
20/2004. (II. 27.) FVM rendelet
a Magyar Takarmánykódex kötelező előírásairól szóló 44/2003. (IV. 26.) FVM rendelet módosításáról
A takarmányok előállításáról, forgalomba hozataláról és felhasználásáról szóló 2001. évi CXIX. törvény 18. §-ának e) és n) pontjaiban foglalt felhatalmazás alapján a következőket rendelem el:
1. §
A Magyar Takarmánykódex kötelező előírásairól szóló 44/2003. (IV. 26.) FVM rendelet (a továbbiakban: R.) 1. §-ának (2) bekezdése a következő n) ponttal egészül ki:
[A Magyar Takarmánykódex (Codex Pabularis Hungaricus) I. kötetének kötelező előírásait e rendelet következő mellékleteiben adom ki:]
"n) az állatok fehérje ellátásának javítására használható egyes (bizonyos) termékekről és azok minősítésének elveiről szóló 14. számú melléklet."
2. §
Ez a rendelet a Magyar Köztársaság és az Európai Közösségek és azok tagállamai között társulás létesítéséről szóló, Brüsszelben 1991. december 16-án aláírt Európai Megállapodás tárgykörében, a Megállapodást kihirdető 1994. évi I. törvény 3. §-ával összhangban az Európai Közösségek következő jogszabályaival összeegyeztethető szabályozást tartalmaz:
1. a Tanács 70/524/EGK irányelve a takarmány-adalékanyagokról, valamint az azt módosító, a Tanács 84/587/EGK, 96/51/EK irányelve,
2. a Bizottság 71/250/EGK irányelve a takarmányok hatósági ellenőrzésére szolgáló közösségi analitikai módszerek meghatározásáról,
3. a Bizottság 72/199/EGK harmadik irányelve a takarmányok hatósági ellenőrzésére szolgáló közösségi analitikai módszerek meghatározásáról,
4. a Bizottság 78/633/EGK nyolcadik irányelve a takarmányok hatósági ellenőrzésére szolgáló közösségi analitikai módszerek meghatározásáról,
5. a Bizottság 81/715/EGK kilencedik irányelve a takarmányok hatósági ellenőrzésére szolgáló közösségi analitikai módszerek meghatározásáról,
6. a Tanács 83/228/EGK irányelve a fehérje ellátásban alkalmazott bizonyos termékek értékelésére vonatkozó iránymutatások meghatározásáról,
7. a Bizottság 84/4/EGK irányelve a takarmányok hatósági ellenőrzésére szolgáló közösségi analitikai módszerek meghatározásáról szóló 71/393/EGK, 72/199/EGK és 78/633/EGK irányelvek módosításáról,
8. a Bizottság 84/425/EGK tizedik irányelve a takarmányok hatósági ellenőrzésére szolgáló közösségi analitikai módszerek meghatározásáról,
9. a Tanács 87/153/EGK irányelve a takarmányokban lévő adalékanyagok értékelésére vonatkozó iránymutatások meghatározásáról, valamint az azt módosító, a Bizottság 94/40EK, 95/11/EK irányelve,
10. a Bizottság 93/117/EK tizenkettedik irányelve a takarmányok hatósági ellenőrzésére szolgáló közösségi analitikai módszerek meghatározásáról,
11. a Tanács 2001/102/EK irányelve a takarmányban előforduló nemkívánatos anyagokról és termékekről szóló 1999/29/EK irányelv módosításáról,
12. a Bizottság 2002/1/EK irányelve a 94/39/EK irányelvnek a krónikus máj elégtelenség esetén a májműködést segítő takarmányozás tekintetében történő módosításáról,
13. az Európai Parlament és a Tanács 2002/32/EK irányelve a takarmányban előforduló nemkívánatos anyagokról, valamint az azt módosító, a Bizottság 2003/100/EK irányelve,
14. a Bizottság 2002/70/EK irányelve a takarmányok dioxin- és dioxinjellegű PCB-tartalmának meghatározására vonatkozó követelmények megállapításáról,
15. a Bizottság 256/2002/EK rendelete új takarmányadalékanyagok ideiglenes engedélyezéséről, egy takarmány-adalékanyag ideiglenes engedélyének meghosszabbításáról és egy takarmány-adalékanyag végleges engedélyezéséről,
16. a Bizottság 1041/2002/EK rendelete egy új takarmány-adalékanyag ideiglenes engedélyezéséről,
17. a Bizottság 1252/2002/EK rendelete egy új takarmány-adalékanyag ideiglenes engedélyezéséről,
18. az Európai Parlament és a Tanács 1774/2002/EK rendelete a nem emberi fogyasztásra szánt állati melléktermékekre vonatkozó egészségügyi előírások megállapításáról,
19. a Bizottság 1876/2002/EK rendelete egy takarmányadalékanyag új alkalmazásának ideiglenes engedélyezéséről,
20. a Bizottság 2188/2002/EK rendelete takarmányadalékanyagok új alkalmazásainak ideiglenes engedélyezéséről,
21. a Bizottság 2003/7/EK irányelve a takarmányokban a kantaxanthin engedélyezési feltételeinek módosításáról, a 70/524/EGK tanácsi irányelvvel összhangban,
22. a Bizottság 2003/104/EK irányelve a metionin-hidroxi-analóg izopropil észterének engedélyezéséről,
23. a Bizottság 162/2003/EK rendelete egy takarmányadalékanyag engedélyezéséről,
24. a Bizottság 261/2003/EK rendelete takarmány-adalékanyagok új használatának ideiglenes engedélyezéséről,
25. a Bizottság 316/2003/EK rendelete takarmány-adalékanyag állandó engedélyezéséről, és engedélyezett takarmány-adalékanyag új használatának ideiglenes engedélyezéséről,
26. a Bizottság 666/2003/EK rendelete egyes mikroorganizmusok takarmányokban való használatának ideiglenes engedélyezéséről,
27. a Bizottság 668/2003/EK rendelete takarmány-adalékanyag állandó engedélyezéséről,
28. a Bizottság 871/2003/EK rendelete a takarmányokban a mangano-mangán-oxid új adalékanyag állandó engedélyezéséről,
29. a Bizottság 877/2003/EK rendelete a benzol-sav savasság szabályozó használatának ideiglenes engedélyezéséről,
30. a Bizottság 1334/2003/EK rendelete a nyomelemek csoportjába tartozó számos takarmány-adalékanyag engedélyezési feltételeinek módosításáról.
3. §
(1) Az R. 1., 2., 4., 5., 6., 10., 11. és 13. számú melléklete e rendelet 1. számú melléklete szerint módosul.
(2) Az R. e rendelet 2. számú melléklete szerinti 14. számú melléklettel egészül ki.
4. §
(1) E rendelet - a (2) bekezdésben foglaltak kivételével - a kihirdetését követő tizenötödik napon lép hatályba, egyidejűleg az R. 4. számú melléklete II. fejezete 6.1. pontja harmadik sorában az "E 161g H Kantaxantin" alpont sor és az R. 4. számú melléklete II. fejezetének 16. Az állatok fehérje ellátásának javítására használható egyes termékekről szóló alfejezete hatályát veszti.
(2) Az 1. számú melléklet III. fejezetének 13. pontja, valamint az R. e rendelet 2. számú mellékletével megállapított 14. számú mellékletének I. fejezete a Magyar Köztársaságnak az Európai Unióhoz történő csatlakozásáról szóló nemzetközi szerződést kihirdető törvény hatálybalépésének napján lép hatályba.
(3) Az R. 2. számú mellékletének II. táblázata a Magyar Köztársaságnak az Európai Unióhoz történő csatlakozásáról szóló nemzetközi szerződést kihirdető törvény hatálybalépésének napján hatályát veszti.
(4) Az R. e rendelet 1. számú melléklete II. fejezetével és III. fejezetének 1-12. pontjával módosított előírásainak nem megfelelő termékek 2004. április 30-ig forgalmazhatóak.
Dr. Németh Imre s. k.,
földművelésügyi és vidékfejlesztési miniszter
1. számú melléklet a 20/2004. (II. 27.) FVM rendelethez
I. Az R. 1. számú mellékletének módosítása
1. Az R. 1. számú mellékletének III. fejezete a) pontjának harmadik franciabekezdése helyébe a következő rendelkezés lép:
[a) nyersfehérje esetében:]
"- 10% vagy annál kevesebb feltüntetett tartalom esetén 1 abszolút%; (A) "
2. Az R. 1. számú mellékletének III. fejezete b) pontjának első franciabekezdése helyébe a következő rendelkezés lép:
[b) összcukor, redukáló cukrok, szacharóz, laktóz és glükóz (dextróz) esetében:]
"- 20% vagy annál több feltüntetett tartalom esetén 2 abszolút%, (A) "
3. Az R. 1. számú mellékletének III. fejezete b) pontjának harmadik franciabekezdése helyébe a következő rendelkezés lép:
[b) összcukor, redukáló cukrok, szacharóz, laktóz és glükóz (dextróz) esetében:]
"- 5% vagy annál kevesebb feltüntetett tartalom esetén 0,5 abszolút%; (A) "
4. Az R. 1. számú mellékletének III. fejezete c) pontjának harmadik franciabekezdése helyébe a következő rendelkezés lép:
[c) keményítő és inulin esetében:]
"- 10% vagy annál kevesebb feltüntetett tartalom esetén 1 abszolút%; (A)"
5. Az R. 1. számú mellékletének III. fejezete d) pontjának harmadik franciabekezdése helyébe a következő rendelkezés lép:
[d) olajok és nyerszsírok esetében:]
"- 5% vagy annál kevesebb feltüntetett tartalom esetén 0,9 abszolút%; (A)"
6. Az R. 1. számú mellékletének III. fejezete e) pontjának harmadik franciabekezdése helyébe a következő rendelkezés lép:
[e) nyersrost esetében:]
"- 6% vagy annál kevesebb feltüntetett tartalom esetén 0,6 abszolút%; (A)"
7. Az R. 1. számú mellékletének III. fejezete f) pontjának harmadik franciabekezdése helyébe a következő rendelkezés lép:
[f) nedvesség és nyers hamu esetében:]
"- 5% vagy annál kevesebb feltüntetett tartalom esetén 0,5 abszolút%; (A)"
8. Az R. 1. számú mellékletének III. fejezete g) pontja helyébe a következő rendelkezés lép:
"g) összfoszfor, nátrium, kalcium-karbonát, kalcium, magnézium, savszám és könnyű petroléterben oldhatatlan anyag esetében:
- 15% vagy több feltüntetett tartalom, illetve 15 vagy több érték esetén 1,5 abszolút%, (A)
- 15%-nál, illetve 15-nél kevesebb, de 2%-nál, illetve 2-nél több feltüntetett tartalom, illetve érték esetén a feltüntetett tartalom (érték) 10%-a, (R)
- 2% vagy annál kevesebb feltüntetett tartalom, illetve 2 vagy annál kevesebb érték esetén 0,2 abszolút%; (A)"
II. Az R. 2. számú mellékletének módosítása
Az R. 2. számú mellékletének "Megjegyzések és meghatározások" része és I. fejezete helyébe a következő rendelkezések lépnek:
"Megjegyzések és meghatározások:
Az I., II. és III. táblázatban felsorolt értékek 12%-os nedvességtartalomra vonatkoznak.
A III. táblázat értékei közvetlen felhasználásra vonatkoznak.
n.sz.: nincs szabályozva
I.
| Nemkívánatos anyagok, vegyületek | Takarmányozásra szánt termékek | Maximális megengedett mennyiség mg/kg-ban (ppm) (12%-os nedvességtartalom mellett) | |||
| A. Anyagok (ionok vagy elemek) | |||||
| 1. Arzén1 | a) Takarmány-alapanyagok, | 2 | |||
| kivéve: | |||||
| — fűből, szárított lucernából és szárított | 4 | ||||
| lóheréből készült liszt, szárított cukor- | |||||
| gyári répaszelet és melaszos készítmé- | |||||
| nyei | |||||
| — pálmamag pogácsa | 42 | ||||
| — foszfátok és mésztartalmú tengeri algák | 10 | ||||
| — kalcium-karbonát | 15 | ||||
| — magnézium-oxid | 20 | ||||
| — hal, vagy más tengeri állatok feldolgozá- | 152 | ||||
| sából nyert takarmány | |||||
| — tengeri alga liszt és tengeri algából nyert | 402 | ||||
| takarmány | |||||
| b) Teljes értékű takarmányok, | 2 | ||||
| kivéve: | |||||
| — teljes értékű hal és prémes állat takar- | 62 | ||||
| mány | |||||
| c) Kiegészítő takarmányok, | 4 | ||||
| kivéve: | |||||
| - — ásványi takarmányok | 12 | ||||
| 2. Ólom | a) Takarmány-alapanyagok, | 10 | |||
| kivéve: | |||||
| — zöldtakarmány | 40 | ||||
| — foszfátok és mésztartalmú tengeri algák | 15 | ||||
| — kalcium-karbonát | 20 | ||||
| — élesztők | 5 | ||||
| b) Teljes értékű takarmányok | 5 | ||||
| c) Kiegészítő takarmányok, | 10 | ||||
| kivéve: | |||||
| — ásványi takarmányok | 15 | ||||
| 3. Fluor | a) Takarmány-alapanyagok, | 150 | |||
| kivéve: | |||||
| — állati eredetű takarmányok, a tengeri | 500 | ||||
| rákfélék (úgymint tengeri krill) kivételé- | |||||
| vel | |||||
| — foszfátok és tengeri rákfélék, úgymint | 2000 | ||||
| tengeri krill | |||||
| — kalcium-karbonát | 350 | ||||
| — magnézium-oxid | 600 | ||||
| — mésztartalmú tengeri algák | 1000 | ||||
| b) Teljes értékű takarmányok, | 150 | ||||
| kivéve: | |||||
| — teljes értékű szarvasmarha-, juh- és | |||||
| kecske-takarmány | |||||
| 1. laktáció alatt | 30 | ||||
| 2. egyéb | 50 | ||||
| — teljes értékű sertés-takarmány | 100 | ||||
| — teljes értékű baromfi-takarmány | 350 | ||||
| — teljes értékű csirke- és jérce-takarmány | 250 | ||||
| c) Szarvasmarha, juh és kecske ásványi takar- | 20003 | ||||
| mányok | |||||
| d) Egyéb kiegészítő takarmány | 1254 | ||||
| 4. Higany | a) Takarmány-alapanyagok, | 0,1 | |||
| kivéve: | |||||
| — hal és tengeri állatok feldolgozásával | 0,5 | ||||
| előállított takarmányok | |||||
| b) Teljes értékű takarmány, | 0,1 | ||||
| kivéve: | |||||
| — kutyának és macskának szánt teljes érté- | 0,4 | ||||
| kű takarmány- | |||||
| c) Kiegészítő takarmány, | 0,2 | ||||
| kivéve: | |||||
| — kutyának és macskának szánt kiegészítő | n.sz | ||||
| takarmány | |||||
| 5. Nitritek (nátrium-nitritben kifejezve) | a) Halliszt | 60 | |||
| b) Teljes értékű takarmányok, | 15 | ||||
| kivéve: | |||||
| — kedvtelésből tartott állatoknak szánt | n.sz. | ||||
| takarmány, kivéve a madarakat és akvá- | |||||
| riumi halakat | |||||
| 6. Kadmium | a) Növényi eredetű takarmány-alapanyagok | 1 | |||
| b) Állati eredetű takarmány- alapanyagok, | 2 | ||||
| kivéve: | |||||
| — a kedvtelésből tartott állatok takarmá- | n.sz. | ||||
| nyaiban felhasznált takarmányok | |||||
| c) Foszfátok | 105 | ||||
| d) Teljes értékű takarmányok kérődzőknek, | 1 | ||||
| kivéve: | |||||
| — teljes értékű takarmányok borjaknak, | n.sz. | ||||
| bárányoknak, gidáknak | |||||
| e) Teljes értékű takarmányok egyéb állatoknak, | 0,5 | ||||
| kivéve: | |||||
| — kedvtelésből tartott állatok takarmányai | n.sz. | ||||
| f) Ásványi takarmányok | 56 | ||||
| g) Egyéb kiegészítő takarmányok kérődző állatfajoknak | 0,5 | ||||
| B. Anyagok | |||||
| 7. Aflatoxin B1 | a) Összes takarmány-alapanyag | 0,02 | |||
| b) Teljes értékű szarvasmarha-, juh- és kecsketakarmány, | 0,02 | ||||
| kivéve: | |||||
| — tejhasznosítású állatok teljes értékű takarmánya | 0,005 | ||||
| — teljes értékű borjú- és bárány-takarmány | 0,01 | ||||
| c) Teljes értékű sertés- és baromfi-takarmány | 0,02 | ||||
| (növendékek kivételével) | |||||
| d) Egyéb teljes értékű takarmány | 0,01 | ||||
| e) Szarvasmarha, juh és kecske kiegészítő ta- | 0,02 | ||||
| karmány (kivéve tej hasznosítású állatok, | |||||
| borjak és bárányok kiegészítő takarmányait) | |||||
| f) Sertés és baromfi kiegészítő takarmányok | 0,02 | ||||
| (növendékek kivételével) | |||||
| g) Egyéb kiegészítő takarmányok | 0,005 | ||||
| 8. Hidrogén-cianid | a) Takarmány-alapanyagok, | 50 | |||
| (kéksav) | kivéve: | ||||
| — lenmag | 250 | ||||
| — lenmag feldolgozott termékei | 350 | ||||
| — manióka és mandula, valamint feldolgo- | 100 | ||||
| zott termékei | |||||
| b) Teljes értékű takarmányok, | 50 | ||||
| kivéve: | |||||
| — csirkének és jércének szánt teljes értékű | 10 | ||||
| takarmány | |||||
| 9. Szabad gosszipol | a) Takarmány-alapanyagok, | 20 | |||
| kivéve: | |||||
| — gyapotmag | 5000 | ||||
| — kipréselt gyapotmag pogácsa és gyapotmag | 1200 | ||||
| liszt | |||||
| b) Teljes értékű takarmányok, | 20 | ||||
| kivéve: | |||||
| — teljes értékű szarvasmarha-, juh- és kecske- | 500 | ||||
| takarmányok | |||||
| — teljes értékű baromfi- (tojók kivételével) és | 100 | ||||
| borjú-takarmányok | |||||
| — teljes értékű nyúl- és sertés-takarmányok | 60 | ||||
| (malacok kivételével) | |||||
| 10. Teobromin | Teljes értékű takarmányok, | 300 | |||
| kivéve: | |||||
| — teljes értékű felnőtt szarvasmarha-ta- | 700 | ||||
| karmány | |||||
| 11. Mustár-illóolaj | a) Takarmány-alapanyag, | 100 | |||
| (allil-izo-tiocionatban kifejezve) (ITC) | kivéve: | ||||
| — repcemag és feldolgozott termékei | 4000 | ||||
| b) Teljes értékű takarmányok, | 150 | ||||
| kivéve: | |||||
| — teljes értékű takarmány kifejlett kérő- | 1000 | ||||
| dzőknek (kivéve a növendék állatot) | |||||
| — teljes értékű takarmány sertés (kivéve | 500 | ||||
| malacokat) és baromfi részére | |||||
| 12. Vinil-tio-oxazolidon | Baromfi teljes értékű takarmányok. | 1000 | |||
| (VTO) | kivéve: | ||||
| — tojóknak szánt teljes értékű takarmányok | 500 | ||||
| 13. Anyarozs | Darálatlan szemestermények | 1000 | |||
| (Claviceps purpurea) | |||||
| 14. Gyommagvak és termések, amelyek alkaloi- | Minden takarmány | 3000 | |||
| dákat, glikozidokat vagy más toxikus anya- | |||||
| gokat tartalmaznak külön-külön vagy az | |||||
| alábbiakat magába foglaló kombinációban: | |||||
| a) Lolium temulentum L. | 1000 | ||||
| (szédítő vadóc, konkolyperje) | |||||
| b) Lolium remotum | 1000 | ||||
| (len vadóc) Schrank | |||||
| c) Datura stramonium L. | 1000 | ||||
| (csattanó maszlag) | |||||
| 15. Ricinus — Ricinus communis L. (maghéj- | Minden takarmány-alapanyag | 10 | |||
| ban kifejezve) | |||||
| 16. Crotalariaspp. | Minden takarmány-alapanyag | 100 | |||
| 17. Aldrin egyedül vagy | Minden | 0,01 | |||
| együttesen | takarmány-alapanyag | ||||
| dieldrinben | kivéve: | ||||
| 18. Dieldrin kifejezve | — zsírok (állati és növényi) | 0,2 | |||
| 19. Camphechlor (Toxafen) | Minden takarmány | 0,1 | |||
| 20. Klordan (cisz- és transzizomerek és oxiklor- | Minden takarmány, | 0,02 | |||
| dán együtt, klordánban kifejezve) | kivéve: | ||||
| — zsírok (álltai és növényi) | 0,05 | ||||
| 21. DDT (DDT, TDE, DDE izomerek összege | Minden takarmány, | 0,05 | |||
| DDT-ben kifejezve) | kivéve: | ||||
| — zsírok (állati és növényi) | 0,5 | ||||
| 22. Endoszulfán (<A> és <B> izomerek és endoszulfán- | Az összes takarmány | 0,1 | |||
| szulfát összege endoszulfátban kifejezve) | kivéve: | ||||
| — kukorica és annak feldolgozásából nyert ter- | 0,2 | ||||
| mékek | |||||
| — olajos magvak és azok feldolgozásából nyert | 0,5 | ||||
| termékek | |||||
| — teljes értékű hal-takarmányok | 0,005 | ||||
| 23. Endrin (endrin és delta-keto-endrin össze- | Minden takarmány, | 0,01 | |||
| ge endrinben kifejezve) | kivéve: | ||||
| — zsírok (állati és növényi) | 0,05 | ||||
| 24. Heptaklór (a heptaklór és a heptaklór- | Minden takarmány, | 0,01 | |||
| epoxid összege heptaklórban kifejezve) | kivéve: | ||||
| — zsírok (állati és növényi) | 0,2 | ||||
| 25. Hexaklór-benzol | Minden takarmány, | 0,01 | |||
| (HCB) | kivéve: | ||||
| — zsírok (állati és növényi) | 0,2 | ||||
| 26. Hexaklór-ciklohexán (HCH) | |||||
| 26.1. alfa-izomerek | Minden takarmány, | 0,02 | |||
| kivéve: | |||||
| — zsírok (állati és növényi) | 0,2 | ||||
| 26.2. béta izomerek | a) Teljes értékű takarmány, | 0,01 | |||
| kivéve: | |||||
| — takarmány tejelő szarvasmarháknak | 0,005 | ||||
| b) Takarmány- alapanyagok, | 0,01 | ||||
| kivéve | |||||
| — zsírok (állati és növényi) | 0,1 | ||||
| 26.3. gamma-izomer | Minden takarmány, | 0,2 | |||
| kivéve: | |||||
| — zsírok (állati és növényi) | 2,0 | ||||
| 27. Dioxin (a polychlorinált dibenzo-para- | Minden növényi eredetű takarmány-alapanyag, | 0,75 ng WHO- | |||
| dioxinok (PCDD-k) és polychlorinált | beleértve a zöldség olajokat és a járulékos ter- | PCDD/F-TEQ/kg7 | |||
| dibenzo-furan-ok (PCDF-ek) összege a | mékeket | ||||
| Világegészségügyi Szervezet (WHO) toxi- | |||||
| kus egyenértékében kifejezve, felhasználva | Ásványok | 1,0 ng WHO- | |||
| a WHO — TEF-et (toxikus egyenérték fak- | PCDD/F-TEQ/kg7 | ||||
| tor, 1997) | Engedélyezett kötő, csomósodást gátló és per- | 0,75 ng WHO- | |||
| gést elősegítő csoportba tartozó ásványi eredetű | PCDD/F-TEQ/kg7 | ||||
| klinoptilolit, kaolin-agyag, kálcium-szulfát-hid- | |||||
| rát, vermikulit, nátrolit-fonolit, szintetikus-kál- | |||||
| cium-aluminátok | |||||
| Állati zsír, beleértve a tejzsírt és tojás zsírt | 2,0 ng WHO- | ||||
| PCDD/F-TEQ/kg7 | |||||
| Más szárazföldi állat termékek, beleértve a tejet | 0,75 ng WHO- | ||||
| és tejtermékeket, a tojást és tojástermékeket | PCDD/F-TEQ/kg7 | ||||
| Halolaj | 6 ng WHO- | ||||
| PCDD/F-TEQ/kg7 | |||||
| Hal, más vízi állatok, azok termékei és járulékos | 1,25 ng WHO- | ||||
| termékei, kivéve a halolajat és a 20%-nál több | PCDD/F-TEQ/kg7 | ||||
| zsírt tartalmazó hidrolizált halfehérjét8 | |||||
| Takarmánykeverékek, kivéve a prémes állatok | 0,75 ng WHO- | ||||
| takarmányait, a kedvtelésből tartott állatok és | PCDD/F-TEQ/kg7 | ||||
| halak takarmányait | |||||
| Halak és kedvtelésből tartott állatok takarmá- | 2,25 ng WHO- | ||||
| nyai | PCDD/F-TEQ/kg7 | ||||
| 20%-nál több zsírt tartalmazó hidrolizált hal- | 2,25 ng WHO- | ||||
| fehérje | PCDD/F-TEQ/kg7 | ||||
| Anyagok, vegyületek | Takarmányok | Maximális megengedett mennyiség mg/kg-ban | |||
| C. Botanikai szennyezések | Minden takarmány | A felsorolt növények magvai és termései, illetve feldolgozott származékai a takarmányokban csak nyomokban lehetnek jelen | |||
| 28. Kajszibarack — Prunus armeniaca L. | |||||
| 29. Keserű mandula — Prunus dulcis (Mill.) D.A. | |||||
| Webb var. amara (DC.) Focke [= Prunus amygda- | |||||
| lus Batsch var. amara (DC.) Focke] | |||||
| 30. Közönséges bükk — Fagus silvatica (L.) | |||||
| 31. Sárgarepce — Camelina sativa (L.) Crantz | |||||
| 32. Indiai mandukafa Bassia longifolia L. = Illipe | |||||
| malabrorum Engl.) Madhuca indica Gmelin [= Bassia latifolia | |||||
| (Roxb). = Illipe latifolia (Roscb.) F. Mueller) 33. Jatropha curcas L. (nincs magyar neve) | |||||
| 34. Croton tiglium L. (nincs magyar neve) | |||||
| 35. Indiai mustár —Brassica juncea (L.) Czern. és | |||||
| Coss. ssp. integrifolia (West.) Thell. | |||||
| 36. Szareptai mustár—Brassica juncea (L.) Czern. | |||||
| és Coss. ssp. juncea | |||||
| 37. Kínai mustár — Brassica juncea (L.) Czern. és | |||||
| Coss. ssp. juncea var. lutea Batalin | |||||
| 38. Fekete mustár — Brassica nigra (L.) Koch | |||||
| 39. Etiópiai mustár — Brassica carinata A. Braun | |||||
| Lábjegyzet 1A maximális szintek az összes arzénra vonatkoznak. 2A megyei (fővárosi) állategészségügyi és élelmiszer-ellenőrző állomás kérésére a takarmány-előállító üzemnek laboratóriumi vizsgálattal ke megállapítania azt, hogy a takarmány szervetlen arzén tartalma alacsonyabb 2 mg/kg-nál. Ez az elemzés rendkívül fontos a Hizikai fusiforme tengei alga fajokra. 3 P%-onként 125 mg F. 4Fluortartalom/l % foszfor. 5 1% foszforra eső kadmiumtartalom maximális értéke 0,5 mg. 61% foszforra eső kadmiumtartalom maximális értéke 0,75 mg. 7Felső határ koncentrációk: a felső határ koncentrációk kiszámítása azt feltételezi, hogy a különböző rokon vegyületek minden értéke kevesebb vagy egyenlő a meghatározási korláttal. 8A közvetlenül szállított friss hal, mely közbülső feldolgozás nélkül takarmányként kerül felhasználásra a prémes állatok részére, kivételt képez maximum szintek alól. 4,0 ng WHO-PCDD/F-TEQ/kg megengedett a kedvtelésből tartott állatok, állatkerti és cirkuszi állatok közvetlen etetésére szánt friss halaknál. F.zen állatokból (prémes állatok, kedvtelésből tartott állatok, állatkerti és cirkuszi állatok) készült termékek, állati proteinek nem kerülhetnek be a táplálékláncba és azok táplálékként történő felhasználása tilos az élelmiszer-termelő állatoknál." | |||||
| Anyagok, vegyületek | Takarmányok | Maximális megengedett mennyiség mg/kg-ban | |
| C. Botanikai szennyezések | Minden takarmány | A felsorolt növények magvai és termései, illetve feldolgozott származékai a takarmányokban csak nyomokban lehetnek jelen | |
| 28. Kajszibarack — Prunus armeniaca L. | |||
| 29. Keserű mandula — Prunus dulcis (Mill.) D.A. | |||
| Webb var. amara (DC.) Focke [= Prunus amygda- | |||
| lus Batsch var. amara (DC.) Focke] | |||
| 30. Közönséges bükk — Fagus silvatica (L.) | |||
| 31. Sárgarepce — Camelina sativa (L.) Crantz | |||
| 32. Indiai mandukafa Bassia longifolia L. = Illipe | |||
| malabrorum Engl.) Madhuca indica Gmelin [= Bassia latifolia | |||
| (Roxb). = Illipe latifolia (Roscb.) F. Mueller) 33. Jatropha curcas L. (nincs magyar neve) | |||
| 34. Croton tiglium L. (nincs magyar neve) | |||
| 35. Indiai mustár —Brassica juncea (L.) Czern. és | |||
| Coss. ssp. integrifolia (West.) Thell. | |||
| 36. Szareptai mustár—Brassica juncea (L.) Czern. | |||
| és Coss. ssp. juncea | |||
| 37. Kínai mustár — Brassica juncea (L.) Czern. és | |||
| Coss. ssp. juncea var. lutea Batalin | |||
| 38. Fekete mustár — Brassica nigra (L.) Koch | |||
| 39. Etiópiai mustár — Brassica carinata A. Braun | |||
| Lábjegyzet 1A maximális szintek az összes arzénra vonatkoznak. 2A megyei (fővárosi) állategészségügyi és élelmiszer-ellenőrző állomás kérésére a takarmány-előállító üzemnek laboratóriumi vizsgálattal ke megállapítania azt, hogy a takarmány szervetlen arzén tartalma alacsonyabb 2 mg/kg-nál. Ez az elemzés rendkívül fontos a Hizikai fusiforme tengei alga fajokra. 3 P%-onként 125 mg F. 4Fluortartalom/l % foszfor. 5 1% foszforra eső kadmiumtartalom maximális értéke 0,5 mg. 61% foszforra eső kadmiumtartalom maximális értéke 0,75 mg. 7Felső határ koncentrációk: a felső határ koncentrációk kiszámítása azt feltételezi, hogy a különböző rokon vegyületek minden értéke kevesebb vagy egyenlő a meghatározási korláttal. 8A közvetlenül szállított friss hal, mely közbülső feldolgozás nélkül takarmányként kerül felhasználásra a prémes állatok részére, kivételt képez maximum szintek alól. 4,0 ng WHO-PCDD/F-TEQ/kg megengedett a kedvtelésből tartott állatok, állatkerti és cirkuszi állatok közvetlen etetésére szánt friss halaknál. F.zen állatokból (prémes állatok, kedvtelésből tartott állatok, állatkerti és cirkuszi állatok) készült termékek, állati proteinek nem kerülhetnek be a táplálékláncba és azok táplálékként történő felhasználása tilos az élelmiszer-termelő állatoknál." | |||
III. Az R. 4. számú mellékletének módosítása
1. Az R. 4. számú melléklete II. fejezete "6. Színezőanyagok, beleértve a pigmenteket'' alfejezete a "12 Asztaxantinban gazdag Phaffia rhodozyma (ATCC 74219)" pontját követően a következő rendelkezésekkel egészül ki:
| EK szám | Adalékanyag | Kémiai leírás, név | Állatfaj vagy kategória | Maximum életkor | Minimum hatóanyag tartalom teljes értékű takarmány mg/kg-on-ként | Maximum hatóanyag tartalom teljes értékű takarmány mg/kg-on-ként- | Egyéb rendelkezések | Az engedély lejárati ideje |
| E 161g | Kantaxanthin | C40H52O2 | baromfiak, | — | — | 25 | A kantaxanthin más karotinoidokkal | Időkorlátozás |
| kivéve a tojó- | és a xantofillel adott keveréke megen- | nélkül | ||||||
| tyúkot | gedhető, feltéve, hogy a keverék | |||||||
| összes koncentrációja a teljesértékű | ||||||||
| takarmányban a 80 mg/kg-ot nem ha- | ||||||||
| ladja meg. | ||||||||
| tojótyúkok | __ | — | 8 | A kantaxanthin más karotinoidokkal | Időkorlátozás | |||
| és a xantofillel adott keveréke megen- | nélkül | |||||||
| gedhető, feltéve, hogy a keverék | ||||||||
| összes koncentrációja a teljesértékű | ||||||||
| takarmányban a 80 mg/kg-ot nem ha- | ||||||||
| ladja meg. | ||||||||
| pisztráng és lazac | — | — | 25 | Alkalmazása hat hónapos kortól meg- | Időkorlátozás | |||
| engedett. | nélkül | |||||||
| A kantaxanthin xantofillel adott keve- | ||||||||
| réke megengedhető, feltéve, hogy a | ||||||||
| keverék összes koncentrációja a | ||||||||
| teljesértékű takarmányban a | ||||||||
| 100 mg/kg-ot nem haladja meg. | ||||||||
| kutyák, macskák | __ | __ | __ | — ■ | Időkorlátozás | |||
| és díszhalak | nélkül | |||||||
| A közösségi rendeletek alapján enge- | — | minden állatfaj vagy állat kategó- ria, a kutyák és a macskák kivételével | — | —. | — | Csak az alábbiak feldolgozása során | Időkorlátozás | |
| délyezett élelmiszer színezőanyagok, | nyert takarmányokban megengedett: | nélkül | ||||||
| a Patent-kék V-t (E 131), a Brilliant- | a) élelmiszer-hulladékok, vagy | |||||||
| sav zöld BS-t (Lisszamin zöld) | b) más alapanyagok, kivéve a gabona | |||||||
| (E 142) és a kantaxanthint kivéve | magvakat és a manióka lisztet, ezek- | |||||||
| kel a szerekkel denaturálva, vagy a | ||||||||
| technikai előkészítés alatt színezve a | ||||||||
| gyártás során szükséges azonosítható- | ||||||||
| ság biztosítására. | ||||||||
| kutyák | __ | __ | __ | - | Időkorlátozás | |||
| nélkül | ||||||||
| macskák | __ | __ | __ | __ | Időkorlátozás | |||
| nélkül | ||||||||
| A közösségi rendeletek alapján enge- | minden állatfaj | __ | __ | __ | Csak az alábbiak feldolgozása során | Időkorlátozás | ||
| délyezett kanta-xanthin, mint élelmi- | vagy állat kategó- | nyert takarmányokban megengedett: | nélkül | |||||
| szer színezőanyag | ria, a baromfiak, a | a) élelmiszer-hulladékok, vagy | ||||||
| lazac, a pisztráng, | b) más alapanyagok, kivéve a gabona | |||||||
| a kutyák és a | magvakat és a manióka lisztet, ezek- | |||||||
| macskák kivéte- | kel a szerekkel denaturálva, vagy a | |||||||
| lével | technikai előkészítés alatt színezve a | |||||||
| > | gyártás során szükséges azonosítható- | |||||||
| ság biztosítására. | ||||||||
| kutyák | — | __ | — | __ | Időkorlátozás | |||
| nélkül | ||||||||
| macskák | __ | __ | __ | __ | Időkorlátozás | |||
| nélkül | ||||||||
| pisztráng, lazac, | __ | __ | 25 | Csak az alábbiak feldolgozása során | Időkorlátozás | |||
| és baromfiak, ki- | nyert takarmányokban megengedett: | nélkül | ||||||
| véve a tojótyúkot | a) élelmiszer-hulladékok, vagy | |||||||
| b) más alapanyagok, kivéve a gabona | ||||||||
| magvakat és a manióka lisztet, ezek- | ||||||||
| kel a szerekkel denaturálva, vagy a | ||||||||
| technikai előkészítés alatt színezve a | ||||||||
| gyártás során szükséges azonosítható- | ||||||||
| ság biztosítására. | ||||||||
| tojótyúkok | — | — | 8 | Csak az alábbiak feldolgozása során | Időkorlátozás | |||
| nyert takarmányokban megengedett: | nélkül" | |||||||
| a) élelmiszer-hulladékok, vagy | ||||||||
| b) más alapanyagok, kivéve a gabona | ||||||||
| magvakat, és a manióka lisztet, ezek- | ||||||||
| kel a szerekkel denaturálva, vagy a | ||||||||
| technikai előkészítés alatt színezve a | ||||||||
| gyártás során szükséges azonosítható- | ||||||||
| ság biztosítására. |
2. Az R. 4. számú melléklete II. fejezetének "7. Hozamfokozók" alfejezete helyébe a következő rendelkezés lép: "7. Hozamfokozók
| Az adalékanyag nyilvántartási száma | A forgalomba hozatalért felelős cég neve és nyilvántartási száma | Az adalékanyag neve (kereskedelmi elnevezés) | Összetétel, kémiai képlet, leírás | Állatfaj vagy kategória | Maximum életkor | Minimum | Maximum | Egyéb előírások | Az engedélyezési időtartam lejárata |
| adalékanyag tartalom teljes értékű takarmányokban (mg/kg) | |||||||||
| 1. | BASF Aktiengesellschaft a DE RP 1 31401 | Kálium-diformát (Formi™ LHS) | Adalékanyag összetétel: Kálium-diformát, szilárd állapotú: min. 98%, Szilikát: maximum 1.5%, Víz: maximum 0.5% Aktív hatóanyag: Kálium-diformát, szilárd KH(COOH)2 CAS szám: 2064205-1 | malacok (választott) | 2 hónap | 6 000 | 18 000 | 2005. 06. 30. | |
| vágósertések | - | 6 000 | 12 000 | 2005.06.30." | |||||
3. Az R. 4. számú melléklete II. fejezetének "9. Savasság szabályozók" alfejezete a következő rendelkezésekkel egészül ki:
| „Az adalékanyag nyilvántartási száma | Az adalékanyag neve (kereskedelmi elnevezés) | Összetétel, kémiai képlet, leírás | Állatfaj vagy kategória | Maximum életkor | Minimum tartalom | Maximum tartalom | Egyéb előírások | Az engedélyezési időtartam lejárata |
| aktivitási egység (CFU)/teljes értékű takarmány kg-onként | ||||||||
| E210 | Benzol-sav | C7H6O2 | vágósertések | — | 5 000 | 10 000 | — | 2007.05.25." |
4. Az R. 4. számú melléklete II. fejezetének "10. Nyomelemek" alfejezete helyébe a következő rendelkezés lép:
| „Adalékanyag | Alkalmazás | Maximális összes elemtartalom takarmány-keverék mg/kg min. max. | Egyéb rendelkezések | ||||
| EK szám | Megnevezés | Kémiai név/leírás | Állatfajok vagy kategóriák | Állat kora max. | Várakozási idő | a) alkalmazási korlátozások b) takarmánytípusok c) használati utasítások, ajánlások d) előállítástól függő speciális tulajdonságok e) speciális alkalmazások | |
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
| 10. Nyomelemek | |||||||
| E2 H H H H | Jód (J) mint Kalcium-jodát, heptahidrát Kalcium-jodát, vízmentes Kálium-jodid Nátrium-jodid | Ca(JO3)2 • 6H2O Ca(JO3)2 KJ NaJ | lófélék halak egyéb állatfajok és kategóriák | 4 20 10 | |||
| E7 | Molibdén (Mo) mint Ammonium-Molibdenát-4hidrát Nátrium-molibdát | (NH4)6Mo7O24 • 4H2O Na2MoO4 • 2H2O | minden | 2,5 | |||
| E8 H | Szelén (Se) mint Nátrium-szelenát Nátrium-szelenit | Na2SeO4 Na2SeO3 | minden | 0,5 | |||
| EK szám | Elem | Az adalékanyag neve | Összetétel, kémiai képlet, leírás | Az adott elem megengedhető legnagyobb tartalma a teljes takarmányra vetítve (mg/kg), illetve napi bevitelének megengedhető legnagyobb mennyisége (mg/nap) | Egyéb előírások | Az engedélyezési időtartam lejárata | ||||
| E1 | Vas-Fe | Vas-karbonát | FeCO3 | Juh: 500 mg/kg (össz) Kedvtelésből tartott állatok: 1250 mg/kg (össz) Malacok a választást megelőző 1 hetes korig: 250 mg/nap Egyéb állatfajok: 750 mg/kg (össz) | — | Időkorlátozás | ||||
| Vas-klorid-tetrahidrát | FeCl2 • 4H2O | nélkül | ||||||||
| Vas-klorid-hexahidrát | FeCl3 • 6H2O | |||||||||
| Vas-citrát-hexahidrát | Fe3(C6H5O7)2 • 6H2O | |||||||||
| Vas-fumarát | FeC4H2O4 | |||||||||
| Vas-laktát, trihidrát | Fe(C3H5O3)2 • 3H2O | |||||||||
| Vas-oxid | Fe2O3 | |||||||||
| Vas-szulfát-monohidrát | FeSO4H2O | |||||||||
| Aminósavak vas-kelát-hidrátja | Fe(x)1-3 nH2O (x= bármely aminosav anionja, amelyet hidrolizált szója-fehérjéből nyertek) Molekulatömege nem haladhatja meg az 1500-t. | |||||||||
| E3 | Kobalt-Co | Kobalt-acetát-tetrahidrát | Co(CH3COO)2 • 4H2O | 2(össz) | — | Időkorlátozás | ||||
| Elemi-kobalt-karbonát-mono-hidrát | 2CoCO3 • 3Co(OH)2 • H2O | nélkül | ||||||||
| Kobalt-klorid-hexahidrát | CoCl2 • 6H2O | |||||||||
| Kobalt-szulfát-heptahidrát | CoSO4 • 7H2O | |||||||||
| Kobalt-szulfát-monohidrát | CoSO4 • H2O | |||||||||
| Kobalt-nitrát-hexahidrát | Co(NO3)2 • 6H2O | |||||||||
| E4 | Réz-Cu | Réz-acetát-monohidrát | Cu(CH3COO)2 • H2O | Sertések: 1. 12 hetes korig: 170 (ossz) 2. egyéb sertés: 25 (össz) Szarvasmarha: 1. a kérődzés beindulásáig: — tejpótlók: 15 (össz) — egyéb teljesértékű takarmány: 15 (össz) | A címkén, illetve a kísérő okmányon fel kell tüntetni: 1. Juhoknál, amennyiben a takarmány réztartalma meghaladja a 10 mg/kg-ot: „A takarmányban található rézmennyiség bizonyos juh fajtáknál mérgezést okozhat.'' 2. Szarvasmarháknál a kérődzés beindulása után, amennyiben a réz mennyisége kevesebb, mint 20 mg/kg: „A takarmányban található réz-, mennyiség rézhiányt okozhat a | Időkorlátozás | ||||
| Elemi-réz-karbonát-monohidrát | CuCO3 • Cu(OH)2 • H2O | nélkül | ||||||||
| Réz-klorid-dihidrát | CuCl2 • 2H2O | |||||||||
| Réz-metionit | Cu(C5H10NO2S)2 | |||||||||
| Réz-oxid | CuO | |||||||||
| Réz-szulfát-pentahidrát | CuSO4 • 5H2O | |||||||||
| Rézkelátok-aminosav-hidrátja | Cu(x)1-3 nH2O (x= bármely aminósav anionja, amelyet hidrolizált szójafehérjéből nyertek) Molekulatömege nem haladhatja meg az 1500-t | 2. egyéb szarvasmarha: 35 (össz) Juh: 15 (össz) Halak: 25 (össz) Páncélos testűek: 50 (össz) Egyéb fajok: 25 (össz) | ||||||||
| Réz-lizin-szulfát | Cu(C6H13N2O2)2.SO4 | szarvasmarhákban a magas mo-libdén vagy foszfortartalmú legelőkön történő legeltetés során." | 2004.03.31. | |||||||
| E5 | Mangán-Mn | Mangán-karbonát | MnCO3 | Halak: 100 (össz) | — | Időkorlátozás | ||||
| Mangán-klorid-tetrahidrát | Mn Cl2 • 4H2O | Egyéb állatfajok: 150 (össz) | nélkül | |||||||
| Mangán-hidrogénfoszfát-trihidrát | MnHPO4.3H2O | |||||||||
| Mangán-oxid | MnO | |||||||||
| Mangano-oxid | Mn2O3 | |||||||||
| Mangán-szulfát-tetrahidrát | MnSo4 • 4H2O | |||||||||
| Mangán-szulfát-monohidrát | MnSo4 • H2O | |||||||||
| Aminósav-hidrátok-mangán-kelátja | Mn (x)1-3 • nH2O (x= bármely aminósav anionja, amelyet hidrolizált szója-fehérjéből nyertek) Molekulatömege nem haladhatja meg az 1500-t | |||||||||
| Mangano-mangán-oxid | MnO Mn2O3 | |||||||||
| E6 | Cink-Zn | Cink-laktát-trihidrát | Zn(C3H5O3)2 • 3H2O | Kedvtelésből tartott állatok: 250 (össz) Halak: 200 (össz) Tejpótlók: 200 (össz) Egyéb állatfajok: 150 (össz) | ■ — | Időkorlátozás nélkül | ||||
| Cink-acetát-dihidrát | Zn(CH3COO)2 • 2H2O | |||||||||
| Cink-karbonát Cink-klorid-monohidrát | ZnCO3 ZnCL2 • H2O | |||||||||
| Cink-oxid | ZnO Maximum ólomtartalom: 600 mg/kg | |||||||||
| Cink-szulfát-heptahidrát | ZnSO4 • 7H2O | |||||||||
| Cink-szulfát-monohidrát | ZnSO4 • H2O | |||||||||
| Amonósavak-hidrátjának-cink-kelátja | Zn (x)1-3 • nH2O (x= bármely aminósav anionja, amelyet hidrolizált szója-fehérjéből nyertek) Molekulatömege nem haladhatja meg az 1500-t" | |||||||||
5. Az R. 4. számú melléklete II. fejezete "13. Enzimek" alfejezetének 11. pontja a következő rendelkezéssel egészül ki:
| EK szám | Adalékanyag megnevezése, kereskedelmi neve | Kémiai név, leírás | Állatfaj vagy kategória | Maximum életkor | Minimum aktivitási egység teljes értékű takarmány kg-onként | Maximum aktivitási egység teljes értékű takarmány kg-onként | Egyéb rendelkezések | Az engedély lejárati ideje |
| 11 | „Endo-1,4-béta-glükanáz | A Trichoderma longibrachiatum | tojótyúk | — | Endo-l,4-bé- | — | 1. A használati utasításban meg kell adni az adalékanyag és az előkeverék tárolási hő- mérsékletét, eltarthatóságát és pelletálási stabilitását. 2. Ajánlott dózis egy kilog- ramm teljesértékű takarmány- ban: Endo-1,4-béta-glükanáz: 400 U—1 280 U Endo-l,(3)4-béta-glükanáz: 900 U—2 880 U Endo-1,4-béta-xilanáz: 1 300 U—4 160 U 3. Nem keményítő tartalmú poliszaharidokban gazdag (főleg arabinoxilán és béta glükán), pl. több mint 40% búzát, tritikálét vagy árpát tar- talmazó takarmánykeverékben való alkalmazásra. 1. A használati utasításban meg kell adni az adalékanyag és az előkeverék tárolási hőmérsékletét, eltarthatóságát és pelletálási stabilitását. 2. Ajánlott dózis egy kilogramm teljesértékű takarmányban: Endo- 1,4-béta-glükanáz: 400 U—1 600 U Endo-1,(3)4-béta-glükanáz: 900 U—3 600 U Endo-1,4-béta-xilanáz: 1 300 U—5 200 U 3. Nem keményítő tartalmú poliszaharidokban gazdag (főleg arabinoxilán és béta glü-kán), pl. több mint 40% búzát, tritikálét, kukoricát vagy búzát és 20% rozsot tartalmazó takarmánykeverékben való alkalmazásra. | 2007.01.01. |
| EC 3.2.1.4 | (ATCC 74252) | ta-glükanáz: | ||||||
| által termelt | 400 U | |||||||
| Endo-1,(3)4-béta-glükanáz | endo-1,4-béta-glükanáz, | |||||||
| EC 3.2.1.6 | endo-1,(3)4-béta-glükanáz, | Endo-1,(3)4- | — | |||||
| endo-1,4-béta-xilanáz | béta-glüka- | |||||||
| Endo-1,4-béta-xilanáz | készítmények, amelyek minimum | náz: | ||||||
| EC 3.2.1.8 | aktivitása: | 900 U | ||||||
| — | ||||||||
| Endo-1,4-béta-glükanáz: | Endo-l,4-bé- | |||||||
| 8 000 U(1)/g vagy ml | ta-xilanáz: | |||||||
| Endo-1,(3)4-béta-glükanáz: | 1300U | |||||||
| 18 000 U(2)/g vagy ml | ||||||||
| Endo-1,4-béta-xilanáz: | ||||||||
| 26 000 U(3)/g vagy ml | ||||||||
| (1)—(3) 1 U az az enzim mennyi- | ||||||||
| ség, amely 0,1 mikromol glükózt | ||||||||
| szabadít fel egy perc alatt 5,0 pH | ||||||||
| mellett 40 °C-on a | ||||||||
| (1)karboxi-metil-cellulózból | ||||||||
| (2) árpa-béta-glükánból | ||||||||
| (3) búza-xilánból | ||||||||
| 11 folytatása | malac | — | Endo-l,4-bé-ta-glükanáz: 400 U Endo-1,(3)4-béta-glüka-náz: 900 U Endo-1,4-béta-xilanáz: 1 300 U | — | 2007.01.01." | |||
| — |
6. Az R. 4. számú melléklete II. fejezetének "13. Enzimek" alfejezetének 24. pontja a következő rendelkezéssel egészül ki:
| „EK szám | Adalékanyag megnevezése, kereskedelmi neve | Kémiai leírás, név | Állatfaj vagy kategória | Maximum életkor | Minimum aktivitási egység teljes értékű takarmány kg-onként | Maximum aktivitási egység teljes értékű takarmány kg-onként | Egyéb rendelkezések | Az engedély lejárati ideje |
| 24 | (folytatás) | Az Aspergillus niger (CNCM I— 1517) által termelt endo-l,4-béta-xilanáz készítmény és endo-l,3(4)-béta-glükanáz készítmény, amelyek minimum aktivitása: | tojótyúk | 1. A használati utasításban meg kell adni az adalékanyag és az előkeverék tárolási hőmérsékletét, eltarthatóságát és pelletálási stabilitását. | 2006. 10. 01. | |||
| Endo-1,4-béta-xilanáz: 28 000 QXU/g 1 QXU az az enzim mennyiség, amely 1 mikromol (xilóz ekvivalensként mért) oldható cukrot egy perc alatt 5,1 pH mellett 50 °C-on a búza-xilánból felszabadít. Endo-1,3(4)-béta-glükanáz: 140 000 QGU/g 1 QGU az az enzim mennyiség, amely 1 mikromol (glükóz ekvivalensként mért) oldható cukrot egy perc alatt 4,8 pH mellett 50 °C-on az árpa-béta-glükánból felszabadít. | 560 QXU 2800 QGU | 2. Ajánlott dózis egy kilogramm teljes értékű takarmányban: 560 QXU 2800 QGU 3. Nem keményítő tartalmú poliszaharidokban gazdag (főleg arabinoxilán és béta glükán), pl. több mint 20% búzát és/vagy árpát tartalmazó takarmánykeverékben való alkalmazásra. | ||||||
| 24. | (folytatás) | Az Aspergillus niger (CNCM I— 1517) által termelt endo-1,4-béta-xilanáz készítmény és endo-l,3(4)-béta-glükanáz készítmény, amely minimum aktivitása: 28 000 QXU/g 1 QXU az az enzim mennyiség, amely 1 mikromol (xilóz ekvivalensként mért) oldható cukrot egy perc alatt 5,1 pH mellett 50 °C-on a búza-xilánból felszabadít 140 000 QGU/g 1 OGU az az enzim mennyiség, amely 1 mikromol (glükóz ekvivalensként mért) oldható cukrot egy perc alatt 4,8 pH mellett 50 °C-on az árpa-béta-glükánból felszabadít. | brojlerpulyka | 280 QXU 1400 QGU | 840 QXU 4200 QGU | 1. A használati utasításban meg kell adni az adalékanyag és az előkeverék tárolási hőmérsékletét, eltarthatóságát és pelletálási stabilitását. 2. Ajánlott adag egy kilogramm teljes értékű takarmányban: 560 QXU 2800 QGU 3. Nem keményítő tartalmú poliszaharidokban gazdag takarmánykeverékben való alkalmazásra (főleg béta gluká-nok és arabinoszilánok), például több mint 20% búzát és/vagy árpát tartalmazó takarmánykeverékekben. | 2007.02. 28." |
7, Az R. 4. számú melléklete II. fejezetének "13. Enzimek" alfejezetének 25. pontjában "Az engedély lejárati ideje" oszlopban a "2004. 06. 30." szövegrész helyébe az "időkorlátozás nélkül" szövegrész lép.
8. Az R. 4. számú melléklete II. fejezetének "13. Enzimek" alfejezetének 34. pontja a következő rendelkezéssel egészül ki:
| „EK szám | Adalékanyag megnevezése, kereskedelmi neve | Kémiai leírás, név | Állatfaj vagy kategória | Maximum életkor | Minimum aktivitási egység teljes értékű takarmány kg-onként | Maximum aktivitási egység teljes értékű takarmány kg-onként | Egyéb rendelkezések | Az engedély lejárati ideje |
| 1601 | Endo-l,3(4)-béta-glükanáz EG 3.2.1.6 Endo-1,4-béta-xilanáz EC 3.2.1.8 | Az Aspergillus niger (NRRL 25541) által termelt Endo-1,3(4)-béta-glükanáz és Endo-1,4-béta-xilanáz készítmény, amely minimum aktivitása: Endo-l,3(4)-béta-glükanáz 1100 U/g 1 U az az enzim mennyiség, amely 1 mikromol (glükóz ekvivalensként mért) oldható cukrot 1 perc alatt 30 °C-on, 4,0 pH mellett a zabbéta-glükánból felszabadít. Endo-1,4 béta-xilanáz: 1600 U/g 1 U az az enzim mennyiség, amely 1 mikromol (xilóz ekvivalensként mért) oldható cukrot 1 perc alatt 30 °C-on, 4,0 pH mellett a zab-xi-lánból felszabadít. | brojlercsirkék | Endo-1,3(4)-béta-glükanáz 138 U Endo-1,4-béta-xilanáz 200 U | 1. A használati utasításban meg kell adni az adalékanyag és az előkeverék tárolási hőmérsékletét, eltarthatóságát és pelletálási stabilitását. 2. Ajánlott adag egy kilogramm teljes értékű takarmányban: Endo-l,3(4)-béta-glükanáz: 138 U Endo-l,4-béta-xilanáz: 200 U 3. Nem keményítő tartalmú poliszaharidokban gazdag takarmánykeverékben való alkalmazásra (főleg béta gluká-nok és arabinoszilánok), például dietikus gabonamagvakat (például: búzát, árpát, rozsot, tritikálét) tartalmazó takarmánykeverékekben. | Időkorlátozás nélkül" |
9. Az R. 4. számú melléklete II. fejezetének "13. Enzimek" alfejezetének 50. pontja a következő rendelkezéssel egészül ki;
| "EK szám | Adalékanyag megnevezése, kereskedelmi neve | Kémiai leírás, név | Állatfaj vagy kategória | Maximum életkor | Minimum aktivitási egység teljes értékű takarmány kg-onként | Maximum aktivitási egység teljes értékű takarmány kg-onként | Egyéb rendelkezések | Az engedély lejárati ideje |
| 50. | (folytatás) | tenyészkocák | 750 FYT | 1. A használati utasításban meg kell adni az adalékanyag és az előkeverék tárolási hőmérsékletét, eltarthatóságát és pelletálási stabilitását. 2. Ajánlott adag egy kilogramm teljes értékű takarmányban: 750—1000 FYT 3. Több mint 0,25% fitin-kö-tésű foszfort tartalmazó takarmánykeverékben való alkalmazásra. | 2007.02.01." |
10. Az R. 4. számú melléklete II. fejezetének "13. Enzimek" alfejezetének 51. pontja helyébe a következő rendelkezés lép:
| "EK szám | Adalékanyag megnevezése, kereskedelmi neve | Kémiai leírás, név | Állatfaj vagy kategória | Maximum életkor | Minimum aktivitási egység teljes értékű takarmány kg-onként | Maximum aktivitási egység teljes értékű takarmány kg-onként | Egyéb rendelkezések | Az engedély lejárati ideje |
| 51 H | Endo-1,4-béta-xilanáz EC 3.2.1.8 BELFEED | A Bacillus subtilis (LMG S— 15136) által termelt endo-1,4-béta-xilanáz készítmény, amely minimum aktivitása: endo-1,4-béta-xilanáz: folyékony: 100 IU/ml 1 IU az az enzim mennyiség, amely 1 mikromol (xilóz ekvivalensként mért) oldható cukrot szabadít fel egy perc alatt 4,5 pH mellett 30 °C-on a nyírfa-xilánból. | brojler-csirke | 10 IU | 1. A használati utasításban meg kell adni az adalékanyag és az előkeverék tárolási hőmérsékletét, eltarthatóságát és pelletálási stabilitását. 2. Ajánlott dózis egy kilogramm teljes értékű takarmányban: 10 IU 3. Arabinoxilánban gazdag, pl. több mint 40% búzát vagy árpát tartalmazó takarmánykeverékben való alkalmazásra. | 2007.01.01. | ||
| A Bacillus subtilis (LMG S— 15136) által termelt endo-1,4-bé-ta-xilanáz készítmény, amely minimum aktivitása: endo-1,4-béta-xilanáz: szilárd és folyékony: 100 IU/g vagy ml 1 1U az az enzim mennyiség, amely 1 mikromol (xilóz ekvivalensként mért) oldható cukrot szabadít fel egy perc alatt 4,5 pH mellett 30 °C-on a nyírfa-xilánból. | brojlerpulyka | 10 IU | 1. A használati utasításban meg kell adni az adalékanyag és az előkeverék tárolási hőmérsékletét, eltarthatóságát és pelletálási stabilitását. 2. Ajánlott dózis egy kilogramm teljes értékű takarmányban: 10 IU 3. Arabinoxilánban gazdag, pl. több mint 40% búzát vagy árpát tartalmazó takarmánykeverékben való alkalmazásra. | 2007.01.01. | ||||
| 51. | Endo-1,4-béta-xilanáz EC 3.2.1.8 | A Bacillus subtilis (LMG S— 15136) által termelt endo-1,4-bé-ta-xilanáz (EC 3.2.1.8) készítmény, amely minimum aktivitása Endo-1,4-béta-xilanáz: 100 IU/g 1 IU az az enzim mennyiség, amely 1 mikromol (xilóz ekvivalensként mért) redukáló cukrot egy perc alatt 4,5 pH mellett 30 °C-on a nyírfa-xilánból felszabadít. | vágó-sertések | 10 IU | 1. A használati utasításban meg kell adni az adalékanyag és az előkeverék tárolási hőmérsékletét, eltarthatóságát és pelletálási stabilitását. 2. Ajánlott adag egy kilogramm teljes értékű takarmányban: Endo-1,4-béta-xilanáz: 10 IU/kg 3. Arabinoxilánban gazdag takarmánykeverékben való alkalmazásra, (például több mint 40% búzát vagy árpát tartalmazót takarmánykeverékben). | 2007.01.01." |
11. Az R. 4. számú melléklete II. fejezetének "14. Mikroorganizmusok" alfejezete "3. Saccharomyces cerevisiae NCYC Sc 47" sorában a "tejelőtehenek" és "hízómarhák" rész helyébe a következő rendelkezések lépnek:
| Állatfaj vagy kategória | Maximum életkor | Telepképző egység CFU teljes értékű takarmány kg-onként | Egyéb rendelkezések | Az engedély lejárati ideje | |
| minimum tartalom | maximum tartalom | ||||
| tejelőtehenek | 4x108 | 2x109 | A használati utasításban meg kell adni az adalékanyag és az előkeverék tárolási hőmérsékletét, eltarthatóságát és pelletálási stabilitását. A használati utasításban meg kell adni: A Saccharomyces cerevisiae napi adagja 100 kg élőtömegre számítva nem haladhatja meg a 4x108 CFU-t. Minden +100 kg élőtömegre 2x109 CFU-t kell számítani. | 2005.05.31 | |
| hízómarhák | 4x109 | 8x109 | A használati utasításban meg kell adni az adalékanyag és az előkeverék tárolási hőmérsékletét, eltarthatóságát és pelletálási stabilitását. A használati utasításban meg kell adni: A Saccharomyces cerevisiae napi adagja 100 kg élőtömegre számítva nem haladhatja meg a 2x1010 CFU-t. Minden + 100 kg élőtömegre 0,5x1010 CFU-t kell számítani. | Időkorlátozás nélkül" | |
12. Az R. 4. számú melléklete II. fejezetének "14. Mikroorganizmusok" alfejezete a következő ponttal egészül ki:
| "EK szám | Adalékanyag megnevezése, kereskedelmi neve | Kémiai leírás, név | Állatfaj vagy kategória | Maximum életkor | Telepképző egység (CFU) teljes értékű takarmány kg-onként | Egyéb rendelkezések | Az engedély lejárati ideje | |
| minimum tartalom | maximum tartalom | |||||||
| 22. | Enterococcus faecium DSM 7134 | Enterococcus faecium készítmény, amely minimum aktivitása: porformában: 1x10 aktivitási egység (CFU)/adalékanyag grammonként. | malacok vágósertések | — — | 0,5xl09 0,2x109 | 4x109 1x109 | A használati utasításban meg kell adni az adalékanyag és az előkeverék tárolási hőmérsékletét, eltarthatóságát és pelletá-lási stabilitását. | 2007.04. 15." |
| granulált formában: (mikrokapszulázott) 1x1010 aktivitási egység (CFU)/adalékanyag grammonként. | ||||||||
13. Az R. 4. számú melléklete a következő III. fejezettel egészül ki:[1]
IV.Az R. 5. számú mellékletének módosítása
1. Az R. 5. számú mellékletének 2. pontjához tartozó 1. és 2. lábjegyzet helyébe a következő rendelkezések lépnek:
1A nyersfehérje tartalmat a 10. számú melléklet IV. módszerével kell meghatározni.
"2 A nyers zsírtartalmat a 10. számú melléklet XI. számú módszerével kell meghatározni."
2.Az R. 5. számú mellékletének 6. pontja helyébe a következő rendelkezés lép:
"6. A cukortartalmat a 10. számú melléklet XIII. számú módszerével, a keményítőtartalmat a 10. számú melléklet XV. számú módszerével kell meghatározni."
V. Az R. 6. számú mellékletének módosítása
Az R. 6. számú melléklete A. Része I. fejezete 3.3. és 3.4. pontja helyébe a következő rendelkezések lépnek:
"3.3. Szakirányú felsőfokú végzettségnek tekintendő az agrár üzemmérnök, az okleveles agrármérnök, a takarmánygazdálkodási üzemmérnök, az okleveles takarmánygazdálkodási szakmérnök, a mezőgazdasági gépész üzemmérnök, az okleveles mezőgazdasági gépészmérnök, az állatorvos, a gyógyszerész, az okleveles vegyészmérnök, az okleveles agrárvegyész, az élelmiszer-technológus üzemmérnök és az élelmiszer-ipari menedzser.
3.4. Középfokú szakirányú végzettségnek tekintendő a mezőgazdasági gépész- és vegyésztechnikus, valamint az élelmiszer-ipari technikus (malom és keveréktakarmány gyártó)."
VI. Az R. 10. számú mellékletének módosítása
Az R. 10. számú melléklete a következő XXXVII-XLVII. fejezettel egészül ki:
"XXXVII.
A spiramicin diffúzióval történő meghatározása agar táptalajban
1. Cél és alkalmazási terület
A módszer a spiramicin takarmányokban és előkeverékekben történő meghatározására szolgál. A meghatározás alsó méréshatára 1 mg/kg (1 ppm).1
2. Vizsgálati alapelv
A mintát metil-alkohol/foszfát-bikarbonát puffer pH 8-as keverékével extraháljuk. A kivonatot dekantáljuk vagy centrifugáljuk és hígítjuk. Antibiotikus aktivitását a spiramicin Micrococcus luteussal beoltott agar táptalajon végbemenő diffúziójának mérésével határozzuk meg. A diffúziót a mikroorganizmus gátlási zónáinak képződése jelzi. Ezen zónák átmérője egyenes arányban áll az antibiotikum koncentrációnak az alkalmazott antibiotikum koncentrációk tartománya feletti logaritmusával.
3.Mikroorganizmus: Micrococcus luteus ATCC 9341 (NCTC 8340, NCIB 8553)
3.1. A törzstenyészet fenntartása
Oltsunk be ferde táptalajokat (4.1.) tartalmazó csöveket Micrococcus luteussal, majd inkubáljuk 30 °C-on 24 órán át. A tenyészetet hűtőgépben tároljuk, 4 °C körüli hőmérsékleten. Kéthetenként oltsuk át.
3.2. A baktérium-szuszpenzió elkészítése
2-3 ml nátrium-klorid oldat (4.3.) segítségével gyűjtsük be egy nemrégiben elkészített ferde agarról (3.1.) a növekményt. Ezzel a szuszpenzióval oltsunk be 250 ml, Roux lombikban lévő táptalajt (4.1.) és inkubáljuk 30 °C-on 18-20 órán át. Gyűjtsük be a növekményt 25 ml nátrium-klorid oldatban (4.3.), majd keverjük össze. Hígítsuk fel a szuszpenziót nátrium-klorid oldattal (4.3.) 1/10 arányban. A szuszpenziónak 650 nm-en, 1 cm-es cellában nátrium-klorid oldat (4.3.) ellenében mért fényáteresztő képessége 75% körüli kell, hogy legyen. Ez a szuszpenzió 4 °G körüli hőmérsékleten egy hétig tárolható.
4. Táptalajok és reagensek
| 4.1. Alap táptalaj2 | ||
| Hús pepton | 6,0 g | |
| Tripton | 4,0 g | |
| Élesztőkivonat | 3,0 g | |
| Húskivonat | 1,5 g | |
| Glükóz | 1,0 g | |
| Agar | 10,0-20,0 g | |
| Víz | 1 000 ml | |
| pH 6,5-6,6 (sterilizálás után) | ||
| 4.2. Vizsgálati táptalaj3 | ||
| Tripton | 5,0g | |
| Élesztőkivonat | 4,0 g | |
| Húskivonat | 3,0 g | |
| Agar | 10,0-20,0 g | |
| Víz | 1 000 ml | |
| pH 8,0 (sterilizálás után) | ||
| 4.3. 0,8%-os (w/v) nátrium-klorid oldat | ||
| Vízben oldjunk fel 8 g nátrium-kloridot, majd hígítsuk 1 000 ml-re; sterilizáljuk. | ||
| 2 Más módszerek is alkalmazhatók, ha megállapítást nyert, hogy azok hasonló baktérium szuszpenziókat eredményeznek. | ||
| 3 Minden hasonló összetételű és ugyanezeket az eredményeket adó kereskedelmi táptalaj felhasználható. | ||
| 4.4. Foszfát-bikarbonát puffer, pH 8,0 | ||
| Dikálium-hidrogén-foszfát K2HPO4 | 16,7 g | |
| Kálium-dihidrogén-foszfát KH2PO4 | 0,5 g | |
| Nátrium-hidrogén-karbonát NaHCO3 | 20,0 g | |
| Vízzel kiegészítve | 1 000 ml-re | |
4.5. Metilalkohol/foszfát-bikarbonát puffer keverék (4.4.)
50/50 (v/v).
4.6. Standard anyag
Ismert aktivitású spiramicin (NE-ben).
5. Standard oldatok
Oldjuk fel a standard anyag (4.6.) pontosan kimért mennyiségét a keverékben (4.5.), majd hígítsuk fel ugyanazzal a keverékkel, hogy egy 1000 NE/ml spiramicint tartalmazó törzsoldatot kapjunk. Lezárt lombikban 4 °C-on tárolva az oldat 5 napig eltartható.
A törzsoldatnak a keverékkel (4.5.) történő többszöri hígításával készítsük el a következő oldatokat:
| S8 | 1 | NE/ml |
| S4 | 0,5 | NE/ml |
| S2 | 0,25 | NE/ml |
| S1 | 0,125 | NE/ml |
6. A kivonat- és vizsgálati oldatok elkészítése
6.1.Extrakció
Mérjünk ki takarmányok esetében 20,0 g, előkeverékek esetében 1,0-20,0 g mintát. Adjunk hozzá 100 ml-t a keverékből (4.5.), majd rázzuk 30 percig. Centrifugáljuk vagy dekantáljuk, majd a felülúszó oldatot hígítsuk fel a keverékkel (4.5.), hogy megközelítőleg 1 NE/ml spiramicin-tartalmat (= U8) kapjunk.
Azoknál a takarmányoknál, ahol a várható spiramicin-szintek 2,5 mg/kg-nál alacsonyabbak, ott az extrakciót a következők szerint kell elvégezni: Mérjünk ki 20 g mintamennyiséget. Adjunk hozzá 100 ml-t a keverékből (4.5.), majd rázzuk 30 percig. Centrifugáljuk néhány percig, vegyünk ki a felülúszó oldatból 50 ml-t és azt egy rotációs bepárlókészülékben, csökkentett nyomáson, 40 °C-ot meg nem haladó hőmérsékleten pároljuk be körülbelül 4 ml-re. A maradékot hígítsuk fel a keverékkel (4.5.), hogy megközelítőleg 1 NE/ml spiramicin-tartalmat (= U8) kapjunk.
6.2. Vizsgálati oldatok
Az U8 oldatnak a keverékkel (4.5.) történő többszöri hígításával (1+1) készítsünk U4 (várható tartalom: 0,5 NE/ml), U2 (várható tartalom: 0,25 NE/ml) és U1 (várható tartalom: 0,125 NE/ml) oldatokat.
7. A vizsgálat módja
7.1. A vizsgálati táptalaj beoltása
Oltsuk be a vizsgálati táptalajt (4.2.) a baktérium-szuszpenzióval (3.2.), körülbelül 50 °C-on. A vizsgálati táptalajt (4.2.) tartalmazó lemezeken végzett előzetes vizsgálatok segítségével határozzuk meg azt a szükséges baktérium-szuszpenzió mennyiséget, amely a különböző spiramicin koncentrációkkal a legnagyobb és legtisztább gátlási zónákat adja.
7.2. A lemezek elkészítése
Az agardiffúziót a négy standard oldat koncentrációval (S8, S4, S2, S1) és a négy vizsgálati oldat koncentrációval (U8, U4, U2, U1) öntött lemezeken végezzük. A standardnak és a kivonatnak ezt a négy-négy koncentrációját minden egyes lemezen el kell helyezni. Ennek érdekében megfelelő nagyságú lemezeket kell választanunk ahhoz, hogy az agar táptalajon legalább nyolc darab, 10-13 mm átmérőjű, egymástól legalább 30 mm távolságra lévő lyukakat fúrhassunk. A vizsgálat olyan lemezeken végezhető el, amelyek egy üveglapból és egy 200 mm átmérőjű és 20 mm magas csiszolt alumínium vagy műanyag karimagyűrűből állnak.
A lemezekre öntsünk annyi, a 7.1. pont szerint beoltott táptalajt (4.2.), hogy az egy körülbelül 2 mm vastagságú réteget képezzen (200 mm átmérőjű lemez esetében ez 60 ml). Hagyjuk egyenletesen eloszlani, fúrjuk ki a lyukakat, és helyezzük el bennük a vizsgálati és standard oldatok pontosan kimért mennyiségeit (az átmérő függvényében lyukanként 0,10 és 0,15 ml között). Minden koncentrációt legalább négy alkalommal alkalmazzunk, hogy minden meghatározás 32 gátlási zóna értékelésén alapuljon.
7.3. Inkubálás
Inkubáljuk a lemezeket 30 ± 2 °C-on, 16-18 órán át.
8. Kiértékelés
8.1. A gátlási zónák átmérőjét 0,1 mm-es pontossággal mérjük meg. Fél-logaritmikus milliméterpapírra jegyezzük fel minden koncentrációnál a mérések középértékeit, jelezve a koncentrációknál a gátlási zónák átmérőihez viszonyított logaritmusát. Szerkesszük meg mind a standard oldat, mind a kivonat regressziós vonalát, például az alábbiak szerint.
8.2 Határozzuk meg a standard legalacsonyabb szint (SA) regressziós pontját a következő képlet segítségével:
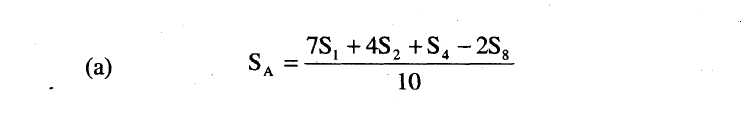
8.3. Határozzuk meg a standard legmagasabb szint (SM) regressziós pontját a következő képlet segítségével:

8.4.Hasonló módon számítsuk ki a regressziós pontokat a kivonat legalacsonyabb szintjére (UA) és a kivonat legmagasabb szintjére (UM) a fenti képletekben az s1, s2, s4 és s8 helyeire u1, u2, u4 és u8 behelyettesítésével.4
8.5.Jegyezzük fel a kiszámított SA és SM értékeket ugyanarra a milliméterpapírra és kössük össze őket, hogy kiadják a standard oldat regressziós vonalát. Hasonló módon jegyezzük fel az UA és UM értékeket és kössük össze őket, hogy kiadják a kivonat regressziós vonalát.
8.6. Ha nem lép fel interferencia, a vonalaknak párhuzamosaknak kell lenniük. Gyakorlati megfontolásból a vonalakat párhuzamosnak tekinthetjük, ha az (SM - SA) és (UM - UA) középértékeitől való eltérése nem haladja meg a 10%-ot.
8.7.Ha a vonalak nem tekinthetők párhuzamosnak, akkor vagy az u1 és s1, vagy az u8 és s8 elhanyagolható, az SA, SM, UA és UM értékek pedig alternatív képletekkel számolhatók ki, hogy megkapjuk az alternatív regressziós vonalakat:
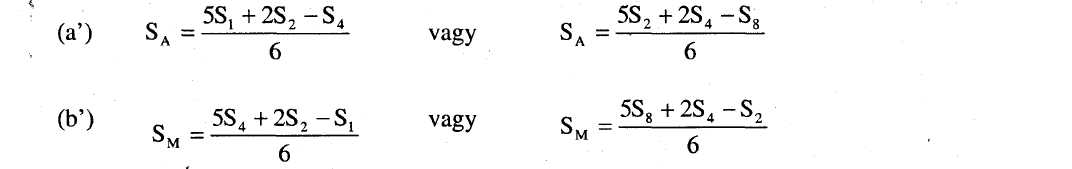
és hasonló módon az UA-ra és UM-ra vonatkozóan. Ugyanaz a párhuzamossági feltétel érvényes ebben az esetben is. Azt a tényt, hogy az eredmény kiszámítása három tényező alapján történt, fel kell tüntetni a zárójelentésen.
8.8 Ha a vonalak párhuzamosnak tekinthetőek, akkor számoljuk ki a relatív aktivitás logaritmusát (log A) a következő képletek egyikének felhasználásával, attól függően, hogy három vagy négy tényező alapján történt a párhuzamosság megállapítása.

8.9.Ha a relatív aktivitás a 0,5 és 2,0 közötti tartományon kívül esik, akkor ismételjük meg a vizsgálatot a kivonat koncentrációin, vagy ahol ez nem lehetséges, ott a standard oldatokon végzett megfelelő módosításokkal. Ha a relatív aktivitást nem tudjuk az előírt tartományon belül tartani, akkor bármely kapott eredményt közelítő eredménynek kell tekinteni, és ezt fel kell tüntetni a zárójelentésen.
8.10.Ha a vonalak nem tekinthetőek párhuzamosnak, meg kell ismételni a meghatározást. Ha a párhuzamosságot még ezután sem sikerül elérni, akkor a meghatározást nem kielégítőnek kell tekinteni.
8.11.Az eredményt a takarmány kilogrammjában lévő spiramicinbázis milligrammos értékében fejezzük ki.
9. Ismételhetőség
Az ugyanazon mintán, ugyanazon analitikus által, párhuzamosan végzett két meghatározás eredménye közötti különbség nem haladhatja meg:
-a 10 mg/kg alatti spiramicinbázis tartalmak esetén a 2 mg/kg-ot, abszolút értékben,
-a 10 és 25 mg/kg közötti tartalmak esetén a legmagasabb érték 20%-át,
-a 25 és 50 mg/kg közötti tartalmak esetén az 5 mg/kg-ot, abszolút értékben,
-az 50 mg/kg feletti tartalmak esetén a legmagasabb érték 10%-át.
XXXVIII.
Virginiamicin meghatározása agar táptalajon történő diffúzióval
1. Cél és alkalmazási terület
A módszer a virginiamicin takarmányokban és előkeverékekben történő meghatározására szolgál. A meghatározás alsó határa 2 mg/kg (2 ppm).5
2. Vizsgálati alapelv
A mintát Tween 80 metilalkoholos oldatával extraháljuk. A kivonatot dekantáljuk vagy centrifugáljuk, majd felhígítjuk. Antibiotikus aktivitását a virginiamicin Micrococcus luteussal beoltott agar táptalajban történő diffúziójának mérésével határozzuk meg. A diffúziót a mikroorganizmus gátlási zónáinak képződése jelzi. Ezeknek a zónáknak az átmérője egyenes arányosnak tekinthető az antibiotikum koncentrációnak az alkalmazott antibiotikum koncentrációk tartományával szembeni logaritmusával.
3. Mikroorganizmus: Micrococcus luteus ATCC 9341 (NCTC 8340, NCIB 8553) 3.1. A törzstenyészet fenntartása
Oltsunk be ferde táptalajokat (4.1.) tartalmazó csöveket Micrococcus luteussal, majd inkubáljuk 30 °C-on 24 órán át. A tenyészetet hűtőgépben tároljuk, 4 °C körüli hőmérsékleten. Kéthetenként oltsuk át.
3.2. A baktérium-szuszpenzió elkészítése
2-3 ml nátrium-klorid oldat (4.3.) segítségével gyűjtsük be egy nemrégiben elkészített ferde agarról (3.1.) a növekményt. Ezzel a szuszpenzióval oltsunk be 250 ml, Roux lombikban lévő táptalajt (4.1.) és inkubáljuk 30 °C-on 18-20 órán át. Gyűjtsük be a növekményt 25 ml nátrium-klorid (4.3.) oldatban, majd keverjük össze. Hígítsuk fel a szuszpenziót nátrium-klorid oldattal (4.3.) 1/10 arányban. A szuszpenziónak a 650 nm-en, 1 cm-es cellában mért fényáteresztő képességének a nátrium-klorid (4.3.) ellenében 75% körülinek kell lennie. Ez a szuszpenzió 4 ÓC körüli hőmérsékleten egy hétig tárolható.
4. Táptalajok és reagensek
| 4.1. Alap- és vizsgálati táptalaj | |
| Hús pepton | 6,0 g |
| Tripton | 4,0 g |
| Élesztőkivonat | 3,0 g |
| Húskivonat | 1,5 g |
| Glükóz | 1,0 g |
| Agar | 10,0-20,0 g |
| Víz | 1000 ml |
| pH 6,5 (sterilizálás után) | |
| 4.2. Foszfát puffer, pH 6 | |
| Kálium-hidrogén-foszfát K2HPO4 | 2,0 g |
| Kálium-dihidrogén-foszfát KH2PO4 | 8,0g |
| Vízzel kiegészítve | 1000 ml-re |
4.3. 0,8%-os (w/v) nátrium-klorid oldat: oldjunk fel vízben 8 g nátrium-kloridot, majd hígítsuk 1000 ml-re; sterilizáljuk.
4.4. Metil-alkohol.
4.5. Foszfát puffer (4.2.) metil-alkohol (4.4.) keveréke: 80/20 (v/v)
4.6. Tween 80 0,5%-os (w/v) metil-alkoholos oldata: oldjunk fel metil-alkoholban 5 g Tween 80-at, majd metil-alkohollal hígítsuk 1000 ml-re.
4.7. Standard anyag: ismert aktivitású virginiamicin.
5. Standard oldatok
Oldjuk fel a standard anyag (4.7.) pontosan kimért mennyiségét metil-alkoholban (4.4.), majd hígítsuk fel metil-alkohollal, hogy 1000 μg/ml virginiamicint tartalmazó törzsoldatot kapjunk. Lezárt lombikban 4 °C-on tárolva az oldat 5 napig eltartható. Ebből a törzsoldatból, a keverékkel (4.5.) történő egymást követő hígításokkal készítsük el a következő oldatokat:
| s8 | 1 | µg/ml |
| s4 | 0,5 | µg/ml |
| s2 | 0,25 | µg/ml |
| s1 | 0,125 | µg/ml |
6. A kivonat és a vizsgálati oldatok elkészítése
6.1. Extrakció
6.1.1. 100 mg/kg vagy az alatti virginiamicin tartalmú termékek.
Mérjünk ki 50 g mintát, adjunk hozzá 200 ml oldatot (4.6.), majd rázzuk 30 percig. Hagyjuk leülepedni vagy centrifugáljuk, majd vegyünk ki a felülúszó oldatból 20 ml-t és azt egy rotációs bepárlóban, 40 °C-ot meg nem haladó hőmérsékleten pároljuk be körülbelül 5 ml-re. A maradékot hígítsuk fel a keverékkel (4.5.), hogy megközelítőleg 1 μg/ml virginiamicin-tartalmat (= u8) kapjunk.
6.1.2. 100 mg/kg-nál nagyobb virginiamicin tartalmú termékek. Mérjünk ki 10,0 g-ot meg nem haladó mintamennyiséget a mintából, amely 1 és 50 mg közötti virginiamicint tartalmaz, adjunk hozzá 100 ml oldatot (4.6), majd rázzuk 30 percig. Hagyjuk leülepedni vagy centrifugáljuk, majd a felülúszó oldatot hígítsuk fel a keverékkel (4.5), hogy megközelítőleg 1 μg/ml virginiamicin tartalmat (= u8) kapjunk.
6.2. Vizsgálati oldatok
Az u8 oldatból a keverékkel (4.5.) történő egymást követő hígítással (1+1) készítsünk u4 (várható tartalom: 0,5 μg/ml), u2 (várható tartalom: 0,25 μg/ml) és u1 (várható tartalom: 0,125 μg/ml) oldatokat.
7. A vizsgálat módja
7.1. A vizsgálati táptalaj beoltása
Oltsuk be a vizsgálati táptalajt (4.1.) a baktérium-szuszpenzióval (3.2.), körülbelül 50 °C-on. A vizsgálati táptalajt (4.1.) tartalmazó lemezeken végzett előzetes vizsgálatokkal határozzuk meg azt a szükséges baktérium-szuszpenzió mennyiséget, amely a különböző virginiamicin koncentrációkkal a legnagyobb és legtisztább gátlási zónákat adja.
7.2. A lemezek elkészítése
Az agardiffúziót a négy standard oldat koncentrációjával (s8, s4, s2, és s1) és a négy vizsgálati oldat koncentrációjával (u8, u4, u2, u1) öntött lemezeken végezzük. A standard és a kivonat ezen négy-négy koncentrációját minden egyes lemezre fel kell vinni. Ezért megfelelő nagyságú lemezeket kell választanunk ahhoz, hogy az agar táptalajban legalább nyolc darab, 10-13 mm átmérőjű, egymástól legalább 30 mm távolságra lévő lyukat alakíthassunk ki. A vizsgálat olyan lemezeken végezhető el, amelyek egy üveglapból és egy annak tetejére helyezett, 200 mm átmérőjű és 20 mm magas csiszolt alumínium vagy műanyag karimagyűrűből állnak.
A lemezekre öntsünk annyi, a 7.1. pont szerint beoltott táptalajt (4.1.), hogy az egy körülbelül 2 mm vastagságú réteget alkosson (200 mm átmérőjű lemez esetében ez 60 ml). Hagyjuk egyenletesen eloszlani, fúrjuk ki a lyukakat és helyezzük el bennük a vizsgálati és standard oldatok pontosan kimért mennyiségeit (az átmérő függvényében lyukanként 0,10 és 0,15 ml közötti mennyiség). Minden koncentrációt legalább négyszer alkalmazzunk, hogy minden meghatározás 32 gátlási zóna kiértékelése alapján történjen.
7.3. Inkubáció
Inkubáljuk a lemezeket 30 ± 2 °C-on, 16-18 órán át.
8. Kiértékelés
8.1.Mérjük meg a gátlási zónák átmérőjét 0,1 mm-es pontossággal. Fél-logaritmikus milliméterpapírra jegyezzük fel az egyes koncentrációknál mért eredmények középértékeit, jelezve a koncentrációk logaritmusát a gátlási zónák átmérőivel összefüggésben. Szerkesszük meg mind a standard oldat, mind a kivonat regressziós vonalát, például az alábbiak szerint.
8.2.Határozzuk meg a standard legalacsonyabb szint (SA) regressziós pontját a következő képlet segítségével: .
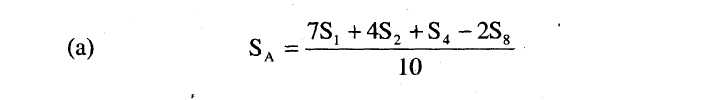
8.3. Határozzuk meg a standard legmagasabb szint (SM) regressziós pontját a következő képlet segítségével:
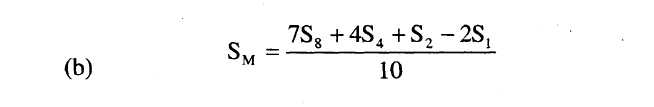
8.4.Hasonló módon számítsuk ki a regressziós pontokat a kivonat legalacsonyabb szintjére (UA) és a kivonat legmagasabb szintjére (UM), a fenti képletekben az s1, s2, s4 és s8 értékek helyett az u1, u2, u4 és u8 értékeket használva.
8.5.Jegyezzük fel a kiszámított SA és SM értékeket ugyanarra a milliméterpapírra és kössük ösz-sze őket, hogy megkapjuk a standard oldat regressziós vonalát. Hasonló módon jegyezzük fel az UA és UM értékeket és kössük össze őket, hogy megkapjuk a kivonat regressziós vonalát.
8.6. Ha interferencia nem lép fel, a vonalaknak párhuzamosnak kell lenniük. Gyakorlati megfontolásból a vonalakat párhuzamosnak tekinthetjük, ha az (SM - SA) és (UM - UA) érték középértékeiktől való eltérése nem haladja meg a 10%-ot.
8.7. Ha a vonalak nem tekinthetőek párhuzamosnak, akkor akár az u1 és s1, akár az u8 és s8 elhagyható, az SA, SM, UA és UM értékek az alternatív képletekkel számolhatók ki, hogy megkapjuk az alternatív regressziós vonalakat:

és hasonló módon az UA-ra és UM-ra vonatkozóan. Ugyanazt a párhuzamossági feltételt kell teljesíte-ni. Azt a tényt, hogy az eredmény három tényező alapján került kiszámításra, fel kell tüntetni a zárójelentésben.
8.8. Ha a vonalak párhuzamosnak tekinthetőek, akkor számoljuk ki a relatív aktivitás logaritmusát (log A) a következő képletek egyikének felhasználásával, attól függően, hogy három vagy négy tényezőből történt-e a párhuzamosság megállapítása.

A mintakivonat aktivitása = a vonatkozó standard aktivitása x A
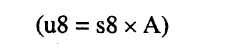
8.9.Ha a relatív aktivitás a 0,5 és 2,0 közötti tartományon kívül esik, akkor ismételjük meg a vizsgálatot a kivonat koncentrációin, vagy ahol ez nem lehetséges, ott a standard oldatokon végzett megfelelő módosításokkal. Ha a relatív aktivitást nem tudjuk az említett tartományon belülre korrigálni, akkor bármely kapott eredményt közelítő eredménynek kell tekinteni, és ezt fel kell tüntetni a zárójelentésben. .
8.10. Ha a vonalak nem tekinthetők párhuzamosnak, ismételjük meg a meghatározást. Ha a párhuzamosságot még ezután sem sikerült elérni, akkor a meghatározást nem kielégítőnek kell tekinteni.
8.11. Az eredményt milligrammban kifejezett virginiamicinre és kilogrammban kifejezett takarmányra vonatkoztatva fejezzük ki.
9. Ismételhetőség
Az ugyanazon mintán, ugyanazon analitikus által párhuzamosan végrehajtott két meghatározás eredménye közötti különbség nem haladhatja meg:
-a 10 mg/kg alatti virginiamicin-tartalom esetén a 2 mg/kg-ot, abszolút értékben,
-a 10 és 25 mg/kg közötti tartalom esetén a legmagasabb érték 20%-át,
-a 25 és 50 mg/kg közötti tartalom esetén az 5 mg/kg-ot, abszolút értékben,
-az 50 mg/kg feletti tartalom esetén a legmagasabb érték 10%-át.
XXXIX.
Cink bacitracin meghatározása agar táptalajon végzett diffúzióval
I. Cél és alkalmazási terület
A módszer a cink bacitracin takarmányokban és előkeverékekben történő meghatározására szolgál. A meghatározás alsó határa 5 mg/kg (5 ppm).8
2. Vizsgálati alapelv
A pH 2-es mintát metil-alkohol/víz/sósav keverékével és nátrium-szulfid oldattal extraháljuk. A nátrium-szulfid hozzáadása a vizsgálat során esetleg interferenciát eredményező oldható réz-sók ki-csapatására szolgál. A kivonatot beállítjuk pH 6,5-re, koncentráljuk (szükség esetén) és felhígítjuk. Antibiotikus aktivitását a cink bacitracin Micrococcus luteussal (flavussal) beoltott agar táptalajon végzett diffúziójának mérésével határozzuk meg. A diffúziót a mikroorganizmus gátlási zónáinak képződése mutatja. Ezeknek a zónáknak az átmérője egyenes arányban áll az antibiotikum koncentrációnak az alkalmazott antibiotikum koncentrációk tartománya feletti logaritmusával.
3. Mikroorganizmus: Micrococcus luteus (flavus) ATCC 10240
3.1. A törzstenyészet fenntartása
Oltsunk be ferde táptalajokat (4.1.) tartalmazó csöveket Micrococcus luteussal (flavus), majd inkubáljuk 30°C-on 24 órán át. A tenyészetet hűtőgépben tároljuk, 4 °C körüli hőmérsékleten. Kéthetenként oltsuk át.
3.2. A baktérium-szuszpenzió elkészítése
2-3 ml nátrium-klorid oldat (4.3.) segítségével gyűjtsük be egy nemrégiben elkészített ferde agarról (3.1.) a növekményt. Ezzel a szuszpenzióval oltsunk be 250 ml, Roux-lombikban lévő táptalajt (4.1.) és inkubáljuk 30 °C-on 18-20 órán át. Gyűjtsük be a növekményt 25 ml nátrium-klorid oldatban (4.3.), majd keverjük össze. Hígítsuk fel a szuszpenziót nátrium-klorid oldattal (4.3.) 1:10 arányban. A szuszpenziónak 650 nm-en, 1 cm-es cellában, a nátrium-klorid oldat (4.3.) ellenében mért fényáteresztő képességének 75% körülinek kell lennie. Ez a szuszpenzió 4 °C körüli hőmérsékleten egy hétig tárolható.
4. Táptalajok és reagensek
| 4.1. Alaptáptalaj | |
| Hús pepton | 6,0 g |
| Tripton | 4,0 g |
| Élesztőkivonat | 3,0 g |
| Húskivonat | 1,5 g |
| Glükóz | 1,0 g |
| Agar | 10,0-20,0 g |
| Víz | 1000 ml |
| pH 6,5-6,6 (sterilizálás után) | |
| 4.2.. Vizsgálati táptalaj11 | |
| Tripton | 10,0 g |
| Élesztőkivonat | 3,0 g |
| Hűskivonat | 1.5 g |
| Glükóz | 1,0 g |
| Agar | 10,0-20,0 g |
| Tween 80 | 1 ml |
| Víz | 1000 ml |
| pH 6,5 (sterilizálás után) |
4.3. 0,8%-os (w/v) nátrium-klorid oldat: oldjunk fel vízben 8 g nátrium-kloridot, majd hígítsuk 1000 ml-re; sterilizáljuk.
4.4. Metil-alkohol/víz/sósav keveréke (4.6): 80/17,5/2,5 (v/v/v).
| 4.5. Foszfát puffer, pH 6,5: | |
| Kálium.hidrogén-foszfát K2HPO2 Kálium-dihidrogén-foszfát KH2PO4 Vízzel kiegészítve | 22,15 g 27,85 g 1 000 ml-re |
4.6. Sósav (d: 1,18-1,19).
4.7. Sósav (0,1 M).
4.8. Nátrium-hidroxid 1 M oldat.
4.9. Nátrium-szulfid, körülbelül 0,5 M oldat.
4.10. Brómkrezol lila oldat 0,04% (w/v): oldjunk fel 0,1 g brómkrezol lilát 18,5 ml 0,01 M nátriumhidroxid oldatban. Töltsük fel vízzel 250 ml-re, majd keverjük össze.
4.11. Standard anyag: ismert aktivitású cink bacitracin (n.e.-ben).
5. Standard oldatok
Mérjünk ki (a jelzett aktivitás szerint) 1 050 n.e.-nek megfelelő standard cink bacitracint (4.11.). Adjunk hozzá 5 ml 0,1 M sósavat (4.7.) és 15 percig hagyjuk állni. Adjunk hozzá 30 ml vizet, foszfát pufferrel (4.5.) (kb. 4 ml) állítsuk be pH 4,5-re, vízzel töltsük fel 50 ml-re, majd alaposan keverjük össze (1 ml = 21 n.e.). Ebből a törzsoldatból, foszfát pufferrel (4.5.) történő egymást követő hígítással készítsük el a következő oldatokat:
| s8 | 0,42 | n.e./ml |
| s4 | 0,21 | n.e./ml |
| s2 | 0,105 | n.e./ml |
| s1 | 0,0525 | n.e./ml |
6. A kivonat és a vizsgálati oldatok elkészítése
6.1. Extrakció
6.1.1.Előkeverékek és ásványi takarmányok
Mérjünk ki 2,0-5,0 g mintát, adjunk hozzá 29,0 ml-t a keverékből (4.4.), valamint 1,0 ml nátriumszulfid oldatot (4.9.), majd rázassuk rövid ideig. Ellenőrizzük, hogy a pH érték 2 körül legyen. Rázzuk 10 percig, majd adjunk hozzá 30 ml foszfát puffert (4.5.), rázzuk 15 percig, majd centrifugáljuk. A felülúszó oldatból vegyünk egy megfelelő aliquot mennyiséget és pH-mérő vagy brómkrezol lila oldat (4.10.) indikátor segítségével, 1 M nátrium-hidroxid oldattal (4.8.) állítsuk be a pH-t 6,5-re. Hígítsuk fel foszfát pufferrel (4.5.), hogy megközelítően 0,42 n.e./ml cink bacitracin tartalmat (= u8) kapjunk.
6.1.2.Fehérje-koncentrátumok
Mérjünk ki 10,0 g mintát, adjunk hozzá 49,0 ml-t a keverékből (4.4.), valamint 1,0 ml nátrium-szulfid oldatot (4.9.), majd rázassuk rövid ideig. Ellenőrizzük, hogy a pH-érték 2 körül legyen. Rázzuk 10 percig, majd adjunk hozzá 50 ml foszfát puffert (4.5.), rázzuk 15 percig, majd centrifugáljuk. A felülúszó oldatból vegyünk egy megfelelő mennyiséget és pH-mérő vagy brómkrezol lila oldat (4.10.) indikátor segítségével, 1 M nátrium-hidroxid oldattal (4.8.) állítsuk be a pH-t 6,5-re. Rotációs bepárlóban, 35 °C-ot meg nem haladó hőmérsékleten pároljuk be körülbelül a mennyiség felét. Hígítsuk fel foszfát pufferrel (4.5.), hogy megközelítőleg 0,42 n.e./ml cink bacitracin tartalmat (= u8) kapjunk.
6.1.3.Egyéb takarmányok
Mérjünk ki 10,0 g mintát (20 grammot 5 mg/kg-os várt cink bacitracin tartalom esetében). Adjunk hozzá 24,0 ml-t a keverékből (4.4.), valamint 1,0 ml nátrium-szulfid oldatot (4.9.), majd 10 percig homogenizáljuk. Adjunk hozzá 25 ml foszfátpuffert (4.5.), rázzuk 15 percig, majd centrifugáljuk. A felülúszó oldatból vegyünk 20 ml-t és pH-mérő vagy brómkrezol lila oldat (4.10.) indikátor segítségével, 1 M nátrium-hidroxid oldattal (4.8.) állítsuk be a pH-t 6,5-re. Rotációs bepárlóban, 35 °C-ot meg nem haladó hőmérsékleten pároljunk be körülbelül 4 ml-re. A maradék anyagot hígítsuk fel foszfátpufferrel (4.5.), hogy 0,42 n.e./ml-es várt cink bacitracin tartalmat (= u8) kapjunk.
6.2. Vizsgálati oldatok
Az u8 oldatból a foszfátpufferrel (4.5.) való egymást követő hígításokkal (1+1) készítsünk u4 (várt tartalom: 0,21 n.e./ml), u2 (várt tartalom: 0,105 n.e./ml) és u1 (várt tartalom: 0,0525 n.e./ml) oldatokat.
7. A vizsgálat módja
7.1. A vizsgálati táptalaj beoltása
Oltsuk be a vizsgálati táptalajt (4.2.) a baktérium-szuszpenzióval (3.2.), körülbelül 50 °C-on. A vizsgálati táptalajjal (4.2.) öntött lemezeken végzett előzetes vizsgálatokkal határozzuk meg azt a szükséges baktérium-szuszpenzió mennyiséget, amely a különböző cink bacitracin koncentrációkkal a legnagyobb és legtisztább gátlási zónákat adja.
7.2. A lemezek elkészítése
Az agardiffúziót a négy standard oldat koncentrációjával (s8, s4, s2, s1) és a négy vizsgálati oldat koncentrációjával (u8, u4, u2, u1 ) öntött lemezeken végezzük. A standardnak és a kivonatnak ezt a négynégy koncentrációját minden egyes lemezre fel kell vinnünk. Ezért megfelelő nagyságú lemezeket kell választanunk ahhoz, hogy az agar táptalajban legalább nyolc darab, 10-13 mm átmérőjű, egymástól legalább 30 mm távolságra lévő lyukat alakíthassunk ki. A vizsgálat olyan lemezeken végezhető el, amelyek egy üveglapból és egy annak tetejére helyezett 200 mm átmérőjű és 20 mm magas csiszolt alumínium vagy műanyag karimagyűrűből állnak. A lemezekre öntsünk annyi, a 7.1. pont szerint beoltott táptalajt (4.2.), hogy az egy körülbelül 2 mm vastagságú réteget alkosson (200 mm átmérőjű lemez esetében ez 60 ml). Hagyjuk egyenletesen eloszlani, fúrjuk ki a lyukakat és helyezzük el bennük a vizsgálati és standard oldatok pontosan kimért mennyiségeit (az átmérő függvényében lyukanként 0,10-0,15 ml). Minden koncentrációt legalább négyszer alkalmazzunk, hogy minden meghatározás 32 gátlási zóna kiértékelése alapján történjen.
7.3. Inkubáció
Inkubáljuk a lemezeket 30 ± 2 °C-on, 16-18 órán át.
8.Kiértékelés
8.1.Mérjük meg a gátlási zónák átmérőjét 0,1 mm-es pontossággal. Fél-logaritmikus milliméterpapírra jegyezzük fel az egyes koncentrációknál mért eredmények középértékeit, jelezve a koncentrációknak a gátlási zónák átmérőihez vonatkoztatott logaritmusát. Szerkesszük meg mind a standard oldat, mind a kivonat regressziós vonalát, például az alábbiak szerint.
8.2.Határozzuk meg a standard legalacsonyabb szint (SA) regressziós pontját a következő képlet segítségével:
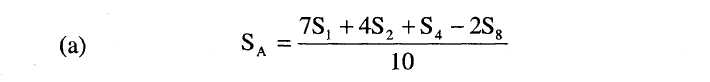
8.3. Határozzuk meg a standard legmagasabb szint (SM) regressziós pontját a következő képlet segítségével:
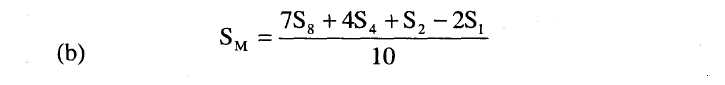
8.4.Hasonló módon számítsuk ki a regressziós pontokat a kivonat legalacsonyabb szintjére (UA) és a kivonat legmagasabb szintjére (UM), a fenti képletekben az s1, s2, s4 és s8 értékek helyett az u1, u2, u4 és u8 értékeket használva.
8.5. Jegyezzük fel a kiszámított SA és SM értékeket ugyanarra a milliméterpapírra és kössük össze őket, hogy megkapjuk a standard oldat regressziós vonalát. Hasonló módon jegyezzük fel az UA és UM értékeket és kössük össze őket, hogy megkapjuk a kivonat regressziós vonalát.
8.6.Ha interferencia nem lép fel, a vonalaknak párhuzamosnak kell lenniük/Gyakorlati megfontolásból a vonalakat párhuzamosnak tekinthetjük, ha az (SM - SA) és (UM - UA) érték középértékeiktől való eltérése nem haladja meg a 10%-ot.
8-.7. Ha a vonalak nem tekinthetőek párhuzamosnak, akkor akár az u1 és s1, akár az u8 és s8 elhagyható, és az SA, SM, UA és UM értékek alternatív képletekkel számolhatók ki, hogy megkapjuk az alternatív regressziós vonalakat:

és hasonló módon az UA-ra és UM-ra vonatkozóan. Ugyanazt a párhuzamossági feltételt kell teljesíteni. Azt a tényt, hogy az eredmény három tényező alapján került kiszámításra, fel kell tüntetni a zárójelentésben.
8.8. Ha a vonalak párhuzamosnak tekinthetőek, akkor számoljuk ki a relatív aktivitás (A) logaritmusát (log A) a következő képletek egyikének felhasználásával, attól függően, hogy három vagy négy tényező alapján történt-e a párhuzamosság megállapítása.

A mintakivonat aktivitása = a vonatkozó standard aktivitása x A
8.9.Ha a relatív aktivitás a 0,5 és 2,0 közötti tartományon kívül esik, akkor ismételjük meg a vizsgálatot a kivonat koncentrációin, vagy ahol ez nem lehetséges, ott a standard oldatokon végzett megfelelő módosításokkal. Ha a relatív aktivitást nem tudjuk az előírt tartományon belülre korrigálni, akkor bármely kapott eredményt közelítő eredménynek kell tekinteni, és ezt fel kell tüntetni a zárójelentésen.
8.10. Ha a vonalak nem tekinthetőek párhuzamosnak, meg kell ismételni a meghatározást. Ha a párhuzamosságot még ezután nem sikerült elérni, akkor a meghatározást nem kielégítőnek kell tekinteni.
8.11. Az eredményt milligrammban kifejezett cink bacitracinre és kilogrammban kifejezett takarmányra vonatkoztatva fejezzük ki.
9. Ismételhetőség
Az ugyanazon mintán, ugyanazon analitikus által párhuzamosan végzett két meghatározás eredménye közötti különbség nem haladhatja meg:
-a 10 mg/kg alatti cink bacitracin tartalom esetén a 2 mg/kg-ot, abszolút értékben,
-a 10 és 25 mg/kg közötti tartalom esetén a legmagasabb érték 20%-át,
-a 25 és 50 mg/kg közötti tartalom esetén az 5 mg/kg-ot, abszolút értékben,
-az 50 mg/kg feletti tartalom esetén a legmagasabb érték 10%-át.
XL.
Metil-benzokvát meghatározása 7-benziloxi-6-butil-3-metoxikarbonil-4-kvinolon
1. Cél és alkalmazási terület
A módszer a metil-benzokvát takarmányokban történő kimutatására szolgál. A meghatározás alsó határértéke 1 mg/kg.
2. Vizsgálati alapelv ;
A metil-benzokvátot a mintából metil-alkoholos metánszulfonsav-oldattal extraháljuk. A kivonatot diklórmetánnal, ioncserélő kromatográfiás módszerrel derítjük, majd ismét diklórmetánnal kezeljük. A metil-benzokvát-tartalom meghatározását fordított fázisú, nagynyomású folyadékkromatográfiás eljárással (HPLC) és UV detektor alkalmazásával végezzük.
3.Reagensek
3.1. Diklórmetán
3.2. Metil-alkohol, HPLC minőségű
3.3. HPLC mobil fázis:
Metil-alkohol (3.2.) és víz (HPLC minőségű) 75 + 25 (v + v) térfogatarányú keveréke.
Szűrjük át egy 0,22 μm-es szűrőn (4.5.), és gázmentesítsük az oldatot (például tízperces ultrahangos kezeléssel).
3.4. Metánszulfonsav-oldat, <O> = 2%
Hígítsunk 20,0 ml metánszulfonsav-oldatot metil-alkohollal (3.2.) 1000 ml-re.
3.5. Sósavoldat, <O> = 10% .
Hígítsunk 100 ml sósavat (P20 c. 1,18 g/ml) vízzel 1000 ml-re.
3.6. Amberlite CG-120 (Na) kationcserélő műgyanta, 100 és 200-as térháló
A gyantát felhasználás előtt előkezeljük: 100 g gyantát mossunk át 500 ml sósavoldattal (3.5.), és állandó keverés mellett hevítsük forrásig forró főzőlapon. Hagyjuk kihűlni, és öntsük le róla a savat. Vákuum mellett szűrjük át egy szűrőpapíron. Mossuk át kétszer a gyantát 500 ml vízzel, majd 250 ml metil-alkohollal (3.2:). További 250 ml metil-alkohollal öblítsük a gyantát, és a szűrőpogácsán keresztül engedett levegővel szárítsuk. A száraz gyantát lezárt üvegben tároljuk.
3.7. Standard anyag: tiszta metil-benzokvát (7-benziloxi-6-butil-3-metoxicarbonil-4-kvinolon)
3.7.1. Metil-benzokvát standard törzsoldat, 500 μg/ml
Mérjünk ki 0,1 mg-os pontossággal, 50 mg standard anyagot (3.7.), oldjuk fel metánszulfonsav-oldattal (3.4.) egy 100 ml-es mérőlombikban, töltsük fel jelig, és keverjük el.
3.7.2. Metil-benzokvát standard közbenső oldat, 50 μg/ml
Öntsünk át 5,0 ml metil-benzokvát standard törzsoldatot (3.7.1.) egy 50 ml-es mérőlombikba, töltsük fel metil-alkohollal (3.2.) jelig, és keverjük el.
3.7.3.Kalibráló oldatok
25 ml-es mérőlombikokba adagoljunk 1,0, 2,0, 3,0, 4,0, illetve 5,0 ml metil-benzokvát standard közbenső oldatot (3.7.2.). A mobil fázissal (3.3.) töltsük fel jelig, és keverjük el. Ezen oldatok 2,0, 4,0, 6,0, 8.0. illetve 10,0 μg/ml metil-benzokvát koncentrációnak felelnek meg. Az oldatokat használat előtt frissen kell elkészíteni.
4. Eszközök
4.1. Laboratóriumi rázógép
4.2. Rotációs, vékonyfilmes bepárló
4.3. Elzárócsappal és egy megközelítőleg 200 m-es tartállyal ellátott, 15 mm belső átmérőjű és 250 mm magas üvegoszlop
4.4. HPLC készülék, változtatható hullámhosszú UV detektorral vagy diódasoros detektorral
4.4.1. Folyadékkromatográfiás oszlop: 300 mm x 4 mm, C18, 10 μm-es töltet, vagy egy ezzel egyenértékű oszlop
4.5. Membránszűrők, 0,22 μm
4.6. Membránszűrők, 0,45 μm
5. A vizsgálat módja
5.1. Általános tudnivalók
5.1.1. Végezzünk vakpróbát egy referenciamintán annak ellenőrzésére, hogy sem metil-benzokvát, sem interferáló anyagok nincsenek jelen.
5.1.2. A mintában találhatóval azonos mennyiségű metil-benzokvát hozzáadásával dúsított referenciaminta analízisével végezzünk visszanyerési próbát. 15 mg/kg-os szintre történő dúsításhoz, adjunk 600 ml standard törzsoldatot (3.7.1.) 20 g referenciamintához, mindezt keverjük el, majd 10 percet várjunk, mielőtt folytatnánk a műveletet az extrakcióval (5.2.).
Megjegyzés: e módszer alkalmazásában a referenciaminta a mintával azonos fajtájú takarmány legyen, és abban az analízis során metil-benzokvát ne legyen kimutatható.
5.2. Extrakció
Mérjünk ki az előkészített mintából 0,01 g pontossággal 20 g-ot, és öntsük át egy 250 ml-es Erlenmeyer lombikba. Adjunk hozzá 100,0 ml metánszulfonsav-oldatot (3.4.), és rázzuk mechanikus rázógépben (4.1.) 30 percig. Szűrjük át az oldatot szűrőpapíron, és tegyük félre a szűrletet a folyadékfolyadék elválasztáshoz (5.3.).
5.3. Folyadék-folyadék elválasztás
Egy 100 ml sósavoldatot (3.5.) tartalmazó 500 ml-es elválasztótölcsérbe öntsünk át 25,0 ml-t az 5.2. lépésben nyert szűrletből. Adjunk hozzá 100 ml diklórmetánt (3.1.) a tölcsérhez, és rázzuk egy percen keresztül. Engedjük, hogy a rétegek elváljanak egymástól, és az alsó réteget (a diklórmetánt) eresszük bele egy 500 ml-es gömblombikba. Ismételjük meg a vizes fázis extrakcióját még kétszer, további 40.40 ml diklórmetánnal, majd vegyítsük az első kivonattal a gömblombikban. A rotációs, vékonyfilmes bepárlókészüléken (4.2.), csökkentett nyomás és 40 °C hőmérséklet mellett teljes száradásig pároljuk be a diklórmetán kivonatot. Oldjuk fel a maradékot 20.25 ml metil-alkoholban (3.2.), zárjuk le az üveget, és tartsuk meg az egész kivonatot az ioncserélő kromatográfiához (5.4.).
5.4. Ioncserélő kromatográfia
5.4.1. A kationcserélő oszlop előkészítése
Az üvegoszlop (4.3.) alsó végébe illesszünk egy kis üvegvatta dugót. Készítsünk a kezelt kationcserélő műgyanta (3.6.) 5,0 grammjából és 50 ml sósavból (3.5.) egy elegyet, majd öntsük bele az üvegoszlopba, majd hagyjuk, hogy leülepedjen. Engedjük ki a felesleges savat, hogy még éppen
ellepje a gyanta felszínét, és addig mossuk az oszlopot vízzel, amíg a szüredéken végzett lakmuszteszt semleges nem lesz. Öntsünk át 50 ml metil-alkoholt (3.2.) az oszlopra, és hagyjuk, hogy leszivárogjon a gyanta felszínén.
5.4.2. Oszlopkromatográfia
Az (5.3.) pontban készített mintakivonatot pipettázzuk óvatosan az oszlop tetejére. A gömblombikot két adag, 5-10 ml mennyiségű metil-alkohollal (3.2.) öblítsük, és öntsük ezen mosófolyadékot is rá az oszlopra. Futtassuk le a kivonatot a gyanta felszínén, és mossuk az oszlopot 50 ml metil-alkohollal, ügyelve arra, hogy az átfolyási sebesség ne haladja meg az 5 ml/percet. Öntsük el a szüredéket. Eluáljuk az oszlopról a metil-benzokvátot 150 ml metánszulfonsav-oldattal (3.4.), majd vegyük fel az eluátumot egy 250 ml-es gömblombikban.
5.5. Folyadék-folyadék elválasztás
5.5.1. Egy 1 l-es elválasztótölcsérbe öntsük át az (5.4.2.) lépésben nyert eluátumot. Az Erlenmeyer-lombikot 5.10 ml metil-alkohollal (3.2.) öblítsük, és a mosófolyadékot öntsük össze az elválasztó tölcsér tartalmával. Adjunk hozzá 300 ml sósavoldatot (3.5.) és 130 ml diklórmetánt (3.1.). Rázzuk, egy percen keresztül, és engedjük, hogy a fázisok elváljanak egymástól. Az alsó réteget (a diklórmetánt) eresszük bele egy 500 ml-es gömblombikba. Ismételjük meg a vizes fázis extrakcióját még kétszer, további 70-70 ml diklórmetánnal, és ezen kivonatokat öntsük össze a gömblombikban lévő első kivonattal.
5.5.2. A rotációs, vékonyfilmes bepárlókészüléken (4.2.), csökkentett nyomás és 40 °C hőmérséklet mellett teljes száradásig pároljuk be a diklórmetán kivonatot. Oldjuk fel a maradékot megközelítőleg 5 ml metil-alkoholban (3.2.), és öntsük az oldatot teljes mennyiségben egy 10 ml-es mérőlombikba. Öblítsük el még kétszer a lombikot 1.2 ml metil-alkohollal, majd öntsük a mérőlombikba. Töltsük fel jelig metil-alkohollal, és keverjük el. Egy aliquot részt szűrjünk át egy membránszűrőn (4.6.). Ezt az oldatot tegyük el a HPLC meghatározáshoz (5.6.).
5.6. HPLC meghatározás 5.6.1. Paraméterek
Az alábbi vizsgálati feltételek csak iránymutatóként szolgálnak, ettől eltérő feltételeket is lehet használni, amennyiben azok ezzel egyenértékű eredményt adnak.
-folyadékkromatográfiás oszlop (4.4.1.),
-HPLC mobil fázis: metil-alkohol és víz keveréke (3.3.),
-átáramlási sebesség: 1-1,5 ml/min,
-észlelési hullámhossz: 265 nm, - befecskendezett mennyiség: 20.50 μl.
A 4,0 μg/ml anyagot tartalmazó kalibráló oldat (3.7.3.) többszöri befecskendezésével ellenőrizzük a kromatográfiás rendszer stabilitását, míg a csúcsok magassága vagy görbe alatti területe, illetve a retenciós idők stabilakká nem válnak.
5.6.2.Kalibrációs görbe
Fecskendezzük be többször egymás után az egyes kalibráló oldatokat (3.7.3.), és mérjük meg az egyes koncentrációkra eső csúcsok magasságát (görbe alatti területét). A kalibráló oldatok átlagos magassága vagy területe legyen az ordináta tengelyen, a megfelelő koncentrációk pedig μg/ml-ben az abszcisszán, és ennek alapján szerkesszünk kalibrációs görbét.
5.6.3. Mintaoldat
Ugyanolyan mennyiségű oldatot használva, mint amennyit a kalibrációs oldatokhoz használtunk, fecskendezzük be a mintakivonatot (5.5.) egymás után többször és határozzuk meg a metilbenzokvát-csúcsok átlagos magasságát (területét).
6. Az eredmények kiszámítása
A mintaoldat által kiváltott metil-benzokvát-csúcsok átlagos magasságát (görbe alatti területét) a kalibrációs görbével egybevetve (5.6.2.), határozzuk meg a mintaoldat koncentrációját μg/ml-ben.
A minta, w (mg/kg)-ben kifejezett metilbenzokvát-tartalma a következő képlet alapján számítható ki:

amelyben:
c = a mintaoldat metil-benzokvát-koncentrációja μg/ml-ben,
m = a minta tömege grammban.
7. Az eredmények validálása
7.1. Azonosítás
Az analizált anyag azonosítását párhuzamos kromatográfiával vagy diódasoros detektorral lehet megerősíteni, amely során a mintakivonat és a 10,0 μg/ml metil-benzokvátot tartalmazó kalibráló oldat (3.7.3.) spektrumát hasonlítjuk össze.
7.1.1. Párhuzamos kromatográfia
A mintakivonatot a standard közbenső oldat (3.7.2.) megfelelő mennyiségének hozzáadásával dúsítjuk. A hozzáadott metil-benzokvát mennyiségének a mintakivonatban található metil-benzokvát becsült mennyiségével azonosnak kell lennie.
A hozzáadott mennyiség és a kivonat hígításának figyelembe vétele után csak a metilbenzokvát-csúcs magassága emelkedhet. A csúcs szélességének, a legnagyobb magasság felénél, az eredeti szélesség megközelítőleg 10%-án belül kell lennie.
7.1.2. Diódasoros kimutatás
Az eredményeket az alábbi kritériumok alapján kell értékelni:
a) a minta, valamint a standard spektrumának, a kromatogram csúcsán rögzített legnagyobb elnyelési hullámhossza, az észlelő rendszer felbontási képessége alapján meghatározott tűréshatáron belül meg kell egyezzen; a diódasoros észlelési módszernél ez tipikus esetben megközelítőleg 2 nm-en belül van;
b) 220 és 350 nm között a mintának, valamint a standardnak a kromatogram görbe csúcsán rögzített spektruma, a spektrum ezen részein nem térhet el egymástól a relatív abszorpciós tartomány 10 és 100%-a között; ez a kritérium akkor teljesül, ha ugyanazok a maximális értékek vannak jelen, és a két spektrum egyetlen észlelési ponton sem tér el a standard analizált anyag abszorpciójának 15%-ánál nagyobb mértékben egymástól;
c) 220 és 350 nm között a mintakivonat által kiváltott görbe felszálló ága, csúcsa és leszálló ága, a spektrumnak ezen a részein a relatív abszorpció 10 és 100%-a közötti tartományban nem térhet el egymástól; ez a kritérium akkor teljesül, ha ugyanazok a maximális értékek vannak jelen, és a két spektrum egyetlen észlelési ponton sem tér el a csúcs abszorpciós spektrumától 15%-nál nagyobb mértékben.
Ha ezen kritériumok közül egy nem teljesül, az analizált anyag jelentétét nem sikerült megerősíteni.
7.2. Ismételhetőség
Két azonos mintán végzett, párhuzamos meghatározás eredménye közötti különbség 4-20 mg/kg metil-benzokvát-tartalom esetén, nem haladhatja meg a magasabb érték 10%-át.
7.3. Visszanyerés
Dúsított referenciaminta esetében legalább 90%-os visszanyerési arányt kell elérni. 8. Kollaborációs vizsgálat eredményei
Tíz laboratóriumban, öt minta elemzésére került sor. Minden egyes mintán kettős elemzést végeztek.
| Eredmények | ||||||
| Vak | 1. liszt | 1. pellet | 2. liszt | 2. pellet | ||
| Középérték mg/kg | n.k. | 4,50 | 4,50 | 8,90 | 8,70 | |
| Sr (mg/kg) | — | 0,30 | 0,20 | 0,60 | 0,50 | |
| CVr (%) | — | 6,70 | 4,40 | 6,70 | 5,70 | |
| SR (mg/kg) | — | 0,40 | 0,50 | 0,90 | 1,00 | |
| CVR (%) | — | 8,90 | 11,10 | 10,10 | 11,50 | |
| Visszanyerés (%) | — | 92,00 | 93,00 | 92,00 | 89,00 | |
| n.k. = nem kimutatható Sr = ismételhetőség szórása CVr = ismételhetőség változékonysági együtthatója Sr = reprodukálhatóság szórása CVr = reprodukálhatóság változékonysági együtthatója | ||||||
XLI.
Avoparcin meghatározása agar-diffúzióval
1. Cél és alkalmazási terület
A módszer avoparcin meghatározására szolgál takarmányokban és előkeverékekben. A meghatározás alsó határa 2 mg/kg (2 ppm). Poliéter típusú gyógyszerek zavarhatják a meghatározást.
2. A módszer elve
A mintát aceton-víz-sósav keverékével extraháljuk. Antibiotikus aktivitását az avoparcin Bacillus subtilis-szel beoltott agar táptalajban történő diffúziójának mérésével határozzuk meg. A diffúziót a mikroorganizmus gátlási zónáinak képződése jelzi. Ezeknek a zónáknak az átmérője egyenes arányosnak tekinthető az antibiotikum koncentráció logaritmusával az alkalmazott gyógyszer koncentráció tartományban.
3. Mikroorganizmus: Bacillus subtilis ATCC 6633 (NC1B 8054)
3.1.A törzstenyészet fenntartása
Oltsunk be a ferde táptalajokat (4.1.) tartalmazó csöveket Bacillus subtilis-szel, majd inkubáljuk 30 °C-on 24 órán át. A tenyészetet hűtőszekrényben tároljuk, 4 °C körüli hőmérsékleten. Havonta oltsuk át.
3.2. A baktérium-szuszpenzió elkészítése
2-3 ml steril víz segítségével gyűjtsük be egy nemrégiben elkészített ferde agarról (3.1.) a növekményt. Ezzel a szuszpenzióval oltsunk be 300 ml, Roux lombikban lévő táptalajt (4.1.) és inkubáljuk 30 °C -on 3-5 napon át. Gyűjtsük a növekményt 15 ml etanolba (4.2.), a spóraképződés mikroszkóp alatt történt ellenőrzése után jól keverjük össze. Ez a szuszpenzió 4 °C -on legalább öt hónapig eltartható.
4. Táptalajok és reagensek
| 4.1. Táptalaj | |
| Pepton | 6,0 g |
| Tripton | 4,0 g |
| Élesztőkivonat | 3,0 g |
| Húskivonat | 1,5 g |
| Glükóz | 1,0 g |
| Agar | 15,0 g |
| Víz | 1000 ml |
| pH = 6,5 (sterilizálás után) |
4.2.Etanol 20% (v/v): 200 ml etanolt hígítsunk 800 ml vízzel
4.3.Sósav, d=1,18-1,19
12 Más módszerek is alkalmazhatók, ha megállapítást nyert, hogy azok hasonló baktérium szuszpenziókat eredményeznek.
4.4. Nátriumhidroxid, 2 mólos oldat
| 4.5. Foszfát puffer, 0,1 mólos | |
| Kálium-dihidrogén-foszfát, KH2PO4: Vízzel 1000 ml-re pH beállítás 4,5-re | 13,6 g |
4.6.Aceton-víz-sósav (4.3.): 65/32,5/2,5 (v/v/v) keverék
4.7.Standard anyag: ismert aktivitású avoparcin-szulfát
5. Standard oldatok
Oldjunk fel kb. 10 mg pontosan kimért standard anyagot (4.7.) foszfát pufferben és ezzel a pufferrel hígítsuk, hogy 10 μg/ml avoparcint tartalmazó törzsoldatot kapjunk. Lezárt lombikban 4 °C-on tárolva az oldat 7 napig stabil marad.
5.1. Előkeverékekhez
Ebből a törzsoldatból, a pufferrel (4.5.) történő egymást követő hígításokkal készítsük el a következő oldatokat:
| s8 | 4,0 μg/ml |
| s4 | 2,0 μg/ml |
| s2 | 1,0 μg/ml |
| s 1 | 0,5 μg/ml |
5.2. Takarmányokhoz
A törzsoldatból, a pufferrel (4.5.) történő egymást követő hígításokkal készítsük el a következő oldatokat:
| s8 | 2,0 μg/ml |
| s4 | 1,0 μg/ml |
| s2 | 0,5 μg/ml |
| s 1 | 0,25 μg/ml |
6. A kivonat és a vizsgálati oldatok elkészítése
6.1. Előkeverékek
Mérjünk be 100 ml-es mérőlombikba annyi mintát, hogy 10-100 mg avoparcint tartalmazzon. Adjunk hozzá 60 ml keveréket (4.6.) és rázassuk 15 percig mechanikus rázógéppel. Állítsuk be a pH-t, ha szükséges, 2,0-ra sósavval (4.3.). A keverékkel (4.6.) töltsük jelig és jól rázzuk össze. Szűrjük le megfelelő szűrőpapíron (pl. Whatman No. 1.), a szűrlet első 5 ml-ét dobjuk el. A szűrlet egy aliquot részének pH-ját nátriumhidroxid oldattal (4.4.) állítsuk be 4,5-re. Ezt az oldatot hígítsuk pufferrel (4.5.), hogy az avoparcin koncentráció várhatóan 4 μg/ml legyen (=U8). Ebből az oldatból pufferrel (4.5.) egymást követő hígításokkal készítsük el a következő oldatokat: U4 (várható tartalom: 2 μg/ml), U2 (várható tartalom: 1 μg/ml), U1 (várható tartalom: 0,5 μg/ml).
6.2. Takarmányok
Mérjünk be 50 g mintát és rázassuk 100 ml keverékkel (4.6.) 30 percig mechanikus rázógéppel. A kivonatot centrifugáljuk (zárt centrifuga csövekben). A tiszta kivonatból vegyünk ki egy aliquot részt (ld. az alábbi táblázatot), állítsuk be a pH-t nátriumhidroxid oldattal (4.4.) 4,5-re. Hígítsuk pufferrel (4.5.), hogy az U8-nak megfelelő koncentrációt adjon (ld. az alábbi táblázatot).
Ebből az oldatból pufferrel (4.5.) egymást követő hígításokkal készítsük el a következő oldatokat: U4 (várható tartalom: 1 μg/ml), U2 (várható tartalom: 0,5 μg/ml), U1 (várható tartalom: 0,25 μg/ml),
| Becsült avoparcin szint (mg/kg ) | 5 | 7,5. | 10 | 15 | 20 | 40 |
| Bemért minta (±0,1 g) | 50 | 50 | 50 | 50 | 50 | 50 |
| A keverék térfogata (4.6.) (ml) | 100 | 100 | 100 | 100 | 100 | 100 |
| A centrifugált kivonat térfogata (ml) | 20 | 15 | 20 | 15 | 20 | 10 |
| Végtérfogat (ml): U8 | 25 | 25 | 50 | 50 | 100 | 100 |
| Várható U8 koncentráció (μg/ml) | 2 | kb. 2 | 2 | kb. 2 | 2 | 2 |
7. Vizsgálati eljárás
7.1,A vizsgálati táptalaj beoltása
Oltsuk be a vizsgálati táptalajt (4.1.) a baktérium-szuszpenzióval (3.2.), 50-60 °C-on. A vizsgálati táp-talajt (4.1.) tartalmazó lemezeken végzett előzetes vizsgálatokkal határozzuk meg azt a szükséges baktérium-szuszpenzió mennyiséget, amely a különböző avoparcin koncentrációkkal a legnagyobb és legtisztább gátlási zónákat adja.
7.2.A lemezek elkészítése
7.2.1.Az agardiffúziót a négy standard oldat koncentrácjával (S8, S4, S2, és S1) és a négy vizsgálati oldat koncentrációjával (U8, U4, U2, U1) öntött lemezeken végezzük. A standard és a kivonat ezen négynégy koncentrációját minden egyes lemezre fel kell vinni. Ezért megfelelő nagyságú lemezeket kell választanunk ahhoz, hogy az agar táptalajban legalább nyolc darab, 10-13 mm átmérőjű, egymástól legalább 30 mm távolságra lévő lyukat alakíthassunk ki. A vizsgálat olyan lemezeken végezhető el, amelyek egy üveglapból és egy annak tetejére helyezett, 200 mm átmérőjű és 20 mm magas csiszolt alumínium vagy műanyag karimagyűrűből állnak.
7.2.2. A lemezekre öntsünk annyi, a 7.1 pont szerint beoltott táptalajt (4.1.), hogy az egy körülbelül 2 mm vastagságú réteget alkosson (200 mm átmérőjű lemez esetében ez 60 ml). Hagyjuk egyenletesen eloszlani, fúrjuk ki a lyukakat és helyezzük el bennük a vizsgálati és standard oldatok pontosan kimért mennyiségeit (az átmérő függvényében lyukanként 0,10 és 0,15 ml közötti mennyiség). Minden koncentrációt legalább négyszer alkalmazzunk, hogy minden meghatározás 32 gátlási zóna kiértékelése alapján történjen.
7.3. Inkubáció
Inkubáljuk a lemezeket 30 °C-on, 16-18 órán át.
8. Kiértékelés
8.1.Mérjük meg a gátlási zónák átmérőjét 0,1 mm-es pontossággal. Fél-logaritmikus milliméterpapíron ábrázoljuk az egyes koncentrációknál mért eredmények középértékeit, a koncentrációk logaritmusát a gátlási zónák átmérőinek függvényében. Szerkesszük meg mind a standard oldat, mind a kivonat egyenesét, például az alábbiak szerint.
8.2.Határozzuk meg a standard legalacsonyabb szint (SA) egyenes pontját a következő képlet segítségével: *

8.3. Határozzuk meg a standard legmagasabb szint (Sa) egyenes pontját a következő képlet segítségével:
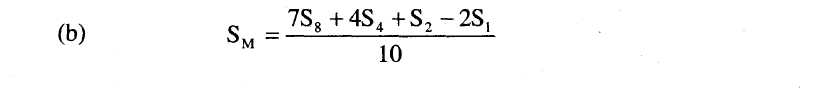
8.4.Hasonló módon számítsuk ki az egyenes pontjait a kivonat legalacsonyabb szintjére (Ua) és a kivonat legmagasabb szintjére (Um), a fenti képletekben az S1, S2, S4, S8 értékek helyett az U1, U2, U4 és U8 értékeket használva.
8.5. Ábrázoljuk a kiszámított Sa és Sm értékeket ugyanarra a milliméterpapírra és kössük össze őket, hogy megkapjuk a standard oldat egyenesét Hasonló módon ábrázoljuk az Ua és Um értékeket és kössük össze őket, hogy megkapjuk a kivonat egyenesét.
8.6.Ha interferencia nem lép fel, a vonalaknak párhuzamosnak kell lenniük. Gyakorlati megfontolásból a vonalakat párhuzamosnak tekinthetjük, ha az (Sm - Sa) és (Um - Ua) érték középértékeiktől való eltérése nem haladja meg a 10%-ot.
8.7. Ha a vonalak nem tekinthetőek párhuzamosnak, akkor akár az U1 és S1, akár az U8 és S8 elhagyható, az Sa, Sm, Ua és Um értékek az alábbi alternatív képletekkel számolhatók ki, hogy megkapjuk az alternatív regressziós egyenest:
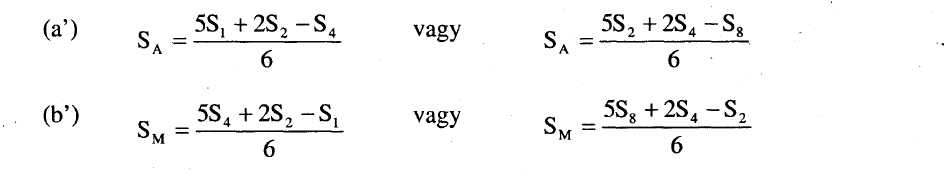
és hasonlóan UA-ra és UM-re. Ugyanazt a párhuzamossági feltételt kell teljesíteni. Azt a tényt, hogy az eredmény három szint alapján került kiszámításra, fel kell tüntetni a jegyzőkönyvben.
8.8. Ha a vonalak párhuzamosnak tekinthetőek, akkor számoljuk ki a relatív aktivitás logaritmusát (log A) a következő képletek egyikének felhasználásával, attól függően, hogy három vagy négy tényezőből történt-e a párhuzamosság megállapítása.

Valódi aktivitás = feltételezett aktivitás x relatív aktivitás
8.9.Ha a relatív aktivitás a 0,5 és 2,0 közötti tartományon kívül esik, akkor ismételjük meg a vizsgálatot a kivonat koncentrációin, vagy ahol ez nem lehetséges, ott a standard oldatokon végzett megfelelő módosításokkal. Ha a relatív aktivitást nem tudjuk az említett tartományon belülre korrigálni, akkor bármely kapott eredményt közelítő eredménynek kell tekinteni, és ezt fel kell tüntetni a jegyzőkönyvben.
8.10.Ha a vonalak nem tekinthetőek párhuzamosnak, ismételjük meg a meghatározást. Ha a párhuzamosságot még ezután sem sikerült elérni, akkor a meghatározást nem kielégítőnek kell tekinteni.
9. Ismételhetőség
Az ugyanazon mintán, ugyanazon analitikus által párhuzamosan végrehajtott két meghatározás eredménye közötti különbség nem haladhatja meg:
-a 2-10 mg/kg avoparcin tartalom esetén a 2 mg/kg-ot, abszolút értékben,
-a 10 és 25 mg/kg közötti tartalom esetén a legmagasabb érték 20%-át,
-a 25 és 50 mg/kg közötti tartalom esetén az 5 mg/kg-ot, abszolút értékben,
-az 50 mg/kg feletti tartalom esetén a legmagasabb érték 10%-át.
XLII.
Monenzin Na meghatározása agar-diffúzióval
1.Cél és alkalmazási terület
A módszer monenzin-Na meghatározására szolgál takarmányokban és előkeverékekben. A meghatározás alsó határa 10 mg/kg (10 ppm).
2.A módszer elve
A mintát 90%-os metanollal extraháljuk. Antibiotikus aktivitását a monenzin-Na Bacillus subtili-sel beoltott agar táptalajban történő diffúziójának mérésével határozzuk meg. A diffúziót a mikroorganizmus gátlási zónáinak képződése jelzi. Ezeknek a zónáknak az átmérője egyenes arányosnak tekinthető az antibiotikum koncentráció logaritmusával az alkalmazott gyógyszer koncentráció tartományban. A vizsgálati rendszer érzékenysége nátrium jelenlétében csökken.
3.Mikroorganizmus: Bacillus subtilis ATCC 6633 (NCIB 8054)
3.1.A törzstenyészet fenntartása
Oltsunk be a ferde táptalajokat (4.1.) tartalmazó csöveket Bacillus subtilis-szel, majd inkubáljuk 30 °C-on 24 órán át. A tenyészetet hűtőszekrényben tároljuk, 4 °C körüli hőmérsékleten. Havonta oltsuk át.
3.2. A baktérium-szuszpenzió elkészítése13
2-3 ml steril víz segítségével gyűjtsük be egy nemrégiben elkészített ferde agarról (3.1.) a növekményt. Ezzel a szuszpenzióval oltsunk be 300 ml, Roux lombikban lévő táptalajt (4.1.), és inkubáljuk 30 °C-on 3-5 napon át. Gyűjtsük a növekményt 15 ml 20%-os etanolba (4.3.), a spóraképződés mikroszkóp alatt történt ellenőrzése után jól keverjük össze. Ez a szuszpenzió 4 °C-on legalább öt hónapig eltartható.
4. Táptalajok és reagensek
| 4.1. Táptalaj14 | |
| Tripton | 10,0 g |
| Élesztőkivonat | 3,0 g |
| Húskivonat | l,5g |
| Glükóz | 1,0 g |
| Agar (minőségtől függően) | 10,0-20,0 g |
| Víz | 1000 ml |
| pH = 6,5 (sterilizálás után) | |
| 4.2. Értékmérő táptalaj | |
| Glükóz | 10,0 g |
| Élesztőkivonat | 2,5 g |
| Dikálium-hidrogén-foszfát K2HPO4 | 0,69 g |
| Kálium-dihidrogén-foszfát KH2PO4 | 0,45 g |
| Agar (minőségtől függően) | 10,0-20,0 g |
| Víz | 1000 ml |
| pH = 6 (sterilizálás után) |
4.3. Etanol 20% (v/v): 200 ml etanolt hígítsunk 800 ml vízzel
4.4. Vízmentes metanol
4.5.Metanol 90% (v/v): 900 ml metanolt (4.4.) 100 ml vízzel hígítunk 4.6. Metanol 50% (v/v): 500 ml metanolt (4.4.) 500 ml vízzel hígítunk
4.7.Alumíniumoxid, granulált (ALCOA F 20 mesh) Aktivált alumínium ugl (Lancaster vagy egyenértékű)
4.8.Standard anyag: ismert aktivitású monenzin-Na
5.Eszközök
5.1.Rotadeszt 250 ml-es gömblombikkal
5.2.Kromatográfiás üvegoszlop (Belső átmérő: 25 mm, hossz: 400 mm, 2 mm-es nyitott véggel.)
5.3.Kromatográfiás üvegoszlop (Belső átmérő: 11 mm, hossz: kb. 300 mm, 2 mm-es nyitott véggel.)
6.Standard oldatok
Oldjunk fel kb. 10 mg pontosan kimért standard anyagot (4.8.) metanolban és ezzel hígítsuk, hogy 800 μg/ml monenzin-Na-ot tartalmazó törzsoldatot kapjunk. Lezárt lombikban 4 °C-on tárolva az oldat két hétig stabil marad.
Ebből a törzsoldatból 50%-os metanollal (4.6.) történő egymást követő hígításokkal készítsük el a következő oldatokat:
| s8 | 8,0 μg/ml |
| s4 | 4,0 μg/ml |
| s2 | 2,0 μg/ml |
| S1 | 1,0 μg/ml |
7. A kivonat elkészítése
7.1.Extrakció
7.1.1.Előkeverékek
Mérjünk be 2 g mintát, adjunk hozzá 100 ml 90%-os metanolt (4.5.), homogenizáljuk és centrifugáljuk néhány percig. Hígítsuk a felsőfázist 50%-os metanollal (4.6.), hogy a monenzin-Na koncentráció várhatóan 8 μg/ml legyen (=U8).
7.1.2.Takarmányok, amelyekben a monenzin-Na koncentráció nem kisebb 50 ppm-nél
Mérjünk be 10-20 g mintát, adjunk hozzá 100 ml 90%-os metanolt (4.5.), homogenizáljuk 15 percig és hagyjuk leülepedni. Helyezzünk egy vattadugót az üvegcső (5.2.) vékony végébe, és töltsünk bele alumínium-oxidot (4.7.), óvatosan tömörítve, amíg a töltet magassága eléri a 75-80 mm-t. Dekantáljuk a kivonatot az alumínium-oxidos kolonnára. 30 ml szűrletet hígítsunk vízzel 50 ml-re. Ezt hígítsuk 50%-os metanollal (4.6.), hogy a monenzin-Na koncentráció várhatóan 8 μg/ml legyen (=U8).
7.1.3.Takarmányok, amelyekben a monezin-Na koncentráció kisebb, mint 50 ppm (alsó határ 10 ppm)
Mérjünk be 10-20 g mintát, adjunk hozzá 100 ml 90%-os metanolt (4.5.), és homogenizáljuk 15 percig. Centrifugáljuk, amíg tiszta oldatot kapunk. 20 ppm monezin-Na-ot tartalmazó minta esetén 40 ml-t vegyünk ki a felsőfázisból. 10 ppm monezin-Na-ot tartalmazó minta esetén 80 ml-t vegyünk ki a felsőfázisból, vákumban pároljuk szárazra, 40 °C alatti hőmérsékleten, a maradékot oldjuk fel 10 ml 90%-os metanolban (4.5.). Helyezzünk egy vattadugót az üvegcső (5.3.) vékony végébe, és töltsünk bele alumínium-oxidot (4.7.), óvatosan tömörítve, amíg a töltet magassága eléri a 75-80 mm-t. A metanolban feloldott bepárlási maradékot dekantáljuk az alumínium-oxid kolonnára és gyűjtsük a szűrletet. Mossuk át a kolonnát 10 ml 90%-os metanollal (4.5.), és egyesítsük a szűrlettel. Az oldatot vákumban pároljuk szárazra, 40 °C alatti hőmérsékleten, a maradékot oldjuk fel 10 ml vízmentes metanolban (4.4.), és egészítsük ki vízzel 20 ml-re. Az oldatot legalább öt percig centrifugáljuk minimum 4000 rpm sebességgel. Ezt hígítsuk 50%-os metanollal (4.6.), hogy a monenzin-Na koncentráció várhatóan 8 μg/ml legyen (=U8).
7.2. Vizsgálati oldatok
Az U8 oldatból 50%-os metanollal (4.6.) egymást követő hígításokkal (1+1) készítsük el a következő oldatokat: U4 (várható tartalom: 4 μg/ml), U2 (várható tartalom: 2 μg/ml), U1 (várható tartalom: 1 μg/ml).
8. Vizsgálati eljárás
8.1. A vizsgálati táptalaj beoltása
Oltsuk be a vizsgálati táptalajt (4.2.) a baktérium-szuszpenzióval (3.2.), 50-60 °C-on. A vizsgálati táptalajt (4.1.) tartalmazó lemezeken végzett előzetes vizsgálatokkal határozzuk meg azt a szükséges baktérium-szuszpenzió mennyiséget, amely a különböző monezin-Na koncentrációkkal a legnagyobb és legtisztább gátlási zónákat adja.
8.2.A lemezek elkészítése
8.2.1. Az agardiffúziót a négy standard oldat koncentrácjával (S8, S4, S2, és S1) és a négy vizsgálati oldat koncentrációjával (U8, U4, U2, U1) öntött lemezeken végezzük. A standard és a kivonat ezen négy-négy koncentrációját minden egyes lemezre fel kell vinni. Ezért megfelelő nagyságú lemezeket kell választanunk ahhoz, hogy az agar táptalajban legalább nyolc darab, 10-13 mm átmérőjű, egymástól legalább 30 mm távolságra lévő lyukat alakíthassunk ki. A vizsgálat olyan lemezeken végezhető el, amelyek egy üveglapból és egy annak tetejére helyezett, 200 mm átmérőjű és 20 mm magas csiszolt alumínium vagy műanyag karimagyűrűből állnak.
8.2.2. A lemezekre öntsünk annyi, a 8.1. pont szerint beoltott táptalajt (4.2.), hogy az egy körülbelül 2 mm vastagságú réteget alkosson (200 mm átmérőjű lemez esetében ez 60 ml). Hagyjuk egyenletesen eloszlani, fúrjuk ki a lyukakat és helyezzük el bennük a vizsgálati és standard oldatok pontosan kimért mennyiségeit (az átmérő függvényében lyukanként 0,10 és 0,15 ml közötti mennyiség). Minden koncentrációt legalább négyszer alkalmazzunk, hogy minden meghatározás 32 gátlási zóna kiértékelése alapján történjen.
8.3.Inkubáció
Inkubáljuk a lemezeket 35-37 °C-on, 18 órán át.
9. Kiértékelés,
9.1.Mérjük meg a gátlási zónák átmérőjét 0,1 mm-es pontossággal. Fél-logaritmikus milliméterpapíron ábrázoljuk az egyes koncentrációknál mért eredmények középértékeit, a koncentrációk logaritmusát a gátlási zónák átmérőinek függvényében. Szerkesszük meg mind a standard oldat, mind a kivonat egyenesét, például az alábbiak szerint:
9.2.Határozzuk meg a standard legalacsonyabb szint (Sa) egyenes pontját a következő képlet segítségével:
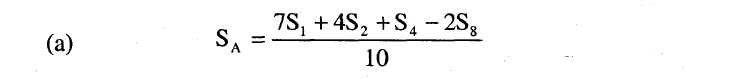
Határozzuk meg a standard legmagasabb szint (Sa) egyenes pontját a következő képlet segítségével:
9.3. Hasonló módon számítsuk ki az egyenes pontjait a kivonat legalacsonyabb szintjére (Ua) és a kivonat legmagasabb szintjére (Um), a fenti képletekben az S1, S2, S4, S8 értékek helyett az U1, U2, U4 és U8 értékeket használva.
9.4. Ábrázoljuk a kiszámított Sa és Sm értékeket ugyanarra a milliméterpapírra és kössük össze őket, hogy megkapjuk a standard oldat egyenesét. Hasonló módon ábrázoljuk az Ua és Um értékeket és kössük össze őket, hogy megkapjuk a kivonat egyenesét.
9.5. Ha interferencia nem lép fel, a vonalaknak párhuzamosnak kell lenniük. Gyakorlati megfontolásból a vonalakat párhuzamosnak tekinthetjük, ha az (Sm - Sa) és (Um - Ua) érték középértékeiktől való eltérése nem haladja meg a 10%-ot.
9.6. Ha a vonalak nem tekinthetőek párhuzamosnak, akkor akár az U1 és S1, akár az U8 és S8 elhagyható, az Sa, Sm, Ua és Um értékek az alábbi alternatív képletekkel számolhatók ki, hogy megkapjuk az alternatív regressziós egyenest:

és hasonlóan UA-ra és UM-re. Ugyanazt a párhuzamossági feltételt kell teljesíteni. Azt a tényt, hogy az eredmény három szint alapján került kiszámításra, fel kell tüntetni a jegyzőkönyvben.
9.7. Ha a vonalak párhuzamosnak tekinthetőek, akkor számoljuk ki a relatív aktivitás logaritmusát (log A) a következő képletek egyikének felhasználásával, attól függően, hogy három vagy négy tényezőből történt-e a párhuzamosság megállapítása.

9.8.Ha a relatív aktivitás a 0,5 és 2,0 közötti tartományon kívül esik, akkor ismételjük meg a vizsgálatot a kivonat koncentrációin, vagy ahol ez nem lehetséges, ott a standard oldatokon végzett megfelelő módosításokkal. Ha a relatív aktivitást nem tudjuk az említett tartományon belülre korrigálni, akkor bármely kapott eredményt közelítő eredménynek kell tekinteni, és ezt fel kell tüntetni a jegyzőkönyvben.
9.9. Ha a vonalak nem tekinthetőek párhuzamosnak, ismételjük meg a meghatározást. Ha a párhuzamosságot még ezután sem sikerült elérni, akkor a meghatározást nem kielégítőnek kell tekinteni.
10. Ismételhetőség
Az ugyanazon mintán, ugyanazon analitikus által párhuzamosan végrehajtott két meghatározás eredménye közötti különbség nem haladhatja meg:
-a 10 és 25 mg/kg közötti tartalom esetén a legmagasabb érték 20%-át,
- a 25 és 50 mg/kg közötti tartalom esetén az 5 mg/kg-ot, abszolút értékben,
-az 50 mg/kg feletti tartalom esetén a legmagasabb érték 10%-át.
XLIII.
Karbonátok meghatározása
1. Cél és alkalmazási terület
Ez a módszer lehetővé teszi a karbonátok, hagyományosan kalcium-karbonátban kifejezett mennyisé-gének meghatározását a legtöbb takarmányban. Bizonyos esetekben (például a vas-karbonát esetében) speciális módszert kell alkalmazni.
2. Vizsgálati alapelv
A karbonátokat sósavban bontjuk szét; a kibocsátott szén-dioxidot egy kalibrált csőben gyűjtjük össze, és térfogatát összehasonlítjuk az azonos körülmények között a kalcium-karbonát a.r. ismert mennyisége által kibocsátott mennyiséggel.
3.Reagensek
3.1. Sósav a.r., d: 1,10.
3.2. Kalcium-karbonát, a.r.
3.1. Kb. 0,1 N kénsav, metilvörös indikátorral megszínezve.
4. Eszközök
Scheibler-Dietrich készülék (lásd az ábrát) vagy annak megfelelő készülék.
5. A vizsgálat módja
5.1.A minta karbonát-tartalmától függően mérjünk ki egy részmintát az alábbiak szerint:
-0,5 g az 50-100% karbonát-tartalmú termékek esetén, kalcium-karbonátban kifejezve; - 1 g a 40-50% karbonát-tartalmú termékek esetén, kalcium-karbonátban kifejezve;
-2-3 g egyéb termékek esetében.
5.2.Helyezzük a részmintát a készülék speciális lombikjába (4), amelyet 10 ml sósavat (3.1.) tartalmazó, törhetetlen anyagból készült, kisméretű csővel szereltek fel, és kapcsoljuk a lombikot a készülékhez. Fordítsuk el a háromfázisú csapot (5), hogy a cső (1) a külső résszel kapcsolódjon. A színezett kénsavval (3.3.) töltött és kalibrált csőhöz (1) kapcsolt mozgatható cső (2) segítségével igazítsuk a folyadék szintjét a nulla jelhez. Az (1) és a (3) csövek összekapcsolásához fordítsuk el a csapot (5), és ellenőrizzük, hogy a szint nullán áll-e.
A lombik (4) megdöntésével folyassunk lassan sósavat (3.1.) a részmintára. A (2) cső leengedésével egyenlítsük ki a nyomást. Rázzuk a (4) lombikot egészen addig, amíg a szén-dioxid kibocsátás teljesen meg nem szűnik.
Az.(l) és a (2) csőben lévő folyadék egyenlő szintre igazításával állítsuk vissza a nyomást. Néhány perc múlva, amikor a gáz mennyisége állandósul, olvassuk le az eredményt.
Végezzünk ellenőrző vizsgálatot ugyanilyen körülmények között 0,5 g kalcium-karbonáton (3.2.).
6. Az eredmények kiszámítása
A kalcium karbonát formájában, grammban kifejezett karbonát-tartalom a mintára vonatkoztatva, százalékosan a következő képlet segítségével számítható ki:

ahol:
V = a részminta által kibocsátott szén-dioxid, ml-ben.
T = 0,5 g CaCO3 a.r. által kibocsátott szén-dioxid, ml-ben.
W = a részminta grammban kifejezett tömege.
7. Észrevételek
7.1. Ha a részminta 2 g-nál nehezebb, először töltsünk 15 ml desztillált vizet a (4) lombikba, és keverjük össze a vizsgálat megkezdése előtt. Az ellenőrző vizsgálat esetében ugyanezt a vízmennyiséget használjuk.
7.2. Ha a használt készülék űrtartalma eltér a Scheibler-Dietrich készülékétől, a mintából és a kontrollanyagból vett részt és az eredmények kiszámítását annak megfelelően kell korrigálni.
A szén-dioxid meghatározására szolgáló Scheibler-Dietrich készülék
XLIV.
Ureáz-aktivitás meghatározása szója-készítményekben
1.Cél és alkalmazási terület
Szójabab és a szójakészítmények mintáinak ureáz-aktivitás meghatározása.
2.A módszer elve
A szójabab és a szójakészítmények mintáinak ureáz-aktivitását azzal az ammónia-nitrogén mennyiséggel mérjük, amelyet 1 g minta 30 °C-on a pufferolt karbamid oldatból percenként felszabadít.
3. Vegyszerek
3.1. Karbamid, a.r.
3.2.Dinátrium-hidrogén-foszfát (Na2HPO4.2H2O)
3.3.Kálium-dihidrogén-foszfát (KH2PO4)
3.4.Sósav
3.5.Nátrium-hidroxid
3.6.Kálium-hidrogén-karbonát
3.7.Metilnarancs-indikátor
Vegyszerek előkészítése
3.8.Foszfát-puffer oldat
4,45 g dinátrium-hidrogén-foszfátot és 3,40 g kálium-dihidrogén-foszfátot 1000 cm3-es mérőlombikban desztillált vízben feloldunk, jelig töltjük, összerázzuk.
3.9.Karbamidos foszfát-pufferoldat (frissen készítve)
30 g karbamidot 1000 cm3-es mérőlombikban foszfát-pufferoldatban feloldunk, jelig töltjük, összerázzuk. Az oldat pH-jának 6,9-7,0 között kell lenni.
3.10.0,1 mólos sósavoldat
3.10.1. Közelítően 0,1 mólos sósavoldat készítése
A 37%-os (m/m) sósavból 8,25 cm3-t 1000 cm3-re hígítunk desztillált vízzel normál lombikban, összerázzuk.
3.10.2. A közelítően 0,1 mólos sósavoldat beállítása kálium-hidrogén-karbonátra
3.10.2.1.Az előkészített kálium-hidrogén-karbonátból lemérünk analitikai mérlegen 2 g körüli mennyiséget 0,2 mg pontossággal, majd visszaméréssel 0,2-0,25 g-ot veszteség nélkül titráló lombikba teszünk.
3.10.2.2.A bemért sót 30 cm3 desztillált vízben oldjuk, 1 csepp metilnarancs-indikátort adunk hozzá és a 0,1 mólos sósavoldattal addig titráljuk, míg az oldat narancsszínű lesz. Ezután a szén-dioxid eltávolítására két percig forraljuk, majd gyorsan lehűtjük és az általában visszasárgult oldatot teljes lehűlés után átmeneti színűre titráljuk.
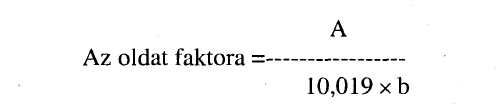
ahol
A = a bemért kálium-hidrogén-karbonát tömege, mg-ban b = a fogyott sósavoldat, cm3-ben
Az egyes faktorokat külön-külön számítjuk ki; azoknak 0,2%-nál nagyobb eltérést nem szabad mutatni. Ha az eltérés nagyobb, a mérést meg kell ismételni.
A faktor ismeretében hígítással, vagy sósav hozzáadásával az oldatot pontosan 1,000 faktorúra kell beállítani.
3.11. 0,1 mólos nátrium-hidroxid
3.11.1.A közelítően 0,1 mólos nátrium-hidroxid készítése
Lehetőség szerint karbonátmentes nátrium-hidroxidból 4 g-ot oldunk desztillált vízben 1000 crn3-re, szobahőmérsékletre hűtjük, jelig töltjük, összerázzuk.
3.11.2.Az 0,1 mólos nátrium-hidroxid oldat faktorának beállítása 0,1 mólos sósavoldatra
Titráló lombikba 20,00 cm3 0,1 mólos sósavoldatot mérünk és metilnarancs-indikátor jelenlétében a beállítandó 0,1 mólos nátrium-hidroxiddal átmeneti színűre titráljuk.
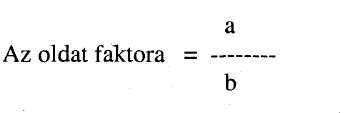
ahol:
a = a bemért 0,1 mólos sósavoldat mennyisége, cm3-ben
b = a fogyott 0,1 mólos nátrium-hidroxid oldat mennyisége, cm3-ben
A faktor ismeretében az oldatot, ha kell hígítással, vagy nátrium-hidroxid hozzáadásával, pontosan 0,1 mólosra állítjuk.
Az egyes faktorokat külön-külön számítjuk ki; azoknak 0,2%-nál nagyobb eltérést nem szabad mutatni. Ha az eltérés nagyobb, a mérést meg kell ismételni.
3.12. Kálium-hidrogén-karbonát előkészítése
A kálium-hidrogén-karbonátot finoman elporítjuk és óraüvegen vékony rétegben szétterítjük, széndioxiddal töltött tömény kénsavas exszikkátorban egy napig állni hagyjuk. Másnap az összetapadt port újból megtörjük és még egy napra exszikkátorba helyezzük. A finom, nem tapadó port légmentesen zárt edényben tároljuk.
4.Eszközök
4.1.pH-mérő készülék, 0,02 pH érzékenységi!, mágneses keverővel, titrálóeszközökkel
4.2.Vízfürdő, 30 °C-ra állítható hőfokszabályozóval és hőmérővel
4.3.Mélyhűtő, jég készítéséhez
4.4.Csiszolt dugós kémcsövek, 150 x 18 mm (20 cm ) méretűek
5. Vizsgálati eljárás 5.1. A minta elkészítése
Az előkészített15 mintából 10 g körüli mennyiséget úgy darálunk meg, hogy átessen a 0,2 mm-es szitán. Ezután a mintát homogenizáljuk, majd 0,2 g-ot* bemérünk 1 mg pontossággal csiszolt dugós kémcsőbe.

ureáz aktivitásra van beállítva. Magasabb aktivitású anyagoknál a bemérést 50 mg-ra lehet csökkenteni.
5.2. Meghatározás
A bemért mintához a kémcső körkörös mozgatása közben hozzáadunk 10 cm3 karbamidos foszfát-pufferoldatot úgy, hogy a minta teljesen átnedvesedjen, azonnal bedugjuk és azonnal betesszük 30 °C-os vízfürdőbe, pontosan 30 percre. Ezután 10 cm 0,1 mólos sósavat adunk hozzá, azonnal jeges vízbe tesszük, majd mérés előtt 20 °C-ra való beállítás után veszteség nélkül az elektród típusától függő méretű és formájú titráló edénybe öntjük, a kémcsövet 2 x 2,5 cm3 desztillált vízzel átöblítjük és azonnal 0,1 mólos nátrium-hidroxiddal titráljuk pH 4,7-ig.
5.3. Vakpróba
Az előkészített és mért mintához 10 cm3 0,1 mólos sósavat és 10 cm3 karbamidos pufferoldatot adunk és 30 percig jeges vízben hagyjuk állni. Ezután 20 °C-ra állítjuk és maradék nélkül átvisszük az elektród típusától függő méretű és formájú titráló edénybe, a kémcsövet 2 x 2,5 cm3 desztillált vízzel átöblítjük. Az oldatot 0,1 mólos nátrium-hidroxiddal pH 4,7-ig titráljuk.
6. Az eredmény kiszámítása és kifejezése
Szójaminták ureázaktivitását az alábbi képlet szerint számítjuk és

ahol
a = a meghatározásnál fogyott 0,1 mólos nátrium-hidroxid mennyisége, cm3-ben b = a vakpróbánál fogyott 0,1 mólos nátrium-hidroxid mennyisége, cm3-ben m = a vizsgálathoz bemért minta mennyisége, g-ban
Az ureázaktivitás értéke két párhuzamos meghatározás középértéke, amelyet kéttizedes pontossággal adunk meg.
7. A vizsgálat ismételhetősége
Azonos mintából végzett két párhuzamos meghatározás eredménye között a megengedett legnagyobb eltérés 0,2 cm3 0,1 mólos nátrium-hidroxid fogyás.
XLV.
A szabad és az összes gosszipol meghatározása
1. Cél és alkalmazási terület
Ez a módszer lehetővé teszi a szabad gosszipol, az összes gosszipol és a kémiailag rokon anyagok mennyiségi meghatározását a gyapotmagban, a gyapotmaglisztben és a gyapotmag-pogácsában, valamint az ezen anyagokat tartalmazó összetett takarmányokban, amennyiben a gosszipolból több mint 20 ppm van jelen.
2. Vizsgálati alapelv
A gosszipolt 3-aminopropán-1-ol jelenlétében, vagy propán-2-ol és hexán keverékével (a szabad gosszipol meghatározása esetén) vagy dimetil-formamiddal (az összes gosszipol meghatározása esetén) extraháljuk. A gosszipol anilinnel gosszipol-dianilinné alakul, amelynek optikai sűrűségét 440 nm-nél mérjük. .
3.Reagensek
3.1. Propán-2-ol-hexán keverék: keverjünk Össze 60 térfogategységnyi propán-2-ol a.r.-t 40 térfogategységnyi n-hexánnal.
3.2. A-oldószer: tegyünk 1 literes mérőlombikba kb. 500 ml propán-2-ol-hexán keveréket (3.1.), 2 ml 3-aminopropán-1-ol-t, 8 ml jégecetet, valamint 50 ml vizet. Töltsük fel térfogatra a propán-2-ol-hexán keverékkel (3.1.). Ez a reagens egy hétig stabil.
3.3. B-oldószer: pipettázzunk 2 ml 3-aminopropán-l-ol-t és 10 ml jégecetet egy 100 ml-es mérőlombikba. Hűtsük le szobahőmérsékletre, és töltsük fel térfogatra N,N-dimetil-formamiddal. Ez a reagens egy hétig stabil.
3.4.Anilin a.r.: ha a vakpróba optikai sűrűsége meghaladja a 0,022-et, pároljuk le az anilint cinkpor fölött, és öntsük el a párlat első és utolsó 10-10%-át. Lehűtve és barna, lezárt üveglombikban tárolva a reagens több hónapig eláll.
3:5. Gosszipol-A standard oldat: tegyünk 27,9 mg gosszipol-acetátot egy 250 ml-es mérőlombikba. Oldjuk fel, és töltsük fel térfogatra az A-oldószerrel (3.2.). Pipettázzunk 50 ml-t ebből az oldatból egy 250 ml-es mérőlombikba, és töltsük fel térfogatra az A-oldószerrel. Ennek az oldatnak a gosszipol-koncentrációja 0,02 mg/ml. Használat előtt hagyjuk egy órán át szobahőmérsékleten állni.
3.6. Gosszipol-B standard oldat: tegyünk 27,9 mg gosszipol-acetátot egy 50 ml-es mérőlombikba. Oldjuk fel, és töltsük fel térfogatra a B-oldószerrel (3.3.). Ennek az oldatnak a gosszipol-koncentrációja 0,5 mg/ml.
Ha fénytől védve tároljuk, a gosszipol-A és -B standard oldat 24 órán át stabil marad.
4. Eszközök
4.1.Keverőgép (kb. 35 fordulat/perc)
4.2.Spektrofotométer
5. A vizsgált módja
5.1. A vizsgálati minta
5.1.1. A vizsgálati minta mennyisége a minta feltételezett gosszipol-tartalmától függ. Jobb kisebb mennyiségű mintával dolgozni, és a szűrlet viszonylag nagyobb mennyiségű aliquot részét használni, hogy elegendő mennyiségű gosszipolt nyerjünk, amely lehetővé teszi a precíz fotometriás mérést. A gyapotmag, a gyapotmagliszt és a gyapotmag-pogácsa szabad gosszipoltartalmának mérésére a minta mennyisége ne haladja meg az 1 g-ot, összetett takarmányok vizsgálata során a minta mennyisége akár 5 g is lehet. A szűrlet 10 ml-nyi aliquot része a legtöbb esetben megfelelő, ez kb. 50.100 μg gosszipolt tartalmaz. Az összes gosszipoltartalom meghatározásához a minta mennyisége 0,5 és 5 g között legyen, így a szűrlet 2 ml-nyi aliquot része kb. 40.200 μg gosszipolt fog tartalmazni.
5.1.2. A vizsgálatokat kb. 20 °C-os szobahőmérsékleten kell végezni.
5.2. A szabad gosszipoltartalom meghatározása
5.2.1.Tegyük a vizsgálandó mintát egy 250 ml-es, csiszolatos lombikba, amelynek az alján üvegzúzalékot helyeztünk el. Pipettázzunk 50 ml A-oldószert (3.2.) a lombikba, zárjuk le a lombikot, és keverjük egy órán át a keverőben. Szűrjük át egy száraz szűrőn, és gyűjtsük össze a szűrletet egy kis, csiszolatos lombikban. Szűrés közben fedjük be a tölcsért egy óraüveggel. Pipettázzunk azonos aliquot mennyiségű, 50.100 μg gosszipolt tartalmazó szűrletet két, 25 ml-es mérőlombikba (A- és B-jelű). Ha szükséges, töltsük fel a mintát 10 ml-re az A-oldószerrel (3.2.). Ezután töltsük fel térfogatra az A-jelű lombikot propán-2-ol-hexán keverékkel (3.1.). Ezt az oldatot referenciaoldatként használjuk, mellyel szemben a mintaoldatot mérjük.
5.2.2. Pipettázzunk 10-10 ml A-oldószert (3.2.) két másik, 25 ml-es mérőlombikba (C- és D-jelű). Töltsük fel a C-jelű lombik tartalmát térfogatra propán-2-ol-hexán keverékkel (3.1.). Ezt az oldatot referenciaoldatként használjuk, mellyel szemben a vakpróba oldatot mérjük.
5.2.3.Adjunk 2-2 ml anilint (3.4.) a D- és a B-jelű lombikhoz. Hevítsük ezeket 30 percen át forró vízfürdő fölött, hogy tartalmuk elszíneződjön. Hűtsük le szobahőmérsékletűre, és töltsük fel tartalmukat térfogatra propán-2-ol-hexán keverékkel (3.1.), homogenizáljuk, majd hagyjuk állni egy órán át.
5.2.4. Határozzuk meg a vakpróba oldat (D) optikai sűrűségét a referenciaoldattal (C) összehasonlítva, és a mintaoldat (B) optikai sűrűségét a referenciaoldattal (A) összehasonlítva egy spektrofotométerrel, 440 nm-nél, 1 cm-es üvegküvettákat használva.
5.2.5.Vonjuk ki a vakpróba oldat optikai sűrűségét a mintaoldat optikai sűrűségéből (= korrigált optikai sűrűség). Ebből az értékből a 6. pontban leírtak szerint számítsuk ki a szabad gosszipoltartalmat.
5.3. Az összes gosszipoltartalom meghatározása
5.3.1. Tegyünk egy 1.5 mg gosszipolt tartalmazó mintát egy 50 ml-es mérőlombikba, és adjunk hozzá 10 ml B-oldószert (3.3.). Készítsünk ezzel párhuzamosan egy vakpróbás oldatot is úgy, hogy 10 ml B-oldószert (3.3) teszünk egy másik, 50 ml-es mérőlombikba. Hevítsük a két lombikot forró vízfürdő fölött 30 percen át. Hűtsük le a lombikokat szobahőmérsékletűre, és töltsük fel tartalmukat térfogatra propán-2-ol-hexán keverékkel (3.1.). Homogenizáljuk, és hagyjuk ülepedni 10-15 percig, utána szűrjük át, majd gyűjtsük össze a szűrletet csiszolatos lombikokba.
5.3.2.A mintaszűrletből pipettázzunk 2-2 ml-t két, 25 ml-es mérőlombikba, és a vakpróba szűrletből is pipettázzunk 2-2 ml-t két másik, 25 ml-es lombikba. Mindkét sorozatban az egyik lombikot töltsük fel 25 ml-re a propán-2-ol-hexán keverékkel (3.1.). Ezeket az oldatokat használjuk referenciaoldatként.
5.3.3. A másik két lombik tartalmához adjunk 2-2 ml anilint (3.4.). Hevítsük ezeket 30 percen át forró vízfürdő fölött, hogy tartalmuk elszíneződjön. Utána hűtsük le őket szobahőmérsékletűre, töltsük fel
tartalmukat propán-2-ol-hexán keverékkel 25 ml-re (3.1.), homogenizáljuk, és hagyjuk állni egy órán át.
5.3.4. Határozzuk meg az optikai sűrűséget az 5.2. pontban a szabad gosszipoltartalomra vonatkozóan ismertetett módon. Ebből az értékből számítsuk ki az összes gosszipoltartalmat a 6. pont alapján.
6. Az eredmények kiszámítása
Az eredmény kiszámítható akár a fajlagos optikai sűrűségből (6.1.), akár egy kalibrációs görbe segítségével (6.2.).
6.1. A fajlagos optikai sűrűségből
A fajlagos optikai sűrűség, a leírt feltételek esetén az alábbiak szerint alakul:

ahol:
E = az 5.2 pont szerint meghatározott korrigált optikai sűrűség p = a minta mennyisége, g-ban a = a szűrlet mennyisége, ml-ben
6.2. Kalibrációs görbéből 6.2.1. Szabad gosszipoltartalom
6.2.1.1. Készítsünk elő két, egyenként 5 db 25 ml-es mérőlombikból álló sorozatot. Pipettázzunk mindkét sorozat lombikjaiba 2,0, 4,0, 6,0, 8,0 és 10,0 ml gosszipol-A standard oldatot (3.5.). Töltsük fel valamennyi lombikot 10 ml-re az A-oldószerrel (3.2.). Egészítsük ki mindkét sorozatot egy-egy,. 25 ml-es, de csak 10 ml A-oldószert (3.2.) tartalmazó lombikkal (vakpróba).
6.2.1.2.Töltsük fel 25 ml-re az egyik sorozat lombikjait (beleértve az egyik vakpróba lombikját is) propán-2-ol.hexán keverékkel (3.1.) (referenciasorozat).
6.2.1.3. Adjunk a másik sorozat mindegyik lombikjának tartalmához 2 ml anilint (3.4.), beleértve a másik vakpróba lombikját is. Hevítsük ezeket a lombikokat 30 percen át forró vízfürdő fölött, hogy tartalmuk elszíneződjön. Ezután hűtsük le őket szobahőmérsékletűre, töltsük fel térfogatra propán-2-ol-hexán keverékkel (3.1.), homogenizáljuk, és hagyjuk állni egy órán át (standard sorozat).
6.2.1.4. Határozzuk meg a standard oldatok optikai sűrűségét az 5.2. pont szerint, összehasonlítva a referencia-sorozat oldataival. Szerkesszük meg a kalibrációs görbét úgy, hogy az optikai sűrűséget a gosszipol μg-ban kifejezett mennyiségének függvényében ábrázoljuk.
6.2.2. Összes gosszipoltartalom
6.2.2.1. Készítsünk elő 6 db 50 ml-es mérőlombikot. Tegyünk az első lombikba 10 ml-t a B-oldószerből (3.3.), a többibe pedig sorban 2,0, 4,0, 6,0, 8,0 és 10,0 ml gosszipol-B standard oldatot (3.6.). Töltsük fel valamennyi lombik tartalmát 10 ml-re a B-oldószerrel (3.3.). Hevítsük a lombikokat forró vízfürdő fölött 30 percen át. Hűtsük le őket szobahőmérsékletűre, töltsük fel térfogatra a propán-2-ol-hexán keverékkel (3.1.), és homogenizáljuk a tartalmukat.
6.2.2.2. Az elkészített oldatokból tegyünk 2-2 ml-t a két sorozatban előkészített, 6-6 db 25 ml-es mérőlombikba. Töltsük fel az első sorozat lombikjainak tartalmát 25 ml-re propán-2-ol-hexán keverékkel (3.1.) (referencia-sorozat).
6.2.2.3.A másik sorozat mindegyik lombikjába tegyünk 2 ml anilint (3.4.). Hevítsük a lombikokat forró vízfürdő fölött 30 percen át. Ezután hűtsük le őket szobahőmérsékletűre, töltsük fel térfogatra pro-pán-2-ol-hexán keverékkel (3.1.), homogenizáljuk, és hagyjuk állni egy órán át (standard-sorozat).
6.2.2.4. Határozzuk meg a standard oldatok optikai sűrűségét az 5.2. pont szerint, összehasonlítva a referencia-sorozat oldataival. Szerkesszük meg a kalibrációs görbét úgy, hogy az optikai sűrűséget a gosszipol μg-ban kifejezett mennyiségének függvényében ábrázoljuk.
6.3 Ismételhetőség
Az ugyanazon a mintán végzett két párhuzamos meghatározás eredménye közötti különbség nem haladhatja meg:
- az 500 ppm-nél kevesebb gosszipolt tartalmazó minták esetében a 15%-ot, relatív értékben,
- az 500 ppm-nél nem kevesebb és 750 ppm-nél nem több gosszipolt tartalmazó minták esetében a 75 ppm-et, abszolút értékben,
- a 750 ppm-nél több gosszipolt tartalmazó minták esetében a 10%-ot, relatív értékben.
XLVI.
A tilozin meghatározásának módszere agar diffúzióval
1. Cél és alkalmazási terület
Ez a módszer a takarmányok, a koncentrátumok és az előkeverékek tilozin-tartalmának meghatározását teszi lehetővé, amennyiben a tilozin 2 ppm-nél nagyobb mennyiségben van jelen.
2. Vizsgálati alapelv
A mintát 80 °C-ra előmelegített, 8 pH-jú foszfátpuffer-oldattal kezeljük, majd metil-alkohollal extraháljuk. Centrifugálás után a kivonatot hígítjuk, és antibiotikum-aktivitását a tilozin, Sarcina luteá-val beoltott agartáptalajban való diffúziójának mérésével határozzuk meg. A diffúziót a mikroorganizmus jelenlétében gátlási zónák kialakulása jelzi. E zónák átmérője egyenesen arányos az antibio-tikum-koncentráció logaritmusával.
3.Mikroorganizmus: Sarcina lutea ATCC No 9341
3.1.Az eredeti törzs fenntartása
Oltsunk be Sarcina lutea-val egy ferde agarcsövet, amelyet a (4.1.) táptalajból vettünk, a pH-t állítsuk 7,0-re. Inkubáljuk egy éjszakán át kb. 35 °C-on. Tegyük a tenyészetet hűtőbe, és havonta oltsuk át vele a ferde agart.
3.2.A baktérium-szuszpenzió elkészítése
3.2.1. Kb. 2.3 ml fiziológiás konyhasóoldattal (4.4.) mossuk le a baktériumokat egy frissen készített ferde agarcsőről (3.1.). Oltsunk be ezzel a szuszpenzióval egy Roux-palackot, amely 250 ml, pH 7,0-re beállított táptalajt (4.1.) tartalmaz. Inkubáljuk 35 °C-on 24 órán át, majd vegyük fel a baktériumokat 25 ml fiziológiás konyhasóoldatban (4.4.). Homogenizáljuk, és hígítsuk fel ezt a szuszpenziót úgy, hogy 650 nm-nél, kb. 75%-os fényáteresztést kapjunk.
3.2.2.Hűtőben tárolva ez a szuszpenzió egy hétig használható.
3.2.3. A meghatározáshoz használt alaptáptalajjal (4.1.) öntött lemezeken végzett előzetes vizsgálatokkal állapítsuk meg az inokulumnak azt a mennyiségét, amely az alkalmazott különböző tilozin-koncentrációk esetén a lehető legnagyobb, még tiszta gátlási zónákat adja. A táptalajt 48-50 °C-on kell beoltani.
4. Táptalajok és reagensek
| 4.1. Alaptáptalaj a meghatározáshoz16 | |
| Glükóz | 1 g |
| Triptines pepton | 10 g |
| Húskivonat | 1,5 g |
| Élesztőkivonat | 3g |
| Agar, minőségtől függően | 10.20 g |
| Desztillált vízzel feltöltve | 1 000 ml-re |
| 16 Bármilyen hasonló összetételű és ugyanazt az eredményt adó, a kereskedelmi forgalomban kapható táptalaj használható. | |
Közvetlenül használat előtt állítsuk a pH-t az eredeti törzs fenntartásához és baktériumszuszpenzió elkészítéséhez 7,0-re, a meghatározáshoz 8,0-ra.
| 4.2. Foszfátpuffer-oldat, pH 8 | |
| Kálium-dihidrogén-foszfát KH2PO4 a. r. | 0,523 g |
| Dikálium-hidrogén-foszfát KH2PO4 a. r. | 16,730 g |
| Desztillált vízzel feltöltve | 1 000 ml-re |
| 4.3. Foszfátpuffer-oldat, pH 7 | |
| Kálium-dihidrogén-foszfát KH2PO4 A. R. Dikálium-hidrogén-foszfát KH2PO4 A. R. Desztillált víz | 5,5 g 13,6 g ad 1000 ml |
4.4.Steril fiziológiás konyhasóoldat
4.5. Tiszta metil-alkohol
4.6.40%-os metil-alkohol
4.7.Foszfátpuffer-oldat (4.2.) /tiszta metanol 60/40 térfogategység arányú keveréke
4.8.Standard anyag: ismert aktivitású tilozin
5. Standard oldatok
5.1. Szárítsuk a standard anyagot (4.8.) 3 órán át 60 °C-on egy vákuumkemencében (5 mm higany). Mérjünk ki belőle 10-50 mg-ot egy mérőlombikba, oldjuk fel 5 ml metil-alkoholban (4.5.), és hígítsuk az oldatot pH 7-es foszfátpuffer-oldattal (4.3.), hogy 1000 μg/ml tilozin-bázis koncentrációt kapjunk.
5.2.A (4.7.) keverékkel történő hígítással készítsünk ebből a törzsoldatból egy 2 μg/ml tilozin-bázist tartalmazó S8 standard munkaoldatot.
5.3.Ezután, a (4.7.) keverék felhasználásával végzett sorozatos hígítással (1 + 1) készítsük el a következő koncentrációjú oldatokat:
| S4 | 1 μg/ml |
| S2 | 0,5 μg/ml |
| S1 | 0,25 μg/ml |
6.Extrahálás
6.1.Mérjünk ki koncentrátumok esetén 10 g, előkeverékek és takarmányok esetén 20 g vizsgálati mintát. Adjunk a mintához 60 ml, előzetesen 80 °C-ra melegített, pH 8-as foszfátpuffer-oldatot (4.2.), és homogenizáljuk 2 percen át (háztartási keverőgéppel vagy Ultra-turrax-szal, stb.).
6.2.Hagyjuk állni 10 percen át, adjunk hozzá 40 ml metil-alkoholt (4.5.), és homogenizáljuk 5 percig, Centrifugáljuk, majd vegyünk egy aliquot részt a kivonatból, és hígítsuk fel a (4.7.) oldattal úgy, hogy 2 μg/ml-es feltételezett tilozin-koncentrációt kapjunk (=U8). Ezután, a (4.7.) keverékkel végzett sorozatos hígítással (1 + 1) készítsük el az U4, U2 és U1 koncentrációjú oldatokat.
6.3. 10 ppm-nél alacsonyabb tartalom esetén pároljuk a mintát szárazra egy rotációs bepárlókészüléken, 35 °C-on, majd oldjuk fel a maradékot 40%-os metil-alkoholban (4,6.).
7.Meghatározási módszer
7.1.A táptalaj be oltása
A meghatározáshoz használt, pH 8,0-re beállított alaptáptalajt (4.1.) oltsuk be 48-50 °C-on a baktérium-szuszpenzióval (3.2.).
7.2.A tálcák előkészítése
7.2.1. Az agardiffúziót tálcákon hajtjuk végre, a standard oldat 4 koncentrációját (S8, S4, S2, S1) és a kivonat 4 koncentrációját (U8, U4, U2, U1) felhasználva. A standard oldat és a kivonat 4-4 koncentrációját minden tálcára fel kell vinni.
7.2.2. Ezért olyan nagyságú tálcát használjunk, amely lehetővé teszi, hogy a rajta elhelyezett agaron legalább 8, egyenként 10-13 mm átmérőjű lyukat fúrhassunk. Számítsuk ki a beoltott táptalaj (7.1.) szükséges mennyiségét úgy, hogy kb. 2 mm vastagságú, egyenletes réteget kapjunk. Leginkább olyan tálcák felelnek meg a célnak, amelyek tökéletesen vízszintes, 200 mm átmérőjű és 20 mm magas alumínium vagy műanyag karimagyűrűvel ellátott sík üveglemezből állnak.
7.2.3. A lyukakba pipettázzunk 0,10-0,15 ml pontosan kimért antibiotikum-oldatot, a lyukak átmérőjétől függően.
7.2.4.Minden egyes minta esetében legalább négyszer ismételjük meg a diffúziót, hogy minden egyes meghatározás összesen 32 gátlási zóna értékelésén alapuljon.
7.3.Inkubálás
Inkubáljuk a tálcákat 35-37 °C-on egy éjszakán át.
8.Értékelés
8.1. Mérjük meg a gátlási zónák átmérőjét, lehetőleg kivetítő segítségével. Jegyezzük fel a mérési eredményeket féllogaritmikus papíron, a koncentrációk logaritmusát a gátlási zónák átmérőjének függvényében ábrázolva. Rajzoljuk meg a standard oldat és a kivonat vonalát. Ha nem áll fenn interferencia, a két vonal párhuzamos lesz.
8.2. A relatív aktivitás logaritmusát a következő képlet segítségével számíthatjuk ki:
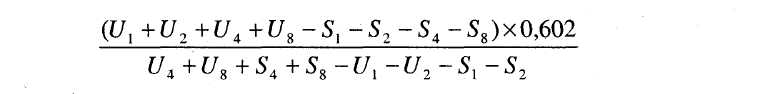
valódi aktivitás = feltételezett aktivitás x relatív aktivitás
9. Ismételhetőség
Az ugyanazon a mintán végzett két párhuzamos meghatározás eredménye közötti különbség relatív értékben kifejezve nem haladhatja meg a 10%-ot.
XLVII.
A flavofoszfolipol agar táptalajon, diffúzió útján történő meghatározása
1. Cél és alkalmazási terület
A módszer a takarmányokban, koncentrátumokban és előkeverékekben található flavofoszfolipol meghatározására szolgál. A meghatározás alsó méréshatára 1 mg/kg (1 ppm).
2. Vizsgálati alapelv
A mintát hígított metil-alkohollal, vísszafolyós hűtő alatt hevítve extraháljuk. Centrifugálás után a kivonatot ioncserélő gyantával történő kezeléssel derítjük (szükség esetén), majd hígítjuk. Antibiotikus aktivitását a flavofoszfolipol, Staphylococcus aureusszal beoltott agar táptalajon végbemenő diffúziójának mérésével határozzuk meg. A diffúziót a mikroorganizmus gátlási zónáinak képződése jelzi. E zónák átmérője egyenes arányos az antibiotikum-koncentrációnak az alkalmazott antibiotikumkoncentrációk tartománya feletti logaritmusával.
3.A mikroorganizmus: staphylococcus aureus atcc 6538 P
3.1.A törzstenyészet fenntartása
Oltsunk Staphylococcus aureusszal ferde agar táptalajt (4.1.). 37 °C-on, 24 órán keresztül inkubáljuk, tároljuk hűtőszekrényben kb. 4 °C-on, és havonta oltsuk át ferde agarra.
3.2.A baktérium-szuszpenzió elkészítése17
Tegyünk félre két, törzstenyészetet tartalmazó (3.1.) csövet és hetente oltsuk át őket. 37 °C-on, 24 órán keresztül inkubáljuk és tároljuk hűtőszekrényben kb. 4 °C-on. A vizsgálat megkezdése előtt 24 órával oltsunk be ezzel a szaporulattal kettő-négy, ferde agar táptalajt (4.1.) tartalmazó csövet. 37 °C-on, 16-18 órán keresztül inkubáljuk. Készítsük el a szaporulat szuszpenzióját nátrium-klorid oldatban (4.3.). A szuszpenziónak, 578 nm-en mérve, 1 cm-es mérőcellában, nátrium-klorid oldat ellenében (4.3.) körülbelül 40%-os fényáteresztésűnek kell lennie.
4. Táptalajok és reagensek
| 4.1. Alaptáptalaj | |
| Húspepton | 6,0 g |
| Tripton | 4,0 g |
| Élesztőkivonat | 3,0 g |
| Húskivonat | 1,5 g |
| Glükóz | 1,0 g |
| Agar | 15,0 g |
| Víz | 1000 ml |
| Sterilizálás utáni pH: 6,5 | |
| 4.2. Vizsgálati táptalaj | |
| 4.2.1. Alapréteg19 | |
| Húspepton | 6,0 g |
| Élesztőkivonat | 3,0 g |
| Húskivonat | 1,5 g |
| Agar | 10,0 g |
| Víz | 1000 ml |
| Sterilizálás utáni pH: 6,5 |
4.2.2. Oltási réteg
Mint a 4.1. pontban, 2,0 g szilikon habzásgátló emulzió20 hozzáadásával.
4.3. 0,4%-os (w/v) nátrium-klorid oldat: oldjunk fel 4 g nátrium-kloridot a.t. vízben és hígítsuk fel 1000 ml-re; sterilizáljuk.
4.4.Tiszta metil-alkohol.
4.5.50%-os (v/v) metil-alkohol: hígítsunk fel 500 ml metil-alkoholt (4.4) 500 ml vízzel.
4.6.80%-os (v/v) metil-alkohol: hígítsunk fel 800 ml metil-alkoholt (4.4) 200 ml vízzel.
4.7.Trisz-hidroximetil-aminometán, a.t.
4.8. Kálium-klorid 1,5%-os (w/v) metil-alkoholos oldata: oldjunk fel 1,5 g a.t. kálium-kloridot 20 ml vízben, töltsük fel metil-alkohollal (4.4) 100 ml-re.
4.9. Kationcserélő: Dowex 50 W x 8, 20.50 szemnagyságú, Na-típusú (Serva kat. 41600-as számú) vagy ezzel egyenértékű.
4.10.Anioncserélő: Dowex 1x2, 50.100 szemnagyságú, Cl-típusú (Serva kat. 41010-es számú) vagy ezzel egyenértékű. Használat előtt tartsa 12-14 óráig 80%-os metil-alkoholban (4.6.).
4.11.Üvegvatta.
4.12: pH indikátorpapír (pH 6,6.8,1).
4.13.Aszkorbinsav.
4.14.Standard anyag: ismert aktivitású flavofoszfolipol.
5.Eszközök
5.1". Kromatográfiás üvegcső, belső átmérője 9 mm, hossza 150-200 mm, szűkített alsó részénél elzárócsappal, felső részénél üvegcsiszolattal -ami összeköti a csepegtetőtölcsérrel (5.2.)-ellátott.
5.2. 250 ml-es csepegtetőtölcsér, elzárócsappal és üvegcsiszolattal.
5.3.250 ml-es Erlenmeyer-lombik üvegcsiszolattal.
5.4.Visszafolyós hűtő üvegcsiszolattal.
6.Standard oldatok
6.1. Oldjunk fel egy pontosan kimért mennyiségű standard anyagot (4.14.) 50%-os metil-alkoholban (4.5.), és hígítsuk fel úgy, hogy 100 μg/ml flavofoszfolipolt tartalmazó törzsoldatot kapjunk. Lezárt lombikokban, 4 °C-on tárolva az oldat két hónapig stabil marad.
6.2.Ezen törzsoldatból, 50%-os metil-alkohollal (4.5.) történő többszöri hígítással készítsük el a következő oldatokat:
| S8 | 0,2 μg/ml |
| S4 | 0,1 μg/ml |
| S2 | 0,05 μg/ml |
| S1 | 0,025 μg/ml |
7. A kivonat elkészítése
7.1. Extrahálás
7.1.1. Koncentrátumok, előkeverékek és ásványi takarmányok
Mérjünk ki 2,0-5,0 g mintát és adjunk hozzá kb. 150 mg aszkorbinsavat (4.13.). Homogenizáljuk 150 ml 50%-os metil-alkohollal (4.5.) egy Erlenmeyer-lombikban (5.3.) és kb. 400 mg trisz-hidroximetil-aminometán (4.7.) segítségével állítsuk be a pH-t 8,1-8,2-re. Ellenőrizzük a pH-t indikátorpapírral (4.12.). Hagyjuk állni 15 percig, azután újra állítsuk be a pH-t trisz-hidroximetil-aminometánnal (4.7.) 8,1-8,2-re, majd forraljuk 10 percig visszafolyós hűtő alatt (5.4.), állandó keveréssel. Hagyjuk lehűlni, centrifugáljuk a keveréket és dekantáljuk a kivonatot.
7.1.2. Egyéb takarmányok
Mérjünk ki egy legalább 30 μg flavofoszfolipolt tartalmazó, 5,0-30,0 g tömegű mintát. Homogenizáljuk 150 ml 50%-os metil-alkohollal (4.5.) egy Erlenmeyer-lombikban (5.3.), és kb. 400 mg trisz-hidroximetil-aminometán (4.7.) segítségével állítsuk be a pH-t 8,1.8,2-re. Ellenőrizzük a pH-t indikátorpapírral (4.12.). Hagyjuk állni 15 percig, azután újra állítsuk be a pH-t trisz-hidroximetil-aminometánnal (4.7.) 8,1-8,2-re, majd forraljuk 10 percig visszafolyós hűtő alatt (5.4.), állandó keveréssel Hagyjuk kihűlni, centrifugáljuk a keveréket és dekantáljuk a kivonatot.
7.2.Derítés (a vizsgálat ezen szakasza koncentrátumok, előkeverékek és ásványi takarmányok esetében elhagyható)
7.2.1. Keverjünk össze 110 ml kivonatot 11 g kationcserélővel (4.9.), forraljuk egy percig visszafolyós hűtő alatt (5.4.), állandó keveréssel. Válasszuk le a kationcserélőt centrifugálással vagy szűréssel. Keverjünk össze 100 ml kivonatot 150 ml metil-alkohollal (4.4.), majd tároljuk az oldatot
12-15 óráig 4 °C-on. Szűrjük le még hidegen a flokkulens anyagot.
7.2.2. Helyezzünk egy üvegvatta dugót (4.11.) az üvegcső aljára (5.1.), öntsünk a csőbe 5 ml anioncserélőt (4.10.) és mossuk az oszlopot 100 ml, 80%-os metil-alkohollal (4.6.). A csepegtetőtölcsér (5.2.) segítségével öntsünk át az oszlopba legalább 100 ml szűrletet, amely várhatóan 16 μg flavofoszfolipolt tartalmaz (30 g-os takarmányminta esetében, 1 ppm-nél 200 ml-t). Adott esetben, mielőtt az oszlopba töltenénk, hígítsuk a szürletet 80%-os metil-alkohollal (4.6.), hogy a várt 16 μg/100ml koncentrációjú flavofoszfolipol-tartalmat kapjuk. Állítsuk be az átfolyási sebességet kb. 2 ml/perc értékre. Öntsük el a szüredéket. Ezután mossuk az oszlopot 50 ml, 80%-os metil-alkohollal (4.6.) és öntsük el a szüredéket.
7.2.3. Eluáljuk a flavofoszfolipolt kálium-klorid metil-alkoholos oldatával (4.8.), az átfolyási sebességet kb. 2 ml/perc értéken tartva. Vegyünk fel 50 ml eluátumot egy mérőlombikban, adjunk hozzá 30 ml vizet és keverjük össze. Ezen oldat flavofoszfolipol-tartalmának 0,2 μg/ml-nek kell lennie
(=U8).
7.3. Vizsgálati oldatok
7.3.1. Adott esetben (pl. a derítés kihagyása esetén), hígítsuk a 7.1.1. pontban kapott kivonatot 50%-os metil-alkohollal (4.5.), hogy a várt 0,2 μg/ml koncentrációjú flavofoszfolipol-tartalmat kapjuk (=U8).
7.3.2. Az U8 oldatból, 50%-os metil-alkohollal (4.5.) történő többszöri hígítás útján (1 + 1 arányban) készítsük el az U4 (várt tartalom: 0,1 μg/ml), az U2 (várt tartalom: 0,05 μg/ml) és az U1 (várt tartalom: 0,025 μg/ml) oldatokat.
8.A vizsgálat módja
8.1.A vizsgálati táptalaj beoltása
Oltsuk be baktérium-szuszpenzióval (3.2.) a vizsgálati táptalajt (4.2.2.) kb. 50 °C-on. A vizsgálati táptalajt (4.2.2.) tartalmazó tálcákon végzett előzetes vizsgálatokkal határozzuk meg a bakterium-szuszpenziónak azt a mennyiségét, amely a legnagyobb és legtisztábban látható gátlási zónákat adja a flavofoszfolipol különböző koncentrációival (kb. 30 ml/liter).
8.2.A tálcák előkészítése
8.2.1. Az agaron végbemenő diffúziót a négyféle koncentrációjú standard oldatot (S8, S4, S2, S1) és a négyféle koncentrációjú vizsgálati oldatot (U8, U4, U2, Ul) tartalmazó tálcákon végezzük. Ezt a négyféle koncentrációban meglévő kivonatot és standard oldatot minden egyes tálcán el kell helyezni. Ezért olyan tálcákat válasszunk, amelyek elég nagyok ahhoz, hogy az agar táptalajon legalább nyolc, egymástól minimum 30 mm távolságra lévő, egyenként 10-13 mm átmérőjű lyuk elférjen rajtuk. A vizsgálat elvégezhető olyan üveglemez tálcákon is, amelyeket egy 200 mm átmérőjű és 20 mm magas megmunkált alumínium vagy műanyag karimagyűrű szegélyez.
8.2.2. Öntsünk a tálcákra annyi táptalajt (4.2.1.), hogy az kb. 1,5 mm vastagságú réteget képezzen (45 ml-t egy 200 mm átmérőjű tálcára). Engedjük egyenletesen eloszlani, helyezzünk rá egy újabb, a 8.1. pontban leírt módon beoltott táptalaj (4.2.2.) réteget, hogy kb. 1 mm vastag réteget képezzen (30 ml-t egy 200 mm átmérőjű tálcán). Engedjük ismét egyenletesen eloszlani, fúrjunk lyukakat, és helyezzük el bennük a pontosan kimért mennyiségű vizsgálati és standard oldatokat (az átmérőtől függően 0,10 és 0,15 ml közötti mennyiséget lyukanként).
8.2.3. Minden egyes koncentrációt legalább négyszer kell felvinni, így mindegyik meghatározás 32 gátlási zóna értékelésén alapulhat.
8.3.Inkubálás
Inkubálja a tálcákat 16-18 órán át 28-30 °C-on.
9.Értékelés
9.1.Mérjük meg a gátlási zónák átmérőjét, 0,1 mm-es pontossággal. Az egyes koncentrációkon végzett mérések átlagát vigyük fel egy féllogaritmikus függvénydiagram-papírra, amely a gátlási zónák átmérőjével arányosan mutatja a koncentrációk logaritmusát. Szerkesszük meg mind a standard oldat, mind a kivonat regressziós egyenesét, például az alábbiak szerint.
9.2.A következő képlet segítségével határozzuk meg a standard legalacsonyabb értékéhez (SL) tartozó regressziós pontot:
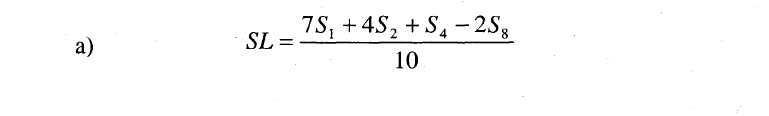
9.3. A következő képlet segítségével határozzuk meg a standard legmagasabb értékéhez (SH) tartozó regressziós pontot:
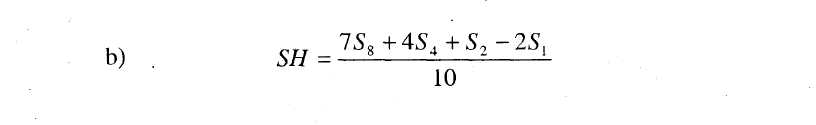
9.4. Hasonló módon, határozzuk meg a kivonat legalacsonyabb (UL) és legmagasabb értékéhez (UH) tartozó regressziós pontokat, úgy, hogy a fenti képletben az S1, S2, S4 és S8 jelöléseket az U1, U2, U4 és U8 jelölésekkel helyettesítjük.
9.5.Vigyük fel a kiszámított SL és SH értékeket ugyanarra a függvény diagram-papírra és kössük össze őket, hogy megkapjuk a standard oldathoz tartozó regressziós egyenest. Hasonló módon vigyük fel az UL és UH értékeket és kössük össze őket, hogy megkapjuk a kivonathoz tartozó regressziós egyenest.
9.6. Amennyiben nem áll fenn semmilyen interferencia, a vonalaknak párhuzamosan kell futniuk. Gyakorlati szempontból a vonalak párhuzamosnak tekinthetőek, ha az (SH-SL) és (UH-UL) értékek legfeljebb 10%-os mértékben térnek el átlagértéküktől.
9.7.Amennyiben a vonalak nem futnak párhuzamosan, akkor vagy az U1 és S1, vagy az U8 és S8 figyelmen kívül hagyható, az SL, SH, UL és UH pedig más képleteket alkalmazva számítható ki, amelyek alternatív regressziós egyeneseket adnak meg:
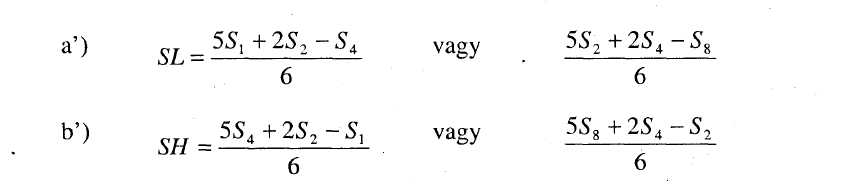
és ehhez hasonlóan az UL és UH esetében. Az alternatív regressziós egyenesek párhuzamosságát az előzőhöz hasonlóan kell ellenőrizni. A zárójelentésben meg kell említeni, hogy az eredmény három érték alapján lett kiszámítva.
9.8. Ha a vonalak párhuzamosnak tekinthetőek, a következő képlet segítségével számítsuk ki a relatív aktivitás logaritmusát (lg A).


Tényleges aktivitás = feltételezett aktivitás x relatív aktivitás.
9.9. Ha a vonalak nem tekinthetőek párhuzamosnak, ismételjük meg a meghatározást. Ha a párhuzamosságot ezután sem sikerül elérni, számoljuk ki a relatív aktivitás logaritmusát (lg A) a c) képlet segítségével. A kapott eredmény azonban csak hozzávetőlegesnek tekinthető, és ezt fel kell tüntetni a zárójelentésben.
10. Ismételhetőség
Ugyanazon analitikus által, ugyanazon a mintán végzett két meghatározás eredménye közötti különbség nem haladhatja meg: - a 0,5 mg/kg értéket, abszolút értékben, 1-2 mg/kg flavofoszfolipol-tartalmom esetén,
-a 25%-ot, 2-10 mg/kg közötti koncentráció legmagasabb értékeire vonatkoztatva,
-a 20 %-ot, 10-25 mg/kg közötti koncentráció legmagasabb értékeire vonatkoztatva,
-az 5 mg/kg értéket, abszolút értékben, 25-50 mg/kg közötti koncentráció esetén,
-a 10%-ot, 50 mg/kg feletti koncentráció legmagasabb értékeire vonatkoztatva."
VII. A R. 11. számú mellékletének módosítása
A R. 11. számú melléklete a következő IV. fejezettel egészül ki:
"IV.
A takarmányok dioxin tartalmának megállapítására szolgáló vizsgálatokhoz szükséges hatósági minták vételét az A. Rész, a minták vizsgálatra való előkészítését, valamint a minta vizsgálatára vonatkozó követelményeket a B. Rész tartalmazza.
A.Rész
Mintavételi módszerek egyes takarmányok dioxintartalmának (PCDD/PCDF) hatósági ellenőrzéséhez és a dioxinszerű PCB-k meghatározásához
1. A vizsgálatok céljára a takarmányokból e mellékletben foglaltak szerint kell mintát venni. Az így kapott összesített minták azon tételek vagy résztételek reprezentatív mintáinak tekintendők, amelyből vételezték őket. A nemkívánatos anyagok maximális szintjeire vonatkozó követelményeknek való megfelelést a laboratóriumi mintákban meghatározott szintek alapján kell megállapítani.
2.A takarmány tétellel azonos minta vizsgálati eredményének elfogadása
Az ellenőrző laboratóriumnak két párhuzamos mérésben kell elvégezni a minta analízisét, ha az első analízis eredménye kevesebb, mint 20%-kal van a maximális szint alatt vagy felett, és ki kell számítani az eredmények átlagát. A tételazonos mintavizsgálat eredményét akkor lehet elfogadni, ha az első analízis eredménye több, mint 20%-kal van a maximális szint alatt, vagy párhuzamos analízis esetén a vizsgálati eredmény átlaga megfelel az előírt maximális szintnek.
B.Rész
Egyes takarmányok dioxintartalmának (PCDD/PCDF) hatósági ellenőrzésekor és dioxinszerű PCB-tartalmuk meghatározásakor végzett minta-előkészítésre és az alkalmazott analitikai módszerekre vonatkozó követelmények
1. Célkitűzések és alkalmazási terület
1.1. Ezeket a követelményeket kell alkalmazni a takarmány-alapanyagokban és a takarmányokban a dioxinok (poliklórozott dibenzopdioxinok (PCDD), poliklórozott dibenzofuránok (PCDF) és a dioxinszerű poriklórozott bifenilek (PCB-k) mennyiségének meghatározására.
1.2.A dioxinok jelenlétét a takarmányokban olyan stratégia alapján kell vizsgálni, amely tartalmaz egy szűrőmódszert azoknak a mintáknak a kiválasztására, amelyekben a dioxinok és dioxinszerű PCB-k mennyisége kevesebb, mint 30-40%-kal van az adott szint alatt vagy felett. E jelentős dioxintartalmú mintákban a dioxinok koncentrációját egy megerősítő módszerrel kell meghatározni.
1.3. A szűrőmódszerek olyan módszerek, amelyekkel kimutatható a dioxinok és a dioxinszerű PCB-k jelenléte az adott szinten. E módszerek nagy mintaáteresztő képességűek, és arra használják őket, hogy nagyszámú minta közül kiszűrjék a lehetséges pozitívakat. Úgy vannak kialakítva, hogy segítségükkel elkerülhetőek legyenek a téves negatív eredmények.
1.4. A megerősítő módszerek teljes vagy kiegészítő információkat szolgáltató módszerek, amelyek lehetővé teszik a dioxinok és dioxinszerű PCB-k azonosítását és egyértelmű mennyiségi meghatározását az adott szinten.
2.Háttér
2.1.Mivel a környezeti és a biológiai minták (ezen belül a takarmány-alapanyag és a takarmány minták is) általában különböző rokon dioxinvegyületek komplex keverékeit tartalmazzák, a kockázatfelmérés megkönnyítése érdekében kidolgozták a Toxikus Egyenértékűségi Faktorok (TEF-ek) koncepcióját. ATEF-ek a 2,3,7,8-helyen szubsztituált PCDD-ket és PCDF-eket tartalmazó keverékek koncentrációjának kifejezésére, újabban pedig a dioxinszerű aktivitással rendelkező egyes nem orto és mono-orto, klórral szubsztituált PCB-knek a 2,3,7,8-TCDD toxikus egyenértékben (TEQ) való kifejezésére is szolgálnak.
2.2. Az egyes anyagok adott mintában mért koncentrációit megszorozzuk a hozzájuk tartozó TEF-értékkel, majd összeadjuk őket, és így megkapjuk a dioxinszerű komponensek TEQ-ben kifejezett összes koncentrációját.
2.3. A "felsőhatár" koncepciójánál a számszerűsítési határt kell használni minden egyes nem számszerűsített rokonvegyület TEQ-hez való hozzájárulásának kiszámításához.
2.4. Az "alsóhatár" koncepciójánál nullát kell használni minden egyes nem számszerűsített rokon vegyület TEQ-hez való hozzájárulásának kiszámításához.
2.5. A "középsőhatár" koncepciójánál a számszerűsítési határ felét kell használni minden egyes nem számszerűsített rokonvegyület TEQ-hez való hozzájárulásának kiszámításához.
3. A minta előkészítésére vonatkozó minőségbiztosítási követelmények
3.1. Az elemzés céljára szolgáló minták elkészítését illetően a takarmányok hatósági ellenőrzésére szolgáló közösségi elemzési módszereket kell alkalmazni.
3.2.Emellett a következő előírásokat kell betartani:
- a mintákat üveg-, alumínium-, polipropilén vagy polietilén edényekben kell tárolni és szállítani; a papírpor maradékokat el kell távolítani a mintatartóból; az üvegeszközöket dioxin jelenlétére előzetesen ellenőrzött oldószerekkel kell kiöblíteni,
- vakelemzés szükséges az egész analitikai eljárás elvégzésével, a mintát azonban ki kell hagyni belőle,
- az extrakcióhoz használt minta tömegének olyannak kell lennie, hogy megfeleljen az érzékenységre vonatkozó követelményeknek.
4. A laboratóriumokra vonatkozó követelmények
4.1. A laboratóriumok demonstrálják egy módszer teljesítményét az adott szint tartományában, pl. az adott szint felénél, egyszeresénél és kétszeresénél, az ismételt analízisekben elfogadható variációs koefficienssel. Az elfogadási kritériumokat részletesen az 5. pont tartalmazza.
4.2.Egy megerősítő módszer esetében a számszerűsítési határnak az adott szint körülbelül egyötödénél kell lennie annak érdekében, hogy az adott szint tartományában elfogadható variációs koefficienseket kapjunk.
4.3. Belső minőségellenőrzési intézkedésként rendszeresen kell végezni vak kontrollvizsgálatokat és beállító kísérleteket vagy kontrollminta-elemzést (lehetőleg hitelesített referencia anyagon).
4.4. Adott analízisek vonatkozásában a kompetencia igazolásának legjobb módja a laboratóriumok szakmai felkészültségét felmérő, laboratóriumok közötti vizsgálatokban való sikeres részvétel. A laboratóriumok közötti vizsgálatokban való sikeres részvétel, pl. a talaj- vagy a szennyvízminták terén, nem feltétlenül bizonyítja az élelmiszer- vagy a takarmányminták területén való jártasságot, amelyek szennyezettségi szintje alacsonyabb. Kötelező így folyamatosan résztvenni olyan, laboratóriumok közötti vizsgálatokban, amelyek a dioxinok és a dioxinszerű PCB-k meghatározására irányulnak a megfelelő takarmány/élelmiszer mátrixokban.
4.5. A laboratóriumokat egy elismert, és az ISO Guide 58-cal összhangban működő testület akkreditálja annak biztosítása céljából, hogy analitikai minőségbiztosítást alkalmazzanak. A laboratóriumokat az EN/ISO/IEC 17025:2000 szabvány szerint kell akkreditálni.
5. A dioxinok és dioxinszerű PCB-k esetében használt analitikai eljárásokra vonatkozó követelmények
Az analitikai eljárások elfogadására vonatkozó alapkövetelmények
5.1.Nagy érzékenység és alacsony kimutatási határ
5.1.1. A PCDD-k és PCDF-ek vonatkozásában a kimutatási határnak a pikogramm ( 10-12 g) TEQ tartományban kell lennie, mivel egyes vegyületek nagyon mérgezőek ezek közül. Ismert, hogy a PCB-k nagyobb koncentrációban fordulnak elő, mint a PCDD-k és a PCDF-ek. A legtöbb PCB rokonvegyület esetében már elegendő a nanogrammos 10-9 g) érzékenység is.
5.1.2. A mérgezőbb dioxinszerű PCB rokonvegyületek (és különösen a nem orto szubsztituált rokonvegyületek) meghatározásához azonban ugyanolyan érzékenységre van szükség, mint a PCDD-k és PCDF-ek esetében.
5.2.Nagy szelektivitás (specifikusság)
Meg kell tudni különböztetni a PCDD-ket, a PCDF-eket és dioxinszerű PCB-ket az igen sokféle, velük együtt extrahálódó és a mérést esetleg zavaró más vegyülettől, amelyek több nagyságrenddel magasabb koncentrációban lehetnek jelen, mint az analízis tárgyát képező vegyületek. A gázkromatográfiás/tömegspektrometriás (GC/MS) módszerekhez a különböző rokonvegyületeket, így pl. a mérgező (pl. a tizenhét 2,3,7,8-helyen szubsztituált PCDD és PCDF és a dioxinszerű PCB-k) és nem mérgező rokonvegyületeket meg kell tudni különböztetni egymástól. A biológiai tesztekkel szelektíven meg kell tudni határozni a TEQ értékeket, mint a PCDD-k, a PCDF-ek és a dioxinszerű PCB-k összegét.
5.3. Nagy pontosság (megbízhatóság és precizitás)
5.3.1.A meghatározásnak érvényes és megbízható becslést kell adnia a minta valódi koncentrációjáról. A nagy pontosságra (a mérés pontossága: egy mérés eredménye és a mérés valódi vagy kijelölt értéke közötti eltérés mértéke) azért van szükség, hogy elkerüljük egy mintaanalízis eredményének visszautasítását a becsült TEQ-érték gyenge megbízhatósága miatt. A pontosságot a megbízhatósággal (egy hitelesített anyagban az adott komponensre mért átlagérték és a hitelesített érték közötti különbség, ez utóbbi százalékában kifejezve) és a precizitással (a precizitást általában standard eltérésként számítjuk, amely magában foglalja a megismételhetőséget és a reprodukálhatóságot is, és azt fejezi ki, hogy az előírt körülmények között többször elvégezve a kísérletet a kapott eredmények mennyire egyeznek meg egymással) fejezzük ki.
5.3.2. A szűrőmódszerek biológiai tesztekből és GC/MS módszerekből állhatnak, a megerősítő módszerek nagy felbontású gázkromatográfiós/nagy felbontású tömegspektrometriás (HRGC/HRMS) módszerek. Az összes TEQ érték esetében a következő kritériumoknak kell megfelelni:
| Szűrőmódszer | Megerősítő módszer | |
| Téves negatívok aránya | < 1% | |
| Megbízhatóság | - 20%-tól + 20%-ig | |
| Variációs koefficiens | < 30% | < 15% |
6. A szűrési és megerősítési célra alkalmazott GC/MS módszerekre vonatkozó különleges követelmények
6.1. Az analitikai eljárás hitelesítéséhez a 13 C-mal jelölt, a 2,3,7,8-helyen klórral szubsztituált belső PCDD/F standardok (illetve a 13 C-mal jelölt belső dioxinszerű PCB standardok, ha dioxinszerű PCB-ket kell meghatározni) hozzáadását az analitikai módszer legelején kell elvégezni, pl. az extrakció előtt. A tetraklórozottól az oktaklórozottig terjedő PCDD/F-ek homológ csoportjai mindegyikéből legalább egy rokonvegyület (illetve, ha dioxinszerű PCB-ket kell meghatározni, akkor a dioxinszerű PCB-k homológ csoportjai mindegyikéből legalább egy rokonvegyületet) kell hozzáadni (alternatív lehetőség: legalább egy rokonvegyület kerül hozzáadásra a PCDD/F-ek és a dioxinszerű PCB-k nyomon követéséhez használt, tömegspektrometriával kiválasztott ionregisztráló funkcióként). Egyértelműen előnyösebb a megerősítő módszerek esetében, ha mind a 17 13 C-mal jelölt 2,3,7,8-helyen szubsztimált belső PCDD/F standardot, illetve (ha dioxinszerű PCB-ket kell meghatározni) mind a 12 13 C-mal jelölt belső dioxinszerű PCB standardot használjuk.
Megfelelő kalibrációs oldatok alkalmazásával a relatív válaszfaktorokat is meg kell határozni azoknál a rokonvegyületeknél, amelyek 13 C-mal jelölt analógját nem adtuk hozzá a standardhoz.
6.2. A növényi eredetű takarmányok és a kevesebb, mint 10% zsírt tartalmazó állati eredetű takarmányok esetében a belső standardokat kötelező az extrakció előtt hozzáadni. A 10%-nál magasabb zsírtartalmú állati eredetű takarmányok esetében a belső standardokat az extrakció előtt vagy a
zsírextrakció után is hozzáadhatjuk. Az extrakció hatékonyságát megfelelően hitelesíteni kell attól függően, hogy a belső standardokat melyik szakaszban adjuk a mintákhoz, valamint, hogy az eredményeket termékre vagy zsírra adjuk meg.
6.3. A GC/MS analízis előtt egy vagy két visszanyerési (helyettes) standardot is hozzá kell adni.
6.4. A visszanyerést ellenőrizni kell. A megerősítő módszerek esetében az egyes belső standardok visszanyerésének a 60-120%-os tartományban kell lennie. Az egyes rokonvegyületek vonatkozásában, különösen egyes hepta- és oktaklorozott dibenzodioxinok és dibenzofuránok esetében, ennél alacsonyabb vagy magasabb visszanyerés is elfogadható azzal a feltétellel, hogy a TEQ-értékhez való hozzájárulásuk nem haladhatja meg (csak PCDD/F-re alapozott) az összes TEQ-érték 10%-át. A szűrőmódszerek esetében a visszanyerésnek a 30-140%-os tartományban kell lennie.
6.5. A dioxinokat a zavaró klórozott vegyületekből, így pl. a PCB-ktől és a klórozott difenil-éterektől megfelelő kromatográfiás eljárással kell elválasztani (lehetőleg florisil-, aluminium-oxid,
illetve szénoszlopon).
6.6. Megfelelő az izomerek gázkromatográfiás elválasztása (< 25% a csúcsok közötti eltérés az 1,2,3,4,7,8-HxCDF és az l,2,3,6,7,8HxCDF között).
6.7. A meghatározást az EPA 1613 módszer B. módosítása szerint kell elvégezni, amely a tetraklórozottól az oktaklórozottig terjedő dioxinok és furánok izotóphígításos HRGC/HRMS-sel vagy más, egyenértékű teljesítménykritériumokkal rendelkező módszerrel történő meghatározására vonatkozik.
6.8. A felsőhatár és az alsóhatár szintje közötti különbség nem haladhatja meg a 20%-ot olyan tartományokban, amelyek dioxin szennyezettsége a maximális tartományban vagy afelett van. A maximális szintnél jóval alacsonyabb szennyezettségű takarmányok esetében a különbség 25-40%-os tartományban mozoghat.
7. Analitikai szűrőmódszerek
7.1. A szűrőmódszerek használatánál különböző analitikai célok teljesíthetőek: egyszerű szűrés és kvantitatív meghatározás
7.1.1. A szűrés
A minták eredményeit összehasonlítjuk egy, az adott szintet mutató referenciaminta eredményével. A referenciánál kisebb eredményt adó mintákat negatívnak minősítjük, a nagyobb eredményt adókat pedig feltételezett pozitívaknak.
7.1.1.1. Minden kísérletsorozatnak tartalmaznia kell egy vak- és egy referencia mintát, amelyeket egyidejűleg azonos körülmények között extrahálunk és tesztelünk. A referencia mintának egyértelműen magasabb eredményt kell adnia, mint a vaknak.
7.1.1.2. Az adott szint ellenőrzésére további, az adott szint felének és kétszeresének megfelelő referencia mintákat is alkalmazni kell annak igazolására, hogy a teszt az adott tartományban megfelelő teljesítményű.
7.1.1.3.Amikor más mátrixokat tesztelünk, a referencia minta vagy referencia minták megfelelőségét is bizonyítani kell, lehetőleg olyan minták felhasználásával, amelyek HRGC/HRMS-sel mért TEQ-értéke hasonló a referencia mintáéhoz, vagy másképpen egy vakmintával, ami erre a szintre van beállítva.
7.1.1.4. Mivel a biológiai tesztekben nem használhatók belső standardok, nagyon fontosak az ismételhetőségi tesztek ahhoz, hogy meg tudjuk állapítani az egy kísérletsorozaton belüli standard eltérést. A variációs koefficiensnek 30% alatt kell lennie.
7.1.1.5. A biológiai tesztek esetében meg kell határozni a célvegyületeket, az esetleges zavaró hatásokat és az elfogadható vakszintek maximumát.
7.1.2. A kvantitatív meghatározás
A kvantitatív megközelítéshez standard hígítási sorok, két vagy három sorozatban párhuzamosan végzett tisztítás és mérés, valamint vak és visszanyerési kontrollok szükségesek. Az eredmények kifejezhetők TEQ-ben, feltételezve, hogy a jelért felelős vegyületek megfelelnek a TEQ-alapelvnek. Ezt TCDD (vagy egy dioxin/furán standard keverék) használatával végezhetjük, amellyel felveszünk egy kalibrációs görbét, hogy ez alapján ki tudjuk számolni az extraktumra és így a mintára jellemző TEQ-szintet. Ezt azután korrigáljuk a vakmintára számított TEQ-szinttel (hogy figyelembe vegyük az alkalmazott oldószerekből és vegyszerekből származó szennyezéseket), valamint egy viszanyerésre számított TEQ-szinttel (amelyet egy, kérdéses szint körüli értékű minőségellenőrző minta TEQ szintjéből számolunk). Fontos megjegyezni, hogy a nyilvánvaló visszanyerési veszteség egy része mátrixhatásokból, illetve biológiai tesztekben kapott TEF-értékek és a WHO által rögzített hivatalos TEF-értékek különbségéből is eredhet.
7.2. A szűrésre használt analitikai módszerekre vonatkozó követelmények
7.2.1 A szűréshez használhatók a GC/MS analitikai módszerek és biológiai tesztek. A GC/MS módszerek vonatkozásában a 6. pontban megállapított követelményeket kell alkalmazni. A sejtalapú biológiai tesztek vonatkozásában a 7.3. pontban, a reagenskészlet-alapú biológiai tesztek vonatkozásában a 7.4. pontban megállapított különleges követelményeket kell alkalmazni.
7.2.2. Egy nagyszámú, a maximális vagy az akciós szint alatti vagy feletti mintákat tartalmazó sorozatnál szükség van a téves pozitív és téves negatív eredmények számával kapcsolatos információkra a megerősítő módszerrel meghatározott TEQ-tartalommal összehasonlításban. A téves negatívak tényleges arányának 1 % alatt kell lennie. A téves pozitív minták arányának elég alacsonynak kell lenni ahhoz, hogy előnyös legyen a szűrőmódszer alkalmazása.
7.2.3. A pozitív eredményeket mindig meg kell erősíteni egy megerősítő analitikai módszerrel (HRGC/HRMS). Továbbá HRGC/HRMS-sel meg kell erősíteni a széles TEQ- tartományból származó mintákat (a negatív minták kb. 2-10%-a). A biológiai tesztek és a HRGC/HRMS eredményeinek összefüggésével kapcsolatos információkat is meg kell adni.
7.3.A sejtalapú biológiai tesztekre vonatkozó különleges követelmények
7.3.1. A biológiai tesztek végzése során minden teszt lefuttatásához szükség van egy TCDD vagy dioxin/furán keverék referenciakoncentráció-sorozatra (teljes dózisválasz görbe, ahol R2 > 0,95). Szűrési célokra azonban egy kibővített, alacsony szintű minták mérésére alkalmas alacsony-szint görbét alkalmazhatunk.
7.3.2. Egy minőségellenőrzési lapon rögzíteni kell a biológiai tesztek eredményeinek megállapításához az adott időtartam alatt használt TCDD referencia-koncentrációt (körülbelül a mennyiségi meghatározás határértékének háromszorosa). Alternatív lehetőséget jelenthet egy referencia mintának a TCDD kalibrációs vonalhoz viszonyított relatív válasza, mivel a sejtek válasza sok tényezőtől függhet.
7.3.3. Minden típusú referenciaanyaghoz fel kell venni egy minőségellenőrzési (QC) diagramot, amelyet ellenőrizni kell annak érdekében, hogy az eredmények összhangban vannak-e a meghatározott iránymutatásokkal.
7.3.4 Különösen a kvantitatív számítások esetén, az alkalmazott hígítási minta indukciójának a válaszgörbe lineáris szakaszára kell esnie. A válaszgörbe lineáris szakasza feletti mintákat fel kell hígítani, és újra meg kell mérni. Ajánlott tehát egyszerre legalább két vagy három hígítást mérni.
7.3.5.Az egyes hígítási minták három párhuzamos sorozatban való mérése esetén a százalékos standard eltérés nem lehet 15% felett, három független kísérlet között pedig nem haladhatja meg a 30%-ot.
7.3.6. A kimutatási határt az oldószeres vak- vagy a háttér által adott válasz standard eltérésének háromszorosában lehet megállapítani. Egy másik megközelítés szerint egy, a napi kalibrációs görbéből számolt háttér (az indukciós faktor az oldószeres vak ötszöröse) feletti választ alkalmazunk. A számszerűsítési határt az oldószeres vak- vagy a háttér által adott válasz standard eltérésének öthatszorosában lehet megállapítani, vagy alkalmazhatunk egy egyértelműen a napi kalibrációs görbéből számolt és a háttérjelnél nagyobb jelet (az indukciós faktor az oldószeres vak 10-szerese).
7.4. A reagenskészlet-alapú biológiai tesztekre vonatkozó különleges követelmények
7.4.1.Követni kell a gyártó mintakészítésre és analízisre vonatkozó utasításait.
7.4.2.A szavatossági idő lejárta után a reagenskészleteket nem szabad felhasználni.
7.4.3. Nem alkalmazhatók a más reagenskészletekben történő felhasználásra készített anyagok vagy összetevők.
7.4.4. A reagenskészleteket a megadott tárolási hőmérsékleti tartományban kell tartani, és a megadott működési hőmérsékleten kell felhasználni.
7.4.5. Az immuntesztek kimutatási határát úgy kapjuk meg, hogy a 10 ismétlésben mért vak alapján kapott átlag és a standard eltérés háromszorosának összegét elosztjuk a lineáris regressziós egyenlet meredekségével.
7.4.6. A laboratóriumban végzett tesztekhez referencia standardokat kell használni, hogy biztosítsuk azt, hogy a standardra adott válasz elfogadható tartományba essen.
8. Az eredményekről készült jelentés
8.1. Amennyiben az alkalmazott analitikai eljárás lehetővé teszi, az analitikai eredményeknek tartalmazniuk kell az egyes PCDD/F- és PCB-rokon vegyületek szintjét, és amelyeket ki kell fejezni alsó határral, felső határral és középső határral is ahhoz, hogy az eredményekről készült jelentés a lehető legtöbb információt tartalmazza, és így lehetővé tegye az eredmények különleges követelmények szerinti értelmezését.
8.2.A jelentésben meg kell adni a minta lipidtartalmát, és a lipidextrakcióhoz alkalmazott módszert is.
8.3. Az egyes belső standardok visszanyerését meg kell adni abban az esetben, ha a visszanyerések kívül esnek a 6. pontban említett tartományon, vagy ha az eredmények meghaladják a maximális szintet, vagy egyéb esetekben kérés alapján.
Megjegyzés:
Eddig még nincs bizonyíték arra vonatkozólag, hogy a kereskedelmi forgalomban kapható
reagenskészlet-aiapú biológiai tesztek kellően érzékenyek és megbízhatóak ahhoz, hogy az
élelmiszerekben vagy takarmányokban a szükséges szinteken dioxinok jelenlétére lehessen szűrni
velük."
VIII. Az R. 13. számú mellékletének módosítása
Az R. 13. számú mellékletének függelékében a "Májműködés segítése idült máj elégtelenség esetén" sor helyébe a következő rendelkezés lép:
| [Különleges táplálási cél | Alapvető termék jellemzők | Állatfajok vagy -kategóriák | A címkén garantálandó jellemzők | Az alkalmazás javasolt időtartama | Egyéb rendelkezések] |
| „Máj működés segítése krónikus máj elégtelenség esetén | Jó minőségű fehérje, mérsékelt fehérjetartalom, magas esszenciális zsírsavtartalom és magas könnyen emészthető szénhidráttartalom | Kutya | - Fehérjeforrás(ok) - Esszenciális zsírsavtartalom - Könnyen emészthető szénhidrátok, megfelelő esetben előkezelésük megjelölésével - Nátrium - Összes réz | Kezdetben legfeljebb 6 hónapig. | A csomagolóeszközön, címkén, illetve a kísérő okmányon feltüntetendő: „Alkalmazás, illetve az alkalmazás időtartamának meghosszabbítása előtt állatorvos véleményének kikérése javasolt." A használati utasításban feltüntetendő: „Folyamatos ivóvízellátást kell biztosítani."" |
| Jó minőségű fehérje, mérsékelt fehérjetartalom és magas esszenciális zsírsavtartalom | Macska | - Fehérjeforrás(ok) - Esszenciális zsírsavtartalom - Nátrium - Összes réz |
2. számú melléklet a 20/2004. (II. 27.) FVM rendelethez
"14. számú melléklet a 44/2003. (IV. 26.) FVM rendelethez
Az állatok fehérje ellátásának javítására használható bizonyos termékek és engedélyezésük alapelvei
I.
1. Az állatok fehérje ellátásának javítására használható bizonyos termékek forgalomba hozatala és felhasználása
1.1. A II. fejezet táblázatában (a továbbiakban: táblázatban) felsorolt termékek a növényi eredetű fehérjeforrásokkal nem pótolható fehérjeigény kielégítésére szolgálnak. A táblázatban foglalt előírások nem érintik e rendelet mellékleteiben és más jogszabályokban a takarmányokra vonatkozóan megadott határértékeket.
1.2. A táblázatban felsorolt termékcsoportok valamelyikéhez tartozó, állatok fehérje ellátásának javítására használható bizonyos termék vagy ilyen terméket tartalmazó takarmány akkor hozható forgalomba, illetve használható fel, ha az állatok fehérje ellátásának javítására használható bizonyos termék szerepel a táblázatban, és megfelel az ott előírt feltételeknek.
1.3. A táblázatba akkor kerülhet felvételre valamely állatok fehérje ellátásának javítására használható bizonyos termék, ha
a) rendelkezik az állatok számára hasznos tápláló értékkel,
b) rendeltetésszerű használatuk mellett nincs káros hatásuk az emberi vagy állati egészségre, illetve a környezetre,
c) a takarmányban ellenőrizhető.
1.4. Állatok fehérje ellátásának javítására használható bizonyos terméknek a táblázatba történő felvétele érdekében a 2. pont szerinti dokumentációt kell készíteni, amelyet meg kell küldeni a tagállamoknak, a Bizottságnak, és ha konzultációt kérnek, a Bizottság által létrehozott tudományos bizottságok tagjainak.
1.5.A dokumentáció kezelése során be kell tartani az üzemi és üzleti titokra vonatkozó előírásokat.
1.6.Nem tekinthető üzleti vagy üzemi titoknak:
1. a termék neve és összetétele,
2. a mikroorganizmusra és a szubsztrátra vonatkozó információk,
3. a termék fiziko-, kémiai és biológiai tulajdonságai,
4. a termékre vonatkozó gyógyszertani, toxikológiai és ökotoxikológiai adatok értékelése,
5. a terméknek a takarmány összetevőjeként való ellenőrzésére szolgáló analitikai módszerek.
1.7. A táblázatban felsorolt valamely, az állatok fehérje ellátásának javítására használható bizonyos termék felhasználását a minisztérium ideiglenesen felfüggesztheti vagy korlátozhatja, amennyiben új információk, vagy meglévő információk újraértékelésének eredményeként megalapozottan megállapítható, hogy veszélyt jelent közvetlenül az állat, illetve közvetve az ember egészségére vagy a környezetre. Erről a minisztérium értesíti az Európai Bizottságot, és a többi tagállamot.
1.8. A táblázatban felsorolt termékek csomagolóeszközén, címkéjén, illetve ömlesztve vagy tartályban való szállítás esetén a kísérő okmányon fel kell tüntetni a termékre vonatkozóan a táblázatban felsorolt adatokat.
2. Az állatok fehérje ellátásának javítására használható bizonyos termékek engedélyezésének alapelvei
2.1.A dokumentáció elkészítéséhez a 3. pont szerinti vizsgálatokat kell elvégezni. A dokumentációnak lehetővé kell tenni az állatok fehérje ellátásának javítására használható bizonyos termékek értékelését a mai ismereteknek megfelelően, valamint meg kell felelnie az 1.3. pontban meghatározott feltételeknek.
2.2.Meg kell adni
a) a mikroorganizmus azonosságának és a táptalaj összetételének megállapításához,
b) a termékek előállításának eljárása, a főbb tulajdonságok, a kiszerelés, a felhasználási feltételek, a meghatározási módszerek és termék táplálkozástani tulajdonságainak meghatározásához,
c) a termék tűréshatárának a célfajokon keresztül történő értékesítéséhez,
d) a termék használata során közvetlenül vagy közvetve keletkező, az embereket vagy a környezetet fenyegető kockázatok felméréséhez szükséges adatokat. Az e célból előírt toxikológiai vizsgálatok a termék jellegétől, az érintett állatfajtól és a termék kísérleti állatokon megfigyelt anyagcseréjétől függnek.
2.3. A dokumentációnak részletes jelentéseket kell tartalmaznia, amelyeket e melléklet szerinti sorrendben és számokkal kell szerepeltetni, és csatolni kell egy összefoglalást. Bármely vizsgálat hiányát meg kell indokolni. A referenciaként hivatkozott kiadványokat mellékelni kell.
2.4. Amennyiben a benyújtott adatok nem elegendőek a táblázatba történő felvételhez, további adatok benyújtása írható elő.
2.5. Valamely termék előállítási eljárásában vagy felhasználásának feltételeiben történő minden változást közölni kell, és szükség esetén kiegészítő dokumentációt kell készíteni.
3.A dokumentáció elkészítéséhez szükséges vizsgálatok
3.1. Mikroorganizmus, táptalaj és előállítási eljárás, a termék főbb tulajdonságai, kiszerelés és felhasználási feltételek, valamint ellenőrzési módszerek
3.1.1.Mikroorganizmus
3.1.1.1. Osztályozás, eredet, morfológia, biológiai tulajdonságok, bármilyen genetikai módosítás.
3.1.1.2. Ártalmatlanság, az erjesztőn kívüli esetleges további fennmaradás és a környezetre gyakorolt esetleges hatások.
3.1.1.3. A tenyésztett törzsek állandósága és tisztasági foka. Ezen ismertető jegyeket ellenőrző módszerek.
3.1.2. Táptalaj és előállítási eljárás
3.1.2.1. A szubsztrátum, a hozzáadott anyagok stb. összetétele.
3.1.2.2. Előállítási, szárítási és tisztítási eljárás. A mikroorganizmus életképtelenné tétele. A színtenyészet összetétele állandóságának ellenőrzésére használt módszerek és az előállítás során fellépő bármely kémiai, fizikai vagy biológiai szennyeződés kimutatása kiderítésére alkalmazott módszerek. 3.1.2.3. A használatra való előkészítés technikai folyamatai.
3.1.3. A termék főbb tulajdonságai
3.1.3.1. Fizikai és fiziko-kémiai tulajdonságok: makro- és mikromorfológia, részecskeméret, sűrűség, faj súly, nedvszívó képesség, oldhatóság, elektrosztatikus tulajdonságok stb.
3.1.3.2. Kémiai összetétel és jellemzők:
3.1.3.2.1. Nedvesség-, nyersfehérje-, nyers zsír-, nyers rost-, nyers hamu-, szénhidráttartalom. Ezen tartalmak ingadozásának határai.
3.1.3.2.2. Összes ammónium-, amid-, nitrát és nitrit, nitrogén, nukleinsav- és fehérjetartalom. Az összes és szabad aminosavak, valamint a purin- és pirimidin-bázisok minőségi és mennyiségi összetétele.
3.1.3.2.3. Összes lipid minőségi és mennyiségi összetétele: zsírsavak, nem szappanosíthó anyagok, lipidben oldható pigmentek, foszfolipidek.
3.1.3.2.4. A szénhidrát tartalom összetétele.
3.1.3.2.5. A szervetlen alkotórészek minőségi és mennyiségi összetétele.
3.1.3.2.6. A vitaminok minőségi és mennyiségi összetétele.
3.1.3.2.7. Az egyéb alkotórészek mennyiségi és minőségi összetétele: adalékanyagok, szubsztrátok és oldószerek szermaradványi, a szubsztrátok anyagcseréjének, a táptalajnak, az előállítási eljárásnak az egyéb, esetlegesen káros szermaradványai, a szubsztrátok anyagcseréjének, a táptalajnak, az előállítási eljárásnak egyéb, esetlegesen káros szermaradványai.
3.1.3.3. A termék mikrobiológiai szennyeződése.
3.1.3.4. A termék viselkedése és állandósága a tárolás során, önmagában és forgalomban lévő takarmányokhoz keverve.
3.1.4. Kiszerelés és felhasználási feltételek
3.1.4.1. A termék tervezett kereskedelmi elnevezései.
3.1.4.2. A termék forgalmazása során tervezett kiszerelései.
3.1.4.3. A termék tervezett felhasználása a takarmányozásban. Tervezett koncentrációk a teljes értékű takarmányokban, valamint az érintett állatfajok napi takarmányadagban tervezett mennyiségében.
3.1.5. A meghatározás módszerei
3.1.5.1. A termék meghatározásának mennyiségi és minőségi módszerei a teljes értékű és kiegészítő takarmányokban. Ezeknek a módszereknek a leírásakor mellékelni kell a különleges jellegzetességekre, érzékenységre, a kimutatási méréshatárokra, hibahatárra és más anyagok által okozott lehetséges interferenciára vonatkozó adatokat. A különböző tervezett kiszerelések szerinti termékmintákat rendelkezésre kell bocsátani.
3.2. A termék táplálkozásiam tulajdonságaira vonatkozó vizsgálatok 3.2.1. A fehérjeérték meghatározása
3.2.1.1. Kémiai, biokémiai és mikrobiológiai vizsgálatok.
3.2.1.2. Kísérleti állatokon végzett vizsgálatok, a referenciafehérjékkel összehasonlítva.
3.2.2. A célfajokra vonatkozó vizsgálatok
3.2.2.1. A következő vizsgálatokat minden egyes célfaj esetében el kell végezni egy olyan kontrollcsoporttal történő összehasonlításban, amely ugyanolyan táplálkozásiam arányok mellett, egyenlő mennyiségű fehérjenitrogént, kérődzők esetében összes nitrogént tartalmazó általánosan elfogadott táplálékot kap:
a) a termék energia és fehérje-kiegészítő értéke a napi takarmányadagban, az állatok különböző fiziológiai stádiumai (pl. növekedési periódus, vemhesség, tojásrakás) esetében tervezett felhasználási feltételek mellett,
b) a termék hatása a növekedés ütemére, a takarmányhasznosulás arányára, a megbetegedési és elhullási arányra, a tervezett felhasználási feltételek mellett,
c) a termék napi felvételének optimális szintjei,
d) a termék hatása az állati eredetű élelmiszerek technológiai érzékszervi vagy egyéb tulajdonságaira a tervezett felhasználási feltételek mellett.
3.2.3. Kísérleti feltételek a takarmány felhasználására előirányzott állatfajokon végzendő vizsgálatok során részletes leírást kell adni az elvégzett vizsgálatokról, a következő adatokkal:
a) az állatok faja, fajtája, kora és neme, azonosítási eljárás,
b) a vizsgálati és kontrollcsoportok száma; az egyes csoportokba tartozó állatok száma (a statisztikai elemzéshez elegendő, a statisztikai paramétereknek megfelelő számú állat),
c) a napi takarmányadag mennyisége, összetétele és beltartalma és ennek analízise,
d) a kísérletek helyszíne, fiziológiai állapot és állategészségügyi feltételek, az állattartás körülményei (a közösségi gyakorlatnak megfelelően),
e) a vizsgálat pontos időtartama, valamint az elvégzett elemzések időpontja,
f) a kísérlet során felmerült kedvezőtlen hatások és jelentkezésük időpontja.
3.3 A termék takarmányozásban történő felhasználásának biológiai következményeire vonatkozó vizsgálatok
E pont szerinti vizsgálatok célja a termék célfajokon történő biztonságos használatának, valamint az embereket és a környezetet érintő olyan kockázatok felmérésének lehetővé tétele, amelyek a használatból közvetve vagy közvetlenül származhatnak. Az e célból előírt toxikológiai vizsgálatok a termék jellegétől, az érintett állatfajoktól és a termék kísérleti állatokon megfigyelt anyagcseréjétől függnek.
3.3.1. A célfajokra vonatkozó vizsgálatok
A következő vizsgálatokat minden célfajon el kell végezni egy olyan kontrollcsoporttal történő összehasonlításban, amely ugyanolyan táplálkozástani arányok mellett, egyenlő mennyiségű fehérjenitrogént, kérődzők esetében össznitrogént tartalmazó szokásos takarmányt kap:
a) a termék maximális bekeverési aránya a napi takarmányadagban, amely semmiféle kedvezőtlen hatást nem eredményez,
b) a termék lehetséges hatásai a termékenységre és szaporodásra, amennyiben célszerű,
c) a termék elfogyasztásából eredő hatások az emésztő rendszer flórájának mikroorganizmusaira, valamint a kórokozók emésztési szakaszban történő megtelepedésére, a tervezett felhasználási feltételek mellett,
d) a termék állati eredetű élelmiszerekben esetlegesen megjelenő maradékanyagainak (szubsztrátumok, táptalaj, oldószerek, szennyeződések) vizsgálata a tervezett felhasználási feltételek mellett,
e) a terméknek az ürülékben esetlegesen megjelenő maradékanyagainak (szubsztrátumok, táptalaj, oldószerek, szennyeződések) vizsgálata a tervezett felhasználási feltételek mellett.
3.3.2. A kísérleti állatokra vonatkozó vizsgálatok
3.3.2.1. Metabolizmus
A termék lebomlása az állatban: felszívódás, felhalmozódás, biológiai átalakítás, kiválasztás.
3.3.2.2. Mutagenitás
A szennyeződések (különösen mikotoxinok és baktériumok) vagy a termék maradékanyagai (szubsztrátumok, táptalaj, oldószerek) okozta lehetséges mutagenitás vizsgálata, ideértve az anyagcsere-aktivációval "in-vitro" végzett vizsgálatokat is.
3.3.2.3. Toxikológiai vizsgálatok
A következő vizsgálatokat olyan kontrollcsoportokkal összehasonlítva kell elvégezni, amelyek ugyanolyan táplálkozástani arányok mellett egyenlő mennyiségű fehérjenitrogént tartalmazó szokásos takarmányt kapnak. A toxikus hatásokat ki kell vizsgálni a kiváltó okok és lefolyások tisztázása, valamint annak megállapítása érdekében, hogy ezek a hatások nem táplálkozástani aránytalanságból vagy a termék étrenden belüli túladagolásából következnek.
3.3.2.3.1.Szubkrónikus toxicitás (legalább 90 nap)
3.3.2.3.1.1. Általában ezeket a vizsgálatokat két állatfajon kell elvégezni, amelyek közül az egyiknek "rágcsálónak kell lennie. A terméket legalább kétféle bekeverési, szinten kell a napi takarmányadaghoz hozzáadni. Ezeket lehetőség szerint úgy kell megválasztani, hogy egy hatás nélküli és egy kedvezőtlen hatásokkal járó szintet lehessen meghatározni. Az állatcsoportoknak mindkét nemből megfelelő számú egyedet kell tartalmaznia. Minden esetben egy kontrollcsoportot is be kell vonni a vizsgálatokba.
3.3.2.3.1.2. Minden vonatkozó biológiai adatot megfelelő időközönként rögzíteni kell, különös tekintettel a növekedési ütemre, a takarmány fogyasztásra, hematológiára, vizeletvizsgálatra, biokémiai paraméterekre, elhullásra, szervek súlyára, a főbb szervek és szövetek általános és hisztopatológiájára. Az eredményeket teljes részletességgel kell közölni és amennyire csak lehetséges, statisztikai értékelést is tartalmazniuk kell.
3.3.2.3.2.Krónikus toxicitás
3.3.2.3.2.1. Általában a krónikus toxicitás vizsgálatokat két állatfajon kell elvégezni, amelyek közül az egyiknek rágcsálónak kell lennie. A terméket legalább kétféle bekeverési szinten kell a napi fejadaghoz hozzáadni. A kísérleteket patkányokon legalább két éven át, egereken 80 héten át kell folytatni. Az állatcsoportoknak mindkét nemből megfelelő számú egyedet kell tartalmazniuk. Minden esetben egy kontrollcsoportot is be kell vonni a vizsgálatokba.
3.3.2.3.2.2. A 3.3.2.3.1. pontban említett biológiai vizsgálatokat lehetőség szerint az állatok egy kisebb mellékcsoportján (a fő csoporttól elválasztott és attól függésben lévő csoport) kell elvégezni, a kísérlet folyamán megfelelő időközönként, valamint a túlélő állatokon a kísérlet végén.
3.3.2.3.3. Karcinogenitás
A karcinogenitás értékelésekor különös figyelmet kell szentelni a megjelenés időpontjára, a megfigyelt tumorok szövettani típusára és elterjedtségére. A tumorok elterjedtségére, illetve a betegségek elterjedtségére vagy előrehaladására vonatkozó bármely hatást a kontrollcsoportokkal összehasonlítva kell értékelni, amint azt a 3.3.2.3. pontban leírtak tartalmazzák. Az eredményeket teljes részletességgel kell közölni és azoknak, amennyire csak lehetséges, statisztikai értékelést is kell tartalmazniuk.
3.3.2.4. Egyéb vizsgálatok
A szaporodásra vonatkozó vizsgálatokat legalább két utódnemzedéken keresztül kell folytatni, illetve össze lehet vonni őket embriótOxicitás-vizsgálatokkal, ideértve a teratogenitás-vizsgálatokat is. Különös figyelmet kell fordítani a termékenységre, a termékenyítő képességre és az alom születés utáni fejlődésének megfigyelésére. Bármely egyéb, tudományosan ellenőrizhető, illetve mérhető adatokat eredményező (pl. közvetítő toxicitás) vizsgálat elvégezhető.
3.3.2.5. Kísérleti állatokra vonatkozó vizsgálatok kísérleti körülményei
Részletes leírást kell adni az elvégzett vizsgálatokról, amely a következő adatokat tartalmazza:
a) az állatok faja, fajtája, törzse és neme,
b) a vizsgálati és kontrollcsoportok száma; az egyes csoportokba tartozó állatok száma (a statisztikai elemzéshez elegendő, a statisztikai paramétereknek megfelelő számú állat),
c) a termék bekeverési szintjei, napi takarmányadag minőségi és mennyiségi összetétele és ennek elemzése,
d) általános tartási körülmények a vizsgálati időszak alatt,
e) a vizsgálat pontos időtartama, valamint az egyes elvégzett vizsgálatok időpontja, f) a különféle vizsgálati csoportokban az elhullások mértéke és időbeli alakulása,
g) a kísérlet folyamán felmerült klinikai tünetek és patológiai módosulások, valamint jelentkezésük időpontja.
3.3.3. A környezetre vonatkozó vizsgálatok
A terméknek a célfajok salakanyagaiban megtalálható lehetséges maradékanyagainak jellegétől (szubsztrátumok, táptalaj, oldószerek, szennyeződések) függően, adatok kérhetők ezeknek a maradékanyagoknak a trágyában, talajban és vízben való viselkedéséről, valamint a talaj biológiájára, növények fejlődésére és a vízi életre kifejtett hatásukról.
3.4. Egyéb vizsgálatok
A termék jellegétől és felhasználási feltételeitől függően adatok kérhetők az allergiás hatásokra, a bőr, valamint a szem nyálkahártyáinak, a légző- és emésztőszervek irritációjára vonatkozóan, a termék kezelésével kapcsolatos lehetséges veszélyek felmérésének és megelőzésének érdekében.
II.[2]
Lábjegyzetek:
[1] Hatályon kívül helyezte a 10/2007. (II. 20.) FVM rendelet 4. § (1) bekezdés b) pontja. Hatálytalan 2007.03.07.
[2] Hatályon kívül helyezte e rendelet 4. § (3) bekezdése. Hatálytalan a Magyar Köztársaságnak az Európai Unióhoz történő csatlakozásáról szóló nemzetközi szerződést kihirdető törvény hatálybalépésének napjától a 2004. évi XXX. törvény alapján 2004.05.01-től.