontját teljes mértékben alkalmazni kell.
A kérelmezőket arra ösztönzik, hogy ahol csak lehetséges, legalább 100-szoros túladagolást alkalmazzanak a vizsgálati csoportnál és következetesen csökkentsék az előírt végpontok számát. Erre a célra az adalékanyag koncentrált formája alkalmazható. A koncentrációt a jelenlevő hordozó mennyiségének csökkentésével kell beállítani, azonban a hatóanyag(ok)/vegyület(ek) arányának az egyéb fermentációs termékben azonosnak kell lennie a végső termékbeli aránnyal. Enzimek esetében az étrendnek tartalmaznia kell a megfelelő szubsztrátum(okat).
A II. melléklet 3.1.2. alpontját teljes mértékben alkalmazni kell minden mikroorganizmus, valamint olyan enzimek tekintetében, amelyek közvetlen katalitikus hatással vannak a bélflóra alkotóelemeire, vagy amelyek egyéb módon befolyásolják a bélflórát.
Ahol új expozíció vagy az expozíció mértékében alapvető növekedés tapasztalható a mikroorganizmusok vonatkozásában, a kommenzalista bélflóra káros hatásának hiányát bizonyítandó, kiegészítő vizsgálatokat kell végezni. A kérődzőknél a bélflóra-mikroorganizmusok közvetlen megszámlálására csak az (in vitro illékony zsírsav-koncentrációbeli változásként, a propionát koncentráció csökkenésként vagy csökkent cellulolízis kedvezőtlen változásaként mért) kérődzőműködés kedvezőtlen változása esetén van szükség.
4.2.3.2. Az adalékanyag biztonságos használatára vonatkozó vizsgálatok a fogyasztók tekintetében
1. Nincs szükség az anyagcserével és a maradékanyaggal kapcsolatos vizsgálatokra.
2. Toxikológiai vizsgálatok a II. melléklet 3.2.2. alpontjának megfelelően.
Az enzimek és a mikroorganizmusok a teljes adalékanyagnak csak egy részét teszik ki, amely a legtöbb esetben egyéb, a fermentálási folyamatból származó egyéb összetevőket is tartalmazhat. Következésképpen vizsgálat keretében kell megbizonyosodni arról, hogy az adalékanyag nem tartalmaz mutagén vagy egyéb anyagot, amely ártalmas lehet a takarmánnyal táplált vagy ezen az adalékanyaggal kezelt vizet fogyasztó állatokból származó termékek fogyasztóira.
Az emlősök (beleértve az embert) közvetlen vagy közvetett táplálékbeviteli célját szolgáló legtöbb élő baktériumot azonban olyan élő szervezetek csoportjából válogatják ki, amelynek hosszú időn át tartó biztonságos használatát bizonyították vagy olyan csoportból, amelyben a toxicitási kockázat jól meghatározott. Hasonlóképpen, az enzimek előállítására állandó jelleggel alkalmazott mikroorganizmusokhoz kapcsolódó kockázat jól ismert és jelentősen csökkent a modern termelési eljárásoknak köszönhetően. Ennélfogva a mikrobiális eredetű enzimek és a hosszú időn át tartó, bizonyítottan biztonságosan használt mikroorganizmusok esetében és ahol a fermentálás folyamata pontosan körülírt és jól ismert, a toxicitási (pl. orális toxicitási, illetve genotoxicitási) vizsgálatokra nincs szükség. Az élő, valamint az enzimtermelésre alkalmazott szervezetek vonatkozásában azonban egyaránt a II. melléklet 2.2.2.2. szakaszát kell alkalmazni.
Amikor új enzimtermelő szervezetről vagy annak alkalmazásáról van szó és a (termelő) szervezet biológiai szempontból nem elégségesen ismert ahhoz, hogy a toxikus anyagcseretermékek esetleges képződését kizárják, élő mikroorganizmusokat és enzimeket tartalmazó adalékanyaggal végzett genotoxicitási és orális toxicitási vizsgálatokat kell bevezetni. Ebben az esetben a genotoxicitási, beleértve a mutagenitási és szubkrónikus orális toxicitási, vizsgálatok szerint kell eljárni. Ajánlott e vizsgálatokat sejtmentes fermentációs táptalajjal, vagy szilárd fázisú fermentáció esetén a megfelelő kivonattal végezni.
4.2.3.3. A felhasználókat és dolgozókat érintő, az adalékanyag biztonságos használatára vonatkozó vizsgálatok
A II. melléklet 3.3. alpontját teljes mértékben alkalmazni kell, kivéve:
- az olyan enzimeket és mikroorganizmusokat, amelyek feltételezetten légúti szenzitizáló hatásúak, kivéve ha rendelkezésre áll ezzel ellentétes, meggyőző bizonyíték. Következésképpen semmilyen közvetlen vizsgálat nem szükséges.
- a termék kiszerelése (pl. mikrokapszula) miatt néhány vagy minden vizsgálat szükségtelenné válhat. Ebben az esetben a megfelelő bizonyítékokat kell benyújtani.
4.2.3.4. Az adalékanyag használatának biztonságára vonatkozó vizsgálatok a környezet tekintetében
A II. melléklet 3.4. alpontját teljes mértékben alkalmazni kell minden, nem bél eredetű mikroorganizmus tekintetében vagy ha ezek nem fordulnak mindenhol elő a környezetben.
4.2.4. IV. szakasz: az adalékanyag hatékonyságára vonatkozó vizsgálatok
A II. melléklet IV. szakaszát teljes mértékben alkalmazni kell.
1. Az állatok tenyésztését, teljesítményét vagy jólétét kedvezően befolyásoló adalékanyagok és az "egyéb állattenyésztési adalékanyagok" funkcionális csoport esetében.
A hatást a célfajonkénti vagy kategóriánkénti összefüggésben lehet csak kimutatni. Az adalékanyag jellemzőitől függően az intézkedések eredményessége a teljesítményjellemzők (takarmány hatékonysága, napi átlagos testsúlygyarapodás, fokozott mértékű állattenyésztés), a hasított test összetétele, a csorda teljesítménye, a szaporodási paraméterek vagy az állatjóllét függvénye. A hatásmódot rövid távú hatékonysági vizsgálatokkal, vagy a megfelelő végpontot mérő laboratóriumi vizsgálatokkal bizonyítani kell.
2. Az állattenyésztés környezeti következményeit kedvezően befolyásoló adalékanyagok
E környezetet kedvezően befolyásoló adalékanyagok tekintetében (pl. csökkent nitrogén vagy foszforkiürülés vagy csökkent metánképződés, ízetlenség), a célfajok vonatkozásában három, az állatokra nézve kiemelkedően hasznos hatású, rövid távú vizsgálattal bizonyítható a hatékonyság. A vizsgálatok során figyelembe kell venni az adalékanyagra adott esetleges adaptív választ.
4.2.5. V. szakasz: forgalomba hozatalt követő felügyeleti terv
E szakaszt az 1831/2003/EK rendelet 7. cikke (3) bekezdésének g) pontja alapján kell alkalmazni.
5. KOKCIDIOSZTATIKUMOK ÉS HISZTOMONOSZTATIKUMOK
5.1. I. szakasz: a dokumentáció összefoglalása
A II. melléklet I. szakaszát teljes mértékben alkalmazni kell.
5.2. II. szakasz: az adalékanyag azonossága, jellemzői és felhasználási feltételei; elemzési módszerek
A II. melléklet II. szakaszát teljes mértékben alkalmazni kell.
5.3. III. szakasz: az adalékanyagok biztonságosságára vonatkozó vizsgálatok
5.3.1. Az adalékanyag biztonságos használatára vonatkozó vizsgálatok a célállatok tekintetében
A II. melléklet 3.1. alpontját teljes mértékben alkalmazni kell.
5.3.2. Az adalékanyag biztonságos használatára vonatkozó vizsgálatok a fogyasztók tekintetében
A II. melléklet 3.2. alpontját teljes mértékben alkalmazni kell.
5.3.3. A felhasználókat és dolgozókat érintő, az adalékanyag biztonságos használatára vonatkozó vizsgálatok
A II. melléklet 3.3. alpontját teljes mértékben alkalmazni kell.
5.3.4. Az adalékanyag biztonságos használatára vonatkozó vizsgálatok a környezet tekintetében
A II. melléklet 3.4. alpontját teljes mértékben alkalmazni kell.
5.4. IV. szakasz: az adalékanyag hatékonyságára vonatkozó vizsgálatok
Ezen adalékanyagok megvédik az állatot az Eimeria spp., illetőleg a Histomonas meleagridis támadás következményétől. Fontos az adalékanyag egyedi (pl. fajkontrollált), valamint profilaktikus (mint pl. a megbetegedés és a halálozási arány, az oocisztaszám és a károsodási mérték csökkenése) hatásainak bizonyítása. Adott esetben a hatékonysági vizsgálatok során olyan adatokat is fel kell jegyezni, amelyek lehetővé teszik a növekedéssel és a takarmányátalakítással (hízó- és tojóbaromfi, valamint nyúl) kapcsolatos zavaró hatások, valamint a tojások termékenységére és keltetésére (tenyészbaromfi) gyakorolt hatások értékelését.
Az előírt hatékonysági adatok megállapításához a célállatokon egy három szakaszból álló kísérletet kell végezni:
- mesterséges izolált és kevert fertőzések
- felhasználási körülményeket szimuláló természetes és mesterséges fertőzések
- tényleges felhasználási körülmények az üzemi kísérletekben
A mesterséges izolált és kevert fertőzéses (pl. tojóbaromfi ketreces) vizsgálatok a paraziták elleni relatív hatékonyság bizonyítását célozzák és nem szükséges megismételni őket. A szimulált felhasználási körülmények (pl. baromfi kifutós kísérletek, nyúlketreces kísérletek) vizsgálatához három jellemző eredményre van szükség. Három üzemi kísérletet kell elvégezni, a természetes fertőzés egyik válfaja mellett.
5.5. V. szakasz: forgalomba hozatalt követő felügyeleti terv
A II. melléklet e szakaszát az 1831/2003/EK rendelet 7. cikke (3) bekezdésének g) pontja alapján kell alkalmazni.
6. A FŐBB FAJOKON VÉGZETT VIZSGÁLATOK EREDMÉNYEINEK EXTRAPOLÁLÁSA A KEVÉSBÉ JELENTŐS FAJOKRA
A kevésbé jelentős fajokat e rendelet 1. cikkének (2) bekezdése határozza meg.
Olyan faj esetén, amely fiziológiai tulajdonságait tekintve rokonságot mutat egy olyan fajjal, amelynél az adalékanyag alkalmazását már engedélyezték, az engedélyezett felhasználástól való indokolt eltérés tekintetében rendszerint egy kevésbé részletes beadványt is elfogadnak.
Az alábbi követelményeket kizárólag a főbb fajokra már engedélyezett adalékanyagnak a kevésbé jelentős fajokra vonatkozó engedélyezési kérelmére kell alkalmazni. Új adalékanyag engedélyezési kérelmére kizárólag a kevésbé jelentős fajok tekintetében minden szakaszt teljes mértékben kell alkalmazni, az adalékanyag kategóriájától és funkcionális csoportjától függően (lásd a III. melléklet vonatkozó különleges követelményeit).
6.1. I. szakasz: a dokumentáció összefoglalása
A II. melléklet I. szakaszát teljes mértékben alkalmazni kell.
6.2. II. szakasz: az adalékanyag azonossága, jellemzői és felhasználási feltételei; elemzési módszerek
A II. melléklet II. szakaszát a következőképpen kell alkalmazni:
- a konkrét engedélyjogosulthoz kötött adalékanyagokra a II. szakaszt teljes mértékben kell alkalmazni
- az egyéb adalékanyagokra a 2.1.2, 2.1.3, 2.1.4, 2.1.4.2, 2.2, 2.3.1, 2.3.2, 2.4.1, 2.4.2, 2.4.4, 2.5, 2.6 bekezdést kell alkalmazni.
6.3. III. szakasz: az adalékanyag használatának biztonságára vonatkozó vizsgálatok
6.3.1. Az adalékanyag biztonságos használatára vonatkozó vizsgálatok a célállatok tekintetében
6.3.1.1. A célfajokra vonatkozó toleranciavizsgálatok
Az egyes takarmány-adalékanyagok kategóriáira és funkcionális csoportjaira vonatkozó követelményeket kell alkalmazni.
Kevésbé jelentős fajok tekintetében akkor nem szükséges a tolerancia vizsgálata, ha az adalékanyag vizsgálata során a vonatkozó, fiziológiai szempontból hasonló jelentősebb fajok tekintetében a biztonsági határ szélesnek (legalább tízes faktorúnak) bizonyult.
Amennyiben három jelentősebb fajban (beleértve az együregű gyomrú és a kérődző állatokat) a biztonsági határ hasonlónak és szélesnek bizonyult, nincs szükség további toleranciavizsgálatra a fiziológiai szempontból nem hasonló, kevésbé jelentős fajok (pl. lovak vagy nyulak) esetében. Ha szükség van a toleranciavizsgálatra, a kevésbé jelentős fajok (kivéve a nyulak) tekintetében a vizsgálat időtartamának a növekedésben levő állatoknál legalább 28, a felnőtt állatok esetében legalább 42 naposnak kell lennie. Nyulak esetében a következő vizsgálati időtartamokat kell alkalmazni: hízónyúl: 28 nap; nőstény tenyésznyúl esetében: egy ciklus (az első megtermékenyítéstől a második elválasztási időszak végéig). Szopósnyúl és elválasztott nyúl esetében (születés után egy héttel kezdődően) 49 napos időszakot elegendőnek tekintik a toleranciavizsgálat céljára és tartalmaznia kell az eredményeket az elválasztásig a tenyésznyulakra vonatkozóan. A (lazacféléktől eltérő) halak esetében 90 napos vizsgálati időszak szükséges.
6.3.2. Az adalékanyag biztonságos használatára vonatkozó vizsgálatok a fogyasztók tekintetében
6.3.2.1. Anyagcsere-vizsgálatok
Az egyes takarmány-adalékanyagok kategóriáira és funkcionális csoportjaira vonatkozó követelményeket kell alkalmazni.
Nincs szükség anyagcsere-vizsgálatra akkor, ha az adalékanyagnak a használatát már engedélyezték egy, az engedélyeztetni kívánt, kevésbé jelentős fajhoz fiziológiás szempontból hasonló fajra. Fiziológiás hasonlóság hiányában az in vitro vizsgálatra épülő metabolittartalom elemzések (pl. jelölt anyag segítségével májsejtekben végzett vizsgálatok) összehasonlítása elfogadható az anyagcserebeli hasonlóság megállapításához.
A kevésbé jelentős fajokban meg kell határozni az adalékanyag anyagcsereútját akkor, ha a kevésbé jelentős faj fiziológiás szempontból nem hasonló a jelentős fajhoz.
6.3.2.2. Maradékanyagok vizsgálata
A élelmezési célra alkalmas szövetekben és termékekben levő jelölő-maradékanyag mennyiségi meghatározására csak abban az esetben van szükség, ha az anyagcsereút hasonló vagy meghatározott. Valamennyi egyéb esetben a II. melléklet 3.2.1.2. szakaszát teljes mértékben alkalmazni kell.
6.3.2.3. A fogyasztói biztonság felmérése
Javaslat a maximális maradékanyag-határértékekre (MRL-ek)
Az MRL-értékek meghatározhatók feltételezve azt, hogy a kevésbé jelentős fajok élelmezési célra alkalmas szöveteiben semmilyen jelentős különbség nem tapasztalható a maradékanyag tekintetében a jelentős fajokhoz viszonyítva.
Az MRL-értékek állatcsoportokon belül a következőképpen vetíthetők ki:
- a főbb növekedésben levő kérődzőkét az összes növekedésben levő kérődzőkére,
- a tejelő tehenek tejéről az egyéb tejelő kérődzők tejére,
- disznókról valamennyi együregű gyomrú emlősre, kivéve a lovakat,
- csirkékről vagy pulykákról az egyéb baromfira,
- a tojótyúkokról az egyéb tojószárnyasokra, és
- a lazacfélékről az egyéb halakra.
A lovakra vonatkozóan az MRL-értékek kivetíthetőek akkor, ha egy főbb kérődzőfaj és egy főbb együregű gyomrú emlősfaj MRL-értékei rendelkezésre állnak.
Ha a különböző anyagcsere-kapacitást és szöveti összetételt képviselő jelentősebb fajokban, mint amilyenek a marhafélék (vagy kecskefélék), a disznó és a csirke (vagy a szárnyas) esetében számolt MRL-ek azonos értékűek, azonos MRL-érték rendelhető a juhfélékhez, a lófélékhez és a nyulakhoz, amely azt jelenti, hogy a extrapolálás minden élelmiszer-termelő állatra elvégezhető, kivéve a halfélékre. Tekintettel az Állatgyógyászati Termékek Bizottságának a lazacfélék és egyéb közönséges halfélék MRL-értékeinek meghatározásáról szóló iránymutatására (10) amely már lehetővé teszi az izom MRL-értékeinek extrapolálását a lazacfélék és egyéb közönséges halfélék MRL-értékeire, és ha a kiinduló anyag jelölőanyagként elfogadható az izom és a bőr MRL-értékeinek vonatkozásában, ebből valamennyi élelmiszer-termelő állat MRL-értékeire lehet következtetni.
Valamennyi élelmiszer-termelő állat esetében a élelmezési célra alkalmas szövetekre és termékekre vonatkozóan elemzési módszereknek kell rendelkezésre állniuk.
6.3.3. A felhasználókat és dolgozókat érintő, az adalékanyag biztonságos használatára vonatkozó vizsgálatok
A II. melléklet 3.3. alpontját teljes mértékben alkalmazni kell.
6.3.4. Az adalékanyag használatának biztonságára vonatkozó vizsgálatok a környezet tekintetében
A fiziológiai szempontból összehasonlítható főbb fajok környezeti kockázatbecsléséből következtetni lehet a környezeti kockázatbecslésre. A nyulaknál alkalmazandó adalékanyagokra a teljes szakaszt alkalmazni kell, szem előtt tartva az adalékanyagok egyes kategóriáira és funkcionális csoportjaira vonatkozó követelményeket.
6.4. IV. szakasz: az adalékanyag hatékonyságára vonatkozó vizsgálatok
Abban az esetben, amikor az adalékanyagot már engedélyezték egy fiziológiai szempontból ugyanazon funkció alapján összehasonlított fajra és ahol az adalékanyag hatásmódja ismert vagy bizonyított, az ezzel megegyező hatásmód bizonyítéka a hatékonyság alátámasztására szolgálhat a kevésbé jelentős fajok esetében. Ahol e kapcsolat nem bizonyítható, a hatékonyság bizonyításában a II. melléklet IV. szakasza általános szabályainak megfelelően kell eljárni. Egyes esetekben alkalmas lehet az azonos termelési fázisban levő állatok (pl. tejtermelésre használt kecske- és juhfélék) kombinált vizsgálata. A szignifikanciát minden vizsgálatban meg kell határozni (P ≤ 0,1) , vagy ahol lehetséges, metaelemzéssel (P ≤ 0,05).
Ha bizonyítani kell a hatékonyságot, a hatékonysági vizsgálatok időtartamának meg kell felelnie a fiziológiai szempontból hasonló főbb fajok összehasonlítható termelési szakaszainak. Egyéb esetekben, a minimális vizsgálati periódusra a II. melléklet 4.4. alpontja és a IV. melléklet vonatkozó előírása érvényes.
6.5. V. szakasz: forgalomba hozatalt követő felügyeleti terv
A II. melléklet e szakaszát az 1831/2003/EK rendelet 7. cikke (3) bekezdése g) pontjának rendelkezései alapján kell alkalmazni.
7. KEDVTELÉSBŐL TARTOTT ÁLLATOK ÉS NEM ÉLELMISZER-TERMELŐ ÁLLATOK
A kedvtelésből tartott és egyén nem élelmiszer-termelő állatokat e rendelet 1. cikkének (1) bekezdése határozza meg.
7.1. I. szakasz: a dokumentáció összefoglalása
A II. melléklet I. szakaszát teljes mértékben alkalmazni kell.
7.2. II. szakasz: az adalékanyag azonossága, jellemzői és felhasználási feltételei; elemzési módszerek
A II. melléklet II. szakaszát a következőképpen kell alkalmazni:
- a konkrét engedélyjogosulthoz kötött adalékanyagokra a II. szakaszt teljes mértékben kell alkalmazni
- egyéb adalékanyagokra a 2.1.2, 2.1.3, 2.1.4, 2.1.4.2, 2.2, 2.3.1, 2.3.2, 2.4.1, 2.4.2, 2.4.4, 2.5, 2.6. pontot kell alkalmazni.
7.3. III. szakasz: az adalékanyag biztonságosságára vonatkozó vizsgálatok
7.3.1. Az adalékanyag biztonságos használatára vonatkozó vizsgálatok a célállatok tekintetében
Az egyes takarmány-adalékanyagok kategóriáira és funkcionális csoportjaira vonatkozó követelményeket kell alkalmazni. Ahol a tolerancia vizsgálatára van szükség, ennek időtartama legalább 28 nap kell, hogy legyen.
Ha három jelentősebb fajban (beleértve az együregű gyomrú és a kérődző állatokat) a biztonsági határ hasonlónak és szélesnek bizonyult, nincs szükség a tolerancia vizsgálatára.
7.3.2. Az adalékanyag biztonságos használatára vonatkozó vizsgálatok a fogyasztók tekintetében
Ezt az alpontot általában nem kérik. Figyelembe kell venni a tulajdonos biztonságát.
7.3.3. A felhasználókat és dolgozókat érintő, az adalékanyag biztonságos használatára vonatkozó vizsgálatok
A II. melléklet 3.3. alpontját teljes mértékben alkalmazni kell.
7.3.4. Az adalékanyag használatának biztonságára vonatkozó vizsgálatok a környezet tekintetében
A II. melléklet 3.4. alpontját teljes mértékben alkalmazni kell mindegyik adalékanyag tekintetében.
7.4. IV. szakasz: az adalékanyag hatékonyságára vonatkozó vizsgálatok
Az egyes takarmány-adalékanyagok kategóriáira és funkcionális csoportjaira vonatkozó követelményeket kell alkalmazni.
Abban az esetben, amikor az állatokon végzett vizsgálatban használt adalékanyagot már engedélyezték egy fiziológiai szempontból hasonló egyéb fajra, nincs szükség további hatékonysági vizsgálatra, amennyiben az adalékanyag hatása és hatásmódja a várttal megegyező. Ha az adalékanyagot még nem engedélyezték, a várt hatás, vagy a hatásmód az előző engedélyezésekétől eltérő, a hatékonyság bizonyításában a II. melléklet IV. szakasza általános szabályainak megfelelően kell eljárni.
A hosszú távú hatékonysági vizsgálatoknak legalább 28 naposnak kell lenniük.
7.5. V. szakasz: forgalomba hozatalt követő felügyeleti terv
A II. melléklet e szakaszát az 1831/2003/EK rendelet 7. cikke (3) bekezdése g) pontjának rendelkezései alapján kell alkalmazni.
8. ÉLELMISZEREKBEN KORÁBBAN ENGEDÉLYEZETT TAKARMÁNY-ADALÉKANYAGOK
8.1. I. szakasz: a dokumentáció összefoglalása
A II. melléklet I. szakaszát teljes mértékben alkalmazni kell.
8.2. II. szakasz: az adalékanyag azonossága, jellemzői és felhasználási feltételei; elemzési módszerek
A II. melléklet II. szakaszát a következőképpen kell alkalmazni:
- a konkrét engedélyjogosulthoz kötött adalékanyagokra a II. szakaszt teljes mértékben kell alkalmazni
- egyéb adalékanyagokra a 2.1.2, 2.1.3, 2.1.4, 2.1.4.2, 2.2, 2.3.1, 2.3.2, 2.4.1, 2.4.2, 2.4.4, 2.5, 2.6 bekezdést kell alkalmazni.
8.3. III. szakasz: az adalékanyagok biztonságosságára vonatkozó vizsgálatok
Az élelmiszer-adalékanyag biztonságos használatára vonatkozó legfrissebb hivatalos értékelést csatolni kell és bármilyen, ezen értékelést követően keletkezett adattal ki kell egészíteni.
Az élelmiszer-adalékanyagként vagy takarmány-összetevőként az Európai Unióban mindenfajta megszorítás nélkül engedélyezett adalékanyagokra általában nem szükséges a fogyasztók és a dolgozók biztonságára vonatkozó vizsgálat.
A II. melléklet 3.1., 3.2. és 3.3. alpontját ezen anyagoknak a biztonságos használatára vonatkozó adatok ismeretében kell alkalmazni. Ennek megfelelően, az ilyen, az élelmiszerekben is alkalmazott anyagok a következőképpen osztályozhatók:
- nem meghatározott ADI (minden felső beviteli határérték egyértelmű feltüntetése nélkül, nagyon alacsony toxicitású anyagokhoz hozzárendelve),
- meghatározott ADI vagy UL, vagy
- hozzárendelt ADI-vel nem rendelkező (olyan anyagokra kell alkalmazni, amelyek esetében a rendelkezésre álló információ nem elegendő ezek biztonságos alkalmazásának megállapításához).
8.3.1. Az adalékanyag biztonságos használatára vonatkozó vizsgálatok a célállatok tekintetében
Ha az adalékanyag alkalmazási szintje a takarmányban azonos az élelmiszerben levővel, a célfajokra vonatkozó biztonságos használat in vivo toxikológiai adatok alapján értékelhető, a kémiai szerkezetnek és a célfaj adalékanyagot metabolizáló kapacitásának figyelembevételével. Ha a takarmányban jóval magasabb az alkalmazási érték, mint a megfelelő élelmiszerben, a célfajban az anyag természetétől függő toleranciavizsgálatot kell végezni.
8.3.2. Az adalékanyag biztonságos használatára vonatkozó vizsgálatok a fogyasztók tekintetében
Ha a takarmány-adalékanyagként való alkalmazás a fogyasztói expozíció megnövekedésével jár, vagy az élelmiszerben való alkalmazástól eltérő anyagcseretermékek képződnek, további toxikológiai és maradékanyag-vizsgálati adatra van szükség.
8.3.2.1. Meghatározott ADI-vel nem rendelkező adalékanyagok
Nincs szükség a biztonság értékelésére a fogyasztók tekintetében, kivéve ha az adalékanyagnak a takarmánnyal való együttes alkalmazásakor más anyagcseretermékek képződnek, mint amikor az adalékanyag az élelmiszer része.
8.3.2.2. Meghatározott ADI-vel vagy UL-lel rendelkező adalékanyagok
A fogyasztói biztonság értékelésekor figyelembe kell venni a takarmány használatából eredő további expozíciót, vagy a célfajoknál való alkalmazásból adódó anyagcseretermékeknek köszönhető specifikus expozíciót. Ez elvégezhető a maradékanyagra vonatkozó irodalmi adatok alapján is.
Azokban az esetekben, ahol maradékanyag-vizsgálatot kell végezni, a követelmény a nem kezelt csoport és a legmagasabb javasolt dózisszint mellett kezelt csoport szöveteiben és termékeiben levő maradékanyagszint összehasonlítására korlátozódik.
8.3.2.3. Engedélyezett ADI-vel nem rendelkező adalékanyagok
Világosan meg kell jelölni az okot, amiért az ADI nem engedélyezett. Ha ez gondot okoz, és az adalékanyagnak a takarmányban való alkalmazása következtében jelentősen megnő a fogyasztói expozíció, teljes toxikológiai felmérésre van szükség.
A takarmányhasználatból eredő további expozíciót a maradékanyagokra vonatkozó irodalmi adatok alapján lehet értelmezni.
Azokban az esetekben, ahol maradékanyag-vizsgálatot kell végezni, a követelmény a nem kezelt csoport és a legmagasabb javasolt dózisszint mellett kezelt csoport szöveteiben és termékeiben levő maradékanyagszint összehasonlítására korlátozódik.
8.3.3. A felhasználókat és dolgozókat érintő, az adalékanyag biztonságos használatára vonatkozó vizsgálatok
A II. melléklet 3.3. alpontját teljes mértékben alkalmazni kell.
A takarmány-adalékanyag biztonságos használatához szem előtt kell tartani ezen élelmiszerekben alkalmazott anyagokkal való bánáshoz szükséges óvintézkedéseket.
8.3.4. Az adalékanyag használatának biztonságára vonatkozó vizsgálatok a környezet tekintetében
A II. melléklet 3.4. alpontját teljes mértékben alkalmazni kell.
8.4. IV. szakasz: az adalékanyag hatékonyságára vonatkozó vizsgálatok
Ha az adalékanyagnak a takarmányban való alkalmazásakor a várt hatás megegyezik az élelmiszerben való alkalmazáskor megszokott hatással, nincs szükség a hatékonyság további bizonyítására. A hatékonyságot illetően a követelményeket máskülönben a II. melléklet IV. szakasza tartalmazza.
8.5. V. szakasz: forgalomba hozatalt követő felügyeleti terv
A II. melléklet e szakaszát az 1831/2003/EK rendelet 7. cikke (3) bekezdése g) pontjának rendelkezései alapján kell alkalmazni.
9. AZ ENGEDÉLYEZÉS MEGVÁLTOZTATÁSA
Az előző engedélyezésekhez benyújtott adatok értékelésére alapozva az 1831/2003/EK rendelet 13. cikkének (3) bekezdése szerinti kérelemhez készített dokumentáció csak a következő elvárásoknak kell eleget tegyen.
Már meglevő engedélyeztetési rendeletben foglalt fogalmaknak, mint pl. az azonosítás, az adalékanyagnak vagy felhasználási körülményeinek a jellemzése, a módosításához benyújtott kérelem, igazolnia kell, hogy e módosítások nem károsak a célfajokra, a fogyasztókra, a felhasználóra és a környezetre. Ebben a tekintetben az adalékanyag akkor tekinthető azonosnak, ha a hatóanyag(ok)/reagens(ek) ugyanazok és a használati feltételek azonosak, a tisztasága lényegében azonos és semmilyen, aggodalomra okot adó új összetevőt nem adtak hozzá. E termékekre vonatkozóan mentesítő kérelmet lehet benyújtani, mivel rendszerint nincs szükség az adalékanyagnak a célfajban, a fogyasztókban és a környezetben történő biztonságos használatát bizonyító vizsgálatoknak a megismétlésére.
A kérelemnek az alábbi követelményeket kell tartalmaznia:
1. az I. mellékletet teljes mértékben alkalmazni kell - beleértve a kérelmezett módosítás részleteit;
2. a II. melléklet II. szakaszát teljes mértékben alkalmazni kell;
3. adatokkal kell alátámasztani, hogy az adalékanyag vegyi és biológiai jellemzői a bevezetett termékével lényegében azonosak;
4. a biológiai egyenértékűséget, adott esetben, pontos leírással vagy specifikus vizsgálatokról megjelent irodalmi adatokkal igazolni kell. Ahol a biológiai egyenértékűség nem teljes mértékben bizonyított, igazolni kell, hogy a maradékanyagszintnek az MRL-értékek alá csökkenéséhez szükséges idő összhangban van az MRL-értékkel;
5. bizonyítani kell, hogy a jelenlegi tudás fényében az adalékanyag biztonságos marad a célfajra, a dolgozókra és a környezetre nézve a jóváhagyott körülmények között;
6. a forgalomba hozatalt követő felügyeleti terv eredményét jelenteni kell, amennyiben az engedélyeztetés ilyen felügyeleti tervre vonatkozó követelményhez kötött; és
7. a módosítási kérelmet igazoló specifikus adatokat a II. melléklet III., IV. és V. szakaszának vonatkozó részeivel összhangban kell benyújtani.
10. ENGEDÉLYEK MEGÚJÍTÁSA
Az 1831/2003/EK rendelet 14. cikke szerinti, az engedély megújításához szükséges kérelem a következő elvárásoknak kell eleget tegyen:
10.1. I. szakasz: a dokumentáció összefoglalása
A II. melléklet I. szakaszát teljes mértékben alkalmazni kell. Be kell nyújtani a takarmány-adalékanyagnak a forgalomba hozatalára vonatkozó eredeti közösségi engedély másolatát, vagy az engedély legutóbbi megújítását. A naprakész dokumentációnak tartalmaznia kell a legfrissebb követelményeket és az eredeti engedélyeztetéstől számított összes változat felsorolását, vagy az engedély legutóbbi megújítását. A kérelmezőnek be kell nyújtania a dokumentáció összefoglalását, részletezve a kérelem célját és az azonosságra és biztonságosságra vonatkozóan bármilyen új információt, a legutóbbi engedélyeztetéstől vagy engedély megújításától számítva.
10.2. II. szakasz: az adalékanyag azonossága, jellemzői és felhasználási feltételei; elemzési módszerek
A II. melléklet II. szakaszát a következőképpen kell alkalmazni:
- a konkrét engedélyjogosulthoz kötött adalékanyagokra a II. szakaszt teljes mértékben kell alkalmazni
- az egyéb adalékanyagokra a 2.1.2, 2.1.3, 2.1.4, 2.1.4.2, 2.2, 2.3.1, 2.3.2, 2.4.1, 2.4.2, 2.4.4, 2.5, 2.6 bekezdést kell alkalmazni.
Bizonyítani kell, hogy az adalékanyag összetétele, tisztasága és aktivitása nem változott vagy módosult számottevően az engedélyezett adalékanyaghoz viszonyítva. A gyártási eljárásban eszközölt minden módosítást rögzíteni kell.
10.3. III. szakasz: az adalékanyagok biztonságosságára vonatkozó vizsgálatok
Bizonyítékokkal igazolni kell, hogy az aktuális ismeretek tükrében, az adalékanyag a jóváhagyott feltételek mellett a célállatokra, a fogyasztókra, a kezelőkre és a környezetre nézve biztonságos marad. A forgalomba hozatal engedélyezése vagy az engedély legutóbbi meghosszabbítása óta eltelt időszakra vonatkozóan el kell készíteni a biztonsági dokumentáció frissítését, amely a következő adatokat tartalmazza:
- a célállatokat, a kezelőket és a környezetet érintő káros hatásokról szóló jelentések, beleértve a baleseteket (addig ismeretlen hatásokat, bármilyen típusú súlyos hatásokat, ismert hatások gyakoribb előfordulását). A káros hatásról szóló jelentésben le kell írni a hatás jellegét, az érintett egyedek és szervezetek számát, a következményeket, a felhasználási körülményeket, az okozati összefüggések feltárását,
- a korábban ismeretlen kölcsönhatásokról és keresztszennyezésekről szóló jelentések,
- adott esetben, a maradékanyag ellenőrzésére vonatkozó adatok,
- az adalékanyag biztonságára vonatkozó bármely egyéb információ,
- az adalékanyag használatának veszélye az állatokra, az emberre és a környezetre nézve.
Ha e tényezők valamelyikéről nem szolgáltatnak további információt, akkor ennek okát világosan meg kell határozni.
A forgalomba hozatalt követő felügyeleti terv eredményét jelenteni kell, ha az előző engedélyezés ilyen felügyeleti tervre vonatkozó követelményhez kötött.
Az 1831/2003/EK rendelet 14. cikke (2) bekezdésének d) pontjában előírtak szerint ahol az engedélyezés megújítása az eredeti engedélyezési feltételek, többek között a jövőbeni felügyelet, módosítására vagy kiegészítésére irányuló javaslatot tartalmaz, a módosítási javaslatot támogató különleges adatokat a II. melléklet III., IV. és V. szakasza vonatkozó részeinek megfelelően kell benyújtani.
11. A 70/524/EGK IRÁNYELVBEN KORÁBBAN ENGEDÉLYEZETT EGYES ADALÉKANYAGOK ÚJRAÉRTÉKELÉSE
E 11. pontban szereplő adalékanyagok a 70/524/EGK rendelet által engedélyezett anyagok, amelyeket az 1831/2003/EK rendelet 10. cikkének (2) bekezdésében foglaltaknak megfelelően újra kell értékelni és amelyek a következő csoportokba sorolhatók:
- antioxidánsok,
- aroma- és ízesítőanyagok,
- emulgeáló és stabilizáló anyagok, sűrítőanyagok és zselésítő anyagok,
- színezőanyagok, beleértve a festékeket,
- tartósítószerek,
- vitaminok, provitaminok és olyan kémiailag jól meghatározott anyagok, amelyeknek hasonló hatása van,
- nyomelemek,
- kötő- és csomósodásgátló anyagok, valamint koagulánsok,
- savasságot szabályozó anyagok, és
- radionuklid kötőanyagok.
Ezen adalékanyagok használatakor fellépő kockázat szintjének és minőségének a felmérését az egyéb adalékanyagokéhoz hasonlóan kell végezni. A hosszú ideig tartó biztonságos használatnak köszönhetően azonban az irodalomban közzétett vizsgálati adatok felhasználásával, e rendelet rendelkezései által előírtaknak megfelelően igazolható, hogy az adalékanyag a jóváhagyott feltételek mellett a célállatokra, a fogyasztókra, a kezelőkre és a környezetre nézve biztonságos marad.
11.1. I. szakasz: a dokumentáció összefoglalása
A II. melléklet I. szakaszát teljes mértékben alkalmazni kell.
11.2. II. szakasz: az adalékanyag azonossága, jellemzői és felhasználási feltételei; elemzési módszerek
A II. melléklet II. szakaszát a következőképpen kell alkalmazni:
- a konkrét engedélyjogosulthoz nem kötött adalékanyagokra a 2.1.2, 2.1.3, 2.1.4, 2.1.4.2, 2.2, 2.3.1, 2.3.2, 2.4.1, 2.4.2, 2.4.4, 2.5, 2.6 bekezdést kell alkalmazni,
- a konkrét engedélyjogosulthoz kötött egyéb adalékanyagokra a II. szakaszt teljes mértékben kell alkalmazni.
11.3. III. szakasz: az adalékanyagok biztonságosságára vonatkozó vizsgálatok
Amennyiben az adalékanyagot a célfaj, a fogyasztók, a felhasználók és a dolgozók, valamint a környezet biztonságára vonatkozóan értékelték, a kérelmezőnek be kell nyújtania az előző engedélyezéshez kötött hatékonysági vizsgálatok összefoglalását, valamint a legutóbbi engedélyeztetéstől számított bármilyen új információt. Amennyiben az anyagnak takarmány-adalékanyagként való alkalmazására vonatkozóan nincs előírásos biztonságossági értékelés, a tudományos közleményekből származó vizsgálatok és adatok felhasználhatóak erre a célra, feltéve hogy egyenértékűek azokkal, amelyeket az új kérelem tartalmazhat. Ellenkező esetben teljes körű biztonságossági vizsgálatot kell végezni.
11.4. IV. szakasz: az adalékanyag hatékonyságára vonatkozó vizsgálatok
Az 1831/2003/EK rendelet 5. cikke (3) bekezdésében meghatározott hatékonysági elvárásoknak való megfelelőség, adott esetben egyéb, vizsgálatoktól eltérő módszerrel készült, főként az adalékanyag hosszú távú használatra vonatkozó dokumentációs anyaggal is bizonyítható.
11.5. V. szakasz: forgalomba hozatalt követő felügyeleti terv
A II. melléklet e szakaszát az 1831/2003/EK rendelet 7. cikke (3) bekezdése g) pontjának rendelkezései alapján kell alkalmazni.
(1) A xenobiotikum olyan vegyi anyag, amely nem természetes alkotórésze az ezen anyagnak kitett szervezetnek. Magában foglalhat olyan anyagokat is, amelyek a szokásosnál sokkal magasabb koncentrációban vannak jelen.
(2) E rendelet alkalmazásában a "silózási folyamat" azt a folyamatot jelenti, amelynek során a szerves anyag természetes lebomlását a természetes fermentálódásból és/vagy a szilázs-adalékanyag hozzáadásából származó, anaerob körülmények között végbemenő savasodás szabályozza.
(3) A FLAVIS-rendszerben (az EU aromaanyagokat kezelő információs rendszere), a 2232/96/EK európai parlamenti és tanácsi rendelet (HL L 299., 1996.11.23., 1. o.) értelmében egy értékelési program elfogadásához szükséges intézkedések megállapításáról szóló, 2000. július 18-i 1565/2000/EK bizottsági rendeletben (HL L 180., 2000.7.19., 8. o.) használt adatbázisban a kémiailag meghatározott aromaanyagokra alkalmazott azonosítószám.
(4) ET-sz.: Európa Tanács szám, amely a botanikailag meghatározott aromatermékekhez használatos az Európa Tanács 1. sz. jelentésében: "Natural sources of flavourings" (Aromaanyagok természetes forrásai), I. kötet, Strasbourg, 2000, illetve a későbbi kötetek.
(5) CAS-szám (CAS-sz.) A Chemical Abstracts Service nyilvántartási száma, vegyi anyagok széles körben használt egyedi azonosítója.
(6) E rendelet e szakaszának alkalmazásában: "toxikológiai jelentőségű anyag" az az anyag, amelyet megtűrt napi vagy heti bevitel (TDI vagy TWI), egy ADI (elfogadható napi bevitel) jellemez, vagy használata korlátozott, vagy az élelmiszerekben felhasználandó aromaanyagokra és az előállításukhoz szükséges alapanyagokra vonatkozó tagállami jogszabályok közelítéséről szóló, 1988. június 22-i 88/388/EGK tanácsi irányelvben meghatározott aktív elven alapszik, vagy pedig nemkívánatos anyag.
(7) Meghatározásuk az Európa Tanács 1. sz. "Aromaanyagok természetes forrásai" című jelentésének 4. mellékletében olvasható, I. kötet, Strasbourg, 2000.
(8) JECFA (FAO/WHO, 1996, Adalékanyag sorozat 35, IPCS, WHO Genf) a megfelelő küszöbértéket a célállatok tekintetében az állat súlyának és a bevitt takarmánynak a függvényében kell megállapítani.
(9) HL L 180., 2000.7.19., 8. o.
(10) Útmutató megjegyzés a lazacfélék és egyéb közönséges halfélék maximális maradékanyag-határértékeinek meghatározásához. Európai Gyógyszerértékelő Ügynökség. Állatgyógyászati termékek értékelő egysége. EMEA/CVMP/153b/97-VÉGLEGES.
IV. MELLÉKLET
Célállatok kategóriái és fogalommeghatározásai, valamint a hatékonysági vizsgálatok minimális időtartamának feltüntetése
1. Táblázat. Állatkategóriák: Sertés
| Kategória | Az állatkategória fogalommeghatározása | Hozzávetőleges időtartam (tömeg/életkor) | Hosszú távú hatékonysági vizsgálatok minimális időtartama | ||
| Időtartam/életkor | Életkor | Tömeg | |||
| Malac (szopós malac) | Fiatal sertésféle, amely kocák tejét fogyasztja | Születéstől | 21–42 naposig | 6–11 kg-ig | 14 nap |
| Malac (elválasztott) | Fiatal sertésféle, amely már túl van a szopási időszakon, és amelyet szaporítás vagy hústermelés céljára nevelnek | 21–42 naptól | 120 naposig | 35 kg-ig | 42 nap |
| Malac (szopós malac és elválasztott malac) | Fiatal sertésféle, születésétől kezdve, amelyet szaporítás vagy hústermelés céljára nevelnek | Születéstől | 120 naposig | 35 kg-ig | 58 nap |
| Hízósertés | Sertésféle, amely túl van az elválasztási időszakon, és amelyet hústermelésre szánnak, a vágóhídra szállítás napjáig | 60–120 naptól | 120–250 naposig (vagy a helyi szokásnak megfelelően) | 80–150 kg (vagy a helyi szokásnak megfelelően) | Vágási súly eléréséig, de minimum 70 napig |
| Tenyészkoca | Nőstény sertésféle, amelyet legalább egyszer megtermékenyítettek/pároztattak | Az első megtermékenyítéstől | A megtermékenyítéstől a második elválasztási időszak végéig (két ciklus) | ||
| Koca, a malacokban megjelenő előny céljából | Nőstény sertésféle, amelyet legalább egyszer megtermékenyítettek/pároztattak | Az ellés előtt legalább két héttel, az elválasztási időszak végéig. | |||
2. Táblázat. Állatkategóriák: Baromfi
| Kategória | Az állatkategória fogalommeghatározása | Hozzávetőleges időtartam (tömeg/életkor) | Hosszú távú hatékonysági vizsgálatok minimális időtartama | ||
| Időtartam | Életkor | Tömeg | |||
| Hízócsirke | Hízlalásra nevelt állat | A kikeléstől | 35 naposig | ~1 600 g-ig (2 kg-ig) | 35 nap |
| Tojástermelés céljából nevelt csirke | Emberi fogyasztásra szánt tojás termelése vagy tenyésztés céljából nevelt nőstény állat | A kikeléstől | ~16 hetesig (20 hetesig) | — | 112 nap (ha a hatékonysági adatok nem állnak rendelkezésre a hízócsirkékre vonatkozóan) |
| Tojótyúk | Tojástermelés céljából tartott termékeny nőstény állat | 16–21 héttől | ~13 hónaposig (18 hónaposig) | 1 200 g-tól (fehér) 1 400 g-tól (barna) | 168 nap |
| Hízópulyka | Hizlalás céljából nevelt pulyka | A kikeléstől | ~14 hetesig (20 hetesig) ~16 hetesig (24 hetesig) | Tojók: ~7 000 g-ig (10 000 g-ig) Hímek: ~12 000 g-ig (20 000 g-ig) | 84 nap |
| Tenyészpulyka | Tenyésztés céljára tartott nőstény és hím állatok | Teljes időtartam | 30 hetestől ~ 60 hetesig | Tojók: ~15 000 g-tól Hímek: ~30 000 g-tól | Legalább hat hónap |
| Tenyésztés céljából nevelt pulyka | Tenyésztés céljára nevelt fiatal nőstény és hím állatok | A kikeléstől | 30 hetesig | Tojók: ~15 000 g-ig Hímek: ~30 000 g-ig | Teljes időtartam (ha a hatékonysági adatok nem állnak rendelkezésre a hízópulykákra vonatkozóan) |
3. Táblázat. Állatkategóriák: Szarvasmarha (házi szarvasmarhafélék, beleértve a Bubalus és Bison fajokat)
| Kategória | Az állatkategória fogalommeghatározása | Hozzávetőleges időtartam (tömeg/életkor) | Hosszú távú hatékonysági vizsgálatok minimális időtartama | ||
| Időtartam | Életkor | Tömeg | |||
| Tenyészborjú | Szaporítás vagy hústermelés céljából nevelt borjú | Születéstől | 4 hónaposig | 60–80 kg-ig (145 kg-ig) | 56 nap |
| Hízóborjú | Borjúhústermelés céljából nevelt borjú | Születéstől | 6 hónaposig | 180 kg-ig (250 kg-ig) | A levágásig, de legalább 84 napig |
| Hízómarha | Szarvasmarhaféle, amely túl van az elválasztási időszakon, és amelyet hústermelésre szánnak, a vágóhídra szállítás napjáig | A kérődzés teljes kifejlődésétől | 10–36 hónaposig | 350–700 kg-ig | 168 nap |
| Tejelő tehén tejtermelés céljára | Nőstény szarvasmarhaféle, amely legalább egy borjat ellett | 84 nap (a teljes laktációs időszakról be kell számolni) | |||
| Tehén szaporítás céljára | Nőstény szarvasmarhaféle, amelyet legalább egyszer megtermékenyítettek/pároztattak | Az első megtermékenyítéstől a második elválasztási időszak végéig | Két ciklus (ha a szaporítási paraméterekre van szükség). | ||
4. Táblázat. Állatkategóriák: Juh
| Kategória | Az állatkategória fogalommeghatározása | Hozzávetőleges időtartam (tömeg/életkor) | Hosszú távú hatékonysági vizsgálatok minimális időtartama | ||
| Időtartam | Életkor | Tömeg | |||
| Tenyészbárány | Későbbi szaporítás céljára nevelt bárány | Születéstől | 3 hónaposig | 15–20 kg | 56 nap |
| Húsbárány | Bárányhústermelés céljára nevelt bárány | Születéstől | 6 hónaposig (vagy idősebb korig) | 55 kg-ig | A vágási súlyig, de legalább 56 napig |
| Tejelő juh (tejtermelés céljára) | Olyan juh, amely legalább egy bárányt ellett | 84 nap (a teljes laktációs időszakról be kell számolni) | |||
| Nőstény juh szaporítás céljára | Olyan nőstény juh, amelyet legalább egyszer megtermékenyítették/pároztattak | Az első megtermékenyítéstől a második elválasztási időszak végéig | Két ciklus (ha a szaporítási paraméterekre van szükség) | ||
5. Táblázat. Állatkategóriák: Kecske
| Kategória | Az állatkategória fogalommeghatározása | Hozzávetőleges időtartam (tömeg/életkor) | Hosszú távú hatékonysági vizsgálatok minimális időtartama | ||
| Időtartam | Életkor | Tömeg | |||
| Tenyészkecske | Későbbi szaporítás céljára nevelt fiatal kecske | Születéstől | 3 hónaposig | 15–20 kg | Legalább 56 |
| Húskecske | Kecskehústermelés céljára nevelt fiatal kecske | Születéstől | 6 hónaposig | Legalább 56 nap | |
| Tejelő kecske (tejtermelés céljára) | Olyan kecske, amely legalább egy gidát ellett | 84 nap (a teljes laktációs időszakról be kell számolni) | |||
| Szaporítás céljára tartott kecske | Olyan nőstény kecske, amelyet legalább egyszer megtermékenyítették/pároztattak | Az első megtermékenyítéstől a második elválasztási időszak végéig | Két ciklus (ha a szaporítási paraméterekre van szükség) | ||
6. Táblázat. Állatkategóriák: Halak
| Kategória | Az állatkategória fogalommeghatározása | Hozzávetőleges időtartam (tömeg/életkor) | Hosszú távú hatékonysági vizsgálatok minimális időtartama | ||
| Időtartam | Életkor | Tömeg | |||
| Lazac és pisztráng | 200–300 g | 90 nap, vagy a kezdeti testtömeg megkétszerezéséig. | |||
| Lazac és pisztráng | Tenyészállomány | Az ívási időszakhoz lehető legközelebb | 90 nap | ||
7. Táblázat. Állatkategóriák: Nyúl
| Kategória | Az állatkategória fogalommeghatározása | Hozzávetőleges időtartam (tömeg/életkor) | Hosszú távú hatékonysági vizsgálatok minimális időtartama | ||
| Időtartam | Életkor | Tömeg | |||
| Szopósnyúl és elválasztott nyúl | Születés után egy héttel kezdődik | 56 nap | |||
| Hízónyúl | Hústermelés céljára nevelt nyúl | Az elválasztási időszak után | 8–11 hetesig | 42 nap | |
| Nőstény tenyésznyúl (szaporítás céljára) | Olyan nőstény nyúl, amelyet legalább egyszer megtermékenyítették/pároztattak | Az első megtermékenyítéstől a második elválasztási időszak végéig | Két ciklus (ha a szaporítási paraméterekre van szükség) | ||
| Nőstény tenyésznyúl (a fiatal nyulakban megjelenő előny céljából) | Olyan nőstény nyúl, amelyet legalább egyszer megtermékenyítettek | Az első megtermékenyí-téstől | Legalább 2 héttel az ellés előtt, az elválasztási időszak végéig (pl. mikroorganizmus termék) | ||
8. Táblázat. Állatkategóriák: Ló
| Kategória | Az állatkategória fogalommeghatározása | Hozzávetőleges időtartam (tömeg/életkor) | Hosszú távú hatékonysági vizsgálatok minimális időtartama | ||
| Időtartam | Életkor | Tömeg | |||
| Ló | Minden kategória | 56 nap | |||
Lábjegyzetek:
[1] A dokumentum eredetije megtekinthető CELEX: 32008R0429 - https://eur-lex.europa.eu/legal-content/HU/ALL/?uri=CELEX:32008R0429&locale=hu