
32009R0152[1]
A Bizottság 152/2009/EK rendelete ( 2009. január 27.) a takarmányok hatósági ellenőrzése során alkalmazott mintavételi és vizsgálati módszerek megállapításáról
A BIZOTTSÁG 152/2009/EK RENDELETE
(2009. január 27.)
a takarmányok hatósági ellenőrzése során alkalmazott mintavételi és vizsgálati módszerek megállapításáról
(EGT-vonatkozású szöveg)
1. cikk
A takarmányok hatósági ellenőrzésére, különös tekintettel az alkotóelemek, többek között a géntechnológiával módosított szervezeteket (GMO-kat) tartalmazó, azokból álló, illetve azokból előállított anyagok, az 1831/2003/EK rendelet ( 1 ) szerinti takarmány-adalékanyagok és a 2002/32/EK európai parlamenti és tanácsi irányelv ( 2 ) szerinti nemkívánatos anyagok meghatározására szolgáló mintavételezést az I. mellékletben meghatározott módszereknek megfelelően kell elvégezni, a mikrobiológiai szennyeződés ellenőrzésére szolgáló mintavétel kivételével.
Az I. mellékletben foglalt mintavételi módszer alkalmazandó a takarmányoknál előforduló peszticid-szermaradványok 2005. február 23-i 396/2005/EK európai parlamenti és tanácsi rendelet ( 3 ) szerinti meghatározására irányuló ellenőrzés, valamint a 619/2011/EU rendeletnek való megfelelés ellenőrzése során.
2. cikk
Az analitikai vizsgálat céljára szolgáló minták előkészítését és az eredmények megadását a II. mellékletben előírt módszerek szerint kell végrehajtani.
3. cikk
A takarmányok hatósági ellenőrzésére szolgáló analitikai vizsgálatokat a III. mellékletben (Analitikai módszerek a takarmány-alapanyagok és takarmánykeverékek összetételének ellenőrzésére), a IV. mellékletben (Analitikai módszerek a takarmányokban lévő engedélyezett adalékanyagok szintjének ellenőrzésére), az V. mellékletben (Analitikai módszerek a takarmányokban lévő nemkívánatos anyagok ellenőrzésére) és a VI. mellékletben (A takarmányok hatósági ellenőrzése során az állati eredetű alkotóelemek meghatározására szolgáló analitikai módszerek) előírt módszerek szerint kell végrehajtani.
4. cikk
A baromfitakarmány-keverék energiaértékét a VII. mellékletnek megfelelően kell kiszámítani.
5. cikk
Megerősítés céljából a VIII. mellékletben a már nem engedélyezett adalékanyagok takarmányokban való illegális jelenlétének ellenőrzésére előírt analitikai módszereket kell használni.
6. cikk
A 71/250/EGK, a 71/393/EGK, a 72/199/EGK, a 73/46/EGK, a 76/371/EGK, a 76/372/EGK, a 78/633/EGK, a 81/715/EGK, a 84/425/EGK, a 86/174/EGK, a 93/70/EGK, a 93/117/EK, a 98/64/EK, az 1999/27/EK, az 1999/76/EK, a 2000/45/EK, a 2002/70/EK és a 2003/126/EK irányelv hatályát veszti.
A hatályon kívül helyezett irányelvekre történő hivatkozásokat az e rendeletre történő hivatkozásoknak kell tekinteni, és a IX. mellékletben található megfelelési táblázatokkal összhangban kell értelmezni.
7. cikk
Ez a rendelet az Európai Unió Hivatalos Lapjában való kihirdetését követő huszadik napon lép hatályba.
Ezt a rendeletet 2009. augusztus 26-tól kell alkalmazni.
Ez a rendelet teljes egészében kötelező és közvetlenül alkalmazandó valamennyi tagállamban.
I. MELLÉKLET
MINTAVÉTELI MÓDSZEREK
1. CÉL ÉS ALKALMAZÁSI TERÜLET
A takarmányok hatósági ellenőrzésére szolgáló mintavételezést az alábbiakban leírt módszerek szerint kell elvégezni. Az így nyert mintákat a mintavételi tételek reprezentatív részének kell tekinteni.
A reprezentatív mintavétel célja kisebb mennyiséget venni a tételből oly módon, hogy e mennyiség bármely jellemző tulajdonságának meghatározásával az adott tulajdonságot illetően a teljes tételre érvényes középértéket kapjunk. A mintavételezés során a tétel különböző részeiből ismételten elemi mintákat kell venni. Ezen elemi mintákat össze kell keverni annak érdekében, hogy az így nyert egyesített mintából reprezentatív osztással reprezentatív végső mintákat lehessen kialakítani.
Ha a mintavételre kerülő takarmányadagok szemrevételezés vagy egyéb releváns információk alapján minőségi eltérést mutatnak a takarmány ugyanazon tételének többi részéhez képest, a szóban forgó adagokat el kell különíteni és altételként kell kezelni őket. Amennyiben a takarmányt nem lehet különálló altételekre osztani, azt egyetlen tételként kell mintavételezni. Ebben az esetben erről a tényről említést kell tenni a mintavételi jegyzőkönyvben.
Ha az e rendelet előírásai alapján mintavételezett takarmányról megállapítják, hogy nem felel meg az uniós követelményeknek, továbbá az egy azonos osztályú vagy megnevezésű takarmánytétel részét képezi, abból kell kiindulni, hogy a tétel többi része sem biztonságos, kivéve, ha a részletes átvizsgálás során nem bizonyítható, hogy a tétel többi része nem felel meg az uniós követelményeknek.
A mintavétel kiterjedhet a 767/2009/EK európai parlamenti és tanácsi rendelet ( 4 ) 11. cikkének (3) bekezdésével összhangban a takarmányipari vállalkozók által távközlő eszköz révén értékesítésre kínált takarmányokra is. A távközlő eszköz révén értékesítésre kínált takarmány mintavételére főszabály szerint az e mellékletben meghatározott pontok vonatkoznak. A távértékesítési minták vételezésének konkrét szempontjait a 11. pont ismerteti.
2. FOGALOMMEGHATÁROZÁSOK
- Tétel: azonosított mennyiségű takarmány, amely közös jellemzőkkel rendelkezik, úgymint eredet, fajta, a csomagolás típusa, csomagoló, feladó, címkézés, valamint - előállítási folyamat esetében - az egyetlen üzemben egységes előállítási paraméterek mellett előállított termelési egység vagy - amennyiben azokat egymás után folyamatosan állítják elő és együtt tárolják - több ilyen egység.
- Mintavételi tétel: egy tétel, illetve a tétel vagy vizsgálati tétel egy azonosított része.
- Lepecsételt minta: pecséttel oly módon ellátott minta, hogy a pecsét felsértése vagy eltávolítása nélkül ne lehessen hozzáférni a mintához.
- Elemi minta: a mintavételi tétel egy pontjából vett mennyiség.
- Egyesített minta: ugyanazon mintavételi tételből vett elemi minták egyesítésével nyert minta.
- Redukált minta: az egyesített minta olyan része, amelyet az utóbbiból reprezentatív redukálási folyamattal nyernek.
- Végső minta: az egyesített minta (összekevert), a redukált minta vagy a homogenizált egyesített minta egy része, az ellenőrzés típusától függően (lásd a 9.4. pontot).
- Laboratóriumi minta: a laboratórium részére szánt minta (a laboratóriumba érkező minta), amely lehet végső, redukált vagy egyesített minta.
- Távértékesítési minta: távközlő eszköz révén értékesítésre kínált takarmánytételből vagy -adagból vett minta.
3. ÁLTALÁNOS RENDELKEZÉSEK
- A mintavételezést az illetékes hatóságok által erre a feladatra felhatalmazott személyek végzik.
- Távértékesítési minta esetében az illetékes hatóság távközlő eszköz révén bekér a takarmányból egy bizonyos mennyiséget a takarmányipari vállalkozótól.
- A mintát pecséttel kell ellátni úgy, hogy a pecsét felsértése vagy eltávolítása nélkül ne lehessen hozzáférni a mintához.
A biztonsági pecsétnek egyértelműen azonosíthatónak és jól láthatónak kell lennie.
- A minta azonosítása: a mintát letörölhetetlenül meg kell jelölni és azonosítani kell oly módon, hogy egyértelmű kapcsolata a mintavételi jegyzőkönyvvel megállapítható legyen.
- Valamennyi egyesített mintából vagy redukált mintából a következő végső mintákat kell venni: egyet az ellenőrzéshez (hatósági ellenőrzés), egyet pedig a takarmányipari vállalkozó részére (védelmi minta) kell venni. Adott esetben referencia céljából lehet még egy végső mintát venni. Amennyiben a teljes egyesített minta homogenizált, a végső mintákat a homogenizált egyesített mintából kell venni, ha ez az eljárás nem ütközik az adott tagállam takarmányipari vállalkozói jogokra vonatkozó jogszabályaiba.
- Az (EU) 2017/625 rendelet 15. cikkének (1) és (2) bekezdésével összhangban, amennyiben a hatósági mintavételezés elvégzéséhez szükséges, a takarmányipari vállalkozók - az illetékes hatóságok kérésére - kötelesek:
- hozzáférést biztosítani az illetékes hatóságok személyzete számára az ellenőrzésük alá tartozó felszerelésekhez, beleértve szükség esetén a megfelelő mintavételi felszerelést és egyéni védőeszközöket is;
- segíteni az illetékes hatóságok személyzetét és együttműködni vele a mintavétel lehetővé tétele érdekében, beleértve a takarmány hozzáférhetővé tételét az illetékes hatóságok személyzete számára.
4. Eszközök
4.1. A mintavételi eszközöket olyan anyagokból kell készíteni, amelyek nem szennyezhetik a mintavételre kerülő termékeket. A többszöri használatra szánt eszközöknek könnyen tisztíthatónak kell lenniük a keresztszennyeződések elkerülése érdekében.
4.2. Szilárd takarmányok mintavételére ajánlott eszközök
4.2.1. Kézi mintavétel
4.2.1.1. Lapos fenekű, függőleges oldalú mintavételező lapát.
4.2.1.2. Nyílással vagy rekeszekkel rendelkező mintavételi szúrcsap. A mintavételi szúrcsap méretének összhangban kell lennie a mintavételi tétel jellemzőivel (a tartály mélysége, a göngyöleg nagysága stb.), valamint a takarmány részecskeméretével.
Amennyiben a mintavételi szúrcsapon több nyílás található, e nyílásokat rekeszekkel kell elválasztani, vagy a szúrcsapnak lépcsősen elhelyezett, szekvenciális nyílásokkal kell rendelkeznie annak biztosítására, hogy a mintavételezés a szúrcsap egymás mellett elhelyezkedő különböző pontjain történjen.
4.2.2. Gépi mintavétel
Mozgásban lévő takarmányok mintavételére megfelelő gépi eszközök használhatók. A gépi eszköz akkor tekinthető megfelelőnek, ha alkalmas legalább az anyagáram teljes keresztmetszetének mintavételezésére.
A (magas áramlási sebességgel) mozgásban lévő takarmányok esetében automata mintavevőket lehet használni.
4.2.3. Mintaosztó
Ha lehetséges és indokolt, a minta megközelítően egyenlő részekre való felosztására tervezett eszközt kell használni a redukált minták reprezentatív elkészítéséhez.
5. AZ ELEMI MINTÁK SZÁMÁRA VONATKOZÓ MENNYISÉGI KÖVETELMÉNYEK
- Az elemi minták számára vonatkozóan az 5.1. és 5.2. pontban leírt mennyiségi követelmények alkalmazandók az 500 tonnát meg nem haladó tömegű és a reprezentatív módon mintavételezhető tételek esetében. A bemutatott mintavételi eljárás érvényes a mintavételi tétel előírt maximális méreténél nagyobb mennyiségekre is, feltéve, hogy figyelmen kívül hagyják az elemi mintáknak az 5.1.1., az 5.1.3., és az 5.1.5 pont alábbi táblázataiban feltüntetett maximális számát, hogy az elemi minták számának meghatározása az eljárás megfelelő részénél megadott négyzetgyökös képlettel történik (lásd az 5.3. pontot), valamint hogy az egyesített minta minimális méretét arányosan növelték. Ez nem zárja ki egy nagy tétel kisebb altételekre történő felosztását, amelyet követően minden egyes altételt az 5.1. és 5.2. pontban leírt eljárással összhangban kell mintavételezni.
- A mintavételi tételnek akkorának kell lennie, hogy minden egyes alkotórészéből mintát lehessen venni.
- Igen nagy tömegű (> 500 tonna) tételek és altételek, valamint azon tételek esetében, amelyek szállítása vagy tárolása nem teszi lehetővé az e pont 5.1. és 5.2. pontja szerinti mintavételi eljárást, az 5.3. pontban bemutatott eljárást kell alkalmazni.
- Távértékesítési minták esetében az illetékes hatóság általában nem ismeri annak a tételnek a méretét, amelyből a mennyiséget kéri. Ezért az 5.1. és 5.2. pontban említett eljárás nem alkalmazható. Ebben az esetben a 11. pontban ismertetett eljárást kell alkalmazni.
- Ha a takarmányipari vállalkozót kötelező monitoringrendszer keretében jogszabályok kötelezik az e rendeletben foglaltaknak való megfelelésre, a vállalkozó a működési jellemzők figyelembevétele érdekében - az illetékes hatóság engedélyét követően - eltérhet az e pontban megállapított mennyiségi követelményektől, feltéve, hogy az illetékes hatóság számára kielégítő módon bizonyította a mintavételi eljárás egyenértékűségét a reprezentativitás tekintetében.
- Különleges esetekben, amikor (a csomagolási forma, a szállítás és a tárolás módja stb. miatt) a mennyiségi követelmények szerinti mintavételi módszer nem alkalmazható a tételen bekövetkező elfogadhatatlan gazdasági kár miatt, alternatív mintavételi módszert lehet alkalmazni, feltéve, hogy az a lehetőségekhez képest reprezentatív, továbbá azt teljeskörűen leírják és dokumentálják.
5.1. A takarmányban egyenletesen eloszló anyagok és termékek ellenőrzése kapcsán az elemi mintákra vonatkozó mennyiségi követelmények
5.1.1. Ömlesztett szilárd takarmány
| A mintavételi tétel tömege | Az elemi minták minimális száma |
| ≤ 2,5 tonna | 7 |
| > 2,5 tonna | √ (20 * a mintavételi tételt kitevő tonnák száma) (*1) , de legfeljebb 40 elemi minta |
| (*1) Amennyiben az eredmény törtszám, fel kell kerekíteni a legközelebbi egész számra. | |
5.1.2. Ömlesztett folyékony takarmány
| A mintavételi tétel tömege | Az elemi minták minimális száma |
| ≤ 2,5 tonna vagy ≤ 2 500 liter | 4 (*1) |
| > 2,5 tonna vagy > 2 500 liter | 7 (*1) |
| (*1) Amennyiben nem lehet homogén folyadékot elérni, növelni kell az elemi minták számát. | |
5.1.3. Kiszerelt takarmány
A (szilárd és folyékony) takarmányt zsákokba, göngyölegekbe, dobozokba, hordókba stb. lehet csomagolni, amelyek az alábbi táblázatban egységek néven szerepelnek. A nagy egységek (≥ 500 kg vagy liter) esetében a mintavételezésnek az ömlesztett takarmányokra érvényes előírásokkal összhangban kell történnie (lásd az 5.1.1. és az 5.1.2. pontot).
| A mintavételi tétel tömege | Azon egységek minimális száma, amelyekből (legalább) egy elemi mintát kell venni (*1) |
| 1 –20 egység | 1 egység (*2) |
| 21 –150 egység | 3 egység (*2) |
| 151 –400 egység | 5 egység (*2) |
| > 400 egység | √ (a mintavételi tételt kitevő egységek számának) (*3) egy negyede, legfeljebb 40 egység |
| (*1) Ha egy egység felbontása befolyásolhatja a vizsgálatot (pl. romlandó nedves takarmányoknál), az elemi mintának a felbontatlan egységnek kell lennie. (*2) Az 1 kg-ot vagy 1 litert meg nem haladó egységek esetében az elemi mintának egy eredeti egység tartalmának kell lennie. (*3) Amennyiben az eredmény törtszám, fel kell kerekíteni a legközelebbi egész számra. | |
5.1.4. Takarmánytömbök és nyalósók
A 25 egységből álló mintavételi tételenként legalább egy, legfeljebb 4 tömböt vagy nyalósót kell mintaként venni.
Az egyenként 1 kg-ot meg nem haladó tömegű blokkok és tömbök esetében az elemi minta egy blokk vagy egy tömb.
5.1.5. Szálastakarmány/zöldtakarmány
| A mintavételi tétel tömege | Az elemi minták minimális száma (*1) |
| ≤ 5 tonna | 5 |
| > 5 tonna | √ (5 (*2) a mintavételi tételt kitevő tonnák száma) (**), de legfeljebb 40 elemi minta |
| (*1) Elismert tény, hogy bizonyos helyzetekben (pl. silótakarmányok esetében) a tétel elfogadhatatlan mértékű sérülése nélkül nem lehet teljesíteni az elemi minták számára vonatkozó előírást. Ekkor alternatív módszerhez lehet folyamodni, útmutató készült az ilyen típusú tételek mintavételezéséhez, amely itt érhető el: https://food.ec.europa.eu/system/files/2016-10/animal-feed-guidance_documents_691_2013_en.pdf. (*2) Amennyiben az eredmény törtszám, fel kell kerekíteni a legközelebbi egész számra. | |
5.2. A takarmányban nem egyenletesen eloszló anyagok és termékek ellenőrzése kapcsán az elemi mintákra vonatkozó mennyiségi követelmények
Az elemi minták számára vonatkozóan itt ismertetett követelményeket az alábbi esetekben kell alkalmazni:
- takarmány-alapanyagokban előforduló aflatoxinok, anyarozs, más mikotoxinok és káros botanikai szennyeződések ellenőrzése,
- alkotóelemek, többek között a géntechnológiával módosított anyagok okozta keresztszennyeződés, illetve a takarmány-alapanyagokban feltételezhetően egyenetlenül eloszló anyagok ellenőrzése.
Amennyiben az ellenőrzést végző hatóságnak erős a gyanúja, hogy ez az egyenetlen eloszlás valamely alkotóelem, illetve a takarmánykeverékben található valamely anyag okozta keresztszennyeződés nyomán is fellép, az alábbi táblázatban feltüntetett mennyiségi követelményeket lehet alkalmazni.
| A mintavételi tétel tömege | Az elemi minták minimális száma |
| < 80 tonna | Lásd az 5.1. pont szerinti mennyiségi követelményeket. Az elemi minták szükséges számát meg kell szorozni 2,5-tel. |
| ≥ 80 tonna | 100 |
5.3. Az igen nagy méretű tételek esetében az elemi mintákra vonatkozó mennyiségi követelmények
A nagy méretű mintavételi tételek (> 500 tonna) esetében az elemi minták szükséges száma = 40 elemi minta + √tonna a takarmányban egyenletesen eloszló anyagok és termékek ellenőrzése esetén, vagy 100 elemi minta + √tonna a takarmányban feltételezhetően nem egyenletesen eloszló anyagok és termékek ellenőrzése esetén.
6. AZ EGYESÍTETT MINTÁRA VONATKOZÓ MENNYISÉGI KÖVETELMÉNYEK
Mintavételi tételenként egy egyesített minta szükséges.
| A takarmány jellege | Az egyesített minta minimális tömege (*1) (*2) | |
| 6.1. | Ömlesztett takarmány | 4 kg |
| 6.2. | Kiszerelt takarmányok | 4 kg (*3) |
| 6.3. | Folyékony vagy félfolyékony takarmányok | 4 liter |
| 6.4. | Takarmánytömbök vagy nyalósók | |
| 6.4.1. | 1 kg-nál nagyobb egyedi tömeg | 4 kg |
| 6.4.2. | legfeljebb 1 kg-os egyedi tömeg | négy eredeti tömb vagy nyalósó tömege |
| 6.5. | Szálastakarmány/zöldtakarmány | 4 kg (*4) |
| (*1) A nagy értékű megmintázott takarmány esetében az előírtnál kisebb mennyiségű egyesített mintát lehet venni azzal a feltétellel, hogy mindezt a mintavételi jegyzőkönyvben ismertetik és dokumentálják. (*2) A takarmányok hatósági ellenőrzése során alkalmazott mintavételi és vizsgálati módszereknek a géntechnológiával módosított anyagok – amelyek esetében az engedélyezési eljárás függőben van vagy amelyek esetében az engedély lejárt – jelenléte tekintetében történő megállapításáról szóló, 2011. június 24-i 619/2011/EU bizottsági rendelet (HL L 166., 2011.6.25., 9. o.) előírásainak megfelelően a géntechnológiával módosított anyagok jelenlétének ellenőrzésére szolgáló egyesített mintának legalább 35 000 magot/szemet kell tartalmaznia. Ez azt jelenti, hogy a kukorica esetében az egyesített minta tömege legalább 10,5 kg, a szójabab esetében pedig 7 kg. Más magvak és szemek, mint például az árpa, a köles, a zab, a rizs, a rozs, a búza és a repcemag tekintetében az egyesített minta 4 kg-os tömege több mint 35 000 magnak/szemnek felel meg. (*3) Kiszerelt takarmányok esetében is előfordulhat, hogy az egyedi kiszerelési egységek méretének függvényében az egyesített minta tömege nem éri el a 4 kg-ot. (*4) A kis fajlagos tömegű szálastakarmányok/zöldtakarmányok (pl. széna, szalma) tekintetében az egyesített minta minimális tömegének 1 kg-nak kell lennie. | ||
7. A VÉGSŐ MINTÁKRA VONATKOZÓ MENNYISÉGI KÖVETELMÉNYEK
Végső minták
Legalább egy végső minta elemzése szükséges. Az analizálásra szolgáló végső minta nem lehet kisebb a következőknél:
| Szilárd takarmányok | 500 g (*1) (*2) (*3) (*4) |
| Folyékony vagy félfolyékony takarmányok | 500 ml (*1) |
| (*1) A 619/2011/EU rendelet előírásainak megfelelően a géntechnológiával módosított anyagok jelenlétének ellenőrzésére szolgáló végső mintának legalább 10 000 magot/szemet kell tartalmaznia. Ez azt jelenti, hogy a kukorica esetében a végső minta tömege legalább 3 000 g, a szójabab esetében pedig 2 000 g. Más magvak és szemek, mint például az árpa, a köles, a zab, a rizs, a rozs, a búza és a repcemag tekintetében a végső 500 g-os minta tömege több mint 10 000 magnak/szemnek felel meg. (*2) Amennyiben az egyesített minta tömege lényegesen kevesebb, mint 4 kg vagy liter (lásd a 6. fejezet lábjegyzeteit), az előírtnál kisebb mennyiségű végső mintát lehet venni azzal a feltétellel, hogy mindezt a mintavételi jegyzőkönyvben ismertetik és dokumentálják. (*3) Hüvelyesek, gabonafélék és fán termő héjas gyümölcsűek mintavételezésekor a peszticid-szermaradványok meghatározásához vett végső minta tömegének – a növényi és állati eredetű termékekben és azok felszínén található peszticid-szermaradványok hatósági ellenőrzésére szolgáló közösségi mintavételi módszerek megállapításáról és a 79/700/EGK irányelv hatályon kívül helyezéséről szóló, 2002. július 11-i 2002/63/EK bizottsági irányelv (HL L 187., 2002.7.16., 30. o.) rendelkezéseivel összhangban – legalább 1 kg-nak kell lennie. (*4) (****) Szemrevételezéses vagy mikroszkópos vizsgálat esetén a vizsgálatra szánt végső minta mennyisége 1 kg. | |
8. IGEN NAGY MÉRETŰ TÉTELEK, ILLETVE OLY MÓDON TÁROLT VAGY SZÁLLÍTOTT TÉTELEK MINTAVÉTELEZÉSE, AMELY NEM TESZI LEHETŐVÉ A MINTAVÉTELEZÉSÉT A TÉTEL EGÉSZÉBEN
8.1. Általános elvek
Amennyiben egy tétel szállítási vagy tárolási módja nem teszi lehetővé, hogy elemi mintákat vegyünk a tétel valamennyi részéből, a mintavételezésre lehetőleg akkor kell sort keríteni, amikor a tétel mozgásban van.
Takarmányok tárolására szolgáló nagy méretű raktárak esetében a raktárak üzemeltetőit ösztönözni kell olyan berendezések felszerelésére, amelyek segítségével (automatikusan) mintát lehet venni az ott tárolt teljes tételből.
Az e pontban meghatározott mintavételi eljárások alkalmazása esetén a takarmányipari vállalkozó vagy annak képviselője tisztában van a mintavételi eljárással. Ha a takarmányipari vállalkozó vagy annak képviselője kétségbe vonja a mintavételi eljárást, lehetővé kell tennie az illetékes hatóság számára, hogy a vállalkozó saját költségén a tétel minden részéből mintát tudjon venni.
8.2. Hajóval szállított nagy tételek
8.2.1. A hajóval szállított nagy tételek dinamikus mintavételezése
A hajóval szállított nagy tételek mintavételezésére lehetőleg akkor kell sort keríteni, amikor az mozgásban van (dinamikus mintavételezés).
A mintavételezést rakterenként (fizikailag elkülöníthető egységenként) kell végezni. A rakterek kiürítése azonban egymás után, részlegesen történik, ezért a kezdeti fizikai elkülönítés a tárolólétesítménybe történő áthelyezést követően már nem áll fenn. A mintavételezést ennek megfelelően vagy a kezdeti fizikai elkülönítés, vagy a tárolólétesítménybe történő áthelyezést követő elkülönítés függvényében lehet elvégezni.
A hajó rakterének kiürítése néhány napot is igénybe vehet. A mintavételezést rendszerint a kiürítés teljes időtartama alatt, szabályos időközönként kell végezni. Hatósági ellenőr jelenléte azonban nem mindig lehetséges vagy célszerű a teljes kiürítési művelet alatt. A mintavételezést tehát a teljes tétel egy részén (mintavételi tétel) is el lehet végezni. Az elemi minták számának meghatározása a mintavételi tétel méretének figyelembevételével történik.
Ha azonos osztályú vagy megnevezésű takarmánytétel egy részének mintavételezése esetén azt állapítják meg, hogy a tétel szóban forgó része nem felel meg az uniós követelményeknek, abból kell kiindulni, hogy a tétel többi része sem biztonságos, kivéve, ha a részletes átvizsgálás során nem bizonyítható, hogy a tétel többi része nem felel meg az uniós követelményeknek.
Abban az esetben is, ha a hatósági mintát automata mintavevő segítségével veszik, indokolt az ellenőr jelenléte. Amennyiben azonban az automatikus mintavételezés előre beállított paraméterekkel történik, és a csalás bármely formáját megelőzendő a mintákat pecséttel ellátott tárolóedényekbe gyűjtik, az ellenőrnek kizárólag a mintavételezés megkezdésekor és befejezésekor, valamint akkor kell jelen lennie, amikor a tárolóedényeket cserélni kell.
8.2.2. A hajóval szállított nagy tételek statikus mintavételezése
Statikus mintavételezés esetén a felülről hozzáférhető tárolók (silók) tekintetében előírt eljárást kell alkalmazni (lásd a 8.4.1. pontot).
A mintavételezést a tétel/raktér hozzáférhető részén (felülről) kell elvégezni. Az elemi minták számának meghatározása a mintavételi tétel méretének figyelembevételével történik. Ha azonos osztályú vagy megnevezésű takarmánytétel egy részének mintavételezése esetén azt állapítják meg, hogy a tétel szóban forgó része nem felel meg az uniós követelményeknek, abból kell kiindulni, hogy a tétel többi része sem biztonságos, kivéve, ha a részletes átvizsgálás során nem bizonyítható, hogy a tétel többi része nem felel meg az uniós követelményeknek.
8.3. A raktárakban tárolt nagy tételek mintavételezése
A mintavételezést a tétel hozzáférhető részén kell elvégezni. Az elemi minták számának meghatározása a mintavételi tétel méretének figyelembevételével történik. Ha azonos osztályú vagy megnevezésű takarmánytétel egy részének mintavételezése esetén azt állapítják meg, hogy a tétel szóban forgó része nem felel meg az uniós követelményeknek, abból kell kiindulni, hogy a tétel többi része sem biztonságos, kivéve, ha a részletes átvizsgálás során nem bizonyítható, hogy a tétel többi része nem felel meg az uniós követelményeknek.
8.4. A raktározási létesítmények (silók) mintavételezése
8.4.1. A felülről (könnyen) hozzáférhető silók mintavételezése
A mintavételezést a tétel hozzáférhető részén kell elvégezni. Az elemi minták számának meghatározása a mintavételi tétel méretének figyelembevételével történik. Ha azonos osztályú vagy megnevezésű takarmánytétel egy részének mintavételezése esetén azt állapítják meg, hogy a tétel szóban forgó része nem felel meg az uniós követelményeknek, abból kell kiindulni, hogy a tétel többi része sem biztonságos, kivéve, ha a részletes átvizsgálás során nem bizonyítható, hogy a tétel többi része nem felel meg az uniós követelményeknek.
8.4.2. A felülről nem hozzáférhető (zárt) silók mintavételezése
8.4.2.1. Felülről nem hozzáférhető (zárt), 100 tonnát meghaladó kapacitású silók
Az ilyen típusú silókban tárolt takarmányból nem lehet statikusan mintát venni. Ezért amennyiben el kell végezni a silóban tárolt takarmány mintavételezését, továbbá nincs lehetőség a tétel mozgatására, meg kell állapodni a létesítmény üzemeltetőjével, hogy az értesíteni fogja az ellenőrt a siló kiürítésének időpontjáról annak érdekében, hogy mintát lehessen venni a mozgásban lévő takarmányból.
8.4.2.2. Felülről nem hozzáférhető (zárt), < 100 tonna kapacitású silók
A mintavételi eljárás 50-100 kg közötti mennyiségben a takarmány tartályba történő engedéséből és e mennyiség megmintázásából áll. Az egyesített minta mérete a teljes tételnek, az elemi minták száma pedig a silóból mintavételezési céllal a tartályba engedett takarmány mennyiségének felel meg. Ha azonos osztályú vagy megnevezésű takarmánytétel egy részének mintavételezése esetén azt állapítják meg, hogy a tétel szóban forgó része nem felel meg az uniós követelményeknek, abból kell kiindulni, hogy a tétel többi része sem biztonságos, kivéve, ha a részletes átvizsgálás során nem bizonyítható, hogy a tétel többi része nem felel meg az uniós követelményeknek.
8.5. Nagy méretű zárt tartályokban tárolt ömlesztett takarmány mintavételezése
Az ilyen típusú tételekből sokszor csak a tartályból történő eltávolítás során lehet mintát venni. Bizonyos esetekben erre nincs lehetőség a behozatal vagy az ellenőrzés helyén, ezért a mintavételezésre a tartály kiürítésekor kell sort keríteni.
9. A MINTAVÉTELRE, A MINTÁK ELŐKÉSZÍTÉSÉRE ÉS CSOMAGOLÁSÁRA VONATKOZÓ UTASÍTÁSOK
9.1. Általános megjegyzések
A mintákat indokolatlan késedelem nélkül kell vételezni és előkészíteni, mindvégig ügyelve a szükséges óvintézkedések betartására, amelyek biztosítják azt, hogy a termék ne változzon meg, illetve ne szennyeződjön. A mintákkal közvetlenül érintkező eszközöknek, felületeknek és tartályoknak tisztáknak és szárazaknak kell lenniük.
9.2. Elemi minták
Az elemi mintákat véletlenszerűen és egyenletes eloszlásban kell venni a teljes mintavételezett tételből és azoknak közel azonos nagyságúaknak kell lenniük.
Az elemi minta minimális tömege 100 gramm, illetve 25 gramm a kis fajlagos tömegű szálastakarmányok/zöldtakarmányok esetében.
Amennyiben a 8. fejezetben meghatározott mintavételi eljárásra vonatkozó szabályok alapján kevesebb mint 40 elemi mintát kell venni, az elemi minták méretét az egyesített minta előírt méretének függvényében kell meghatározni (lásd a 6. fejezetet).
Kiszerelt takarmány kis tételeinek mintavételezésekor - amikor is a mennyiségi követelmények szerint korlátozott számú elemi mintát kell venni - az elemi minta egy olyan eredeti egység tartalma, amelynek tartalma nem haladja meg az 1 kg-ot vagy az 1 litert.
Kis egységű (pl.: < 250 g) kiszerelt takarmány mintavételezésekor az elemi minta mérete az egység méretéről függ.
Távértékesítési minták esetében az elemi minta mérete az egység méretétől függ, és egyedi esetekben 100 g-nál vagy 100 ml-nél kisebb mennyiséget is tartalmazhat.
9.2.1. Ömlesztett takarmány
Adott esetben, a mintavételi tétel mozgatása közben (berakodáskor, illetve kirakodáskor) is végre lehet hajtani a mintavételt.
9.2.2. Kiszerelt takarmány
Miután a mintavétel elvégzésére az 5. pont szerint kiválasztottuk az előírt számú egységet, minden egység tartalmának egy részét szúrcsap vagy lapát segítségével ki kell venni. Ha szükséges, az egységek egyenkénti kiürítése után is le lehet venni a mintákat.
9.2.3. Homogén vagy homogenizálható folyékony vagy félfolyékony takarmányok
Miután a mintavétel elvégzésére az 5. pont szerint kiválasztottuk az előírt számú egységet, az egységek tartalmát szükség esetén homogenizálni kell, és minden egyes egységből ki kell venni egy bizonyos mennyiséget.
Az elemi mintákat a tartályok kiürítésekor is lehet vételezni.
9.2.4. Nem homogenizálható, folyékony vagy félfolyékony takarmányok
Miután a mintavétel elvégzésére az 5. pont szerint kiválasztottuk az előírt számú egységet, a különböző rétegekből kell a mintákat levenni.
A tartályok tartalmának kiürítésekor is lehet mintákat venni, ám ebben az esetben az első frakciókat el kell önteni.
A minta összesített űrtartalmának mindkét esetben legalább 10 liternek kell lennie.
9.2.5. Takarmánytömbök és nyalósók
Miután a mintavétel elvégzésére az 5. pont szerint kiválasztottuk a kívánt számú blokkot vagy tömböt, minden blokk vagy tömb egy részét lehet venni. Nem homogén blokk vagy tömb gyanúja esetén a teljes blokkot vagy tömböt fel lehet mintaként használni.
Az egyenként 1 kg-ot meg nem haladó tömegű blokkok és tömbök esetében az elemi minta egy blokk vagy egy tömb.
9.3. Egyesített minták elkészítése
Az egyesített minta kialakítása érdekében az elemi mintákat össze kell keverni.
9.4. Végső minták elkészítése
Az egyesített mintában lévő anyagot gondosan össze kell keverni ( 5 ).
Valamennyi vizsgálati mintát egy, a célnak megfelelő külön tartályba/edénybe kell tenni. Minden szükséges óvintézkedést meg kell tenni a minta összetételének a szállítás vagy tárolás során előforduló esetleges megváltozásának, illetve a minta beszennyeződésének vagy meghamisításának elkerülése érdekében.
9.4.1. Egyenletesen eloszló anyagok
A takarmányban egyenletesen eloszló alkotóelemek vagy anyagok ellenőrzésekor az egyesített minta tömegét reprezentatív módon, lehetőleg mechanikus vagy automata osztóval minimum 2 kg-ra vagy 2 literre lehet csökkenteni (redukált minta) ( 6 ). Peszticid-szermaradványok hüvelyesekben, gabonafélékben és fán termő héjas gyümölcsűekben való jelenlétének ellenőrzésekor a redukált minta minimális tömegének 3 kg-nak kell lennie. Ha a takarmány jellege nem teszi lehetővé az osztó használatát, vagy az osztó nem elérhető, a mintát a negyedeléses módszer segítségével lehet csökkenteni.
Ezt követően az egyesített mintából vagy a redukált mintákból megközelítőleg egyenlő számú (ellenőrzési, védelmi és esetlegesen referenciacélra szánt) végső mintát kell venni a 7. pontban bemutatott mennyiségi követelményeknek megfelelően.
9.4.2. Nem egyenletesen eloszló anyagok
Alkotóelemek, többek között géntechnológiával módosított anyagok, illetve a takarmányban feltételezhetően nem egyenletesen eloszló anyagok ellenőrzésekor az egyesített minta:
i. teljesen homogenizált. Ezt követően a homogenizált egyesített mintából megközelítőleg egyenlő számú (ellenőrzési, védelmi és esetlegesen referenciacélra szánt) végső mintákat kell venni a 7. pont mennyiségi követelményeinek megfelelően; vagy
ii. tömegét mechanikus vagy automata osztóval legalább 2 kg-ra, illetve 2 literre kell csökkenteni ( 7 ). A mintát szükség esetén negyedeléses módszerrel is lehet csökkenteni, de kizárólag abban az esetben, ha a takarmány jellege nem teszi lehetővé az osztó használatát. A géntechnológiával módosított anyagok jelenlétének 619/2011/EU rendelet szerinti ellenőrzésekor a redukált mintának legalább 35 000 magot/szemet kell tartalmaznia annak érdekében, hogy el lehessen érni a hatósági, valamint a védelmi és a referenciacélra szánt végső minták esetében érvényes, minimálisan 10 000 mag/szem számot (lásd a 6. pont (**) jelölésű és a 7. pont (*) jelölésű lábjegyzetét).
A redukált mintából megközelítőleg egyenlő számú végső mintát kell venni a 7. pont mennyiségi követelményeinek megfelelően.
9.5. A minta csomagolása
A tartályokat, illetve csomagokat le kell pecsételni és címkével kell ellátni oly módon, hogy a pecsét felsértése nélkül ne lehessen kinyitni azokat. A pecsétnek magában kell foglalnia a teljes címkét. Egy másik lehetőség szerint a mintát zárható edénybe kell helyezni oly módon, hogy azt a tárolóedény vagy tartály visszafordíthatatlan roncsolása nélkül ne lehessen felnyitni, elkerülve így a tárolóedény vagy tartály esetleges újrafelhasználását.
9.6. A minták laboratóriumba történő küldése
A mintát - a vegyelemző számára szükséges információkkal együtt - indokolatlan késedelem nélkül el kell küldeni az arra a kijelölt vizsgálati laboratóriumba.
10. MINTAVÉTELI JEGYZŐKÖNYV
Minden egyes mintáról jegyzőkönyvet kell vezetni, amely lehetővé teszi az egyes mintavételi tételek és azok méretének egyértelmű azonosítását.
Jegyzőkönyvbe kell foglalni az e rendeletben meghatározott mintavételi eljárástól történő bármely eltérést.
A jegyzőkönyvet elérhetővé kell tenni a hatósági ellenőrző laboratórium, továbbá a takarmányipari vállalkozó és/vagy a takarmányipari vállalkozó által kijelölt laboratórium számára.
11. TÁVÉRTÉKESÍTÉSI MINTA
- Távértékesítési minta esetében távközlő eszközön keresztül kell bekérni a takarmányipari vállalkozótól egy takarmánymennyiséget. Ebben az esetben a takarmány bekérésekor az illetékes hatóságnak nem kell felfednie magát a hivatalos minőségében a takarmányipari vállalkozó előtt, és álnevet is használhat.
- A távértékesítési minta egyesített mintájának és végső mintáinak vételezését a szállítmány átvételét követően az e célra felhatalmazott személyeknek azonnal el kell végezniük. Az egyesített minta előállításához a kapott teljes mennyiségből véletlenszerűen és egyenletesen elosztva megfelelő számú elemi mintát kell venni, és gondosan össze kell keverni/homogenizálni kell az 5., a 9.2. és a 9.3. pontban megállapított elveknek lehető leginkább megfelelő módon. Ha a takarmány egyedi kiszerelési egységekbe van csomagolva, legalább 4 darabot kell kérni, amelyekből legalább egy elemi mintát kell venni. Amennyiben eseti alapon bizonyítást nyer, hogy a kapott egységek különböző tételekből származnak, a mintavétel tárgyát képező egységek számát csökkenteni kell, és az ugyanabból a tételből származó egységekre kell korlátozni. A takarmányban nem egyenletesen eloszló összetevők vagy anyagok távértékesítési mintájának elemzése esetén az elemi minták számának a takarmányban egyenletesen eloszló anyagok elemzett mintáinak számához képest legalább 2,5-szer nagyobbnak kell lennie.
Az egyesített mintából ezután a megfelelő végső mintákat (ellenőrzés, védelem és esetlegesen referencia céljából) a 9.4. pontban meghatározott elvekkel összhangban kell venni, és a mintavételi jegyzőkönyvben fel kell tüntetni, hogy a minta távértékesítési minta. Az illetékes hatóság ezt követően haladéktalanul tájékoztatja a takarmányipari vállalkozót a mintavételről. A takarmányipari vállalkozót arról is értesítik, hogy az illetékes hatóság lehetőség szerint egy (védelmi célú) mintát egy meghatározott helyen, védelmi célból a rendelkezésére bocsát, vagy elküldi a takarmányipari vállalkozónak, vagy a takarmányipari vállalkozó által kijelölt laboratóriumba küldi a hatályos nemzeti szabályoknak megfelelően.
Ha a mintát közvetlenül a hatósági laboratóriumba küldik, a végső mintát a laboratóriumban erre felhatalmazott személyeknek kell elkészíteniük és lepecsételniük vagy az erre felhatalmazott személyek jelenlétében kell elkészíteni és lepecsételni. A távértékesítési minta mintavételi jegyzőkönyvét a végső minták összeállítása után haladéktalanul meg kell küldeni az illetékes hatóságnak, amely tájékoztatja a takarmányipari vállalkozót a mintavételről.
Úgy kell tekinteni, hogy a takarmányipari vállalkozó által az illetékes hatóság részére átadott mennyiség egy azonos osztályú vagy megnevezésű takarmánytétel részét képezi. A 178/2002/EK európai parlamenti és tanácsi rendelet ( 8 ) 15. cikkének megfelelően, ha egy azonos osztályú vagy megnevezésű takarmánytétel egy részének mintavételezése esetén azt állapítják meg, hogy a tétel szóban forgó része nem felel meg az uniós követelményeknek, a távértékesítési minta esetében is abból kell kiindulni, hogy a tétel többi része sem biztonságos, kivéve, ha a részletes kivizsgálást követően (adott esetben helyszíni vizsgálat keretében) nem áll fenn bizonyíték arra, hogy a tétel többi része nem felel meg az uniós követelményeknek.
II. MELLÉKLET
A TAKARMÁNYVIZSGÁLATI MÓDSZEREKRE VONATKOZÓ ÁLTALÁNOS RENDELKEZÉSEK
A. MINTÁK ANALITIKAI VIZSGÁLATRA TÖRTÉNŐ ELŐKÉSZÍTÉSE
1. Cél
Az e mellékletben leírt eljárások az I. mellékletben megállapított rendelkezéseknek megfelelő mintavételezés után, az ellenőrző laboratóriumoknak megküldött minták analitikai vizsgálat céljából történő előkészítésére vonatkoznak.
A laboratóriumi mintákat oly módon kell előkészíteni, hogy a vizsgálati módszerekben előírt kimért mennyiségek homogének és a végső mintákra jellemzőek legyenek.
Az e mellékletben leírt eljárásokon kívül az EN ISO 6498 szabványban előírt minta-előkészítési irányelveket is követni kell.
2. Óvintézkedések
A követendő mintaelőkészítési eljárás függ az alkalmazandó vizsgálati módszertől és az ellenőrizendő alkotóelemektől, illetve anyagoktól. Ezért elsőrendű fontosságú annak biztosítása, hogy a követett minta-előkészítési eljárás megfeleljen az alkalmazott vizsgálati módszernek, valamint az ellenőrizendő alkotóelemeknek, illetve anyagoknak.
Minden szükséges műveletet oly módon kell elvégezni, hogy minimális legyen a minta beszennyeződésének és összetétele megváltozásának lehetősége.
Az őrlést, keverést és rostálást haladéktalanul végre kell hajtani azért, hogy a minta a lehető legrövidebb ideig érintkezzen a levegővel és a fénnyel. Kerülni kell az olyan daráló- és őrlőgépek használatát, amelyek a mintát érzékelhetően felmelegítik.
A különösen hőérzékeny takarmányok esetében a kézi őrlés alkalmazása javasolt. Figyelmet kell fordítani annak biztosítására is, hogy az eszköz maga ne lehessen szennyeződés forrása.
A minta homogenizálása vízzel történő nagysebességű keveréssel előállított iszapként bizonyos esetekben homogénebb részmintákat eredményezett, mint a száraz homogenizálás/őrlés, különösen a heterogén eloszlású vegyi anyagok esetében. Ugyanakkor a megfelelő száraz őrléssel történő homogenizálás is homogén részmintákat biztosíthat.
Bizonyos esetekben, például az anyarozs, a káros botanikai szennyeződések stb. meghatározása során a minta homogenizálása nem őrléssel, hanem a minta megfelelő összekeverésével végezhető el.
Ha az előkészítést nem lehet a minta nedvességtartalmának jelentős megváltozása nélkül elvégezni, akkor a nedvességtartalmat az előkészítés előtt és után is meg kell határozni a III. melléklet A. részében meghatározott módszer szerint.
3. Az eljárás
3.1. Általános eljárás
A vizsgálati aliquotot a végső homogenizált mintából kell venni. Az átlós negyedeléssel történő mintavétel nem ajánlott, mivel magas eloszlási hibát tartalmazó vizsgálati aliquotokat eredményezhet.
3.1.1. Előkészítés nélkül őrölhető takarmányok
- Keverjük össze a végső mintát, és vegyük fel egy megfelelően tiszta, száraz, légmentesen záródó tartályba. Közvetlenül a vizsgálatra szánt mennyiség (vizsgálati aliquot) kimérése előtt keverjük újra össze a teljes homogenitás elérése érdekében.
3.1.2. Szárítás után őrölhető takarmányok
- Amennyiben a vizsgálati módszerekben másképpen nem írják elő, a III. melléklet A. részében említett nedvességmeghatározási módszer 4.3. pontja szerint leírt előzetes szárítási eljárásnak megfelelően szárítsuk a végső mintát 8-12 %-os nedvességtartalmúra. Ezt követően a 3.1.1. pontban leírtak szerint járjunk el.
3.1.3. Folyékony vagy félfolyékony takarmányok
- Vegyük fel a végső mintát egy megfelelően tiszta, száraz, légmentesen záródó tartályba. Közvetlenül a vizsgálatra szánt mennyiség (vizsgálati aliquot) kimérése előtt alaposan keverjük össze a teljes homogenitás elérése érdekében.
3.1.4. Egyéb takarmányok
- Azok a végső minták, amelyek a fent említett eljárások egyikével sem készíthetők elő, bármely egyéb, olyan eljárással kezelhetők, amely biztosítja, hogy a vizsgálatra kimért mennyiségek (vizsgálati aliquotok) homogének és a végső mintákra jellemzőek legyenek.
3.2. A szemrevételezéses vagy mikroszkópos vizsgálat, illetve a teljes egészében homogenizált egyesített minta esetében alkalmazandó külön eljárás
- A szemrevételezéses (mikroszkóp alkalmazása nélküli) ellenőrzés a teljes egyesített vagy végső minta felhasználásával történik.
- Mikroszkópos ellenőrzés esetén a laboratórium redukálhatja az egyesített mintát, vagy tovább redukálhatja a már redukált mintát. A védelem és esetlegesen a referencia céljait szolgáló végső mintákat a hatósági ellenőrzéshez használt végső mintákra érvényes eljárással egyenértékű eljárást követve kell venni.
- Amennyiben a teljes egyesített minta homogenizált, a végső mintákat a homogenizált egyesített mintából kell venni.
- Az anyarozs és a káros botanikai szennyeződések meghatározásához a végső mintát két, egyenlő tömegű, körülbelül 500 grammos részmintára kell osztani. Egy részmintát kell megvizsgálni. Amennyiben a részminták eredménye nem haladja meg a felső határérték 50 %-át (analitikai küszöbérték), a minta megfelel a felső határértéknek. Ha az eredmény meghaladja a felső határérték 50 %-át, egy másik részmintát kell megvizsgálni, és a felső határértéknek való megfelelés ellenőrzésére a két részminta eredményének átlagértékét kell figyelembe venni.
4. A minták tárolása
A mintákat olyan hőmérsékleten kell tárolni, amely összetételüket nem változtatja meg. A vitaminok vagy fényre különösen érzékeny anyagok vizsgálatára szánt mintákat a fény esetleges hátrányos hatását kizáró feltételek mellett kell tárolni.
B. A VIZSGÁLATI MÓDSZEREK SORÁN HASZNÁLT REAGENSEKRE ÉS ESZKÖZÖKRE VONATKOZÓ RENDELKEZÉSEK
1. Amennyiben a vizsgálati módszerekben másképpen nem írják elő, minden analitikai reagensnek analitikai tisztaságúnak (a. t.) kell lennie. Nyomelem-analízis végrehajtásakor a reagensek tisztaságát vakpróbával kell ellenőrizni. Az így kapott eredmények a reagensek esetleges további tisztítását tehetik szükségessé.
2. A vizsgálati módszerek leírásában említett bármely oldatkészítési, hígítási, öblítési vagy mosási művelet esetében, ahol az alkalmazott oldószer vagy hígítószer fajtáját nem tüntetik fel, vizet kell használni. Általános szabályként ioncserélt vagy desztillált vizet kell használni. A vizsgálati módszerekben jelzett különleges esetekben a vizet speciális tisztítási eljárásokkal kell kezelni.
3. Az ellenőrző laboratóriumokban megtalálható alapfelszerelésre való tekintettel a vizsgálati módszerek leírásában csak a speciális vagy speciális használatot megkövetelő eszközöket és készülékeket említik. Ezeknek tisztáknak kell lenniük, különösen a nagyon kis anyagmennyiségek meghatározása esetén.
C. A VIZSGÁLATI MÓDSZEREK ALKALMAZÁSA ÉS AZ EREDMÉNYEK KIFEJEZÉSE
1. Extrahálási eljárás
Számos módszer külön extrahálási eljárást határoz meg. Főszabályként a módszerben említettől eltérő extrahálási eljárást is lehet követni, feltéve, hogy az alkalmazott extrahálási eljárás hatékonysága a vizsgált mátrix tekintetében bizonyítottan egyenértékű a módszerben említett eljárás extrahálási hatékonyságával.
2. Tisztítási eljárás
Számos módszer külön tisztítási eljárást határoz meg. Főszabályként a módszerben említettől eltérő tisztítási eljárást is lehet követni, feltéve, hogy az alkalmazott tisztítási eljárás a vizsgált mátrix tekintetében bizonyítottan a módszerben említett eljárással egyenértékű vizsgálati eredményt ad.
3. Meghatározások száma
Nemkívánatos anyagok vizsgálata esetében, ha az első meghatározás eredménye jelentősen (> 50 %) alacsonyabb, mint az ellenőrizendő előírás, nincs szükség további meghatározásokra, feltéve, hogy a megfelelő minőségügyi eljárásokat alkalmazzák. Ettől eltérő esetben párhuzamos vizsgálatra (második meghatározásra) van szükség ahhoz, hogy kizárjuk a minták belső keresztszennyeződésének, valamint esetleges összecserélődésének veszélyét. A két meghatározás számtani középértékét használjuk a további értékeléshez.
Takarmány-adalékanyagok alsó és felső határértékeinek vizsgálata esetében, ha az első meghatározás eredményei az alsó határérték felett vagy a felső határérték alatt vannak, nincs szükség további meghatározásokra, feltéve, hogy a megfelelő minőségügyi eljárásokat alkalmazzák. Ettől eltérő esetben párhuzamos vizsgálatra (második meghatározásra) van szükség ahhoz, hogy kizárjuk a minták belső keresztszennyeződésének, valamint esetleges összecserélődésének veszélyét. A két meghatározás számtani középértéke használható a további értékeléshez.
Valamely anyag vagy összetevő feltüntetett mennyiségére végzett vizsgálat esetében, ha az első meghatározás eredménye igazolja a feltüntetett mennyiséget, azaz a vizsgálati eredmény a feltüntetett tartalom elfogadható szórási tartományába esik, nincs szükség további meghatározásokra, feltéve, hogy a megfelelő minőségügyi eljárásokat alkalmazzák. Ettől eltérő esetben párhuzamos vizsgálatra (második meghatározásra) van szükség ahhoz, hogy kizárjuk a minták belső keresztszennyeződésének, valamint esetleges összecserélődésének veszélyét. A két meghatározás számtani átlagát kell használni a további értékeléshez (az átlagolt vizsgálati eredmény a feltüntetett tartalom elfogadható szórási tartományába esik-e vagy sem).
Néhány esetben az elfogadható szórási tartományról jogszabályok rendelkeznek, mint például a 767/2009/EK rendelet és az (EU) 2019/4 európai parlamenti és tanácsi rendelet ( 9 ).
4. Az alkalmazott vizsgálati módszer jegyzőkönyvi feltüntetése
A vizsgálati jegyzőkönyvben fel kell tüntetni az alkalmazott vizsgálati módszert.
5. A vizsgálati eredmények jegyzőkönyvi feltüntetése
A vizsgálati eredményt a vizsgálati módszerben meghatározott módon és megfelelő helyiérték-számmal kell kifejezni, és ha szükséges, akkor a végső minta előkészítés előtti nedvességtartalmára kell korrigálni.
A takarmányra vonatkozó uniós jogszabályokban a legtöbb szabályozási szint (pl. felső határérték, alsó határérték) 12 %-os nedvességtartalmú takarmányra vonatkozik. Ezért ezekben az esetekben annak érdekében, hogy a mintán mért vizsgálati eredményt a szabályozási szinthez képest értékelni lehessen, a vizsgálati eredményt először el kell osztani a minta szárazanyag-tartalmával (%-ban), megszorozva 88-cal, a következő képlet szerint:
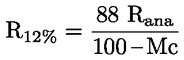
ahol:
Mc : a minta nedvességtartalma (%-ban). 100 - Mc tehát a minta szárazanyag-tartalmát jelöli (%-ban).
Rana : a mintán mért vizsgálati eredmény.
R12 % : eredmény 12 %-os nedvességtartalmú takarmányban; a szabályozási szinthez képest értékelendő.
Emellett, ha a következő feltételek teljesülnek:
- a vizsgálat eredménye jelentősen (> 50 %) alacsonyabb vagy magasabb, mint az ellenőrizendő címkézési információ/termék specifikáció (attól függően, hogy a címkézési információ/specifikáció felső vagy alsó határérték);
- a mintavételnek alávetett takarmány nedvességtartalma ismert, és megállapítható, hogy a nedvességtartalom korrekciója nem változtatja meg az értékelést,
akkor - feltéve, hogy a megfelelő minőségi eljárásokat alkalmazzák, és az elemzés kizárólag a jogszabályi rendelkezések betartásának ellenőrzésére szolgál - a nedvességtartalom korrekciója elhagyható (pl. olyan esetekben, amikor nincs specifikáció vagy szabályozási szint), kivéve, ha értelmezésre van szükség.
Ha az analitikai eredményt korrigálják a nedvességtartalomra, a vonatkozó mérési bizonytalanságot is korrigálni kell ugyanabban az eljárásban.
Az anyarozs vagy káros botanikai szennyeződések szemrevételezéses/mikroszkópos vizsgálattal történő meghatározása esetén nincs szükség a nedvességtartalom korrekciójára.
6. Mérési bizonytalanság és visszanyerési arány nemkívánatos anyagok vizsgálata esetében
A 2002/32/EK szerinti nemkívánatos anyagok tekintetében valamely takarmányozásra szánt termék akkor minősül nem megfelelőnek a megengedett határérték szempontjából, ha a két független meghatározás átlagaként kapott, 12 %-os nedvességtartalmú takarmányra vonatkoztatott vizsgálati eredmény - figyelembe véve a körülbelül 95 %-os konfidenciaszintnek megfelelő 2-es kiterjesztési tényező alkalmazásával számított kiterjesztett mérési bizonytalanságot, és a visszanyerési korrekciót -, meghaladja a megengedett határértéket. Ez azt jelenti, hogy a megfelelés megállapítására a mért koncentrációt alkalmazzák a visszanyerés alapján végzett korrekció, valamint a kiterjesztett mérési bizonytalanság kivonása után. Ez az eljárás csak abban az esetben alkalmazandó, ha az elemzési módszer lehetővé teszi a kiterjesztett mérési bizonytalanság becslését és a visszanyeréssel történő korrekciót (pl. szemrevételezéses/mikroszkópos vizsgálat esetén nem szükséges).
Ha a védelem céljából vett minta vizsgálati eredménye meghaladja a megengedett határértéket (a kiterjesztett mérési bizonytalanság figyelembevétele nélkül), ez az erre vonatkozó nemzeti szabályok hiányában megerősíti az ellenőrzési minta tekintetében megállapított nem megfelelőséget.
A vizsgálati eredményt a következők szerint kell a jegyzőkönyvben feltüntetni (amennyiben az alkalmazott vizsgálati módszer lehetővé teszi a kiterjesztett mérési bizonytalanság becslését):
a) visszanyerésre korrigáltan - amennyiben helyénvaló és releváns -, és ha korrigálták, azt fel kell tüntetni. A visszanyerési arányt meg kell adni, kivéve, ha a torzítás belső korrekciója része az eljárásnak, ahol a torzítás a mért érték és a referenciakoncentráció közötti különbség. Nem szükséges feltüntetni a visszanyerési korrekciót, ha a visszanyerés aránya 90-110 % közötti;
b) "x +/- U" formában, ahol x a vizsgálati eredmény, U a kiterjesztett mérési bizonytalanság, a kiterjesztési tényező 2 ( 10 ), amely körülbelül 95 %-os konfidenciaszintet eredményez.
Ha azonban a vizsgálat eredménye jelentősen (> 50 %) alacsonyabb, mint az ellenőrizendő előírás, és feltéve, hogy megfelelő minőségi eljárásokat alkalmaznak és a vizsgálat célja kizárólag a jogszabályi rendelkezéseknek való megfelelés ellenőrzése, el lehet hagyni a visszanyerés arányának és a kiterjesztett mérési bizonytalanságnak a jegyzőkönyvi feltüntetését (pl. olyan esetekben, amikor nincs specifikáció vagy szabályozási szint), kivéve, ha értelmezésre van szükség.
7. Mérési bizonytalanság és visszanyerési arány takarmány-adalékanyagok tartalmának vizsgálata esetében
Annak ellenőrzése érdekében, hogy a takarmány-adalékanyagok megfelelnek-e az engedélyezett minimális és maximális tartalomnak, úgy kell tekinteni, hogy a takarmány-adalékanyag jelenléte nem felel meg a megállapított minimális és maximális tartalomnak, ha úgy ítélik meg, hogy a 12 %-os nedvességtartalmú takarmányra vonatkoztatott vizsgálati eredmény, két független meghatározás számtani középértékeként:
- meghaladja a maximális tartalmat, figyelembe véve a kiterjesztett mérési bizonytalanságot és a visszanyerési korrekciót. Ez azt jelenti, hogy a megfelelés megállapítására a mért koncentrációt (azaz két meghatározás számtani középértékét) alkalmazzák a visszanyerés alapján végzett korrekció, valamint a kiterjesztett mérési bizonytalanság kivonása után;
- a minimális tartalomnál alacsonyabb, figyelembe véve a kiterjesztett mérési bizonytalanságot és a visszanyerési korrekciót. Ez azt jelenti, hogy a megfelelés megállapítására a mért koncentrációt (azaz két meghatározás számtani középértékét) alkalmazzák a visszanyerés alapján végzett korrekció, valamint a kiterjesztett mérési bizonytalanság hozzáadása után.
Ha a védelem céljából vett minta vizsgálati eredménye meghaladja a megengedett határértéket (a kiterjesztett mérési bizonytalanság figyelembevétele nélkül), ez erre vonatkozó nemzeti szabályok hiányában megerősíti az ellenőrzési minta tekintetében megállapított nem megfelelőséget.
A vizsgálati eredményt a következők szerint kell a jegyzőkönyvben feltüntetni (amennyiben az alkalmazott vizsgálati módszer lehetővé teszi a kiterjesztett mérési bizonytalanság becslését):
a) visszanyerésre korrigáltan - amennyiben helyénvaló és releváns -, és ha korrigálták, azt fel kell tüntetni. A visszanyerési arányt meg kell adni, kivéve, ha a torzítás belső korrekciója része az eljárásnak, ahol a torzítás a mért érték és a referenciakoncentráció közötti különbség. Nem szükséges feltüntetni a visszanyerési korrekciót, ha a visszanyerés aránya 90-110 % közötti;
b) "x +/- U" formában, ahol x a vizsgálati eredmény (két meghatározás számtani középértéke), U a kiterjesztett mérési bizonytalanság, a kiterjesztési tényező 2 ( 11 ), amely körülbelül 95 %-os konfidenciaszintet eredményez.
III. MELLÉKLET
ANALITIKAI MÓDSZEREK A TAKARMÁNY-ALAPANYAGOK ÉS TAKARMÁNYKEVERÉKEK ÖSSZETÉTELÉNEK ELLENŐRZÉSÉRE
A. A NEDVESSÉGTARTALOM MEGHATÁROZÁSA
1. Cél és alkalmazási terület
Ez a módszer lehetővé teszi a takarmányok nedvességtartalmának meghatározását. Illóanyagokat (mint például szerves savakat) tartalmazó takarmányok esetében figyelembe kell venni, hogy a nedvességtartalommal együtt jelentős mennyiségű illóanyag meghatározása történik.
Nem vonatkozik a tejtermékek mint takarmány-alapanyagok és a túlnyomórészt tejtermékekből álló takarmánykeverékek analízisére, az állati és növényi zsírok és olajok és olajos magvak és olajtartalmú gyümölcsök analízisére.
Az olajmagvak nedvességtartalmának meghatározását « »A nedvesség- és az illóanyag-tartalom meghatározása" című EN ISO 665 szabványban előírt módszerrel kell elvégezni azzal a kitétellel, hogy a szójababot a nedvességtartalom meghatározása előtt meg kell őrölni.
2. Vizsgálati alapelv
A mintát, a takarmány jellege szerint, meghatározott körülmények között szárítjuk. A tömegveszteséget méréssel határozzuk meg. Amennyiben magas nedvességtartalmú, szilárd takarmányokkal dolgozunk, előzetes szárítást kell végezni.
3. Eszközök
3.1. Nem nedvszívó anyagból készült, könnyen tisztítható aprítógép, amelynek használata gyors és egynemű őrlést tesz lehetővé anélkül, hogy észrevehető mennyiségű hőtermeléssel járna, a lehető legnagyobb mértékben megakadályozza a külső levegővel történő érintkezést, és megfelel a 4.1.1. és 4.1.2. pontban foglalt követelményeknek (pl. kalapácsos törő vagy vízhűtéses mikroaprítók, összecsukható kúpos malmok, finommozgású-lassú fordulatú vagy fogaskerekes aprítók).
3.2. 1 mg pontosságú analitikai mérleg.
3.3. Nem rozsdásodó fémből vagy üvegből készült, légmentes záródást biztosító fedéllel ellátott száraz tartályok; megfelelő munkafelület, amely lehetővé teszi a vizsgálati minta 0,3 g/cm2 körüli mennyiségben történő terítését.
3.4. Elektromos fűtésű izotermikus, megfelelően szellőztethető szárítószekrény (± 2 °C), amely gyors hőfokszabályozást tesz lehetővé ( 12 ).
3.5. Szabályozható, elektromos fűtésű vákuum szárítószekrény olajszivattyúval és forró, száraz levegő vagy szárítóanyag (pl. kalcium-oxid) bejuttatására alkalmas szerkezettel ellátva.
3.6. Megfelelő szárítóanyagot tartalmazó, vastag, perforált fém- vagy porcelánlemezes exszikkátor.
4. A vizsgálat módja
Megjegyzés: Az ebben a szakaszban leírt műveleteket a mintacsomagok kibontása után azonnal el kell végezni. Az elemzést legalább egyszer meg kell ismételni.
4.1. Előkészítés
4.1.1. A 4.1.2. és a 4.1.3. pont alatt nem szereplő takarmányok
Vegyünk legalább 50 g-ot a mintából. Szükség esetén aprítsuk fel vagy oszlassuk el úgy, hogy a nedvességtartalma egyenletes legyen (lásd a 6. pontot).
4.1.2. Gabona- és darafélék
Vegyünk legalább 50 g-ot a mintából. Őröljük le úgy, hogy a kapott részecskék legalább 50 %-a átjusson egy 0,5 mm-es szembőségű szitán, és ne maradjon vissza 10 %-nál több egy kerek szemű, 1 mm-es szembőségű szitán.
4.1.3. Folyékony vagy kenőcsös állagú takarmányok, túlnyomórészt olajokból és zsírokból álló takarmányok
Mérjünk ki 25 g mintát 10 mg pontossággal, adjunk hozzá megfelelő mennyiségű vízmentes homokot, 10 mg pontossággal kimérve, és keverjük addig, amíg homogén masszát nem kapunk.
4.2. Szárítás
Szárítsunk meg egy tartályt (3.3. pont) fedelével a 103 °C-ra beállított szárítószekrényben 30 perc ± 1 percig. Vegyük ki a tartályt a szárítószekrényből, és hagyjuk lehűlni szobahőmérsékletre az exszikkátorban (3.6. pont),
4.2.1. A 4.2.2. és a 4.2.3. pont alatt nem szereplő takarmányok
Fedelével együtt mérjük le a tartályt 1 mg pontossággal. Mérjünk a lemért tartályba 5 g mintát 1 mg pontossággal, és terítsük el egyenletesen. Helyezzük a tartályt fedél nélkül 103 °C-ra előmelegített szárítószekrénybe. Annak megakadályozása érdekében, hogy a szárítószekrény hőfoka a kívánatos érték alá süllyedjen, a tartályt a lehető leggyorsabban helyezzük a szárítószekrénybe. Hagyjuk száradni négy órán át attól az időponttól számítva, amikor a szárítószekrény hőfoka újból elérte a 103 °C-ot. Nyissuk ki a szárítószekrényt, azonnal helyezzük vissza a tartály fedelét, vegyük ki a tartályt a szárítószekrényből, hagyjuk hűlni 30-45 percen át az exszikkátorban (3.6. pont), és mérjük le 1 mg-os pontossággal.
Túlnyomórészt (> 50 %) állati és növényi eredetű olajokból és zsírokból álló takarmányok esetén szárítsuk további 30 percen át 103 °C-on a szárítószekrényben. A két mérés különbsége nem haladhatja meg a nedvességtartalom 0,1 %-át.
4.2.2. Gabonafélék, liszt, darafélék és derce
Fedelével együtt mérjük le a tartályt 0,5 mg pontossággal. Mérjünk a lemért tartályba 5 g megőrölt mintát 1 mg pontossággal, és terítsük el egyenletesen. Helyezzük a tartályt fedél nélkül 130 °C-ra előmelegített szárítószekrénybe. Annak megakadályozása érdekében, hogy a szárítószekrény hőfoka a kívánatos érték alá süllyedjen, a tartályt a lehető leggyorsabban helyezzük a szárítószekrénybe. Hagyjuk száradni két órán át attól az időponttól számítva, amikor a szárítószekrény hőfoka újból elérte a 130 °C-ot. Nyissuk ki a szárítószekrényt, azonnal helyezzük vissza a tartály fedelét, vegyük ki a tartályt a szárítószekrényből, hagyjuk hűlni 30-45 percen át az exszikkátorban (3.6. pont), és mérjük le 1 mg-os pontossággal.
4.2.3. Több mint 4 % szacharózt vagy laktózt tartalmazó takarmánykeverékek: takarmány-alapanyagok, mint a szentjánoskenyér, a hidrolizált gabonatermékek, a malátamagvak, a szárított répaszelet, a hal- és cukorlevek
Fedelével együtt mérjük le a tartályt 0,5 mg pontossággal. Mérjünk a lemért tartályba 5 g mintát 1 mg pontossággal, és terítsük el egyenletesen. Helyezzük a tartályt fedél nélkül 80-85 °C-ra előmelegített vákuum szárítószekrénybe (3.5. pont). Annak megakadályozása érdekében, hogy a szárítószekrény hőfoka a kívánatos érték alá süllyedjen, a tartályt a lehető leggyorsabban helyezzük a szárítószekrénybe.
Emeljük a nyomást 100 torr-ra, és hagyjuk a mintát száradni ezen a nyomáson négy órán át száraz, forró levegő áramoltatásával vagy szárítóanyag (20 mintához kb. 300 g) felhasználásával. Utóbbi esetben a kívánt nyomás elérésekor kapcsoljuk le a vákuumszivattyút. A szárítási időt attól a pillanattól kell számítani, amikor a szárítószekrény hőmérséklete visszaáll 80-85 °C-ra. Óvatosan állítsuk vissza a szárítószekrényben a légköri nyomást. Nyissuk ki a szárítószekrényt, azonnal helyezzük vissza a tartály fedelét, vegyük ki a tartályt a szárítószekrényből, hagyjuk hűlni 30-45 percen át az exszikkátorban (3.6. pont), és mérjük le 1 mg-os pontossággal. Szárítsuk további 30 percen át a vákuum szárítószekrényben 80-85 °C-on, és mérjük le újra. A két mérés különbsége nem haladhatja meg a nedvességtartalom 0,1 %-át.
4.3. Előszárítás (részleges)
A szilárd anyagok elemzése érdekében a finom őrlést megelőzően részlegesen szárítani kell a 85 %-nál kevesebb szárazanyagot tartalmazó "nedves" takarmányokat (pl. zöldtakarmányok, teljes vegyes takarmányadagok (TMR), (nem) folyékony takarmányok); instabil anyagok esetében a részleges szárítás nem lehetséges.
A részleges szárítás történhet légkeveréses szárítószekrényben, mikrohullámú sütőben vagy fagyasztva szárítással. A fagyasztva szárítással történő részleges szárítás kivételével a cél a takarmány szárítása a minta hőmérsékletének 60 °C alatt tartása mellett, hogy a kémiai összetétel a lehető legkisebb mértékben sérüljön. A 60 °C-ot meghaladó hőmérsékleten történő szárítás kémiai változásokat okoz a takarmányban (pl. fehérjelebomlást). A szárított takarmányt a részleges szárazanyag mérése előtt körülbelül 15 percig szobahőmérsékleten egyensúlyba kell hozni (ekvilibrálni kell) annak érdekében, hogy a lehető legkisebb legyen az őrlés és a tárolás során esetlegesen bekövetkező nedvességváltozás. A 60 °C alatti hőmérsékleten történő szárítás nem távolítja el az összes vizet a takarmányból; ezért a (kezdeti) részleges szárítás nem jelenti a takarmány teljes szárazanyag-tartalmát. A szárítást követően a részmintát őrölik és elemzik a részlegesen száraz minta (végső) szárazanyagára (a maradék 3-15 %-os nedvességtartalomra), amikor más kémiai összetevőket meghatároznak.
Ezért a szárazanyag meghatározására kétlépcsős eljárás javasolt. Először határozzuk meg a részleges szárazanyag-tartalmat (ha nem éri el a szárazanyag 85 %-át), majd határozzuk meg az őrölt vizsgálati minta fennmaradó szárazanyag-tartalmát, majd szorozzuk meg a részleges szárazanyagot a fennmaradó szárazanyag-tartalommal a teljes szárazanyag-tartalom meghatározása érdekében.
5. Az eredmények kiszámítása
A minta százalékosan kifejezett nedvességtartalmát (X) az alábbi képletek segítségével számítjuk ki:
5.1. Szárítás előszárítás nélkül
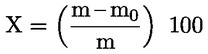
ahol:
m = a vizsgálati minta eredeti tömege, grammban,
m0 = a száraz vizsgálati minta tömege, grammban.
5.2. Szárítás előszárítással ( 13 )
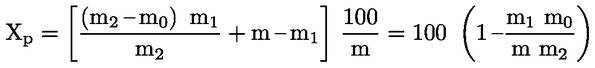
ahol:
m = a vizsgálati minta eredeti tömege, grammban,
m1 = a vizsgálati minta tömege, grammban, előszárítás után,
m2 = a vizsgálati minta tömege, grammban, aprítás vagy őrlés után,
m0 = a száraz vizsgálati minta tömege, grammban.
5.3. Ismételhetőség
Az ugyanazon mintán végzett két párhuzamos meghatározás eredményei közötti különbség nem haladhatja meg a nedvességtartalom abszolút értékének 0,2 %-át, kivéve a kedvtelésből tartott állatok nedves eledelét és a műcsontokat, ahol a különbség nem haladhatja meg a nedvességtartalom abszolút értékének 0,5 %-át.
6. Észrevételek
Ha az aprítás szükségesnek bizonyul, és úgy tűnik, hogy ez befolyásolja a termék nedvességtartalmát, a takarmány-alkotóelemek analízisének eredményét az eredeti minta nedvességtartalmának megfelelően korrigálni kell.
B. AZ ÁLLATI ÉS NÖVÉNYI ZSÍROK ÉS OLAJOK NEDESSÉGTARTALMÁNAK MEGHATÁROZÁSA
1. Cél és alkalmazási terület
Ez a módszer lehetővé teszi az állati és növényi zsírok és olajok víz- és illóanyag-tartalmának meghatározását.
2. Vizsgálati alapelv
A mintát 103 °C-on, a tömegállandóság eléréséig szárítjuk (két egymást követő mérés közötti tömegveszteségnek legfeljebb 1 mg-nak kell lennie). A tömegveszteséget méréssel határozzuk meg.
3. Eszközök
3.1. Rozsdamentes anyagból készült, 8-9 cm átmérőjű és körülbelül 3 cm magas, lapos fenekű edény.
3.2. Rögzített üveggömbbel és a felső végén kiterjedési csővel rendelkező hőmérő, amely körülbelül 80 °C-tól legalább 110 °C-ig beosztással rendelkezik és körülbelül 10 cm hosszú.
3.3. Homokfürdő vagy elektromos főzőlap.
3.4. Hatékony szárítóközeget tartalmazó exszikkátor.
3.5. Analitikai mérleg.
4. A vizsgálat módja
Mérjünk ki mg pontossággal 20 g homogenizált mintát a hőmérőt (3.2. pont) tartalmazó száraz, lemért edénybe (3.1. pont). A hőmérővel folyamatosan kevergetve hevítsük a homokfürdőn vagy a főzőlapon (3.3. pont) úgy, hogy a hőmérséklet körülbelül hét perc alatt érje el a 90 °C-ot.
Csökkentsük a hőt, ügyelve az edény aljáról felszálló buborékok gyakoriságára. A hőmérséklet nem haladhatja meg a 105 °C-ot. Folytassuk az edény alján lévő anyag keverését, amíg a buborékképződés le nem áll.
A teljes kiszárítás érdekében hevítsük az edényt néhányszor 103 °C ± 2 °C-ra, miközben két hevítés között 93 °C-ra hűtjük tartalmát. Ezután hagyjuk szobahőmérsékletre lehűlni az exszikkátorban (3.4. pont), és mérjük le. Ismételjük addig a műveletet, amíg a két egymást követő mérés közötti tömegveszteség már nem haladja meg a 2 mg-ot.
Megjegyzés: A minta tömegének növekedése a többszöri hevítést követően a zsír oxidációját jelzi, ebben az esetben az eredményt a tömeg növekedését közvetlenül megelőző mérés alapján kell kiszámítani.
5. Az eredmények kiszámítása
A mintára vonatkoztatott, százalékosan kifejezett nedvességtartalmat (X) a következő képlettel lehet kiszámítani:
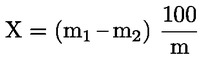
ahol:
m = a vizsgálati minta tömege, grammban;
m1 = az edény hevítés előtti tömege, grammban, a tartalmával együtt;
m2 = az edény hevítés utáni tömege, grammban, a tartalmával együtt.
A 0,05 %-nál kisebb eredményeket a " 0,05 %-nál kisebb« megnevezéssel kell feljegyezni.
Ismételhetőség
Az ugyanazon mintán elvégzett két párhuzamos meghatározás eredményeként kapott nedvességtartalmak eltérésének abszolút értéke nem haladhatja meg a 0,1 %-ot.
C. A NITROGÉNTARTALOM MEGHATÁROZÁSA ÉS A NYERSFEHÉRJE-TARTALOM KISZÁMÍTÁSA
1. Cél és alkalmazási terület
E módszer lehetővé teszi a takarmányok nyersfehérje-tartalmának meghatározását a Kjeldahl-módszer szerint meghatározott nitrogéntartalom alapján ( 14 ).
2. Vizsgálati alapelv
A mintát katalizátor jelenlétében kénsavval roncsoljuk. A savas oldatot nátrium-hidroxid-oldattal lúgosítjuk. Az ammóniát lepároljuk, és ismert mennyiségű kénsavban vesszük fel, amelynek feleslegét nátrium-hidroxid standardoldatával titráljuk.
Alternatív megoldásként a felszabadult ammóniát bőséges bórsavoldatba desztilláljuk, majd sósav- vagy kénsavoldattal titráljuk.
3. Reagensek
3.1. Kálium-szulfát.
3.2. Katalizátor: réz(II)-oxid, CuO vagy réz(II)-szulfát-pentahidrát, CuSO4 5H2 O.
3.3. Granulált cink.
3.4. Kénsav, p20 = 1,84 g/ml.
3.5. Kénsav, normál mérőoldat, c(H2SO4) = 0,25 mol/l.
3.6. Kénsav, normál mérőoldat, c(H2SO4) = 0,10 mol/l.
3.7. Kénsav, normál mérőoldat, c(H2SO4) = 0,05 mol/l.
3.8. Metilvörösindikátor; oldjunk fel 300 mg metilvöröst 100 ml etanolban, σ = 95-96 % (v/v).
3.9. Nátrium-hidroxid-oldat (technikai minőségű is használható) β = 40 g/100 ml (m/v: 40 %).
3.10. Nátrium-hidroxid, normál mérőoldat, c (NaOH) = 0,25 mol/l.
3.11. Nátrium-hidroxid, normál mérőoldat, c (NaOH) = 0,10 mol/l.
3.12. Granulált horzsakő, sósavban mosott és meggyújtott.
3.13. Acetanilid (olvadáspont = 114 °C, N-tartalom = 10,36 %).
3.14. Szacharóz (nitrogénmentes).
3.15. Bórsav (H3BO3).
3.16. Metilvörösindikátor-oldat: oldjunk fel 100 mg metilvöröst 100 ml etanolban vagy metil-alkoholban.
3.17. Brómkrezol-zöld-oldat: oldjunk fel 100 mg brómkrezol-zöldet 100 ml etanolban vagy metil-alkoholban.
3.18. Bórsavas oldat (10 g/l - 40 g/l, a használt eszköztől függően)
Kolorimetriás végpont-meghatározás alkalmazásakor a metilvörös és a brómkrezol indikátort a bórsavas oldathoz kell adni. Ha 1 liter bórsavas oldatot készítünk, a térfogat kiigazítása előtt 7 ml metilvörösindikátor-oldatot (3.16. pont) és 10 ml brómkrezol-zöld oldatot (3.17. pont) kell hozzáadni.
A használt víztől függően a bórsavas oldat pH-ja tételenként változhat. A bórsavoldat pH-értékének 4,3 és 4,7 között kell lennie. Pozitív vakpróbához gyakorta szükség lehet kis mennyiségű lúg hozzáadására.
Megjegyzés: Rendszerint jó eredményt ad körülbelül 3-4 ml NaOH-nak (3.11. pont) 1 liter 10 g/l bórsavas oldathoz való hozzáadása. Tároljuk az oldatot szobahőmérsékleten, és tárolás közben védjük a fénytől és az ammóniagáz-forrásoktól.
3.19. Sósav, normál mérőoldat, c(HCl) = 0,10 mol/l.
Megjegyzés: A normál mérőoldatok más koncentrációi (3.5., 3.6., 3.7., 3.10., 3.11. és 3.19. pont) is használhatók, ha ezt a számításokban korrigáljuk. A koncentrációt mindig négytizedes pontossággal kell megadni.
4. Eszközök
A Kjeldahl-eljárás szerinti roncsolás, desztillálás és titrálás elvégzésére alkalmas készülék.
5. A vizsgálat módja
5.1. Roncsolás
Mérjünk ki 0,001 g pontossággal 1 g mintát, és helyezzük be a roncsoló lombikjába. Adjunk hozzá 15 g kálium-szulfátot (3.1. pont), megfelelő mennyiségű katalizátort (3.2. pont) (0,3-0,4 g réz(II)-oxidot vagy 0,9-1,2 g réz(II)-szulfát-pentahidrátot), 25 ml kénsavat (3.4. pont) és szükség szerint néhány szemcse horzsakövet (3.12. pont), majd keverjük össze.
Először mérsékelten hevítsük a lombikot, szükség esetén időnként keverjük meg annak tartalmát, egészen addig, amíg a massza karbonizálódik és a hab eltűnik; ezt követően erősebben hevítsük, amíg a folyadék erős forrásba nem jön. A hevítés akkor megfelelő, ha a forrásban levő sav a lombik falán lecsapódik. Kerüljük a lombik oldalának túlhevítését és azt, hogy ahhoz szerves részecskék tapadjanak.
Amikor az oldat feltisztult és világoszöld színűvé vált, folytassuk a forralást további két órán át, majd hagyjuk lehűlni az oldatot.
5.2. Lepárlás
Óvatosan adjunk hozzá annyi vizet, amennyi a szulfátok teljes feloldódásához elegendő. Engedjük lehűlni, majd szükség szerint adjunk hozzá néhány szemcsényi cinket (3.3. pont). Folytassuk az 5.2.1. vagy az 5.2.2. pont szerint.
5.2.1. Lepárlás kénsavba
A lepárlókészülék gyűjtőlombikjába mérjünk be pontosan 25 ml kénsavat (3.5. vagy 3.7. pont), a feltételezett nitrogéntartalomtól függően. Adjunk hozzá néhány csepp metilvörösindikátort (3.8. pont).
Kapcsoljuk össze a Kjeldahl-lombikot a lepárlókészülék hűtőjével, és legalább 1 cm mélyen merítsük be a hűtő végét a gyűjtőlombikban lévő folyadékba (lásd az észrevételt, 8.3. pont). Lassan, ammóniaveszteség nélkül öntsünk 100 ml nátrium-hidroxid oldatot (3.9. pont) a Kjeldahl-lombikba (lásd az észrevételt, 8.1. pont). Hevítsük a lombikot az ammónia lepárlódásáig.
5.2.2. Lepárlás bórsavba
Amennyiben a párlat ammóniatartalmának titrálását manuálisan végezzük, az alább megadott eljárást kell alkalmazni. Amennyiben a lepárlóegység teljesen automatikusan a párlat ammóniatartalmát is titrálja, kövessük a gyártó utasításait a lepárlóegység működtetése tekintetében.
Tegyünk a hűtő kifolyójához 25-30 ml bórsavas oldatot (3.18. pont) tartalmazó gyűjtőlombikot úgy, hogy az adagolócső a bórsavas oldat szintje alatt legyen. Állítsuk be a lepárlóegységet 50 ml nátrium-hidroxid oldat (3.9. pont) adagolására. A lepárlóegységet a gyártó utasításainak megfelelően működtessük, és pároljuk le a nátrium-hidroxid oldat hozzáadásával felszabadult ammóniát. Gyűjtsük össze a párlatot a bórsavas fogadóoldatban. A párlat mennyisége (a gőzlepárlás ideje) a minta nitrogéntartalmától függ. Kövessük a gyártó utasításait.
Megjegyzés: Félautomata lepárlóegység esetében a nátrium-hidroxid felesleg hozzáadása és a gőzlepárlás automatikusan történik.
5.3. Titrálás
Folytassuk az 5.3.1. vagy az 5.3.2. pont szerint.
5.3.1. Kénsav
A gyűjtőlombikban levő kénsavfelesleget a végpont eléréséig titráljuk nátrium-hidroxid oldattal (3.10. vagy 3.11. pont) az alkalmazott kénsav-koncentrációtól függően.
5.3.2. Bórsav
A gyűjtőlombik tartalmát a normál sósavmérőoldattal (3.19. pont) vagy normál kénsavmérőoldattal (3.6. pont) titráljuk büretta segítségével, és olvassuk le a használt párlat mennyiségét.
Kolorimetriás végpont-meghatározás alkalmazásakor a végpont elérése akkor történik, amikor a rózsaszín első jele megjelenik a tartalomban. Becsüljük meg a büretta eredményét 0,05 ml pontossággal. Egy megvilágított mágneses keverőlemez vagy fotometrikus detektor segíthet a végpont megjelenítésében.
Ez történhet automatikusan automata titrálóval felszerelt gőzlepárló használatával.
Kövessük a gyártó utasításait az adott lepárló vagy lepárló/titráló működtetésekor.
Megjegyzés: Automata titrálórendszer használatakor a titrálás a lepárlás kezdetekor rögtön megkezdődik és 1 %-os bórsavas oldatot (3.18. pont) használunk.
Teljesen automatikus lepárlórendszer használatakor az ammónia automatikus titrálása potenciometrikus pH-rendszer alkalmazásával végpont-meghatározással is elvégezhető.
Ebben az esetben pH-mérővel felszerelt automata titrálót használunk. A pH-mérőt a szokásos laboratóriumi pH-kalibrálási eljárásokat követve megfelelően kalibrálni kell a 4-7 közötti pH tartományra.
A titrálás pH-végpontjának elérése 4,6-os pH értéknél következik be, ami a titrációs görbe legmeredekebb pontja (inflexiós pont).
5.4. Vakpróba
A reagensek nitrogénmentességének igazolására végezzünk vakpróbát (roncsolás, lepárlás és titrálás) a minta helyett 1 g szacharózt (3.14. pont) használva.
6. Az eredmények kiszámítása
A számításokat a 6.1. vagy a 6.2. pont szerint végezzük.
6.1. Számítás az 5.3.1. pont szerinti titrálás esetén
A tömegszázalékban kifejezett nyersfehérje-tartalom kiszámítása a következő képlettel történik:
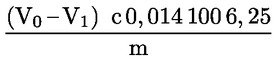
Ahol:
V0 = a vakpróbában használt NaOH (3.10. vagy 3.11. pont) térfogata (ml).
V1 = a minta titrálásához használt NaOH (3.10. vagy 3.11. pont) térfogata (ml).
c = a nátrium-hidroxid (3.10. vagy 3.11. pont) koncentrációja (mol/l).
m = a minta tömege (g).
6.2. Számítás az 5.3.2. pont szerinti titrálás esetén
6.2.1. Sósavas titrálás
A tömegszázalékban kifejezett nyersfehérje-tartalom kiszámítása a következő képlettel történik:
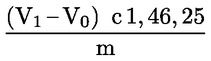
ahol:
m = a vizsgált adag g-ban kifejezett tömege;
c = a normál sósav-mérőoldat (3.19. pont) koncentrációja (mol/l);
V0 = a vakpróbában használt sósav térfogata (ml);
V1 = a vizsgált adaghoz használt sósav térfogata (ml).
6.2.2. Kénsavas titrálás
A tömegszázalékban kifejezett nyersfehérje-tartalom kiszámítása a következő képlettel történik:
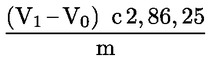
ahol:
m = a vizsgált adag g-ban kifejezett tömege;
c = a normál kénsav-mérőoldat (3.6. pont) koncentrációja (mol/l)
V0 = a vakpróbában használt kénsav (3.6. pont) térfogata (ml)
V1 = a vizsgált adaghoz használt kénsav (3.6. pont) térfogata (ml)
7. A módszer validálása
7.1. Ismételhetőség
Az ugyanazon mintán végzett két párhuzamos meghatározás eredménye közötti különbség nem haladhatja meg:
- a 0,4 %-ot abszolút értékben, 20 %-nál alacsonyabb nyersfehérje-tartalom esetében;
- a magasabb értékhez viszonyított 2,0 %-ot, 20 % és 40 % közötti nyersfehérje-tartalom esetében;
- 40 %-nál magasabb nyersfehérje-tartalom esetén a 0,8 %-ot abszolút értékben.
7.2. Reprodukálhatóság
Az ugyanazon mintán különböző laboratóriumokban végzett két meghatározás eredménye közötti különbség nem haladhatja meg:
- az 1,8 %-ot abszolút értékben, 20 %-nál alacsonyabb nyersfehérje-tartalom esetében;
- a magasabb értékhez viszonyított 9,0 %-ot, 20 % és 40 % közötti nyersfehérje-tartalom esetében;
- 40 %-nál magasabb nyersfehérje-tartalom esetén a 3,6 %-ot abszolút értékben.
7.3. Pontosság
Végezzük el az analízist (roncsolás, lepárlás és titrálás) megfelelő mennyiségű acetaniliden (3.13. pont) (pl. 0,2-0,3 g) 1 g szacharóz (3.14. pont) jelenlétében; 1 g acetanilid 14,80 ml kénsavat (3.5. pont) fogyaszt. A visszanyerés mértékének legalább 99 %-osnak kell lennie.
8. Észrevételek
8.1. A készülék lehet kézi, félautomata vagy automata típusú. Ha az alkalmazott készülék esetében a roncsolási és a lepárlási lépések között mintaátvitelre van szükség, ezt anyagveszteség nélkül kell végrehajtani. Ha a lepárlókészülék lombikja nincs felszerelve csepegtetőtölcsérrel, a nátrium-hidroxidot közvetlenül a lombik és a hűtő összekapcsolása előtt kell hozzáadni, a folyadékot lassan az edény belső oldala mentén folyatva.
8.2. Ha a roncsolt anyag megszilárdul, az 5.1. pontban megadottnál nagyobb mennyiségű kénsavval (3.4. pont) kezdjük újra a meghatározást.
8.3. Alacsony nitrogéntartalmú termékek esetén a gyűjtőlombikba mérendő kénsav (3.7. pont) térfogata szükség esetén 10 vagy 15 ml-re csökkenthető, és vízzel 25 ml-re feltölthető.
8.4. Rutinanalízishez más analitikai módszerek is használhatóak a nyersfehérje meghatározására, de a C. részben leírt Kjeldahl-módszer a referencia-módszer. Minden mátrix esetében egyedileg kell igazolni, hogy az alternatív (pl. DUMAS) módszerrel kapott eredmények egyenértékűek a referencia-módszerrel kapott eredményekkel. Mivel az alternatív módszerrel kapott eredmények - még az egyenértékűség igazolását követően is - eltérhetnek kissé a referencia-módszerel kapott eredményektől, az analitikai jelentésben meg kell említeni a nyersfehérje meghatározására használt analitikai módszert.
D. A KARBAMID MEGHATÁROZÁSA
1. Cél és alkalmazási terület
Ez a módszer lehetővé teszi a kérődzők takarmányában takarmány-adalékanyagként használt karbamid szintjének meghatározását.
2. Vizsgálati alapelv
A mintát vízben szuszpendáljuk egy derítőszerrel. A szuszpenziót leszűrjük. A szűrlet karbamidtartalmát, 4-dimetil-amino-benzaldehid (4-DMAB) hozzáadása után, az optikai sűrűség 420 nm-es hullámhosszon való megmérésével határozzuk meg.
3. Reagensek
3.1. 4-dimetil-amino-benzaldehid-oldat: oldjunk fel 1,6 g 4-DMAB -t 100 ml 96 %-os etanolban, és adjunk hozzá 10 ml sósavat (ρ20 1,19 g/ml). A reagens legfeljebb kéthetes időtartamon keresztül áll el.
3.2. Carrez I. oldat: vízben oldjunk fel 21,9 g cink-acetátot, Zn(CH3COO)2 2H2O és 3 g jégecetet. Töltsük fel vízzel 100 ml-re.
3.3. Carrez II. oldat: vízben oldjunk fel 10,6 g kálium-[hexaciano-ferrát(II)]-ot, K4 Fe (CN)6 3H2O. Töltsük fel vízzel 100 ml-re.
3.4. Aktív szén, amely nem abszorbeálja a karbamidot (ezt ellenőrizni kell).
3.5. Karbamid, 0,1 %-os oldat (w/v).
4. Eszközök
4.1. Billenődobos keverőgép: kb. 35-40 fordulatszám/perc teljesítménnyel.
4.2. Kémcsövek: 160 × 16 mm, csiszolt üvegdugóval.
4.3. Spektrofotométer.
5. A vizsgálat módja
5.1. A minta elemzése
Mérjünk ki a mintából 2 g-ot mg-nyi pontossággal, és 1 g aktív szénnel (3.4. pont) együtt helyezzük egy 500 ml-es mérőlombikba. Adjunk hozzá 400 ml vizet és 5 ml Carrez I. oldatot (3.2. pont), keverjük kb. 30 másodpercig, majd adjunk hozzá 5 ml Carrez II. oldatot (3.3. pont). Keverjük a forgó dobon harminc percig. Töltsük térfogatra vízzel, rázzuk fel, és szűrjük le.
Vegyünk el 5 ml-t az áttetsző, színtelen szűrletekből, öntsük csiszolt üvegdugóval ellátott kémcsövekbe, adjunk hozzájuk 5 ml 4-DMAB oldatot (3.1. pont), és keverjük fel. Helyezzük a kémcsöveket vízfürdőbe 20 °C-on (+/- 4 °C). Tizenöt perc elteltével a spektrofotométer segítségével, 420 nm-en mérjük meg a mintaoldat optikai sűrűségét. Ezt hasonlítsuk össze a vakpróba során a reagensekből készített oldattal.
5.2. Kalibrációs görbe
Vegyünk el a karbamid oldatból (3.5. pont) 1, 2, 4, 5 és 10 ml-t, öntsük 100 ml-es mérőlombikokba, és töltsük fel őket térfogatra vízzel. Vegyünk el 5 ml-t minden egyes oldatból, és mindegyikhez adjunk 5 ml 4-DMAB oldatot (3.1. pont), homogenizáljuk, és a fentiek szerint mérjük meg az optikai sűrűséget összehasonlítva az 5 ml 4-DMAB oldatot és 5 ml vizet tartalmazó karbamidmentes kontrolloldattal. Szerkesszük meg a kalibrációs görbét.
6. Az eredmények kiszámítása
A kalibrációs görbe segítségével határozzuk meg a mintában lévő karbamidmennyiséget.
Az eredményt mg karbamid/kg mintában adjuk meg.
7. A módszer értékelése
7.1. Ismételhetőség
Az ugyanazon mintán, ugyanazon laboratóriumban, ugyanazon elemző által párhuzamosan végzett két meghatározás eredménye közötti különbség nem haladhatja meg:
- 420 nm-en
- a legalább 3 000 mg/kg-ot elérő, de az 5 000 mg/kg-ot el nem érő karbamidtartalom esetén a magasabb érték 50 %-át,
- a legalább 5 000 mg/kg-ot elérő, de a 7 000 mg/kg-ot el nem érő karbamidtartalom esetén a magasabb érték 25 %-át,
- a legalább 7 000 mg/kg-ot elérő karbamidtartalom esetén a magasabb érték 20 %-át,
- 435 nm-en
- a legalább 3 000 mg/kg-ot elérő, de az 5 000 mg/kg-ot el nem érő karbamidtartalom esetén a magasabb érték 40 %-át,
- a legalább 5 000 mg/kg-ot elérő, de a 9 000 mg/kg-ot el nem érő karbamidtartalom esetén a magasabb érték 25 %-át,
- a legalább 9 000 mg/kg-ot elérő karbamidtartalom esetén a magasabb érték 5 %-át.
7.2. Reprodukálhatóság
Az ugyanazon mintán különböző laboratóriumokban és/vagy különböző elemzők által végzett két meghatározás eredménye közötti különbség nem haladhatja meg:
- 420 nm-en
- a legalább 3 000 mg/kg-ot elérő, de a 12 000 mg/kg-ot el nem érő karbamidtartalom esetén a 3 000 mg/kg-ot,
- a legalább 12 000 mg/kg-ot elérő karbamidtartalom esetén a 4 500 mg/kg-ot,
- 435 nm-en
- a legalább 3 000 mg/kg-ot elérő, de a 8 000 mg/kg-ot el nem érő karbamidtartalom esetén a magasabb érték 50 %-át,
- a legalább 8 000 mg/kg-ot elérő karbamidtartalom esetén a magasabb érték 25 %-át.
8. A körvizsgálat eredményei
Uniós laboratóriumok közötti összehasonlításra került sor, amelyben 18 laboratórium vett részt. Öt pozitív, kérődzők takarmányozására szánt takarmánykeverékből vett mintát (amelyek az 1. és a 2. táblázatban ANYAG néven szerepelnek) vizsgáltak meg (1 elemzéssel), mint rejtett azonosságú mintapárt, míg egy kérődzők takarmányozására szánt takarmánykeverék vakmintát csak egyszer elemeztek.
A nemzetközi iránymutatásokban meghatározott megismételhetőségi (r) és reprodukálhatósági (R) határértékekre vonatkozó számításokat a kiugró értékek eltávolítása után végezték el az érvényes értékek varianciaelemzésével.
A módszer teljesítményjellemzőinek kiszámított adatait (ismételhetőség, reprodukálhatóság) a következő táblázatok mutatják be. Egyetlen vizsgált minta - beleértve a vakanyagot is - sem adott hamis pozitív eredményt vagy hamis negatív eredményt.
1. táblázat
A módszer teljesítményjellemzői a karbamidra vonatkozó, λ = 420 nm-en minden anyagban elvégzett mérésnél
| 2. ANYAG | 5. ANYAG | 3. ANYAG | 4. ANYAG | 6. ANYAG | |
| Juh | Szarvasmarha | Juh | Juh | Szarvasmarha | |
| Tömeghányad célértéke (mg kg-1) | 3 000 | 5 000 | 7 001 | 9 036 | 11 000 |
| Átlagos tömeghányad (mg kg-1) | 4 241 | 6 993 | 7 830 | 9 962 | 12 071 |
| A reprodukálhatóság szórása, sR (mg kg-1) | 1 141 | 1 303 | 985 | 994 | 1 711 |
| Megismételhetőségi szórás, sR (mg kg-1) | 723 | 601 | 549 | 712 | 737 |
| A reprodukálhatóság relatív szórása, RSDR (%) | 27 | 19 | 13 | 10 | 14 |
| Megismételhetőségi relatív szórás, RSDr (%) | 17 | 9 | 7 | 7 | 6 |
| Reprodukálhatósági határ, R [R = 2,8 × sR ] | 3 195 | 3 649 | 2 759 | 2 784 | 4 790 |
| Ismételhetőségi határ, r [r = 2,8 × sr ] | 2 024 | 1 684 | 1 536 | 1 994 | 2 064 |
2. táblázat
A módszer teljesítményjellemzői a karbamidra vonatkozó, λ = 435 nm-en minden anyagban elvégzett mérésnél
| 2. ANYAG | 5. ANYAG | 3. ANYAG | 4. ANYAG | 6. ANYAG | |
| Juh | Szarvasmarha | Juh | Juh | Szarvasmarha | |
| Tömeghányad célértéke (mg kg-1) | 3 000 | 5 000 | 7 001 | 9 036 | 11 000 |
| Átlagos tömeghányad (mg kg-1) | 4 101 | 6 467 | 7 890 | 10 062 | 11 642 |
| A reprodukálhatóság szórása, sR (mg kg-1) | 706 | 1 194 | 675 | 745 | 1 378 |
| Megismételhetőségi szórás, sR (mg kg-1) | 570 | 628 | 613 | 196 | 167 |
| A reprodukálhatóság relatív szórása, RSDR (%) | 17 | 18 | 9 | 7 | 12 |
| Megismételhetőségi relatív szórás, RSDr (%) | 14 | 10 | 8 | 2 | 1 |
| Reprodukálhatósági határ, R [R = 2,8 × sR ] | 1 977 | 3 344 | 1 889 | 2 087 | 3 859 |
| Ismételhetőségi határ, r [r = 2,8 × sr ] | 1 596 | 1 759 | 1 715 | 549 | 467 |
9. Észrevételek
9.1. 3 %-ot meghaladó karbamidtartalom esetén, csökkentsük a minta nagyságát 1 g-ra vagy hígítsuk az eredeti oldatot úgy, hogy 500 ml-ben ne legyen több 50 mg karbamidnál.
9.2. Alacsony karbamidtartalom esetén növeljük a minta nagyságát mindaddig, amíg a szűrlet áttetsző és színtelen marad.
9.3. A kollaboratív vizsgálatok fenti eredményei nem mutatnak jelentős precizitásbeli különbséget a 420 nm-en vagy 435 nm-en mért karbamid között.
E. AZ AMINOSAVAK MEGHATÁROZÁSA (A TRIPTOFÁN KIVÉTELÉVEL)
Az aminosavak meghatározására (a triptofán kivételével) használt analitikai módszerek a következők:
- EN ISO 13903 Takarmányok. Az aminosav-tartalom meghatározása
- EN ISO 17180 Takarmányok. A lizin, a metionin és a treonin meghatározása kereskedelmi aminosav-tartalmú termékekben és előkeverékekben ( 15 )
- az alábbi 1-10. pontban leírt elemzési módszer.
1. Cél és alkalmazási terület
Ez a módszer az egyes aminosavakból 10 %-nál kevesebb tartalmazó takarmány-alapanyagok, takarmánykeverékek és előkeverékek ( 16 ) szabad (szintetikus és természetes) és összes (peptidhez kötött és szabad) aminosav-tartalmának meghatározását teszi lehetővé, aminosav-analizátort használatával. A módszer a következő aminosavakra alkalmazható: cisztein, metionin, lizin, treonin, alanin, arginin, aszparaginsav, glutaminsav, glicin, hisztidin, izoleucin, leucin, fenilalanin, prolin, szerin, tirozin és valin.
A módszer nem tesz különbséget az aminosavak különböző sói között, és nem különbözteti meg az aminosavak D és L formáit. A módszer nem érvényes triptofán vagy az aminosavak hidroxi analógjainak meghatározására.
2. Vizsgálati alapelv
2.1. Szabad aminosavak
A szabad aminosavakat híg sósavval extraháljuk. Az együtt extrahált nitrogéntartalmú makromolekulákat szulfoszalicilsavval csapatjuk ki, és szűréssel távolítjuk el. A szűrt oldat kémhatását pH = 2,20 értékre állítjuk be. Az aminosavakat ioncserés kromatográfiával választjuk szét, és ninhidrines reakcióval, 570 nm-en végzett fotometriás detektálással határozzuk meg.
2.2. Összes aminosav
A választott eljárás függ a vizsgált aminosavaktól. A ciszteint és a metionint a hidrolízis előtt ciszteinsavvá, illetve metionin-szulfonná kell oxidálni. A tirozint az oxidálatlan minták hidrolizátumaiból kell meghatározni. Az 1. pontban (Cél és alkalmazási terület) felsorolt összes többi aminosav akár oxidált, akár oxidálatlan mintából meghatározható.
Az oxidálást 0 °C-on végezzük perhangyasav/fenol keverékével. Az oxidálószer-felesleget nátrium-diszulfittal bontjuk le. Az oxidált vagy oxidálatlan mintát sósavval (3.20. pont) hidrolizáljuk 23 órán keresztül. A hidrolizátum kémhatását pH = 2,20 értékre állítjuk be. Az aminosavakat ioncserés kromatográfiával választjuk szét, és ninhidrines reakcióval, 570 nm-en (prolin esetében 440 nm-en) végzett fotometriás detektálással határozzuk meg.
3. Reagensek
Kétszer desztillált vizet vagy ezzel egyenértékű minőségű vizet kell használni (vezetőképessége < 10 μS).
3.1. Hidrogén-peroxid, w (w/w) = 30 %.
3.2. Hangyasav, w (w/w) = 98-100 %.
3.3. Fenol.
3.4. Nátrium-diszulfit.
3.5. Nátrium-hidroxid.
3.6. 5-szulfo-szalicilsav-dihidrát.
3.7. Sósav, sűrűsége megközelítőleg 1,18 g/ml.
3.8. Tri-nátrium-citrát-dihidrát.
3.9. 2,2'-Tiodietanol (tiodiglikol).
3.10. Nátrium-klorid
3.11. Ninhidrin.
3.12. Petroléter, forrásponttartomány 40-60 °C.
3.13. Norleucin vagy más, belső standardként történő használatra alkalmas vegyület.
3.14. Nitrogéngáz (10 ppm-nél kevesebb oxigénnel).
3.15. 1-oktanol.
3.16. Aminosavak.
3.16.1. Az 1. pontban (Cél és alkalmazási terület) felsorolt aminosavak standard anyagai. Tiszta vegyületek kristályvíztartalom nélkül. Használat előtt egy hétig szárítsuk vákuumban P2O5 vagy H2SO4 felett.
3.16.2. Ciszteinsav.
3.16.3. Metionin-szulfon.
3.17. Nátrium-hidroxid-oldat, c = 7,5 mol/l:
Oldjunk fel 300 g NaOH-t (3.5. pont) vízben, és vízzel töltsük fel 1 l-re.
3.18. Nátrium-hidroxid-oldat, c = 1 mol/l:
Oldjunk fel 40 g NaOH-t (3.5. pont) vízben, és vízzel töltsük fel 1 l-re.
3.19. Hangyasav-fenol oldat:
Elegyítsünk 889 g hangyasavat (3.2. pont) 111 g vízzel és adjunk hozzá 4,73 g fenolt (3.3. pont).
3.20. Hidrolízis keverék, c = 6 mol HCl/l 1 g fenol/l tartalommal:
Adjunk egy g fenolt (3.3. pont) 492 ml HCl-hez (3.7. pont), és vízzel töltsük fel 1 l-re.
3.21. Extraháló keverék, c = 0,1 mol HCl/l 2 %-os tiodiglikol-tartalommal: Vegyünk 8,2 ml HCl-t (3.7. pont), hígítsuk megközelítőleg 900 ml vízzel, adjunk hozzá 20 ml tiodiglikolt (3.9. pont), és vízzel töltsük fel 1 l-re (ne keverjük össze közvetlenül a 3.7. és a 3.9. pontot).
3.22. 5-szulfo-szalicilsav ß = 6 %:
Oldjunk fel 60 g 5-szulfo-szalicilsavat (3.6. pont) vízben, és vízzel töltsük fel 1 l-re.
3.23. Oxidáló keverék (perhangyasav-fenol):
Elegyítsünk 0,5 ml hidrogén peroxidot (3.1. pont) 4,5 ml hangyasav-fenol oldattal (3.19. pont) egy kis főzőpohárban. Inkubáljuk 20-30 °C-on egy órán át a perhangyasav keletkezéséhez, majd hűtsük le jeges vízfürdőben (15 percig), mielőtt a mintához adnánk.
Figyelem: Kerüljük a bőrrel való érintkezést, és viseljünk védőruházatot.
3.24. Citrátpuffer, c = 0,2 mol Na+/l, pH 2,20:
Oldjunk fel 19,61 g nátrium-citrátot (3.8. pont), 5 ml tiodiglikolt (3.9. pont), 1 g fenolt (3.3. pont) és 16,50 ml HCl-t (3.7. pont) megközelítőleg 800 ml vízben. Állítsuk be a pH-t 2,20-re. Töltsük fel vízzel 1 literre.
3.25. Eluáló pufferek, a használt analizátor feltételei szerint készítve (4.9. pont).
3.26. Ninhidrin reagens, a használt analizátor feltételei szerint készítve (4.9. pont).
3.27. Aminosavak standardoldata. Ezeket az oldatokat 5 °C alatt kell tárolni.
3.27.1. Aminosavak standard törzsoldata (3.16.1. pont).
c = 2,5 μmol/l, mindegyik sósavban.
Kereskedelmi forgalomban kapható.
3.27.2. Ciszteinsav és metionin-szulfon standard törzsoldata, c = 1,25 μmol/ml.
1 literes mérőlombikban citrátpufferban (3.24. pont) oldjunk fel 0,2115 g ciszteinsavat (3.16.2. pont) és 0,2265 g metionin szulfont (3.16.3. pont), majd az oldatot töltsük fel jelig citrátpufferrel. Legfeljebb 12 hónapig tároljuk 5 °C alatt. Nem használjuk ezt az oldatot, ha a standard törzsoldat (3.27.1. pont) tartalmaz ciszteinsavat és metionin szulfont.
3.27.3. Belső standard pl. norleucin standard törzsoldata, c = 20 μmol/ml.
Mérőlombikban oldjunk fel 0,6560 g norleucint (3.13. pont) citrátpufferben (3.24. pont), majd az oldatot töltsük fel 250 ml-re citrátpufferrel. Legfeljebb 6 hónapig tároljuk 5 °C alatt.
3.27.4. Standard aminosavak kalibrálóoldata hidrolizátumokkal való használatra, c = 5 nmol/50 μl ciszteinsav és metionin szulfon és c = 10 nmol/50 μl más aminosav. Egy 100 ml-es főzőpohárban oldjunk fel 30 ml citrátpufferben (3.24. pont) 2,2 g nátrium-kloridot (3.10. pont). Adjunk hozzá 4,00 ml standard aminosav-törzsoldatot (3.27.1. pont), 4,00 ml ciszteinsav és metionin szulfon standard törzsoldatot (3.27.2. pont) és 0,50 ml-t a belső standard (3.27.3. pont) standard törzsoldatából, ha ilyet használunk. A pH-t állítsuk 2,20-re nátrium-hidroxiddal (3.18. pont).
Öntsük át a teljes mennyiséget egy 50 ml-es mérőlombikba, töltsük fel jelig citrátpufferrel (3.24. pont), és keverjük össze.
Legfeljebb 3 hónapig tároljuk 5 °C alatt.
Lásd még az Észrevételek 9.1. pontját.
3.27.5. Standard aminosavak kalibrálóoldata hidrolizátumokkal való használatra, az 5.3.3.1. pont szerint készítve és a kivonatokkal való használatra (5.2. pont). A kalibrálóoldat a 3.27.4. pont szerint készül, de elhagyjuk a nátrium-kloridot.
Legfeljebb 3 hónapig tároljuk 5 °C alatt.
4. Eszközök
4.1. 100 vagy 250 ml-es visszafolyós hűtővel ellátott gömblombik.
4.2. 100 ml-es, csavaros tetejű, gumi/teflon béléses bórszilikát üveglombik (pl. Duran, Schott) szárítószekrényben történő használatra.
4.3. Intenzív szellőztetésű és ± 2 °C-nál nagyobb pontosságú hőmérséklet-szabályozóval ellátott szárítószekrény.
4.4. pH-mérő (három tizedesjegyig)
4.5. Membránszűrő (0,22 μm).
4.6. Centrifuga.
4.7. Rotációs vákuumbepárló.
4.8. Mechanikus rázógép vagy mágneses keverő
4.9. Aminosav-analizátor vagy HPLC-berendezés ioncserélő oszloppal, készülék ninhidrinhez, utókolonnás derivatizáláshoz és fotometriás detektor.
A kolonna szulfonált polisztirol gyantával van töltve, amely képes az aminosavak egymástól és más ninhidrin-pozitív anyagoktól való elválasztására. A puffer és ninhidrin vonal áramlását ± 0,5 %-os áramlási stabilitású szivattyúk biztosítják a standard kalibrálás és a minta analízisének időszakában.
Néhány aminosav-analizátornál olyan hidrolízises eljárások is alkalmazhatók, amelyekben a hidrolizátum nátriumkoncentrációja k = 0,8 mol/l, és amelyek tartalmazzák az oxidációs lépésből származó valamennyi hangyasavmaradékot. Más analizátorok nem biztosítanak megfelelő elválasztást bizonyos aminosavak esetében, ha a hidrolizátum feleslegben tartalmaz hangyasavat, és/vagy magas benne a nátrium-ion koncentráció. Ebben az esetben a sav térfogatát a hidrolízis után és a pH-beállítás előtt mintegy 5 ml-re való bepárlással csökkentjük. A bepárlást vákuum alatt és maximum 40 °C-on kell végezni.
5. A vizsgálat módja
5.1. A minta előkészítése
Őröljük meg a mintát annyira, hogy az átjusson egy 0,5 mm-es szemméretű szitán. Őrlés előtt a nagy nedvességtartalmú mintákat vagy légszáraz állapotba kell hozni 50 °C-nál nem magasabb hőmérsékleten, vagy liofilizálni kell azokat. A nagy zsírtartalmú mintákat őrlés előtt petroléterrel (3.12. pont) extrahálni kell.
5.2. A szabad aminosavak meghatározása
Az előkészített mintából (5.1. pont) megfelelő mennyiséget (1-5 g) mérjünk be 0,2 mg pontossággal egy Erlenmeyer-lombikba, és adjunk hozzá 100,0 ml extraháló keveréket (3.21. pont). Rázassuk a keveréket 60 percig mechanikus rázógép vagy mágneses keverő segítségével (4.8. pont). Hagyjuk leülepedni az üledéket, és a felülúszó oldatból pipettázzunk 10,0 ml-t egy 100 ml-es főzőpohárba.
Keverés közben adjunk hozzá 5,0 ml szulfoszalicilsavat (3.22. pont), és folytassuk a keverést mágneses keverő segítségével 5 percig. A felülúszó folyadékot szűrjük le, vagy centrifugáljuk a csapadék leválasztása céljából. A kapott oldat 10,0 ml-ét tegyük egy 100 ml-es főzőpohárba, és állítsuk be a pH-t 2,20-ra nátrium-hidroxid-oldattal (3.18. pont), majd töltsük át egy megfelelő térfogatú mérőlombikba citrát-puffer segítségével (3.24. pont), és a lombikot töltsük fel jelig a pufferoldattal (3.24. pont).
Ha belső standardot használunk, adjunk 1,00 ml belső standardot (3.27.3. pont) minden 100 ml végső oldathoz, és a lombikot töltsük fel jelig a pufferoldattal (3.24. pont).
Folytassuk a műveletet a kromatográfiával az 5.4. pont szerint.
Ha a kivonatok vizsgálatára nem ugyanazon a napon kerül sor, akkor azokat 5 °C alatt kell tárolni.
5.3. Az összes aminosav meghatározása
5.3.1. Oxidáció
0,2 mg pontossággal mérjünk ki 0,1-1 g-ot az előkészített mintából (5.1. pont):
- egy 100 ml-es gömblombikba (4.1. pont) nyitott hidrolízishez (5.3.2.3. pont), vagy
- egy 250 ml-es gömblombikba (4.1. pont), ha alacsony nátriumkoncentrációra van szükség (5.3.3.1. pont), vagy
- egy csavaros tetejű 100 ml-es palackba (4.2. pont), (zárt hidrolízishez 5.3.2.4. pont).
A kimért mintamennyiségnek hozzávetőlegesen 10 mg nitrogéntartalommal és legfeljebb 100 mg nedvességtartalommal kell rendelkeznie.
Helyezzük a lombikot/palackot jeges vízfürdőbe, és hűtsük le 0 °C-ra, adjunk hozzá 5 ml oxidációs keveréket (3.23. pont), és keverjük össze egy hajlított végű üvegspatulával. Zárjuk le légmentesen a spatulát is magában foglaló lombikot/palackot, tegyük a lezárt tartályt tartalmazó jeges vízfürdőt 0 °C-os hűtőszekrénybe, és hagyjuk állni 16 órán keresztül. 16 óra múlva vegyük ki a hűtőszekrényből, és a feleslegben lévő oxidáló reagenst 0,84 g nátrium-diszulfit (3.4. pont) adagolásával bontsuk le.
Folytassuk a műveletet az 5.3.2.1. pont szerint.
5.3.2. Hidrolízis
5.3.2.1. Az oxidált minták hidrolízise
Az 5.3.1. pont szerint elkészített oxidált mintához adjunk 25 ml hidrolíziskeveréket (3.20. pont), ügyelve arra, hogy lemossuk az edény falára és a spatulára tapadt mintamaradékot.
Az alkalmazott hidrolízis-eljárástól függően az 5.3.2.3. vagy az 5.3.2.4. pont szerint folytassuk a műveletet.
5.3.2.2. A nem oxidált minta hidrolízise
Mérjünk be egy 100 ml-es vagy egy 250 ml-es gömblombikba (4.1. pont) vagy egy 100 ml-es csavaros fedelű palackba (4.2. pont) 0,1-1 g-ot az előkészített mintából (5.1. pont) 0,2 mg pontossággal. A mintából bemért adagnak hozzávetőlegesen 10 mg nitrogéntartalommal kell rendelkeznie. Óvatosan adjunk hozzá 25 ml hidrolíziskeveréket (3.20. pont), és keverjük őket össze. Az 5.3.2.3. vagy az 5.3.2.4. pont szerint folytassuk a műveletet.
5.3.2.3. Nyitott hidrolízis
Tegyünk 3 üveggyöngyöt a lombikban lévő keverékbe (ami az 5.3.2.1. vagy az 5.3.2.2. pont szerint készült), és forraljuk folyamatos buborékolás mellett, reflux alatt, 23 órán keresztül. Amikor a hidrolízis befejeződött, mossuk ki a hűtőt 5 ml citrát-pufferrel (3.24. pont). Vegyük le a lombikot, és hűtsük le jeges vízfürdőben.
Az 5.3.3. pont szerint folytassuk a műveletet.
5.3.2.4. Zárt hidrolízis
Tegyük az 5.3.2.1. vagy az 5.3.2.2. pont szerint elkészített keveréket tartalmazó palackot egy 110 °C-os szárítószekrénybe (4.3. pont). A (gázfejlődésnek tulajdonítható) nyomás kialakulásának megelőzése és a robbanás elkerülése érdekében az első órában helyezzük a csavaros fedelet az edény nyílására. Ne zárjuk le az edényt a fedővel. Ne zárjuk le az edényt a fedővel. Egy óra elteltével zárjuk le az edényt a fedővel, és hagyjuk a szárítószekrényben (4.3. pont) 23 órán keresztül. Amikor a hidrolízis befejeződött, vegyük ki az edényt a szárítószekrényből, óvatosan nyissuk fel a palack fedelét, és tegyük a palackot jeges vízfürdőbe. Hagyjuk kihűlni.
A pH beállításához alkalmazott eljárástól függően (5.3.3. pont) a palack tartalmát teljes egészében vigyük át egy 250 ml-es főzőpohárba vagy egy 250 ml-es gömblombikba citrát-puffer segítségével (3.24. pont).
Az 5.3.3. pont szerint folytassuk a műveletet.
5.3.3. A pH beállítása
Az aminosav-analizátor (4.9. pont) nátriummal szembeni tűrőképességétől függően az 5.3.3.1. vagy az 5.3.3.2. pont szerint végezzük el a pH beállítását.
5.3.3.1. Alacsony nátriumkoncentrációt igénylő kromatográfiás rendszerek (4.9. pont) esetén:
Tanácsos belső standard törzsoldatot (3.27.3. pont) használni alacsony nátriumkoncentrációt igénylő aminosav-analizátorok alkalmazása esetén (amikor a sav térfogatát csökkenteni kell).
Ebben az esetben bepárlás előtt adjunk 2,00 ml belső standard törzsoldatot (3.27.3. pont) a hidrolizátumhoz.
Adjunk 2 csepp 1-oktanolt (3.15. pont) az 5.3.2.3. vagy az 5.3.2.4. pont szerint nyert hidrolizátumhoz.
Rotációs bepárló segítségével (4.7. pont) csökkentsük a térfogatot 5-10 ml-re vákuum alatt, 40 °C-on. Ha a térfogat véletlenül 5 ml alá csökken, akkor a hidrolizátumot ki kell önteni, és az analízist újra kell kezdeni.
Nátrium-hidroxid-oldattal (3.18. pont) állítsuk be a pH-t 2,20-re, és az 5.3.4. ponttal folytassuk a műveletet.
5.3.3.2. Minden más típusú aminosav-analizátor (4.9. pont) esetén:
Az 5.3.2.3. vagy az 5.3.2.4. pont szerint kapott hidrolizátumokat részlegesen semlegesítsük úgy, hogy keverés közben óvatosan hozzáadunk 17 ml nátrium-hidroxid-oldatot (3.17. pont), mialatt a hőmérsékletet 40 °C alatt tartjuk.
Szobahőmérsékleten állítsuk be a pH-t 2,20-re a 3.17. pont alatti nátrium-hidroxid-oldattal és végül a 3.18. pont alatti nátrium-hidroxid-oldattal. Folytassuk a műveletet az 5.3.4. pont szerint.
5.3.4. Mintaoldat a kromatográfiához
A beállított pH-jú hidrolizátumot (5.3.3.1. vagy 5.3.3.2. pont) maradék nélkül, citrát-pufferrel (3.24. pont) vigyük át egy 200 ml-es mérőlombikba, és töltsük fel jelig a pufferrel (3.24. pont).
Ha még nem használtunk belső standardot, adjunk hozzá 2,00 ml belső standardot (3.27.3. pont), és töltsük fel jelig citrát-pufferrel (3.24. pont). Keverjük össze alaposan.
Folytassuk a műveletet a kromatográfiával (5.4. pont).
Ha a mintaoldatok vizsgálatára nem ugyanazon a napon kerül sor, 5 °C alatt kell őket tárolni.
5.4. Kromatográfia
Kromatográfiás vizsgálat előtt a kivonatot (5.2. pont) vagy a hidrolizátumot (5.3.4. pont) állítsuk be szobahőmérsékletre. Rázzuk fel a keveréket, és szűrjünk át megfelelő mennyiséget egy 0,22 μm-es membránszűrőn (4.5. pont). A kapott tiszta oldatot vessük alá ioncserés kromatográfiának aminosav-analizátor segítségével (4.9. pont).
Az injektálást lehet kézzel vagy automatikusan végezni. Ügyeljünk arra, hogy - ± 0,5 %-os eltéréssel - azonos mennyiségű oldatot tegyünk az oszlopra a standardok és minták analíziséhez, kivéve, amikor belső standardot használunk, valamint, hogy a nátrium-aminosav arányok, amennyire csak lehet, megegyezzenek a standardban és a mintában.
A kalibrálás gyakorisága általában a ninhidrin reagens és az analitikai rendszer stabilitásától függ. A standardot vagy a mintát citrát-pufferrel (3.24. pont) hígítjuk annak érdekében, hogy a standard csúcsterülete a minta aminosav csúcsterületének 30-200 %-a legyen.
Az aminosavak kromatográfiás vizsgálata az alkalmazott analizátor és a használt gyanta típusától függően némiképp eltérhet. A választott rendszernek képesnek kell lennie arra, hogy az aminosavakat elválassza egymástól és a ninhidrin-pozitív anyagoktól. A működési tartományban a kromatográfiás rendszer reakciójának egyenesen arányosnak kell lennie a kolonnára felvitt aminosavak mennyiségével.
A kromatográfiás lépésnél az alábbiakban említett csúcs-mélyponti arányt alkalmazzuk, amikor (a meghatározandó aminosavak vonatkozásában) ekvimoláris oldatot analizálunk. Az ekvimoláris oldatnak az aminosav-analizátor rendszerrel (4.9. pont). pontosan mérhető aminosavak maximális mennyiségének legalább 30 %-át tartalmaznia kell.
A treonin-szerin szétválasztásánál a kromatogramon a két egymást átfedő aminosav közül a kisebb csúcs-mélyponti aránya ne haladja meg a 2:10 értéket (ha csak cisztein, metionin, treonin és lizin meghatározásáról van szó, a szomszédos csúcsok nem megfelelő elválása hátrányosan befolyásolja a meghatározást). Minden más aminosav esetében a szétválasztásnak jobbnak kell lennie, mint 1:10.
A rendszernek el kell tudnia választani a lizint a "mesterséges lizinképződményektől" és az ornitintől.
6. Az eredmények kiszámítása
Minden aminosavra megmérjük a minta és a standard csúcsterületét, és kiszámítjuk a minta egy kg-jában lévő aminosav g-ban kifejezett mennyiségét (X).
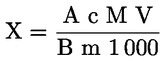
Ha belső standardot használunk, szorozzuk meg -vel.
A = a hidrolizátum vagy a kivonat csúcsterülete
B = a kalibráló standardoldat csúcsterülete
C = a hidrolizátumban vagy a kivonatban lévő belső standard csúcsterülete
D = a belső standard csúcsterülete a kalibráló standard oldatban
M = a meghatározandó aminosav molekulatömege
c = a standard koncentrációja μmol/ml-ben
m = a minta tömege (g) (ha szárított vagy zsírtalanított, akkor az eredeti tömegre korrigálva)
V = ml összes hidrolizátum (5.3.4. pont) vagy ml kivonat (6.1. pont) számított teljes hígítási térfogata
Mind a cisztin, mind a cisztein mint cisztein-sav kerül meghatározásra az oxidált minta hidrolizátumaiban, de cisztinként kerül kiszámításra (C6H12N2O4S2, M 240,30 g/mol), ha M 120,15 g/molt (= 0,5 × 240,30 g/mol) használunk.
A metionin metionin-szulfonként kerül meghatározásra az oxidált minta hidrolizátumaiban, de metioninként kerül kiszámításra a metionin molekulatömegének (M = 149,21 g/mol) használatával.
A hozzáadott szabad metionin extrahálás után metioninként kerül meghatározásra, és a számításhoz ugyanezt a molekulatömeget (M) használjuk.
6.1. A szabad aminosavak (5.2. pont) meghatározása esetén a kivonatok teljes hígítási térfogatát (F) a következőképpen számítjuk ki:
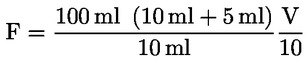
V = a végső kivonat térfogata
7. A módszer értékelése
A módszert nemzetközi összehasonlító vizsgálat során ellenőrizték, amelyet 1990-ben végeztek négy különböző takarmány (kevert sertéstáp, brojlertápkeverék, fehérjekoncentrátum, előkeverék) felhasználásával.
Megjegyzés: A módszert 2003-ban egy második nemzetközi összehasonlító körvizsgálat során tesztelték rejtett azonosságú mintapárok felhasználásával brojlercsirke befejező táp, brojlercsirke indító táp, kukorica-, halliszt- és baromfilisztmintákra. A részleteket lásd az EN ISO 13903 szabványban.
Az 1990-es összehasonlító vizsgálat szélsőértékek kizárásával kapott átlagértékeit és standard szórásait az ebben a pontban található táblázatok mutatják be:
Átlagértékek g/kg-ban
| Referenciaanyag | Aminosav | |||
| Treonin | Ciszt(e)in | Metionin | Lizin | |
| Kevert sertéstáp Takarmány | 6,94 n = 15 | 3,01 n = 17 | 3,27 n = 17 | 9,55 n = 13 |
| Brojlercsirke takarmánykeverék | 9,31 n = 16 | 3,92 n = 18 | 5,08 n = 18 | 13,93 n = 16 |
| Fehérjekoncentrátum | 22,32 n = 16 | 5,06 n = 17 | 12,01 n = 17 | 47,74 n = 15 |
| Előkeverék | 58,42 n = 16 | — | 90,21 n = 16 | 98,03 n = 16 |
| n = a részt vevő laboratóriumok száma | ||||
7.1. Ismételhetőség
Az előző táblázat összehasonlító vizsgálatának megismételhetőségét "laboratóriumon belüli szórásként" kifejezve az alábbi táblázatok mutatják:
Az megismételhetőség variációs együtthatója (%) (CVr)
| Referenciaanyag | Aminosav | |||
| Treonin | Ciszt(e)in | Metionin | Lizin | |
| Kevert sertéstáp Takarmány | 1,9 n = 15 | 3,3 n = 17 | 3,4 n = 17 | 2,8 n = 13 |
| Brojlercsirke takarmánykeverék | 2,1 n = 16 | 2,8 n = 18 | 3,1 n = 18 | 2,1 n = 16 |
| Fehérjekoncentrátum | 2,7 n = 16 | 2,6 n = 17 | 2,2 n = 17 | 2,4 n = 15 |
| Előkeverék | 2,2 n = 16 | — | 2,4 n = 16 | 2,1 n = 16 |
| n = a részt vevő laboratóriumok száma | ||||
7.2. Reprodukálhatóság
A fent említett összehasonlító vizsgálatra vonatkozóan a különböző laboratóriumok közötti szórásértékeket az alábbi táblázat adja meg:
A reprodukálhatóság variációs együtthatója (%) (CVr)
| Referenciaanyag | Aminosav | |||
| Treonin | Ciszt(e)in | Metionin | Lizin | |
| Kevert sertéstáp Takarmány | 4,1 n = 15 | 9,9 n = 17 | 7,0 n = 17 | 3,2 n = 13 |
| Brojlercsirke takarmánykeverék | 5,2 n = 16 | 8,8 n = 18 | 10,9 n = 18 | 5,4 n = 16 |
| Fehérjekoncentrátum | 3,8 n = 16 | 12,3 n = 17 | 13,0 n = 17 | 3,0 n = 15 |
| Előkeverék | 4,3 n = 16 | – | 6,9 n = 16 | 6,7 n = 16 |
| n = a részt vevő laboratóriumok száma | ||||
8. Referenciaanyagok használata
A módszer helyes alkalmazását lehetőség szerint hitelesített referenciaanyagokon végzett ismételt mérésekkel kell ellenőrizni. Hitelesített aminosav-kalibrálóoldattal végzett kalibrálás javasolt.
9. Észrevételek
9.1. Az aminosav-analizátorok közötti különbségek miatt a standard aminosavak (lásd a 3.27.4. és a 3.27.5. pontot) és a hidrolizátumok (lásd az 5.3.4. pontot) kalibrálóoldatainak végső koncentrációit irányadónak kell tekinteni.
A berendezés lineáris reakciójának tartományát valamennyi aminosavra ellenőrizni kell.
A standardoldatot citrát-pufferrel hígítjuk, hogy a csúcsterületek a tartomány közepére essenek.
9.2. Amennyiben nagy teljesítményű folyadékkromatográfiás berendezést használunk a hidrolizátumok elemzéséhez, a gyártó ajánlásainak megfelelően kell a kísérleti körülményeket optimalizálni.
9.3. Ha 1 %-nál több kloridot tartalmazó takarmánykeverékekre vagy előkeverékekre (koncentrátum, ásványi takarmányok, kiegészítő takarmányok) alkalmazzuk a módszert, előfordulhat a metionin alulértékelése, és ezért speciális kezelést kell végezni.
10. Teljesítménykritériumok
A két körvizsgálatból származó (1990-ből lásd a fenti 7. pontban és 2005-ből lásd az EN/ISO 13903 szabványban közölt) eredmények (a tirozin kivételével) összesítése a következő kritériumokat tartalmazza a megismételhetőség és a reprodukálhatóság tekintetében. Az ebből a két laboratóriumközi vizsgálatból származó értékek valószínűleg nem alkalmazhatók a megadottakon kívül más koncentrációtartományokra és mátrixokra.
10.1. Megismételhetőség
Az ugyanazon mintán, ugyanazon laboratóriumban, ugyanazon elemző által párhuzamosan végzett két meghatározás eredménye közötti különbség nem haladhatja meg:
- az összes aminosavra vonatkozóan, glicin, alanin, lizin, prolin, glutaminsav, izoleucin és hisztidin esetén a magasabb érték 6 %-át;
- az összes aminosavra vonatkozóan, treonin, fenil-alanin, metionin, aszparaginsav és leucin esetén a magasabb érték 8 %-át;
- az összes aminosavra vonatkozóan, arginin és valin esetén a magasabb érték 10 %-át;
- a szerin összes aminosavra vonatkozóan a magasabb érték 12 %-át;
- a cisztin összes aminosavra vonatkozóan a magasabb érték 15 %-át.
10.2. Reprodukálhatóság
Az ugyanazon mintán különböző laboratóriumokban és/vagy különböző elemzők által végzett két meghatározás eredménye közötti különbség nem haladhatja meg:
- az összes aminosavra vonatkozóan, glicin, alanin és treonin esetén a magasabb érték 15 %-át;
- az összes aminosavra vonatkozóan, lizin, prolin, fenil-alanin, metionin és aszparaginsav esetén a magasabb érték 20 %-át;
- az összes aminosavra vonatkozóan, glutaminsav és leucin esetén a magasabb érték 22 %-át;
- az arginin összes aminosavra vonatkozóan a magasabb érték 27 %-át;
- az izoleucin összes aminosavra vonatkozóan a magasabb érték 32 %-át;
- az összes aminosavra vonatkozóan valin és szerin esetén a magasabb érték 35 %-át;
- a hisztidin összes aminosavra vonatkozóan a magasabb érték 40 %-át;
- a cisztin összes aminosavra vonatkozóan a magasabb érték 50 %-át.
F. A TRIPTOFÁN MEGHATÁROZÁSA
A triptofán meghatározására használt analitikai módszerek a következők:
- EN ISO 13904 Takarmány. A triptofántartalom meghatározása,
- az alábbi 1-9. pontban leírt elemzési módszer.
1. Cél és alkalmazási terület
A módszer a takarmányokban található összes és a szabad triptofán meghatározására szolgál. Nem különbözteti meg a D- és az L-formákat.
2. Vizsgálati alapelv
Az összes triptofán meghatározásához a mintát alkalikus körülmények között, telített bárium-hidroxid-oldattal hidrolizáljuk, majd 110 °C-ra hevítjük, és ezen a hőmérsékleten tartjuk 20 órán át. Hidrolízis után belső standardot adunk hozzá.
A szabad triptofán meghatározásához a mintát enyhén savas körülmények között extraháljuk belső standard jelenlétében.
A hidrolizátumban vagy a kivonatban található triptofánt és a belső standardot HPLC fluoreszcenciás detektálással határozzuk meg.
3. Reagensek
3.1. Bideszt vagy azzal egyenértékű minőségű vizet kell használni (konduktivitás < 10 μS/cm).
3.2. Standard anyag: vákuumban, foszfor-pentoxid felett szárított triptofán (tisztaság/tartalom ≥ 99 %).
3.3. Belső standard anyag: vákuumban, foszfor-pentoxid felett szárított α-metil-triptofán (tisztaság/tartalom ≥ 99 %).
3.4. Bárium-hidroxid-oktahidrát (BaCO3 képződésének elkerülése érdekében ügyelnünk kell arra, hogy a Ba(OH)2.8 H2O-t ne tegyük ki túlzott mértékben levegő hatásának, mert ez zavarhatja a meghatározást) (lásd az Észrevételek 9.3. pontját).
3.5. Nátrium-hidroxid.
3.6. Ortofoszforsav, w (w/w) = 85 %.
3.7. Sósav, ρ20 1,19 g/ml.
3.8. Metil-alkohol, HPLC-minőségű
3.9. Petroléter, forrásponttartomány 40-60 °C.
3.10. Nátrium-hidroxid-oldat, c = 1 mol/l:
Oldjunk fel 40,0 g NaOH-t (3.5. pont) vízben, és vízzel (3.1. pont) töltsük fel 1 literre.
3.11. Sósav, c = 6 mol/l:
Vegyünk 492 ml HCl-t (3.7. pont), és vízzel töltsük fel 1 literre.
3.12. Sósav, c = 1 mol/l:
Vegyünk 82 ml HCl-t (3.7. pont), és vízzel töltsük fel 1 literre.
3.13. Sósav, c = 0,1 mol/l:
Vegyünk 8,2 ml HCl-t (3.7. pont), és vízzel töltsük fel 1 literre.
3.14. Ortofoszforsav, c = 0,5 mol/l:
Vegyünk 34 ml ortofoszforsavat (3.6. pont), és vízzel (3.1. pont) töltsük fel 1 literre.
3.15. Triptofán (3.2. pont) koncentrált oldata, c = 2,50 μmol/ml:
Egy 500 ml-es mérőlombikban oldjunk fel 0,2553 g triptofánt (3.2. pont) sósavban (3.13. pont), majd az oldatot töltsük fel jelig sósavval (3.13. pont). Tároljuk -18 °C-on maximum négy hétig.
3.16. Koncentrált belső standardoldat, c = 2,50 μmol/ml:
Egy 500 ml-es mérőlombikban oldjunk fel 0,2728 g α-metil-triptofánt (3.3. pont) sósavban (3.13. pont), majd az oldatot töltsük fel jelig sósavval (3.13. pont). Tároljuk -18 °C-on maximum négy hétig.
3.17. A triptofán kalibráló standardoldata és a belső standard:
Vegyünk 2,00 ml-t a triptofán koncentrált oldatából (3.15. pont) és 2,00 ml-t a koncentrált belső standard (α-metil-triptofán) oldatból (3.16. pont). Oldjuk fel vízben (3.1. pont) és metil-alkoholban (3.8. pont) a kész hidrolizátuméval körülbelül azonos térfogatra és körülbelül azonos metil-alkohol koncentrációra (10-30 %).
Ezt az oldatot közvetlenül a felhasználás előtt kell elkészíteni.
Készítés során közvetlen napfényhatástól óvjuk.
3.18. Ecetsav
3.19. 1,1,1-triklór-2-metil-2-propanol.
3.20. Etanolamin w (w/w) > 98 %
3.21. 1 g 1,1,1-triklór-2-metil-2-propanol (3.19. pont) 100 ml metil-alkoholban készített oldata (3.8. pont).
3.22. Mozgófázis a HPLC-hez: 3,00 g ecetsav (3.18. pont) + 900 ml víz (3.1. pont) + az 1,1,1-triklór-2-metil-2-propanol (3.19. pont) metil-alkohollal (3.8. pont) készült oldatának (3.21. pont) (1 g/100 ml) 50,0 ml-e. A pH-t etanolaminnal (3.20. pont) állítsuk 5,00-re. Az oldatot vízzel (3.1. pont) töltsük fel 1 000 ml-re.
4. Eszközök
4.1. HPLC-berendezés spektrofluorometriás detektorral.
4.2. Folyadékkromatográfiás oszlop, 125 mm × 4 mm, C18, 3 μm töltet vagy azzal egyenértékű berendezés.
4.3. pH-mérő.
4.4. Polipropilén lombik, 125 ml térfogatú, széles nyakkal és csavaros zárókupakkal.
4.5. Membránszűrő, 0,45 μm.
4.6. Autokláv, 110 (± 2) °C, 1,4 (± 0,1) bar.
4.7. Mechanikus rázógép vagy mágneses keverő.
4.8. Kémcsőkeverő (vortex).
5. A vizsgálat módja
5.1. A minták elkészítése
Őröljük meg a mintát annyira, hogy az átjusson egy 0,5 mm-es szemméretű szitán. Őrlés előtt a nagy nedvességtartalmú mintákat vagy légszáraz állapotba kell hozni 50 °C-nál nem magasabb hőmérsékleten, vagy liofilizálni kell azokat. A nagy zsírtartalmú mintákat őrlés előtt petroléterrel (3.9. pont) extrahálni kell.
5.2. A szabad triptofán (kivonat) meghatározása
1 mg-os pontossággal mérjünk be az elkészített mintából (5.1. pont) megfelelő mennyiséget (1-5 g) egy Erlenmeyer-lombikba. Adjunk hozzá 100,0 ml sósavat (3.13. pont) és 5,00 ml koncentrált belső standardoldatot (3.16. pont). Rázzuk vagy keverjük 60 percen át mechanikus rázógépen vagy mágneses keverőn (4.7. pont). Engedjük a zagyot leülepedni, majd pipettázzunk a felülúszó oldatból 10,0 ml-t főzőpohárba. Adjunk hozzá 5 ml ortofoszforsavat (3.14. pont). Nátrium-hidroxiddal (3.10. pont) állítsuk a pH-t 3,0-ra. Adjunk hozzá annyi metil-alkoholt (3.8. pont), hogy a végtérfogatra számított metil-alkohol-koncentráció 10 és 30 % között legyen. Vigyük át megfelelő űrtartalmú mérőlombikba, és vízzel hígítsuk a kromatográfiához szükséges térfogatra (körülbelül a kalibráló standardoldat (3.17. pont) térfogatával megegyező térfogatra).
A HPLC oszlopra való felvitel előtt szűrjük át az oldat néhány ml-ét 0,45 μm-es membránszűrőn (4.5. pont). A kromatográfiát az 5.4. pont szerint végezzük.
A standardoldatot és a kivonatokat óvjuk a közvetlen napfényhatástól. Ha a kivonatokat nem lehet a készítés napján analizálni, azok 5 °C-on legfeljebb három napig tárolhatók.
5.3. Az összes triptofán (hidrolizátum) meghatározása
Az elkészített mintából (5.1. pont) mérjünk be 0,2 mg-os pontossággal 0,1-1 g-ot a polipropilén lombikba (4.4. pont). A mintából bemért adagnak hozzávetőlegesen 10 mg nitrogéntartalommal kell rendelkeznie. Adjunk hozzá 8,4 g bárium-hidroxid-oktahidrátot (3.4. pont) és 10 ml vizet. Keverjük össze kémcsőkeverővel (4.8. pont) vagy mágneses keverőn (4.7. pont). A teflon-bevonatú mágnest hagyjuk a keverékben. Mossuk le 4 ml vízzel az edény falát. Helyezzük rá a csavaros zárókupakot, és lazán zárjuk le a lombikot. Helyezzük autoklávba (4.6. pont) forró vízzel, és gőzöljük 30-60 percig. Zárjuk be az autoklávot, és autoklávozzuk 110 (± 2) °C hőmérsékleten 20 órán át.
Az autokláv kinyitása előtt csökkentsük a hőmérsékletet kevéssel 100 °C alá. A Ba(OH)2.8 H2O kikristályosodásának elkerülése érdekében adjunk a meleg keverékhez 30 ml, szobahőmérsékletű vizet. Óvatosan rázzuk vagy keverjük össze. Adjunk hozzá 2,00 ml koncentrált belső standard (α-metil-triptofán) oldatot (3.16. pont). Hűtsük az edényeket víz- vagy jégfürdőben 15 percen át.
Ezután adjunk hozzá 5 ml ortofoszforsavat (3.14. pont). Tartsuk az edényt a hűtőfürdőben, és keverés közben semlegesítsük az oldatot sósavval (3.11. pont), majd állítsuk a pH-t 3,0-ra sósavval (3.12. pont). Adjunk hozzá annyi metil-alkoholt, hogy a végtérfogatra számított metil-alkohol-koncentráció 10 és 30 % között legyen. Vigyük át megfelelő űrtartalmú mérőlombikba, és vízzel hígítsuk a kromatográfiához szükséges térfogatra (például 100 ml-re). Ügyeljünk, hogy a metil-alkohol hozzáadása ne okozzon kicsapódást.
A HPLC oszlopra való felvitel előtt szűrjük át az oldat néhány ml-ét 0,45 μm-es membránszűrőn (4.5. pont). A kromatográfiát az 5.4. pont szerint végezzük.
A standardoldatot és a hidrolizátumokat óvjuk a közvetlen napfényhatástól. Ha a hidrolizátumokat nem lehet a készítés napján analizálni, azok 5 °C-on legfeljebb három napig tárolhatók.
5.4. HPLC-meghatározás
Az izokratikus analízisre vonatkozó alábbi vizsgálati feltételek csak útmutatásként szolgálnak, ettől eltérő feltételeket is lehet használni, feltéve, hogy ezzel egyenértékű eredményt adnak (lásd még az Észrevételek 9.1. és 9.2. pontját):
| Folyadékkromatográfiás oszlop (4.2. pont) | C18, 125 mm × 4 mm, 3 μm-es töltettel, vagy ezzel egyenértékű berendezés. |
| Oszlophőmérséklet: | szobahőmérséklet. |
| Mozgófázis (3.22. pont): | 3,00 g ecetsav (3.18. pont) + 900 ml víz (3.1. pont) + az 1,1,1-triklór-2-metil-2-propanol (3.19. pont) metil-alkohollal (3.8. pont) készült oldatának (3.21. pont) (1 g/100 ml) 50,0 ml-e. A pH-t etanolaminnal (3.20. pont) állítsuk 5,00-re. Töltsük fel vízzel (3.1. pont) 1 000 ml-re. |
| Átáramlási sebesség: | 1 ml/min. |
| Teljes futtatási idő: | körülbelül 34 perc. |
| Kimutatási hullámhossz: | gerjesztés: 280 nm, emisszió: 356 nm. |
| Befecskendezett mennyiség: | 20 μl |
6. Az eredmények kiszámítása
100 g mintában lévő triptofán grammban kifejezett mennyiségét (X) az alábbiak szerint számítjuk.
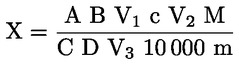
A = a belső standard, standard kalibrálóoldat (3.17. pont) csúcsterülete
B = a triptofán-kivonat (5.2. pont) vagy -hidrolizátum (5.3. pont) csúcsterülete
V1 = a kalibrálóoldathoz (3.17. pont) hozzáadott koncentrált triptofánoldat (3.15. pont) térfogata ml-ben (2 ml)
c = a kalibrálóoldathoz (3.17. pont) hozzáadott koncentrált triptofánoldat (3.15. pont) koncentrációja, μmó/ml-ben (= 2,50)
V2 = az extrahálásnál (5.2. pont) (= 5,00 ml) vagy a hidrolizátumhoz (5.3. pont) (= 2,00 ml) hozzáadott, koncentrált belső standardoldat (3.16. pont) térfogata ml-ben
C = a belső standard, kivonat (5.2. pont) vagy hidrolizátum (5.3. pont) csúcsterülete
D = a triptofán, standard kalibrálóoldat (3.17. pont) csúcsterülete
V3 = a standard kalibrálóoldathoz (3.17. pont) hozzáadott, koncentrált belső standardoldat (3.16. pont) térfogata, ml-ben (= 2,00 ml)
m = a minta tömege, g-ban (szárított és/vagy zsírtalanított minta esetében az eredeti tömegre korrigált)
M = a triptofán molekulatömege (= 204,23 g/mol)
7. Ismételhetőség
Ugyanazon a mintán végzett két párhuzamos meghatározás eredményének különbsége nem haladhatja meg a magasabb érték 10 %-át.
8. A körvizsgálat eredményei
Uniós körvizsgálatra került sor (negyedik összehasonlító vizsgálat), amelynek során három mintát vizsgáltak meg a hidrolízis módszerének hitelesítése céljából, 12 laboratóriumban. Mindegyik mintát többször (5) analizálták. Az eredmények a következő táblázatban szerepelnek.
| 1. minta Sertéstáp | 2. minta L-triptofánnal kiegészített sertéstáp | 3. minta Takarmánykoncentrátum sertések számára | |
| L | 12 | 12 | 12 |
| n Átlag [g/kg] | 50 2,42 | 55 3,40 | 50 4,22 |
| sr [g/kg] | 0,05 | 0,05 | 0,08 |
| r [g/kg] | 0,14 | 0,14 | 0,22 |
| CVr [%] | 1,9 | 1,6 | 1,9 |
| SR [g/kg] | 0,15 | 0,20 | 0,09 |
| R [g/kg] | 0,42 | 0,56 | 0,25 |
| CVR [%] | 6,3 | 6,0 | 2,2 |
| L = az eredményeket beküldő laboratóriumok száma n = a (Cochran- és Dixon-próbával azonosított) kiugró értékek kizárását követően megmaradt egyedi értékek száma sr = az ismételhetőség szórása SR = a reprodukálhatóság szórása r = az ismételhetőség R = a reprodukálhatóság CVr = az ismételhetőség relatív szórása, % CVR = a reprodukálhatóság relatív szórása, % | |||
Egy másik uniós körvizsgálatot is végeztek (harmadik összehasonlító vizsgálat), amelynek során két mintát vizsgáltak meg a szabad triptofán kivonására használt módszer hitelesítése céljából, 13 laboratóriumban. Mindegyik mintát többször (5) analizálták. Az eredmények a következő táblázatban szerepelnek.
| 4. minta Búza és szója keveréke | 5. minta Búza és szója keveréke (= 4. minta), hozzáadott triptofánnal (0,457 g/kg) | |
| L n | 12 55 | 12 60 |
| Átlag [g/kg] | 0,391 | 0,931 |
| sr [g/kg] | 0,005 | 0,012 |
| r [g/kg] | 0,014 | 0,034 |
| CVr [%] | 1,34 | 1,34 |
| SR [g/kg] | 0,018 | 0,048 |
| R [g/kg] | 0,050 | 0,134 |
| CVR [%] | 4,71 | 5,11 |
| L = az eredményeket beküldő laboratóriumok száma n = a (Cochran- és Dixon-próbával azonosított) kiugró értékek kizárását követően megmaradt egyedi értékek száma sr = az ismételhetőség szórása SR = a reprodukálhatóság szórása r = az ismételhetőség R = a reprodukálhatóság CVr = az ismételhetőség relatív szórása, % CVR = a reprodukálhatóság relatív szórása, % | ||
Egy további uniós körvizsgálatot is végeztek, amelynek során négy mintát vizsgáltak meg a triptofán hidrolízisére használt módszer hitelesítése céljából, hét laboratóriumban. Az eredmények az alábbi táblázatban szerepelnek. Mindegyik mintát többször (5) analizálták.
| 1. minta Vegyes sertéstakarmány (CRM 117) | 2. minta Alacsony zsírtartalmú halliszt (CRM 118) | 3. minta Szójaliszt (CRM 119) | 4. minta Fölözött tejpor (CRM 120) | |
| L | 7 | 7 | 7 | 7 |
| n | 25 | 30 | 30 | 30 |
| Átlag [g/kg] | 2,064 | 8,801 | 6,882 | 5,236 |
| sr [g/kg] | 0,021 | 0,101 | 0,089 | 0,040 |
| r [g/kg] | 0,059 | 0,283 | 0,249 | 0,112 |
| CVr [%] | 1,04 | 1,15 | 1,30 | 0,76 |
| SR [g/kg] | 0,031 | 0,413 | 0,283 | 0,221 |
| R [g/kg] | 0,087 | 1,156 | 0,792 | 0,619 |
| CVR [%] | 1,48 | 4,69 | 4,11 | 4,22 |
| L = az eredményeket beküldő laboratóriumok száma n = a (Cochran- és Dixon-próbával azonosított) kiugró értékek kizárását követően megmaradt egyedi értékek száma sr = az ismételhetőség szórása SR = a reprodukálhatóság szórása r = az ismételhetőség R = a reprodukálhatóság CVr = az ismételhetőség relatív szórása, % CVR = a reprodukálhatóság relatív szórása, % | ||||
9. Észrevételek
9.1. Speciális kromatográfiás paraméterek alkalmazása a triptofán és az α-metil-triptofán jobb elválasztását teheti lehetővé.
Izokratikus elúció, majd grádiens oszlopon történő tisztítás:
| Folyadékkromatográfiás oszlop: | C18, 125 mm × 4 mm, 5 μm-es töltettel, vagy ezzel egyenértékű berendezés. | |
| Oszlophőmérséklet: | 32 °C | |
| Mozgófázis: | A: 0,01 mol/l KH2PO4/metil-alkohol, 95 + 5 (V + V). | |
| B: metil-alkohol | ||
| Grádiens program: | 0 perc 100 % A | 0 % B |
| 15 perc 100 % A | 0 % B | |
| 17 perc 60 % A | 40 % B | |
| 19 perc 60 % A | 40 % B | |
| 21 perc 100 % A | 0 % B | |
| 33 perc 100 % A | 0 % B | |
| Átáramlási sebesség: | 1,2 ml/perc | |
| Teljes futtatási idő: | körülbelül 33 perc. | |
9.2. A kromatográfia a HPLC típusától és az oszloptöltetként alkalmazott anyagtól függően változik. A választott rendszernek képesnek kell lennie a triptofán és a belső standard alapvonalon történő elválasztására. Fontos továbbá, hogy a bomlástermékek jól elváljanak a triptofántól és a belső standardtól. A belső standard nélküli hidrolizátumokat is futtatni kell a belső standard alatti alapvonal szennyezettségének ellenőrzése céljából. Fontos, hogy a futtatási idő elég hosszú legyen az összes bomlástermék eluálásához, mert különben a későn eluálódó csúcsok zavarhatják a későbbi kromatográfiás futtatásokat.
A kromatográfiás rendszernek a működési tartományban lineáris választ kell adnia. A lineáris választ a belső standard állandó (a normális) koncentrációjával és a triptofán változó koncentrációival kell mérni. Fontos, hogy mind a triptofán-, mind a belső standard csúcsok nagysága a HPLC/fluoreszcens rendszer lineáris tartományába essen. Ha a triptofán- és/vagy a belső standard csúcs(ok) túl kicsi(k) vagy túl nagy(ok), az analízist meg kell ismételni más mintamérettel és/vagy más végtérfogattal.
9.3. Bárium-hidroxid
Állás hatására a bárium-hidroxid egyre nehezebben oldódik. Ennek következtében a HPLC-meghatározáshoz használt oldat zavaros lesz, ami alacsony triptofánértékeket eredményezhet.
G. A NYERSOLAJOK ÉS A ZSÍROK MEGHATÁROZÁSA
1. Cél és alkalmazási terület
Ez a módszer az állati takarmányokban lévő nyersolajok és zsírok meghatározására szolgál.
A továbbiakban leírt két módszer használata a takarmány jellegétől és összetételétől, valamint az analitikai vizsgálat céljától függ.
Az olajos magvakban és olajtartalmú gyümölcsökben, valamint az olyan takarmányokban található nyersolajok és zsírok meghatározására, amelyekben a nyersolaj-/zsírtartalom meghaladja a 15 %-ot, az extrahálást az A. eljárással, az ismételt extrahálást pedig a B. eljárással (5.3. pont) kell elvégezni.
1.1. A. eljárás - Közvetlenül extrahálható nyersolajok és zsírok
Ez a módszer a növényi eredetű takarmány-alapanyagok esetében alkalmazható, kivéve azokat, amelyek a B. módszer tárgykörébe esnek.
1.2. B. eljárás - Összes nyersolaj és zsír
Ez a módszer az állati eredetű takarmány-alapanyagok és minden takarmánykeverék esetében alkalmazható. Mindazon anyagokra alkalmazandó, amelyekből az olajok és a zsírok teljesen nem extrahálhatók előzetes hidrolízis nélkül (pl. sikér, élesztő, burgonyafehérjék és extrudálás, a pelyhesítés és a melegítés révén készített termékek).
1.3. Az eredmények értelmezése
Mindazon esetekben, amikor magasabb eredményt kapunk a B. eljárással, mint az A. eljárással, a B. eljárással kapott eredményt tekintsük a helyes értéknek.
2. Vizsgálati alapelv
2.1. A. eljárás
A mintát petroléterrel extraháljuk. Az oldószert bepároljuk, a maradékanyagot megszárítjuk, és megmérjük.
2.2. B. eljárás
A mintát melegítés közben sósavval kezeljük. A keveréket lehűtjük, és leszűrjük. A maradékanyagot kimossuk, megszárítjuk és a meghatározást az A. eljárás szerint végezzük el.
3. Reagensek
3.1. Petroléter, forrásponttartomány: 40-60 °C. Brómszáma kisebb legyen 1-nél, és a bepárlási maradékanyaga kisebb legyen, mint 2 mg/100 ml.
3.2. Nátrium-szulfát, vízmentes.
3.3. Sósav, c = 3 mol/l.
3.4. Szűrési segédanyag, pl. Kieselguhr, Hyflo-supercel.
4. Eszközök
4.1. Extrahálókészülék. Amennyiben szifonnal ellátott készülékről van szó (Soxhlet-készülék) a visszafolyás mértéke körülbelül óránként 10 ciklus legyen; ha nem szifonos típust használunk, a visszafolyás mértéke körülbelül percenként 10 ml legyen.
4.2. Extrahálótégely, petroléterben oldható anyagoktól mentes, a 4.1. pont követelményeinek megfelelő porozitással.
4.3. Szárítószekrény, amely lehet 75 ± 3 °C-ra beállított vákuum szárítószekrény, vagy 100 ± 3 °C-ra beállított légkeveréses kemence.
5. A vizsgálat módja
5.1. A. eljárás (lásd a 8.1. pontot)
1 mg pontossággal mérjünk ki 5 g mintát, töltsük át egy extraháló tégelybe (4.2. pont), és fedjük be egy zsírmentes gyapotvattatamponnal.
Tegyük az extraháló tégelyt az extrahálókészülékbe (4.1. pont), és extraháljuk hat órán keresztül petroléterrel (3.1. pont). A petroléter-kivonatot egy horzsakődarabokat tartalmazó, száraz, lemért lombikba gyűjtsükmo ( 17 ).
Pároljuk be az oldószert. A lombikot másfél órán át tartsuk a szárítószekrényben (4.3. pont), hogy a maradékanyag megszáradjon. Hagyjuk kihűlni egy exszikkátorban, és mérjük meg a tömegét. Ismét szárítsuk 30 percig annak biztosítása érdekében, hogy az olajok és zsírok tömege stabilizálódjon (a tömegveszteség a két egymást követő mérés között legfeljebb 1 mg lehet).
5.2. B. eljárás
1 mg pontossággal mérjünk ki 2,5 g mintát (lásd a 8.2. pontot), tegyük egy 400 ml-es főzőpohárba vagy egy 300 ml-es Erlenmeyer-lombikba, adjunk hozzá 100 ml sósavat (3.3. pont) és horzsakődarabokat. Fedjük le a főzőpoharat egy óraüveggel, vagy csatlakoztassunk az Erlenmeyer-lombikhoz egy visszafolyós hűtőt. Lassú lángon vagy főzőlapon enyhén forraljuk fel, és tartsuk így egy órán keresztül. Ne hagyjuk, hogy az anyag odatapadjon az edény falához.
Hűtsük le, és adjunk hozzá annyi szűrési segédanyagot (3.4. pont), amely elég ahhoz, hogy megakadályozza a szűrés folyamán keletkező olaj- vagy zsírveszteséget. Szűrjük át egy nedvesített, zsírmentes, kettős szűrőpapíron. Addig mossuk hideg vízzel a maradékanyagot, amíg semleges szűrletet nem kapunk. Ellenőrizzük, hogy a szűrlet nem tartalmaz olajat vagy zsírokat. Ezek jelenléte azt jelzi, hogy a mintát a hidrolízis előtt petroléterrel extrahálni kell az A. eljárás szerint.
Tegyük a maradékanyagot tartalmazó kettős szűrőpapírt egy óraüvegre, és szárítsuk a légkeveréses szárítószekrényben (4.3. pont) másfél órán keresztül, 100 °C ± 3 °C-on.
Tegyük a száraz maradékanyagot tartalmazó kettős szűrőpapírt egy extrahálótégelybe (4.2. pont), és fedjük le egy zsírmentes üvegvatta tamponnal. Helyezzük az extrahálótégelyt egy extrahálókészülékbe (4.1. pont), és folytassuk a műveletet az 5.1. pont második és harmadik bekezdése szerint.
5.3. A. eljárás és a B. eljárással történő ismételt extrahálás
Az olajos magvakban és olajtartalmú gyümölcsökben, valamint az olyan takarmányokban található nyersolajok és zsírok meghatározására, amelyekben a nyersolaj-/zsírtartalom meghaladja a 15 %-ot, az extrahálást az A. eljárással, az ismételt extrahálást pedig a B. eljárással kell elvégezni.
Ez azt jelenti, hogy a petroléterrel történő extrahálást (A. eljárás) követően a maradékot vagy annak egy részét sósavval újra extraháljuk (B. eljárás). A nyersolaj- és zsírtartalom az A és a B eljárás eredményének összege.
6. Az eredmény kifejezése
A maradékanyag tömegét a minta százalékában fejezzük ki.
7. Ismételhetőség
Ugyanazon a mintán, ugyanazon analitikus által végzett két párhuzamos meghatározás eredménye közötti különbség nem haladhatja meg a:
- az 5 % alatti nyersolaj és zsírtartalmaknál a 0,2 %-ot, abszolút értékben,
- 5-10 % közötti olaj- és zsírtartalom esetében a 4,0 %-ot, a legmagasabb eredményhez viszonyítva,
- a 10 % feletti nyersolaj és zsírtartalmaknál a 0,4 %-ot, abszolút értékben.
8. Észrevételek
8.1. Magas olaj- és zsírtartalmú termékeknél, amelyeket nehéz felaprítani, vagy amelyek nem alkalmasak arra, hogy homogén vizsgálati mintát képezzünk belőlük, a következőképpen járjunk el:
1 mg pontossággal mérjünk ki 20 g mintát, és keverjük össze 10 g vagy annál több vízmentes nátriumszulfáttal (3.2. pont). Extraháljuk petroléterrel (3.1. pont) az 5.1. pont szerint. A kapott kivonatot töltsük fel petroléterrel (3.1. pont) 500 ml-re, és keverjük össze. Helyezzünk 50 ml oldatot egy horzsakődarabokat tartalmazó, száraz, lemért kis lombikba. Pároljuk be az oldószert, szárítsuk, majd az 5.1. pont utolsó bekezdésében leírtak szerint járjunk el.
Távolítsuk el az oldószert az extrahálótégelyben hagyott maradékanyagból, aprítsuk fel a maradékanyagot 1 mm-es finomságúra, tegyük vissza az extrahálótégelybe (ne adjunk hozzá nátriumszulfátot), és az 5.1. pont második és harmadik bekezdésében leírtak szerint folytassuk a műveletet.
Az olaj- és zsírtartalmat a minta százalékában számítsuk ki a következő képlet segítségével:
ahol:
m1 = a maradékanyag tömege grammban az első extrahálás után (a kivonat aliquot része),
m2 = a maradékanyag tömege grammban a második extrahálás után.
8.2. Egyes (például az olajokban és zsírokban szegény) termékek esetében a vizsgálati mintát meg lehet növelni.
8.3. A magas víztartalmú hobbiállat-eledeleket vízmentes nátriumszulfáttal kell keverni a B. eljárás szerinti hidrolízis és extrahálás előtt.
8.4. Az 5.2. pontban hatásosabb lehet forró víz használata hideg víz helyett a maradékanyag szűrés utáni mosásához.
8.5. Az 1,5 órás szárítási időt néhány takarmány esetében meg kell hosszabbítani. El kell kerülni a túlzott szárítást, mivel az alacsony eredményekhez vezethet. Mikrohullámú sütő is használható.
H. A NYERSROST MEGHATÁROZÁSA
1. Cél és alkalmazási terület
Ez a módszer lehetővé teszi a takarmányokban előforduló, hagyományosan nyersrostnak nevezett, savas és lúgos közegben oldhatatlan, zsírmentes szerves anyagok meghatározását.
A módszer nem alkalmazható a lignocellulóz és a növényi szén (túl finom részecskék) esetében.
2. Vizsgálati alapelv
A szükség esetén zsírtalanított mintát többször egymás után meghatározott koncentrációjú kénsav és kálium-hidroxid forrásban lévő oldataival kezeljük. A maradékot zsugorított üvegszűrővel szűréssel elválasztjuk, kimossuk, megszárítjuk, lemérjük és 475-500 °C-on elhamvasztjuk. A hamvasztásból eredő tömegveszteség megegyezik a vizsgálati minta nyersrosttartalmával.
3. Reagensek
3.1. Kénsav, c = 0,13 mol/l.
3.2. Habzásgátló anyag (pl. n-oktanol).
3.3. Szűrési segédanyag (Celite 545 vagy azzal egyenértékű), négy órán át 500 °C-on hevítve (8.6. pont).
3.4. Aceton.
3.5. Petroléter, forráspont-tartomány: 40-60 °C.
3.6. Sósav, c = 0,5 mol/l.
3.7. Kálium-hidroxid oldat, c = 0,23 mol/l.
4. Eszközök
4.1. Kénsavval vagy kálium-hidroxiddal történő roncsolásra szolgáló fűtőegység, amely egy, a szűrőtégely (4.2. pont) csatlakoztatására alkalmas tartóval, illetve egy olyan elvezetőcsővel van felszerelve, amelyhez egy lehetőleg sűrített levegővel töltött folyadékelvezetőhöz és a vákuumhoz kapcsolt csap tartozik. Használat előtt mindennap öt percig melegítsük elő az egységet forrásban lévő vízzel.
4.2. Üveg szűrőtégely 40-90 μm porozitású, olvasztott zsugorított üveg szűrőlappal. Az első használat előtt melegítsük fel 500 °C-ra néhány percre, és hűtsük le (8.6. pont).
4.3. Visszafolyós hűtővel ellátott, legalább 270 ml-es, forralásra alkalmas henger.
4.4. Szárítószekrény, hőfokszabályzóval.
4.5. Tokos kemence, hőfokszabályzóval.
4.6. Extrahálóegység, amelyhez a szűrőtégelyhez (4.2. pont) való tartólemez és a vákuum, illetve folyadék kivezetésére szolgáló csappal ellátott ürítőpipa tartozik.
4.7. Csatlakozógyűrűk a fűtőegység (4.1. pont), a szűrőtégely (4.2. pont) és a henger (4.3. pont) összekapcsolásához, a hideg extrahálóegység (4.6. pont) és a tégely csatlakoztatásához.
5. A vizsgálat módja
Mérjünk ki 1 mg-os pontossággal 1 g-ot az előkészített mintából, és helyezzük a szűrőtégelybe (4.2. pont) (lásd az Észrevételek 9.1., 9.2. és 9.3. pontját), majd adjunk hozzá 1 g szűrési segédanyagot (3.3. pont).
Állítsuk össze a fűtőegységet (4.1. pont) és a szűrőtégelyt (4.2. pont), majd csatlakoztassuk a hengert (4.3. pont) a szűrőtégelyhez. Öntsünk 150 ml forrásban lévő kénsavat (3.1. pont) az összeállított hengerbe és tégelybe, és szükség esetén adjunk hozzá néhány csepp habzásgátló anyagot (3.2. pont).
5 ± 2 perc alatt hozzuk forrásba a folyadékot, és forraljuk erőteljesen pontosan 30 percig.
Nyissuk meg az ürítőpipa csapját (4.1. pont), vákuum alatt szűrjük át a kénsavat a szűrőtégelyen, és mossuk a maradékot egymást követően háromszor, 30 ml-es forróvízadagokkal, ügyelve arra, hogy a maradékot minden mosás után szárazra szűrjük.
Zárjuk el a kivezető csapot, és öntsünk 150 ml forrásban lévő kálium-hidroxid oldatot (3.7. pont) az összeállított hengerbe és tégelybe, majd adjunk hozzá néhány csepp habzásgátló anyagot (3.2. pont). 5 ± 2 perc alatt hozzuk forrásba a folyadékot, és forraljuk erőteljesen pontosan 30 percig. Szűrjük, és ismételjük meg a kénsavas lépésnél alkalmazott mosási műveletet.
Az utolsó mosás és szárítás után szereljük le a tégelyt, és tartalmával együtt csatlakoztassuk a hideg extrahálóegységhez (4.6. pont). Vákuumot használva mossuk a tégelyben lévő maradékot háromszor egymás után 25 ml-es acetonadagokkal (3.4. pont), ügyelve arra, hogy a maradékot minden mosás után szárazra szűrjük.
Szárítsuk a tégelyt tömegállandóságig a szárítószekrényben 130 °C-on. Az egyes szárítások után hűtsük le exszikkátorban, és gyorsan mérjük le. Helyezzük a tégelyt a tokos kemencébe, és 475-500 °C-on hamvasszuk tömegállandóságig (a tömegveszteségnek két egymást követő mérés között 2 mg-nál kisebbnek kell lennie) legalább 30 percen át.
A mérés előtt minden hevítés után hűtsük a tégelyt először a kemencében, majd az exszikkátorban.
Végezzünk vakpróbát minta nélkül. A hamvasztásból adódó tömegveszteség nem haladhatja meg a 4 mg-ot.
6. Az eredmények kiszámítása
A minta százalékosan kifejezett nyersrosttartalma az alábbi képlettel adható meg:
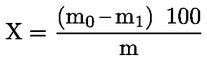
ahol:
m = a minta tömege g-ban;
m0 = a meghatározás során végzett hamvasztás utáni tömegveszteség, g-ban;
m1 = a vakpróba során végzett hamvasztás utáni tömegveszteség, g-ban.
7. Ismételhetőség
Ugyanazon a mintán végzett két párhuzamos meghatározás eredménye közötti különbség nem haladhatja meg:
- 10 %-nál alacsonyabb nyersrosttartalom esetén abszolút értékben a 0,6 %-ot,
- 10 %-os vagy annál magasabb nyersrosttartalom esetén a magasabb érték 6 %-át.
8. Reprodukálhatóság
Az ugyanazon mintán különböző laboratóriumokban végzett két meghatározás eredménye közötti különbség nem haladhatja meg:
- 10 %-nál alacsonyabb nyersrosttartalom esetén abszolút értékben az 1,0 %-ot,
- 10 %-os vagy annál magasabb nyersrosttartalom esetén a magasabb érték 10 %-át.
9. Észrevételek
9.1. A 10 %-nál több nyerszsírt tartalmazó takarmányokat az analízis előtt petroléterrel (3.5. pont) zsírtalanítani kell. Csatlakoztassuk a szűrőtégelyt (4.2. pont) tartalmával együtt a hideg extrahálóegységhez (4.6. pont), és vákuumban mossuk a maradékot egymás után háromszor 30 ml-es petroléteradagokkal, ügyelve arra, hogy a maradék száraz legyen. Csatlakoztassuk a tégelyt tartalmával együtt a fűtőegységhez (4.1. pont), és folytassuk a meghatározást az 5. pont szerint.
9.2. A petroléterrel (3.5. pont) közvetlenül nem extrahálható zsírokat tartalmazó takarmányokat a 8.1. pontban bemutatott módon kell zsírtalanítani, majd savas forralást követően még egyszer zsírtalanítani kell őket. A savas forralás és az ezt követő mosás után csatlakoztassuk a tégelyt tartalmával együtt a hideg extrahálóegységhez (4.6. pont), és mossuk három alkalommal 30 ml acetonnal, amit további három mosás követ 30 ml-es petroléteradagokkal. Vákuumban szűrjük, amíg száraz nem lesz, majd folytassuk az analízist az 5. pontban leírtak szerint, a kálium-hidroxidos kezeléssel kezdve.
9.3. Ha a takarmányok kalcium-karbonátban kifejezve több mint 5 % karbonátot tartalmaznak, akkor csatlakoztassuk a tégelyt (4.2. pont) a lemért mintával együtt a fűtőegységhez (4.1. pont). Mossuk a mintát háromszor 30 ml sósavval (3.6. pont). Az egyes savadagok hozzáadása után, a szűrés előtt körülbelül egy percig hagyjuk állni a mintát. Mossuk egyszer 30 ml vízzel, és folytassuk az 5. pontban leírtak szerint.
9.4. Ha állvány formájú berendezést használunk (több tégely csatlakozik ugyanahhoz a fűtőegységhez), akkor ugyanannak a mintának nem végezhető el két egyedi vizsgálata ugyanabban a sorozatban.
9.5. Ha a forralás után nehéz leszűrni a savas és a lúgos oldatokat, akkor vezessünk sűrített levegőt a fűtőegység ürítőcsövébe, és így folytassuk a szűrést.
9.6. Az üveg szűrőtégelyek élettartamának meghosszabbítása érdekében a hamvasztás hőmérséklete nem haladhatja meg az 500 °C-ot. Ügyeljünk arra, hogy a hevítési és a hűtési ciklusok alatt kerüljük a túlzott, lökésszerű hőhatást.
I. A CUKOR MEGHATÁROZÁSA
1. Cél és alkalmazási terület
A módszer lehetővé teszi a redukálócukor és az invertálás utáni összes cukor glükózban vagy, adott esetben, 0,95-os tényezővel konvertálva, szacharózban kifejezett szintjének meghatározását. A módszer takarmánykeverékek esetében alkalmazható. Egyéb takarmányokra különleges módszerek vonatkoznak. Adott esetben, a laktózt külön meg kell mérni, és figyelembe kell venni az eredmények kiszámításánál.
Ezt a módszert kell alkalmazni a takarmány energiaértékének kiszámításához használt cukortartalom meghatározására.
Abban az esetben, ha a cukortartalmat más célból kell meghatározni, más elemzési módszerek is alkalmazhatók.
2. Vizsgálati alapelv
A cukrot hígított etanolban extraháljuk; az oldatot Carrez I. és II. oldattal derítjük. Az etanol kizárása után az invertálás előtti és utáni mennyiség Luff-Schoorl-módszerrel kerül meghatározásra.
3. Reagensek
3.1. Etanol-oldat 40 % (v/v) sűrűség: 0,948 g/ml, 20 °C-on, fenolftaleinhez semlegesítve.
3.2. Carrez I. oldat: vízben oldjunk fel 21,9 g cink-acetátot, Zn(CH3COO)2 2H2O és 3 g jégecetet. Töltsük fel vízzel 100 ml-re.
3.3. Carrez II. oldat: vízben oldjunk fel 10,6 g kálium-[hexaciano-ferrát(II)]-ot, K4 Fe (CN)6 3H2O. Töltsük fel vízzel 100 ml-re.
3.4. 0,1 %-os (w/v) metilnarancsoldat.
3.5. 4 mol/liter koncentrációjú sósav.
3.6. 0,1 mol/liter koncentrációjú sósav.
3.7. 0,1 mol/liter koncentrációjú nátrium-hidroxid oldat.
3.8. Luff-Schoorl-reagens:
Óvatosan kevergetve öntsük a citromsavoldatot (3.8.2. pont) a nátrium-karbonát-oldatba (3.8.3. pont). Adjuk hozzá a réz-szulfát oldatot (3.8.1. pont), és öntsük fel 1 literig vízzel. Hagyjuk másnapig ülepedni, majd szűrjük.
Ellenőrizzük az így kapott reagens koncentrációját (0,05 mol/liter koncentrációjú Cu; 1 mol/liter koncentrációjú Na2 CO3), lásd az 5.4. pont utolsó bekezdését. Az oldat pH-értékének körülbelül 9,4-nek kell lenni.
3.8.1. Réz-szulfát-oldat: 100 ml vízben oldjunk fel 25 g vasmentes rézszulfátot, Cu SO4 5H2O.
3.8.2. Citromsavoldat: 50 ml vízben oldjunk fel 50 g citromsavat, C6H8O7•H2O.
3.8.3. Nátrium-karbonát-oldat: kb. 300 ml meleg vízben oldjunk fel 143,8 g dehidratált nátrium-karbonátot. Hagyjuk kihűlni.
3.9. 0,1 mol/liter koncentrációjú nátrium-tioszulfát-oldat.
3.10. Keményítőoldat: adjunk 30 ml vízben elkevert 5 g oldható keményítőt 1 liter forrásban lévő vízhez. Forraljuk három percen keresztül, hagyjuk kihűlni, szükség esetén adjunk hozzá 10 mg higany-jodidot tartósítószerként.
3.11. 3 mol/liter koncentrációjú kénsav.
3.12. 30 %-os (w/v) kálium-jodid-oldat:
3.13. Sósavban forralt, vízben mosott és szárított, granulált habkő.
3.14. 3-metilbután-1-ol.
4. Eszközök
Billenődobos keverőgép: kb. 35-40 fordulatszám/perc teljesítménnyel.
5. A vizsgálat módja
5.1. A minta extrahálása
Mérjünk ki mg-os pontossággal 2,5 g-ot a mintából, és helyezzük egy 250 ml-es mérőlombikba. Adjunk hozzá 200 ml etanolt (3.1. pont), és keverjük a keverőgépen egy órán keresztül. Adjunk hozzá 5 ml Carrez I. oldatot (3.2. pont), és kevergessük körülbelül 30 másodpercig. Adjunk hozzá 5 ml Carrez II. oldatot (3.3. pont), és kevergessük egy percig. Töltsük fel térfogatra etanollal (3.1. pont), homogenizáljuk, és szűrjük. Vegyünk el 200 ml-t a szűrletből, és az etanol nagy részének eltávolítása céljából pároljuk térfogatának kb. felére. Helyezzük a bepárlás utáni maradékot mennyiségileg egy 200 ml-es mérőlombikba meleg vízzel, hűtsük, töltsük fel térfogatra vízzel, homogenizáljuk, és szükség esetén szűrjük. Ezen oldatot használjuk a redukálócukor és invertálás utáni összes cukor mennyiségének meghatározására.
5.2. A redukálócukor meghatározása
Pipetta segítségével vegyünk fel legfeljebb 25 ml oldatot, amely 60 mg-nál kevesebb, glükózban kifejezett redukálócukrot tartalmaz. Szükség esetén töltsük fel az oldatot desztillált vízzel 25 ml-re, és határozzuk meg redukálócukor-tartalmát a Luff-Schoorl-módszerrel. Az eredményt a minta glükóztartalmára vonatkoztatva, százalékosan fejezzük ki.
5.3. Az invertálás utáni összes cukor meghatározása
Pipetta segítségével vegyünk fel az oldatból 50 ml-t, és öntsük át egy 100 ml-es mérőlombikba. Adjunk hozzá néhány csepp metilnarancsoldatot (3.4. pont), majd óvatosan és folyamatos kevergetés közben, adjunk hozzá sósavat (3.5. pont), amíg a folyadék határozottan vörössé nem válik. Adjunk hozzá 15 ml sósavat (3.6. pont), merítsük a lombikot gyorsan forró vízfürdőbe, és tartsuk ott harminc percen keresztül. Hűtsük le gyorsan kb. 20 °C-ra, és adjunk hozzá 15 ml nátrium-hidroxid-oldatot (3.7. pont). Töltsük fel vízzel 100 ml-re, és homogenizáljuk. Vegyünk el legfeljebb 25 ml oldatot, amely 60 mg-nál kevesebb, glükózban kifejezett redukálócukrot tartalmaz. Szükség esetén töltsük fel az oldatot desztillált vízzel 25 ml-re, és határozzuk meg redukálócukor-tartalmát a Luff-Schoorl-módszerrel. Az eredményt a glükózra vagy, adott esetben, 0,95-os tényezővel szorozva a szacharózra vonatkoztatva, százalékosan fejezzük ki.
5.4. Titrálás a Luff-Schoorl-módszerrel
Pipetta segítségével vegyünk fel 25 ml Luff-Schoorl-reagenst (3.8. pont), és helyezzük át egy 300 ml-es Erlenmeyer-lombikba; adjunk hozzá pontosan 25 ml-t a derített cukoroldatból. Adjunk hozzá kétszemcsényi habkövet (3.13. pont), hevítsük közepes méretű, szabad lángon, közben kézzel kevergessük, és kb. két perc alatt forraljuk fel a folyadékot. Az Erlenmeyer-lombikot azonnal helyezzük egy 6 cm átmérőjű lyukkal ellátott azbesztes dróthálóra, amely alatt előzőleg lángot gyújtunk. A lángot úgy kell szabályoznunk, hogy az Erlenmeyer-lombiknak csak az alját melegítse. Illesszünk egy visszafolyós hűtőt az Erlenmeyer-lombikhoz. Forraljuk pontosan tíz percig. Hűtsük le azonnal hideg vízben, és kb. öt perc elteltével a következők szerint titráljuk:
Adjunk hozzá 10 ml kálium-jodid-oldatot (3.12. pont), és közvetlenül ezután (a bőséges habképződés veszélye miatt óvatosan) adjunk hozzá 25 ml kénsavat (3.11. pont). Titráljuk nátrium-tioszulfát-oldattal (3.9. pont), amíg fakósárga színt nem kapunk, adjuk hozzá a keményítő-indikátort (3.10. pont), és fejezzük be a titrálást.
Forralás nélkül, 10 ml kálium-jodid-oldat (3.12. pont) és 25 ml kénsav (3.11. pont) hozzáadása után végezzük el ugyanezt a titrálást egy pontosan kimért, 25 ml Luff-Schoorl-reagenst (3.8. pont) és 25 ml vizet tartalmazó keveréken.
6. Az eredmények kiszámítása
A táblázat segítségével határozzuk meg a glükóz mennyiségét mg-ban, amely a két titrálás eredménye közti különbségnek felel meg 0,1 mol/liter koncentrációjú nátrium-tioszulfát mg-jában kifejezve. Az eredményt a minta százalékában fejezzük ki.
7. Különleges vizsgálati módok
7.1. A melaszban gazdag, valamint egyéb, kevésbé homogén összetételű takarmányok esetében mérjünk ki 20 g-ot, és helyezzük egy 1 literes mérőlombikba 500 ml vízzel felöntve. Keverjük forgó dobon egy órán keresztül. Az 5.1. pontban leírtak szerint derítsük Carrez I. oldat (3.2. pont) és Carrez II. oldat (3.3. pont) reagensek használatával, ebben az esetben azonban négyszeres mennyiséget használjunk az egyes reagensekből. Töltsük fel térfogatra 80 %-os (v/v) etanollal.
Homogenizáljuk és szűrjük. Vonjuk ki az etanolt az 5.1. pontban leírtak szerint. Ha nincs jelen dextrinált keményítő, töltsük fel térfogatra desztillált vízzel.
7.2. Melaszok, valamint cukorban gazdag és gyakorlatilag keményítőmentes takarmány-alapanyagok (szentjánoskenyér, szárítottcukorrépa-szeletek stb.) esetében mérjünk ki 5 g-ot, és helyezzük egy 250 ml-es mérőlombikba, adjunk hozzá 200 ml desztillált vizet, és keverjük forgó dobon egy órán keresztül vagy szükség esetén tovább. Az 5.1. pontban leírtak szerint derítsük Carrez I. oldat (3.2. pont) és Carrez II. oldat (3.3. pont) reagensekkel. Töltsük fel térfogatra hideg vízzel, homogenizáljuk, és szűrjük. Az összes cukortartalom meghatározásához az 5.3. pontban leírtak szerint folytassuk a vizsgálatot.
8. Észrevételek
8.1. A habképződés elkerülése érdekében a Luff-Schoorl-reagenssel történő forralás előtt tanácsos (a mennyiségtől függetlenül) kb. 1 ml 3-metilbután-1-ol (3.14. pont) hozzáadása.
8.2. A glükózban kifejezett, invertálás utáni összes cukortartalom és a glükózban kifejezett, redukálócukor-tartalom különbsége 0,95-dal szorozva megadja a szacharóztartalomra vonatkoztatott százalékos arányt.
8.3. A laktóz nélkül számított redukálócukor-tartalom meghatározására két módszer alkalmazható:
8.3.1. Közelítő számításhoz, szorozzuk meg 0,675-tel a laktóztartalmat, amely egy másik elemzési módszerrel került megállapításra, és vonjuk ki a kapott eredményt a redukálócukor-tartalomból.
8.3.2. A laktóz nélkül számított redukálócukor-tartalom pontos kiszámításához ugyanazt a mintát kell használni a két végső meghatározáshoz. Az egyik vizsgálatot az 5.1. pontban foglaltak szerint nyert oldaton végezzük, a másikat pedig a laktóz meghatározása során nyert oldaton az említett célból meghatározott módszer szerint (más típusú cukrok erjesztése és derítés után).
A jelen lévő cukormennyiség meghatározása mindkét esetben a Luff-Schoorl-módszerrel történik, és a glükóz mg-ban kerül kiszámításra. Az egyik értéket kivonjuk a másikból, és az eredményt a minta százalékában fejezzük ki.
Példa
A kiválasztott két mennyiség az egyes meghatározások esetében 250 mg-os mintának felel meg.
Az első esetben 44,2 mg glükóznak megfelelő, 17 ml 0,1 mol/liter koncentrációjú nátrium-tioszulfát oldat fogyott; a második esetben 11 ml, amely 27,6 mg glükóznak felel meg.
A különbség 16,6 mg glükóz.
Ezért a glükózra átszámított (laktóz nélküli) redukálócukor-tartalom a következő
25 ml Luff-Schoorl-reagenshez tartozó értékek táblázata
0,1 mol/liter koncentrációjú Na2 S2 O3 ml-e, kétpercnyi melegítés, tízpercnyi forralás
| Na2 S2 O3 0,1 mol/liter koncentráció | Glükóz, fruktóz, invertcukor C6 H12 O6 | Laktóz C12 H22 O11 | Na2 S2 O3 0,1 mol/liter koncentráció | ||
| ml | mg | különbség | mg | különbség | ml |
| 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 | 2,4 4,8 7,2 9,7 12,2 14,7 17,2 19,8 22,4 25,0 27,6 30,3 33,0 35,7 38,5 41,3 44,2 47,1 50,0 53,0 56,0 59,1 62,2 | 2,4 2,4 2,5 2,5 2,5 2,5 2,6 2,6 2,6 2,6 2,7 2,7 2,7 2,8 2,8 2,9 2,9 2,9 3,0 3,0 3,1 3,1 | 3,6 7,3 11,0 14,7 18,4 22,1 25,8 29,5 33,2 37,0 40,8 44,6 48,4 52,2 56,0 59,9 63,8 67,7 71,7 75,7 79,8 83,9 88,0 | 3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,8 3,8 3,8 3,8 3,8 3,8 3,9 3,9 3,9 4,0 4,0 4,1 4,1 4,1 | 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
J. A LAKTÓZ MEGHATÁROZÁSA
1. Cél és alkalmazási terület
Ez a módszer lehetővé teszi a 0,5 %-nál magasabb laktóztartalmú takarmányok laktózszintjének meghatározását.
2. Vizsgálati alapelv
A cukrokat vízben oldjuk. Az oldatot Saccharomyces cerevisiae élesztővel erjesztjük, amely a laktózt érintetlenül hagyja. Derítés és szűrés után a szűrlet laktóztartalmát Luff-Schoorl-módszerrel határozzuk meg.
3. Reagensek
3.1. Saccharomyces cerevisiae szuszpenzió: oldjunk fel 25 g friss élesztőt 100 ml vízben. A szuszpenzió hűtőben legfeljebb egy hétig áll el.
3.2. Carrez I. oldat: vízben oldjunk fel 21,9 g cink-acetátot, Zn(CH3COO)2 2H2O és 3 g jégecetet. Töltsük fel vízzel 100 ml-re.
3.3. Carrez II. oldat: vízben oldjunk fel 10,6 g kálium-[hexaciano-ferrát(II)]-ot, K4 Fe (CN)6 3H2O. Töltsük fel vízzel 100 ml-re.
3.4. Luff-Schoorl-reagens:
Óvatosan kevergetve öntsük a citromsavoldatot (3.4.2. pont) a nátrium-karbonát-oldatba (3.4.3. pont). Adjuk hozzá a réz-szulfát-oldatot (3.4.1. pont), és töltsük fel egy 1 l-re vízzel. Hagyjuk másnapig ülepedni, majd szűrjük. Ellenőrizzük az így kapott reagens koncentrációját (0,05 mol/liter Cu; 1 mol/liter Na2 CO3). Az oldat pH-értékének körülbelül 9,4-nek kell lenni.
3.4.1. Réz-szulfát-oldat: 100 ml vízben oldjunk fel 25 g vasmentes rézszulfátot, Cu SO4 5H2O.
3.4.2. Citromsavoldat: 50 ml vízben oldjunk fel 50 g citromsavat, C6H8O7O2O.
3.4.3. Nátrium-karbonát-oldat: kb. 300 ml meleg vízben oldjunk fel 143,8 g dehidratált nátrium-karbonátot. Hagyjuk kihűlni.
3.5. Sósavban forralt, vízben mosott és szárított, granulált habkő.
3.6. 30 %-os (w/v) kálium-jodid-oldat:
3.7. 3 mol/liter koncentrációjú kénsav.
3.8. 0,1 mol/liter koncentrációjú nátrium-tioszulfát oldat.
3.9. Keményítőoldat: adjunk 30 ml vízben elkevert 5 g oldható keményítőt 1 liter forrásban lévő vízhez. Forraljuk 3 percig, hagyjuk hűlni, és amennyiben szükséges, adjunk hozzá 10 mg higany-jodidot tartósítószerként.
4. Eszközök
38-40 °C-ra beállított, hőfokszabályzóval ellátott vízfürdő.
5. A vizsgálat módja
Mérjünk ki a mintából 1 g-ot mg-os pontossággal, és ezt a részmintát helyezzük egy 100 ml-es mérőlombikba. Adjunk hozzá 25-30 ml vizet. Helyezzük a lombikot harminc percre forró vízfürdőbe, majd hűtsük le kb. 35 °C-ra. Adjunk hozzá 5 ml élesztő-szuszpenziót (3.1. pont), és homogenizáljuk. Hagyjuk a lombikot két órán keresztül 38-40 °C-on állni a vízfürdőben. Hűtsük le kb. 20 °C-ra.
Adjunk hozzá 2,5 ml Carrez I. oldatot (3.2. pont), és kevergessük harminc másodpercig, majd adjunk hozzá 2,5 ml Carrez II. oldatot (3.3. pont), és újra kevergessük harminc másodpercig. Töltsük fel vízzel 100 ml-re, keverjük össze, és szűrjük. Pipetta segítségével vegyünk fel a szűrletből egy részt, amely nem haladja meg a 25 ml-t, és amely lehetőleg 40-80 mg laktózt tartalmaz, és helyezzük át egy 300 ml-es Erlenmeyer-lombikba. Amennyiben szükséges, töltsük fel 25 ml-re vízzel.
Végezzünk vakpróbát ugyanilyen módon 5 ml élesztő oldattal (3.1. pont). A laktóztartalmat a Luff-Schoorl-módszer szerint, a következőképpen határozzuk meg: adjunk az oldathoz pontosan 25 ml Luff-Schoorl-reagenst (3.4. pont) és kétszemcsényi habkövet (3.5. pont). Közepes méretű, szabad lángon történő melegítés közben kézzel kevergessük, és kb. két perc alatt forraljuk fel a folyadékot. Az Erlenmeyer-lombikot azonnal helyezzük egy 6 cm átmérőjű lyukkal ellátott azbesztes dróthálóra, amely alatt előzőleg lángot gyújtunk. A lángot úgy kell szabályoznunk, hogy az Erlenmeyer-lombiknak csak az alját melegítse. Illesszünk egy visszafolyós hűtőt az Erlenmeyer-lombikhoz. Forraljuk pontosan tíz percig. Hűtsük le azonnal hideg vízben, és kb. öt perc elteltével a következők szerint titráljuk:
Adjunk hozzá 10 ml kálium-jodid-oldatot (3.6. pont), és rögtön utána (a bőséges habképződés veszélye miatt óvatosan) adjunk hozzá 25 ml kénsavat (3.7. pont). Titráljuk nátrium-tioszulfát-oldattal (3.8. pont) amíg fakósárga színt nem kapunk, adjuk hozzá a keményítő-indikátort (3.9. pont), és fejezzük be a titrálást.
Forralás nélkül, 10 ml kálium-jodid-oldat (3.6. pont) és 25 ml kénsav (3.7. pont) hozzáadása után végezzük el ugyanezt a titrálást egy pontosan kimért, 25 ml Luff-Schoorl-reagenst (3.4. pont) és 25 ml vizet tartalmazó keveréken.
6. Az eredmények kiszámítása
A csatolt táblázat segítségével határozzuk meg a laktóz mennyiségét mg-ban, amely a két titrálás eredménye közti különbségnek felel meg, 0,1 mol/liter koncentrációjú nátrium-tioszulfát ml-ében kifejezve.
A dehidratált laktózra vonatkozó eredményt a minta százalékában fejezzük ki.
7. Észrevételek
1. A 40 %-nál több erjeszthető cukrot tartalmazó termékek esetében 5 ml-nél nagyobb mennyiségű élesztő-szuszpenziót (3.1. pont) használjunk.
2. A "csökkentett laktóztartalmú" takarmányban (pl. macskák számára készült tej) a laktózt fruktózzá alakítják át, amely 2 óra alatt nem erjed meg teljesen, ami magasabb vagy álpozitív eredményt eredményez (mivel a kivonatban fruktózmaradványok maradnak).
25 ml Luff-Schoorl-reagenshez tartozó értékek táblázata
0,1 mol/liter koncentrációjú Na2 S2 O3 ml-e, kétpercnyi melegítés, tízpercnyi forralás
| Na2 S2 O3 0,1 mol/liter koncentráció | Glükóz, fruktóz, invertcukor C6 H12 O6 | Laktóz C12 H22 O11 | Na2 S2 O3 0,1 mol/liter koncentráció | ||
| ml | mg | különbség | mg | különbség | ml |
| 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 | 2,4 4,8 7,2 9,7 12,2 14,7 17,2 19,8 22,4 25,0 27,6 30,3 33,0 35,7 38,5 41,3 44,2 47,1 50,0 53,0 56,0 59,1 62,2 | 2,4 2,4 2,5 2,5 2,5 2,5 2,6 2,6 2,6 2,6 2,7 2,7 2,7 2,8 2,8 2,9 2,9 2,9 3,0 3,0 3,1 3,1 | 3,6 7,3 11,0 14,7 18,4 22,1 25,8 29,5 33,2 37,0 40,8 44,6 48,4 52,2 56,0 59,9 63,8 67,7 71,7 75,7 79,8 83,9 88,0 | 3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,8 3,8 3,8 3,8 3,8 3,8 3,9 3,9 3,9 4,0 4,0 4,1 4,1 4,1 | 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
K. A KEMÉNYÍTŐ MEGHATÁROZÁSA
- POLARIMETRIÁS MÓDSZER -
1. Cél és alkalmazási terület
E módszer lehetővé teszi a keményítő és a nagy molekulasúlyú keményítő-bomlástermékek takarmányokban jelen lévő szintjének meghatározását a feltüntetett energiaérték (a VII. melléklet rendelkezései) és a 767/2009/EK rendelet betartásának ellenőrzése céljából.
Ezt a módszert kell alkalmazni a takarmány energiaértékének kiszámításához használt keményítőtartalom meghatározására.
Abban az esetben, ha a keményítőtartalmat más célból kell meghatározni, más elemzési módszerek is alkalmazhatók.
2. Vizsgálati alapelv
A módszer két meghatározásból áll. Az első meghatározás során a mintát híg sósavval kezeljük. Tisztítás és szűrés után az oldat optikai forgatóképességét polarimetriával mérjük.
A második meghatározás során a mintát 40 %-os etanollal extraháljuk. A szűrlet sósavval való savanyítása, tisztítása és szűrése után az optikai forgatóképességet ugyanúgy mérjük, mint az első meghatározás esetében.
A minta keményítőtartalmát úgy kapjuk meg, hogy a két mért érték különbségét megszorozzuk egy ismert együtthatóval.
3. Reagensek
3.1. 25 %-os (w/w) sósav-oldat, sűrűség: 1,126 g/ml.
3.2. 1,13 %-os (w/v) sósav-oldat
A koncentrációt 0,1 mol/liter koncentrációjú nátrium-hidroxid-oldattal végzett titrálással kell ellenőrizni, 94 %-os (v/v) etanolban oldott 0,1 % (w/v) metilvörös jelenlétében. 10 ml semlegesítéséhez 30,94 ml 0,1 mol/liter koncentrációjú NaOH szükséges.
3.3. Carrez I. oldat: vízben oldjunk fel 21,9 g cink-acetátot, Zn(CH3COO)2 2H2O és 3 g jégecetet. Töltsük fel vízzel 100 ml-re.
3.4. Carrez II. oldat: vízben oldjunk fel 10,6 g kálium-[hexaciano-ferrát(II)]-ot, K4 Fe (CN)6 3H2O. Töltsük fel vízzel 100 ml-re.
3.5. 40 %-os (v/v) etanol-oldat, sűrűség: 0,948 g/ml 20 °C-on.
4. Eszközök
4.1. 250 ml-es Erlenmeyer-lombik szabványos üvegcsiszolattal és visszafolyós hűtővel.
4.2. Polariméter vagy szachariméter.
5. A vizsgálat módja
5.1. A minta előkészítése
Őröljük a mintát olyan finomságúra, hogy az teljes egészében átjusson egy 0,5 mm szemméretű, kerek szemű szitán.
5.2. A teljes optikai forgatóképesség (P vagy S) meghatározása (lásd az Észrevételek 7.1. pontját)
Mérjünk ki a megőrölt mintából mg pontossággal 2,5 g-ot, és helyezzük azt egy 100 ml-es mérőlombikba. Adjunk hozzá 25 ml sósavat (3.2. pont), rázzuk össze a vizsgálati minta egyenletes eloszlásának biztosítása céljából, és adjunk hozzá további 25 ml sósavat (3.2. pont). Merítsük a lombikot forrásban levő vízfürdőbe, az első három percben erőteljesen és folyamatosan rázogassuk a csomóképződés megakadályozása céljából. A vízfürdőben elegendő víznek kell lennie ahhoz, hogy a fürdő a lombik behelyezésekor forrásponton maradjon. A lombikot rázogatás közben nem szabad kivenni a vízfürdőből. Pontosan 15 perc elteltével vegyük ki a lombikot a vízfürdőből, adjunk hozzá 30 ml hideg vizet és azonnal hűtsük le 20 °C-ra.
Adjunk hozzá 5 ml-t a Carrez I. oldatból (3.3. pont), és rázogassuk kb. harminc másodpercen át. Ezt követően adjunk hozzá 5 ml-t a Carrez II. oldatból (3.4. pont), és ismét rázzuk kb. harminc másodpercen át. Töltsük fel vízzel térfogatra, keverjük össze és szűrjük meg. Ha a szűrlet nem teljesen tiszta (ami ritkán fordul elő), nagyobb mennyiségű (pl. 10 ml) Carrez I. és II. oldat hozzáadásával ismételjük meg a meghatározást.
Egy 200 mm-es csőben a polariméterrel vagy a szachariméterrel mérjük meg az oldat optikai forgatóképességét.
5.3. A 40 %-os etanolban oldható anyagok optikai forgatóképességének (P' vagy S') meghatározása
Mérjünk ki a mintából mg pontossággal 5 g-ot, helyezzük egy 100 ml-es mérőlombikba, és adjunk hozzá mintegy 80 ml etanolt (3.5. pont) (lásd az Észrevételek 7.2. pontját). Hagyjuk a lombikot állni szobahőmérsékleten 1 órán át; ezalatt hatszor erőteljesen rázzuk össze, hogy a vizsgálati minta alaposan összekeveredjen az etanollal. Töltsük fel térfogatra etanollal (3.5. pont), keverjük össze és szűrjük meg.
A szűrletből pipettázzunk 50 ml-t (= 2,5 g minta) egy 250 ml-es Erlenmeyer-lombikba, adjunk hozzá 2,1 ml sósavat (3.1. pont), és erősen rázzuk össze. Csatlakoztassunk visszafolyós hűtőt az Erlenmeyer-lombikhoz, és merítsük az utóbbit forrásban levő vízfürdőbe. Pontosan 15 perc elteltével vegyük ki az Erlenmeyer-lombikot a vízfürdőből, öntsük át annak tartalmát egy 100 ml-es mérőlombikba, öblítsük kis mennyiségű hideg vízzel, majd hűtsük le 20 °C-ra.
Derítsük Carrez I. oldattal (3.3. pont) és Carrez II. oldattal (3.4. pont), vízzel töltsük fel térfogatra, keverjük össze, szűrjük át, majd mérjük meg az optikai forgatóképességét az 5.2. pont második és harmadik bekezdésében ismertetett módon.
6. Az eredmények kiszámítása
A keményítőtartalmat (%) a következőképpen számítjuk ki:
6.1. Mérés polariméterrel
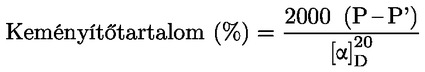
P = a teljes optikai forgatóképesség szögfokban
P' = a 40 %-os (V/V) etanolban oldható anyagok optikai forgatóképessége szögfokban
[Kép #1] = a tiszta keményítő fajlagos optikai forgatóképessége. Az e tényezőre hagyományosan elfogadott számértékek a következők:
Kép #1
+ 185,9° : rizskeményítő
+ 185,7° : burgonyakeményítő
+ 184,6° : burgonyakeményítő
+ 182,7° : búzakeményítő
+ 181,5° : árpakeményítő
+ 181,3° : zabkeményítő
+ 184,0° : takarmánykeverékekben található egyéb keményítő- és keményítőkeverék-fajták.
6.2. Mérés szachariméterrel
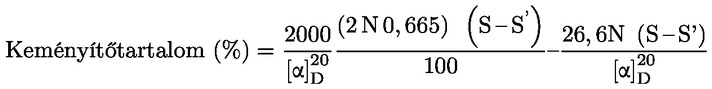
S = teljes optikai forgatóképesség szachariméter-fokban
S' = a 40 %-os (v/v) etanolban oldható anyagok optikai forgatóképessége szachariméter-fokban
N
=
az a szacharózmennyiség (g) 100 ml vízben, amely 100 szachariméter-foknak megfelelő optikai forgatóképességet ad 200 mm-es csőben mérve
16,29 g a francia szachariméterek esetében
26,00 g a német szachariméterek esetében
20,00 g a vegyes szachariméterek esetében
[Kép #2] = a tiszta keményítő fajlagos optikai forgatóképessége (lásd a 6.1. pontot)
Kép #2
6.3. Ismételhetőség
Az ugyanazon a mintán végzett két párhuzamos meghatározás eredményének különbsége 40 %-nál alacsonyabb keményítőtartalom esetén, abszolút értékben nem lehet több mint 0,4, 40 %-os vagy annál nagyobb keményítőtartalom esetén pedig nem haladhatja meg az 1 %-os relatív értéket.
7. Észrevételek
7.1. Ha a minta több mint 6 %-nyi karbonátot tartalmaz kalcium-karbonátra számítva, e karbonátot híg kénsav pontosan megfelelő mennyiségével történő kezeléssel el kell roncsolni a teljes optikai forgatóképesség meghatározását megelőzően.
7.2. Magas laktóztartalmú termékek - pl. tejsavópor vagy sovány tejpor - esetében, 80 ml etanol (3.5. pont) hozzáadását követően, a következőképpen kell eljárni. Csatlakoztassunk visszafolyós hűtőt a lombikhoz, és merítsük az utóbbit 50 °C-os vízfürdőbe 30 percre. Hagyjuk lehűlni, majd folytassuk az analízist az 5.3. pontban leírt módon.
7.3. A következő takarmány-alapanyagokról ismert, hogy a takarmányokban való nagy mennyiségű jelenlétük esetén olyan interferenciákat idéznek elő a keményítőtartalom polarimetriás módszerrel való meghatározásakor, amelyek téves eredményekhez vezethetnek:
- (cukor)répakészítmények, pl. (cukor)répapép, (cukor)répamelasz, (cukor)répapép-melasz, (cukor)répaseprő, (répa)cukor,
- citruspép,
- lenmag; lenmagpogácsa; extrahált lenmag,
- repcemag; repcemagpogácsa; extrahált repcemag; repcemaghéj,
- napraforgómag; extrahált napraforgómag; részlegesen hántolt, extrahált napraforgómag,
- koprapogácsa; extrahált kopra,
- burgonyapép,
- szárított élesztő,
- inulinban gazdag termékek (pl. csicsókaszeletek és csicsókaliszt),
- töpörtyű,
- szójababtermékek.
Ezekben az esetekben a 121/2008/EK bizottsági rendeletben ( 18 ) előírt elemzési módszer alkalmazható. Ez a módszer az 1 %-nál kevesebb keményítőt tartalmazó takarmányok esetében is alkalmazható.
L. A NYERSHAMU MEGHATÁROZÁSA
1. Cél és alkalmazási terület
A módszer lehetővé teszi a takarmányok nyershamutartalmának meghatározását.
2. Vizsgálati alapelv
A mintát 550 °C-on hamvasztjuk; a maradékanyagot megmérjük.
3. Reagensek
20 %-os (w/v) ammónium-nitrát-oldat.
4. Eszközök
4.1. Főzőlap.
4.2. Hőfokszabályzós, elektromos tokos kemence.
4.3. Kvarcból, porcelánból vagy platinából készült négyszögletes (60 × 40 × 25 mm) vagy kör alakú (átmérő: 60-75 mm, magasság: 20-40 mm) hamvasztótégelyek.
5. A vizsgálat módja
Mérjünk ki a mintából 5 g-ot mg-os pontossággal (2,5 g-ot a duzzadóképes anyagból származó termékek esetében), és helyezzük egy hamvasztótégelybe, amelyet előzetesen 550 °C-ra hevítettük, majd lehűtöttük és táráztunk. Helyezzük a tégelyt a főzőlapra, és fokozatosan addig hevítsük, amíg az anyag el nem szenesedik. Hamvasszuk az 5.1. vagy az 5.2. pont szerint.
5.1. Helyezzük a tégelyt az 550 °C-ra beállított, kalibrált tokos kemencébe. Tartsuk ezen a hőmérsékleten egészen addig, amíg széntartalmú részecskéktől mentesnek tűnő fehér, halványszürke vagy vöröses hamut nem nyerünk. Helyezzük a tégelyt egy exszikkátorba, hagyjuk kihűlni, majd azonnal mérjük meg.
5.2. Helyezzük a tégelyt az 550 °C-ra beállított, kalibrált tokos kemencébe. Hamvasszuk 3 órán át. Helyezzük a tégelyt egy exszikkátorba, hagyjuk kihűlni, majd azonnal mérjük meg. Hamvasszuk újra 30 percen keresztül, annak érdekében, hogy a hamu tömege állandó maradjon (két egymást követő mérés közötti tömegveszteség legfeljebb 1 mg lehet).
6. Az eredmények kiszámítása
A maradék tömegét a táratömeg kivonásával számoljuk ki.
Az eredményt a minta százalékában fejezzük ki.
7. Észrevételek
7.1. A nehezen hamvasztható anyagok hamuját legalább három órán keresztül előzetesen hamvasztani kell, le kell hűteni, majd néhány csepp 20 %-os ammónium-nitrát oldatot vagy vizet kell hozzáadni (ügyelve arra, hogy elkerüljük a hamu szétszóródását vagy a csomóképződést). A szárítószekrényben való szárítása után folytassuk a kalcinálást. Szükség szerint ismételjük a műveletet a teljes elhamvadásig.
7.2. A 7.1. pontban ismertetett eljárással szemben ellenálló anyagok esetében a következőképpen járjunk el: háromórás hamvasztás után helyezzük a hamut meleg vízbe, és egy kis méretű, hamumentes szűrőn szűrjük át. Hamvasszuk el a szűrőt és annak tartalmát az eredeti tégelyben. Helyezzük a szűrletet a lehűtött tégelybe, párologtassuk kiszáradásig, hamvasszuk, és mérjük meg.
7.3. Olajok és zsírok esetében mérjünk ki pontosan egy 25 g-os mintát egy megfelelő méretű tégelybe. Hamumentes szűrőpapírcsíkkal meggyújtva, szenesítsük az anyagot. A gyulladás után a lehető legkevesebb vízzel nedvesítsük meg. Szárítsuk, és hamvasszuk az 5. pontban ismertetett módszerrel.
M. A SÓSAVBAN OLDHATATLAN HAMU MEGHATÁROZÁSA
1. Cél és alkalmazási terület
A módszer lehetővé teszi a takarmányokban lévő sósavban oldhatatlan ásványi anyagok szintjének meghatározását. A minta jellegétől függően két módszer alkalmazható.
1.1. A. módszer: a szerves takarmány-alapanyagok és a legtöbb takarmánykeverék esetében alkalmazható.
1.2. B. módszer: olyan ásványi vegyületek és keverékek, valamint összetett takarmányok esetében alkalmazható, amelyeknek az A. módszer szerint meghatározott, sósavban nem oldható anyagtartalma 1 %-nál magasabb.
2. Vizsgálati alapelv
2.1. A. módszer: a mintát hamvasztjuk, a hamut sósavban forraljuk, az oldhatatlan maradékanyagot pedig szűrjük és megmérjük.
2.2. B. módszer: a mintát sósavval kezeljük. Az oldatot szűrjük, a maradékanyagot hamvasztjuk, az így kapott hamut pedig az A. módszer szerint kezeljük.
3. Reagensek
3.1. 3 mol/liter koncentrációjú sósav.
3.2. 20 %-os (w/v) triklór-ecetsav oldat.
3.3. 1 %-os (w/v) triklór-ecetsav oldat.
4. Eszközök
4.1. Főzőlap.
4.2. Hőfokszabályzós, elektromos tokos kemence.
4.3. Kvarcból, porcelánból vagy platinából készült négyszögletes (60 × 40 × 25 mm) vagy kör alakú (átmérő: 60-75 mm, magasság: 20-40 mm) hamvasztótégelyek.
4.4. Hamumentes szűrők.
5. A vizsgálat módja
5.1. A. módszer:
Hamvasszuk a mintát a nyershamu meghatározásánál ismertetett módszer szerint. Annak az analízisnek a során kapott hamut is használhatjuk.
75 ml sósav (3.1. pont) felhasználásával helyezzük a hamut egy 250-400 ml-es főzőpohárba. Lassan forraljuk, és tartsuk forrásban óvatosan tizenöt percig. Szűrjük át a meleg oldatot egy hamumentes szűrőpapíron, és mossuk a maradékot meleg vízzel mindaddig, amíg a savreakció már nem észlelhető. Szárítsuk ki a maradékot tartalmazó szűrőt, és hamvasszuk egy tárázott tégelyben legalább 550 °C és legfeljebb 700 °C hőmérsékleten. Hűtsük le egy exszikkátorban, és mérjük meg.
5.2. B. módszer
Mérjünk le a mintából 5 g-ot mg-nyi pontossággal, és helyezzük egy 250-400 ml-es főzőpohárba. Adjunk hozzá egymást követően 25 ml vizet és 25 ml sósavat (3.1. pont), keverjük össze, és várjunk a pezsgés elállásáig. Adjunk hozzá még 50 ml sósavat (3.1. pont). Várjunk az esetleges gázkibocsátás megszűnéséig, majd helyezzük a főzőpoharat forró vízfürdőbe, és tartsuk ott harminc percig vagy szükség esetén tovább, az esetlegesen jelen lévő összes keményítő teljes hidrolízise érdekében. Még meleg állapotban szűrjük át egy hamumentes szűrőn, és mossuk a szűrőt 50 ml meleg vízben (lásd az Észrevételek 7. pontját). Helyezzük a maradékanyagot tartalmazó szűrőt hamvasztótégelybe, szárítsuk ki, és hamvasszuk legalább 550 °C és legfeljebb 700 °C hőmérsékleten. 75 ml sósav (3.1. pont) felhasználásával helyezzük a hamut egy 250-400 ml-es főzőpohárba; kövessük az 5.1. pont második albekezdésében leírtakat.
6. Az eredmények kiszámítása
A maradék tömegét a táratömeg kivonásával számoljuk ki. Az eredményt a minta százalékában fejezzük ki.
7. Észrevételek
Ha a szűrés problémásnak bizonyul, kezdjük újra az analízist úgy, hogy az 50 ml sósavat (3.1. pont) 50 ml 20 %-os (w/v) triklór-ecetsavval (3.2. pont) helyettesítjük, és a szűrőt 1 %-os meleg triklór-ecetsavban oldatban (3.3. pont) mossuk.
N. AZ ÖSSZES FOSZFOR MEGHATÁROZÁSA
Az összes foszfort az alábbiak szerint kell meghatározni:
- a "Takarmányok. Mintavételi és elemzési módszerek. A kalcium-, nátrium-, foszfor-, magnézium-, kálium-, vas-, cink-, réz-, mangán-, kobalt-, molibdén- és ólomtartalom meghatározása ICP-AES-sel" című EN 15510 szabványban előírt analitikai módszerrel, vagy
- a "Takarmányok. Mintavételi és elemzési módszerek. A kalcium-, nátrium-, foszfor-, magnézium-, kálium-, kén-, vas-, cink-, réz-, mangán- és kobalttartalom meghatározása ICP-AES-sel, nyomás alatti feltárás után" című EN 15621 szabványban előírt analitikai módszerrel, vagy
- az alábbiakban leírt fotometriás eljárással.
FOTOMETRIÁS ELJÁRÁSSAL
1. Cél és alkalmazási terület
Ez a módszer lehetővé teszi a takarmányok összes foszfortartalmának meghatározását. Különösen alkalmas az alacsony foszfortartalmú termékek elemzésére. Bizonyos esetekben (foszforban gazdag termékeknél) gravimetriás módszert használhatunk.
2. Vizsgálati alapelv
A mintát száraz égetéssel (szerves takarmányok esetén) vagy savas feltárással (ásványi vegyületek és folyékony takarmányok esetén) mineralizáljuk, és savas oldatba helyezzük. Az oldatot molibdén-vanadát reagenssel kezeljük. Az így keletkező sárga oldat optikai sűrűségét 430 nm-nél mérjük egy spektrofotométeren.
3. Reagensek
3.1. Kalcium-karbonát.
3.2. Sósav, ρ20 = 1,10 g/ml (közelítőleg 6 mol/liter).
3.3. Salétromsav, ρ20 = 1,045 g/ml.
3.4. Salétromsav, ρ20 = 1,38-1,42 g/ml.
3.5. Kénsav, ρ20 = 1,84 g/ml.
3.6. Molibdén-vanadát reagens: 1 literes mérőlombikban keverjünk össze 200 ml ammónium-heptamolibdát-oldatot (3.6.1. pont), 200 ml ammónium-monovanadát-oldatot (3.6.2. pont) és 134 ml salétromsavat (3.4. pont). Töltsük fel vízzel térfogatra.
3.6.1. Ammónium-heptamolibdát-oldat: oldjunk fel forró vízben 100 g ammónium-heptamolibdátot, (NH4) 6Mo7O24.4H2O. Adjunk hozzá 10 ml ammóniát (d: 0,91 g/ml), és töltsük fel vízzel 1 literre.
3.6.2. Ammónium-monovanadátoldat: oldjunk fel 400 ml forró vízben 2,35 g ammónium-monovanadátot, NH4VO3. Állandó keverés közben, lassan adjunk hozzá 20 ml hígított salétromsavat (7 ml HNO3 (3.4. pont) + 13 ml H2O), és töltsük fel vízzel 1 literre.
3.7. 1 mg/ml foszfortartalmú standardoldat: oldjunk fel 4,387 g kálium-dihidrogén-foszfátot (KH2PO4) vízben. Töltsük fel vízzel 1 literre.
4. Eszközök
4.1. Kvarc, porcelán vagy platina hamvasztótégelyek.
4.2. Elektromos tokos kemence, 550 °C-ra beállított termosztáttal.
4.3. 250 ml-es Kjeldahl-lombik.
4.4. Mérőlombikok és precíziós pipetták.
4.5. Spektrofotométer.
4.6. Kb. 16 mm átmérőjű kémcsövek, 14,5 mm átmérőjűre keskenyedő dugóval; űrtartalom: 25-30 ml.
5. A vizsgálat módja
5.1. Oldatkészítés
A minta jellege szerint készítsünk oldatot az 5.1.1. vagy az 5.1.2. pontban leírtak szerint.
5.1.1. Általános módszer
Mérjünk le kb. 1 g-ot vagy valamivel többet a mintából, 1 mg pontossággal. Tegyük a vizsgálati mintát egy Kjeldahl-lombikba és adjunk hozzá 20 ml kénsavat (3.5. pont), rázzuk össze, hogy az anyag teljesen telítődjön a savval és ne tapadjon semmi a lombik oldalára, hevítsük és tartsuk forrásponton 10 percen át. Hagyjuk kissé lehűlni, adjunk hozzá 2 ml salétromsavat (3.4. pont), lassan hevítsük, majd hagyjuk újra kissé lehűlni, ezt követően adjunk hozzá kevés salétromsavat (3.4. pont), és forraljuk fel újra. Addig ismételjük ezt a műveletet, amíg színtelen oldatot nem kapunk. Hagyjuk hűlni, adjunk hozzá egy kevés vizet, majd dekantáljuk a folyadékot egy 500 ml-es mérőlombikba, és öblítsük ki a Kjeldahl-lombikot forró vízzel. Hagyjuk hűlni, töltsük fel vízzel térfogatra, homogenizáljuk, és szűrjük.
5.1.2. Szerves anyagokat tartalmazó, kalcium- és magnézium-dihidrogén-foszfátoktól mentes minták
Mérjünk kb. 2,5 g-ot a mintából 1 mg pontossággal egy hamvasztótégelybe. Keverjük össze a vizsgálati mintát 1 g kalcium-karbonáttal (3.1. pont), amíg teljesen egyneművé nem válik. Hamvasszuk a szárítószekrényben 550 °C-on, amíg fehér vagy szürke hamut nem kapunk (egy kis szénmaradvány nem számít). Tegyük a hamut egy 250 ml-es főzőpohárba. Adjunk hozzá 20 ml vizet és sósavat (3.2. pont), amíg a pezsgés meg nem szűnik. Adjunk hozzá még 10 ml sósavat (3.2. pont). Tegyük a főzőpoharat homokfürdőbe és hagyjuk teljesen elpárologni, hogy a kvarc szárazzá és oldhatatlanná váljon. Oldjuk fel a maradékot 10 ml salétromsavban (3.3. pont), és forraljuk a homokfürdőben vagy főzőlapon 5 percen át anélkül, hogy teljesen beszárítanánk. Dekantáljuk a folyadékot egy 500 ml-es mérőlombikba, majd öblítsük ki a főzőpoharat néhányszor forró vízzel. Hagyjuk hűlni, töltsük fel vízzel térfogatra, homogenizáljuk, és szűrjük.
5.2. Elszíneződés kialakulása és az optikai sűrűség mérése
Hígítsuk az 5.1.1. vagy az 5.1.2. pont szerint nyert szűrlet aliquot részét úgy, hogy 40 μg/ml-nél nem nagyobb foszforkoncentrációt kapjunk. Tegyünk 10 ml-t ebből az oldatból kémcsőbe (4.6. pont), és adjunk hozzá 10 ml molibdén-vanadát reagenst (3.6. pont). Homogenizáljuk és hagyjuk állni legalább 10 percen át 20 °C-on. Mérjük meg az optikai sűrűséget 430 nm-nél spektrofotométeren egy olyan oldattal szemben, amelyet úgy nyertünk, hogy 10 ml molibdén-vanadát reagenst (3.6. pont) adtunk 10 ml vízhez.
5.3. Kalibrációs görbe
A standardoldatból (3.7. pont) készítsünk egyenként 5, 10, 20, 30 és 40 μg/ml foszfort tartalmazó oldatokat. Vegyünk 10 ml-t ezekből az oldatokból, és adjunk hozzá mindegyikhez 10 ml molibdén-vanadát reagenst (3.6. pont). Homogenizáljuk és hagyjuk állni legalább 10 percen át 20 °C-on. Mérjük meg az optikai sűrűséget az 5.2. pontban leírt módon. Szerkesszük meg a kalibrációs görbét úgy, hogy az optikai sűrűséget a megfelelő foszformennyiség függvényében ábrázoljuk. A 0 és 40 μg/ml közötti koncentrációk esetében a görbe lineáris lesz.
6. Az eredmények kiszámítása
A vizsgálati minta foszfortartalmát a kalibrációs görbe segítségével határozzuk meg.
Az eredményt a minta százalékában fejezzük ki.
Ismételhetőség
Az ugyanazon mintán végzett két párhuzamos meghatározás eredményének különbsége nem haladhatja meg:
- 5 %-osnál alacsonyabb foszfortartalom esetén a magasabb érték 3 %-át,
- az 5 %-os vagy annál magasabb foszfortartalom esetén abszolút értékben a 0,15 %-ot.
O. A KLORIDOKBÓL SZÁRMAZÓ KLÓR MEGHATÁROZÁSA
1. Cél és alkalmazási terület
Ez a módszer lehetővé teszi a vízben oldható kloridokban található, hagyományosan nátrium-kloridban kifejezett klór mennyiségének meghatározását. A módszer minden takarmány esetében alkalmazható.
2. Vizsgálati alapelv
A kloridokat vízben oldjuk. Ha a termék szerves anyagot tartalmaz, tisztítjuk. Az oldatot salétromsavval enyhén savasítjuk, és a kloridokat ezüst-klorid formájában csapjuk ki ezüst-nitrát-oldat felhasználásával. Az ezüst-nitrát felesleget ammónium-tiocianát oldattal, a Volhard-módszer szerint titráljuk.
3. Reagensek
3.1. 0,1 mol/liter koncentrációjú ammónium-tiocianát-oldat.
3.2. 0,1 mol/liter koncentrációjú ezüst-nitrát-oldat.
3.3. Telített ammónium-vasszulfát-oldat (NH4)Fe(SO4)2.
3.4. Salétromsav, sűrűség: 1,38 g/ml.
3.5. Dietiléter.
3.6. Aceton.
3.7. Carrez I. oldat: vízben oldjunk fel 21,9 g cink-acetátot, Zn(CH3COO)2 2H2O és 3 g jégecetet. Töltsük fel vízzel 100 ml-re.
3.8. Carrez II. oldat: vízben oldjunk fel 10,6 g kálium-[hexaciano-ferrát(II)]-ot, K4 Fe (CN)6 3H2O. Töltsük fel vízzel 100 ml-re.
3.9. Klorid-mentes, azokat nem abszorbeáló aktív szén.
4. Eszközök
Billenődobos keverőgép: kb. 35-40 fordulatszám/perc teljesítménnyel.
5. A vizsgálat módja
5.1. Oldatkészítés
A minta jellegétől függően készítsünk oldatot az 5.1.1., az 5.1.2. vagy az 5.1.3. pontban foglaltak szerint.
Ezzel egy időben végezzünk vakpróbát az analizálandó minta kihagyásával.
5.1.1. Szerves anyagtól mentes minták
Mg-os pontossággal mérjünk le a mintából legfeljebb 10 g-ot, amely legfeljebb 3 g klórt tartalmaz kloridok formájában. 400 ml vízzel helyezzük egy 500 ml-es mérőlombikba kb. 20 °C-on. Keverjük harminc percig a keverőgépen, töltsük fel térfogatra, homogenizáljuk, és szűrjük.
5.1.2. Szerves anyagot tartalmazó minták, az 5.1.3. pontban felsorolt termékek kivételével
Mérjünk le a mintából 5 g-ot mg-os pontossággal, és 1 g aktív szénnel együtt helyezzük egy 500 ml-es mérőhengerbe. Adjunk hozzá 400 ml, körülbelül 20 °C-os vizet és 5 ml Carrez I. oldatot (3.7. pont), keverjük 30 másodpercig, majd adjunk hozzá 5 ml Carrez II. oldatot (3.8. pont). Keverjük harminc percig a keverőgépen, töltsük fel térfogatra, homogenizáljuk, és szűrjük.
5.1.3. Főzött takarmányok, lenpogácsák és lenliszt, lenlisztben gazdag termékek, valamint más, nyákban és kolloid anyagokban (például dextrinált keményítőben) gazdag termékek
Készítsük el az oldatot az 5.1.2. pontban foglaltak szerint, de ne szűrjük. Dekantáljuk (ha szükséges, centrifugáljuk), vegyünk el 100 ml-t a felülúszó folyadékból, és öntsük át egy 200 ml-es mérőlombikba. Keverjük össze acetonnal (3.6. pont), és töltsük fel térfogatra ezzel az oldószerrel, homogenizáljuk, és szűrjük.
5.2. Titrálás
Pipettázzunk egy Erlenmeyer-lombikba 25-100 ml-t (a feltételezett klórtartalom szerint) a szűrletből, amelyet az 5.1.1., az 5.1.2. vagy az 5.1.3. pontban foglaltak szerint nyertünk. Az aliquot rész nem tartalmazhat 150 mg-nál több klórt (Cl). Szükség szerint hígítsuk vízzel, legalább 50 ml-ig, adjunk hozzá 5 ml salétromsavat (3.4. pont), 2 ml telített ammónium-vasszulfát-oldatot (3.3. pont) és két csepp ammónium-tiocianát-oldatot (3.1. pont) egy nulla jelig töltött büretta segítségével. Büretta segítségével helyezzük át az ezüst-nitrát oldatot (3.2. pont) úgy, hogy 5 ml-nyi többletet kapjunk. Adjunk hozzá 5 ml dietilétert (3.5. pont), és rázzuk fel erősen, a csapadék koagulálásáig. Titráljuk az ezüst-nitrát többletet az ammónium-tiocianát oldattal (3.1. pont), amíg egy percen át tartó vörösesbarna színárnyalatot nem kapunk.
6. Az eredmények kiszámítása
A nátriumklorid százalékában kifejezett klór mennyiségét (X) a következő képlet segítségével számíthatjuk ki:
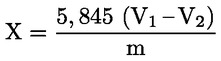
ahol:
V1 = a hozzáadott, 0,1 mol/liter koncentrációjú ezüst-nitrát oldat mennyisége, ml-ben
V2 = a titráláshoz használt, 0,1 mol/liter koncentrációjú ammónium-tiocianát oldat mennyisége, ml-ben.
m = a minta tömege az aliquotban g-ban kifejezve.
Ha a vakpróba azt jelzi, hogy 0,1 mol/liter koncentrációjú ezüst-nitrát oldat került felhasználásra, vonjuk ki ezt az értéket a (V1 - V2) mennyiségből.
7. Észrevételek
7.1. A titrálást potenciometriás vagy amperometriás módszerrel is el lehet végezni.
7.2. Olajban és zsírban rendkívül gazdag termékek esetében először dietiléterrel vagy petroléterrel végezzünk zsírtalanítást.
7.3. Halliszt esetében a titrálás Mohr-módszerrel is végezhető.
P. KARBONÁTOK MEGHATÁROZÁSA
1. Cél és alkalmazási terület
A módszer lehetővé teszi a takarmányokban lévő, hagyományosan kalcium-karbonátban kifejezett karbonátok mennyiségének meghatározását, kivéve a vas-karbonátot tartalmazó takarmányokat.
2. Vizsgálati alapelv
A karbonátokat sósavban bontjuk szét; a kibocsátott szén-dioxidot egy kalibrált csőben gyűjtjük össze, és térfogatát ismert mennyiségű kalcium-karbonát által azonos körülmények között kibocsátott mennyiséggel hasonlítjuk össze.
3. Reagensek
3.1. Sósav, sűrűség: 1,10 g/ml.
3.2. Tiszta kalcium-karbonát.
3.3. Kb. 0,05 mól/liter koncentrációjú kénsav, metilvörösindikátorral megszínezve.
4. Eszközök
Scheibler-Dietrich-készülék (lásd a függelékben szereplő ábrát) vagy annak megfelelő készülék (kalciméter).
5. A vizsgálat módja
A minta karbonáttartalmától függően mérjünk ki egy részmintát az alábbiak szerint:
a) 0,5 g az 50-100 % karbonáttartalmú termékek esetén, kalcium-karbonátban kifejezve;
b) 1 g a 40-50 % karbonáttartalmú termékek esetén, kalcium-karbonátban kifejezve;
c) 2-3 g egyéb termékek esetében.
Az esetlegesen jelen lévő karbonátok lebontása érdekében adjunk sósavat (a fenti 3.1. pont) a részmintához. A szén-dioxid mennyiségét Scheibler-Dietrich-készülékkel vagy annak megfelelő készülékkel (kalciméter) mérjük, és összehasonlítjuk a 0,5 g tiszta kalcium-karbonát által termelt szén-dioxid mennyiségével (a fenti 3.2. pont).
Minden meghatározást azonos körülmények között végezzünk, hogy elkerülhető legyen a hőmérséklet- és nyomáskülönbségekből fakadó korrekció. A meghatározást lehetőleg szabályozott hőmérsékletű helyiségben végezzük.
A Scheibler-Dietrich-készülék használatára vonatkozó eljárást a függelék részletesen ismerteti.
6. Számítás
A tiszta kalcium-karbonátként kifejezett karbonáttartalmat az alábbi képlet segítségével számítjuk ki:
| X = | V ×100 |
| V1 × 2 m |
Ahol:
X = a minta kalcium-karbonátként kifejezett %-os (m/m) karbonáttartalma.
V = a részminta által kibocsátott szén-dioxid, ml-ben.
V1 = 0,5 g CaCO3 által kibocsátott szén-dioxid, ml-ben.
m = a részminta grammban kifejezett tömege.
7. Észrevételek
7.1. Ha a Scheibler-Dietrich-készüléket használjuk, valamint a minta tömege 2 g-nál nagyobb, először töltsünk 15 ml desztillált vizet a lombikba (az alábbi függelékben szereplő ábra 4. pontja), és a vizsgálat megkezdése előtt keverjük össze sósavval (a fenti 3.1. pont). Az ellenőrző vizsgálat esetében ugyanennyi desztillált vizet használjunk.
7.2. Ha a használt készülék űrtartalma eltér a Scheibler-Dietrich-készülékétől, a mintából és a kontrollanyagból vett részt, valamint a számításokat annak megfelelően kell korrigálni.
Függelék
A Scheibler-Dietrich-készülék használatára vonatkozó részletes eljárás
Helyezzük a részmintát a készülék speciális lombikjába (az ábra 4. pontja), amelyet 10 ml sósavat (a fenti 3.1. pont) tartalmazó, törhetetlen anyagból készült, kis méretű csővel szereltek fel, és kapcsoljuk a lombikot a készülékhez. Fordítsuk el a háromfázisú csapot (az ábra 5. pontja) úgy, hogy a kalibrált cső (az ábra 1. pontja) a külső résszel kapcsolódjon. A színezett kénsavval (a fenti 3.3. pont) töltött és a kalibrált csőhöz (az ábra 1. pontja) kapcsolt mozgatható cső (az ábra 2. pontja) segítségével igazítsuk a folyadék szintjét a nulla jelhez. A csövek (az ábra 1. és 3. pontja) összekapcsolásához fordítsuk el a háromfázisú csapot (az ábra 5. pontja), és ellenőrizzük, hogy a szint nullán áll-e.
A speciális lombik (az ábra 4. pontja) megdöntésével folyassunk lassan sósavat (a fenti 3.1. pont) a részmintára. A mozgatható cső (az ábra 2. pontja) leengedésével egyenlítsük ki a nyomást. Rázzuk a speciális lombikot (az ábra 4. pontja) egészen addig, amíg a szén-dioxid-kibocsátás teljesen meg nem szűnik.
A csövekben (az ábra 1. és 2. pontja) lévő folyadék egyenlő szintre igazításával állítsuk vissza a nyomást. Néhány perc múlva, amikor a gáz mennyisége állandósul, olvassuk le az eredményt.
Végezzünk ellenőrző vizsgálatot ugyanilyen körülmények között 0,5 g kalcium-karbonáton (a fenti 3.2. pont).
SCHEIBLER-DIETRICH-KÉSZÜLÉK SZÉN-DIOXID MEGHATÁROZÁSÁRA
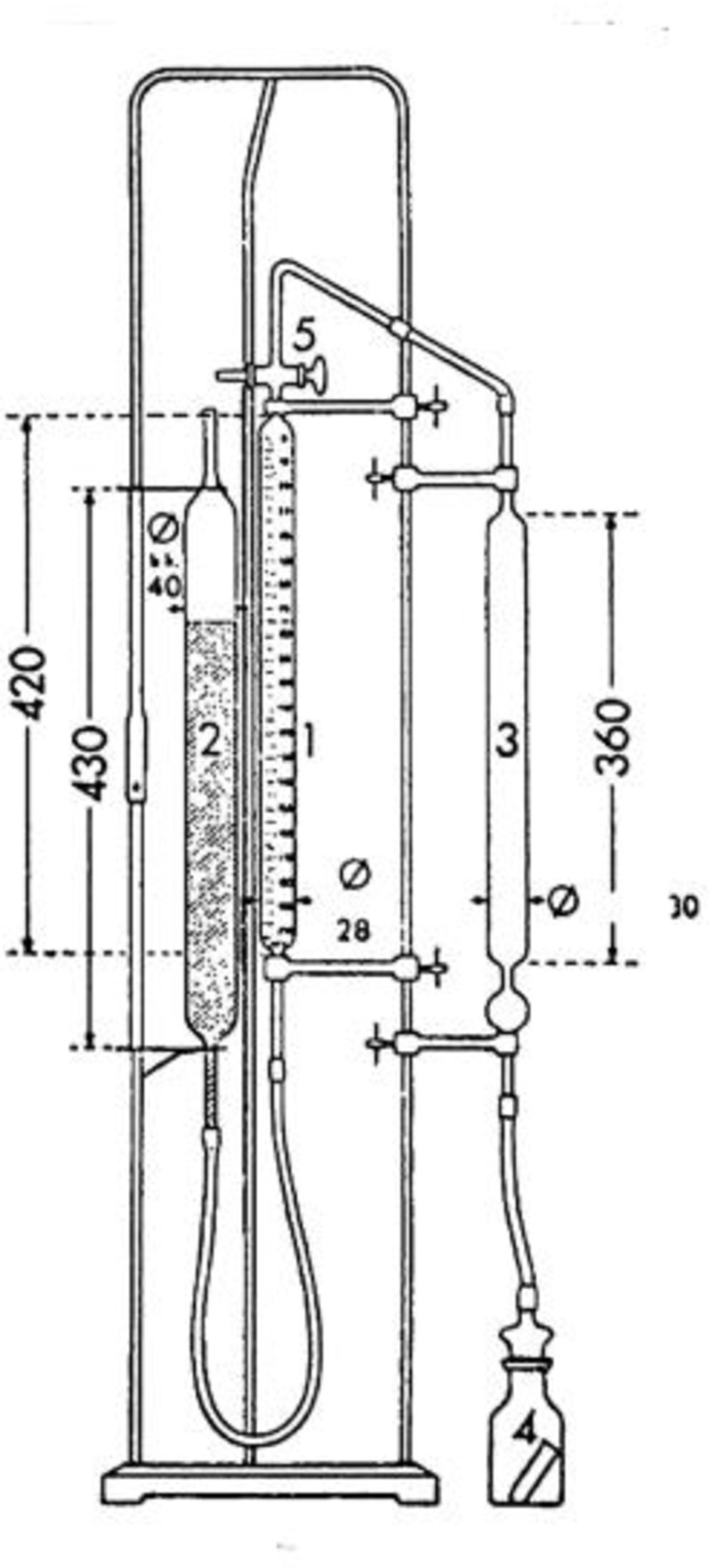
(mm-ben mérve)
IV. MELLÉKLET
ANALITIKAI MÓDSZEREK A TAKARMÁNYOKBAN LÉVŐ ENGEDÉLYEZETT ADALÉKANYAGOK SZINTJÉNEK ELLENŐRZÉSÉRE
A. Az A-vitamin meghatározása
Az A-vitamint az alábbiak szerint kell meghatározni:
- a "Takarmányok. Mintavételi és elemzési módszerek. Az A-, E- és D-vitamin-tartalom meghatározása ( 19 ). Szilárd fázisú extrakciós (SPE-) tisztítás és nagy hatékonyságú folyadékkromatográfiás (HPLC-) módszer" című EN 17547 szabványban előírt analitikai módszerrel, vagy
- fordított fázisú, nagy teljesítményű folyadékkromatográfiával (RP-HPLC) UV vagy fluoreszcens detektor használatával, az alábbi 1-9. pontban leírtak szerint.
1. Cél és alkalmazási terület
Ez a módszer lehetővé teszi a takarmányok A-vitamin- (retinol-) tartalmának meghatározását. Az A-vitamin magában foglalja az ezzel a módszerrel meghatározásra kerülő all-transz-retinil-alkoholt és annak cisz-izomerjeit. Az A-vitamin-tartalmat nemzetközi egység (NE)/kg-ban fejezzük ki. Egy NE 0,300 μg all-transz-A-vitamin-alkohol, 0,344 μg all-transz-A-vitamin-acetát vagy 0,550 μg all-transz-A-vitamin-palmitát aktivitásának felel meg.
A meghatározási határ 2 000 NE A-vitamin/kg.
2. Vizsgálati alapelv
A mintát etanolos kálium-hidroxid oldattal hidrolizáljuk, és az A-vitamint petroléterrel extraháljuk. Az oldószert bepárlással eltávolítjuk, a maradékot metanolban oldjuk, és - szükség esetén - a kívánt koncentrációra hígítjuk. Az A-vitamin-tartalmat fordított fázisú, nagy teljesítményű folyadék-kromatográfiával (RP-HPLC) határozzuk meg, UV- vagy fluoreszcens detektor alkalmazásával. A kromatográfiás paramétereket úgy válasszuk meg, hogy az all-transz-A-vitamin-alkohol és annak cisz-izomerjei ne váljanak szét.
3. Reagensek
3.1. Etanol, σ = 96 %
3.2. Petroléter, forrásponttartomány: 40-60 °C
3.3. Metil-alkohol
3.4. Kálium-hidroxid-oldat, c = 50 g/100 ml
3.5. Nátrium-aszkorbát-oldat, c = 10 g/100 ml (lásd az Észrevételek 7.7. pontját)
3.6. Nátrium-szulfid, Na2S · x H2O (x = 7 - 9)
3.6.1. Nátrium-szulfid-oldat, c = 0,5 mol/l glicerinben, ß = 120 g/l (x = 9-nél) (lásd az Észrevételek 7.8. pontját)
3.7. Fenolftaleinoldat, c = 2 g/100 ml etanolban (3.1. pont)
3.8. 2-propanol
3.9. Mozgófázis a HPLC-hez: metil-alkohol (3.3. pont) és víz keveréke, pl. 980 + 20 (v + v). A pontos arányt az alkalmazott oszlop tulajdonságai határozzák meg.
3.10. Nitrogén, oxigénmentes
3.11. All-transz-A-vitamin-acetát, fokozott tisztaságú, igazolt aktivitású, pl. 2,80 × 106 NE/g
3.11.1. All-transz-A-vitamin-acetát törzsoldat: mérjünk be 0,1 mg pontossággal 50 mg A-vitamin acetátot (3.11. pont) egy 100 ml-es mérőlombikba. Oldjuk fel 2-propanolban (3.8. pont), majd töltsük fel jelig ugyanazzal az oldószerrel. Ennek az oldatnak a névleges koncentrációja 1 400 NE A-vitamin/ml. A pontos A-vitamin-tartalmat az 5.6.3.1. pont szerint kell meghatározni.
3.12. All-transz-A-vitamin-palmitát, fokozott tisztaságú, igazolt aktivitású, pl. 1,80 × 106 NE/g
3.12.1. All-transz-A-vitamin-palmitát törzsoldat: mérjünk be 0,1 mg pontossággal 80 mg A-vitamin-palmitátot (3.12. pont) egy 100 ml-es mérőlombikba. Oldjuk fel 2-propanolban (3.8. pont), majd töltsük fel jelig ugyanazzal az oldószerrel. Ennek az oldatnak a névleges koncentrációja 1 400 NE A-vitamin/ml. A pontos tartalmat az 5.6.3.2. pont szerint kell meghatározni.
3.13. 2,6-Di-terc-butil-4-metilfenol (BHT) (lásd az Észrevételek 7.5. pontját)
4. Eszközök
4.1. Rotációs vákuumbepárló
4.2. Barna üvegedények
4.2.1. 500 ml-es, lapos fenekű vagy Erlenmeyer-lombikok, csiszolt üvegnyakkal
4.2.2. Csiszolt üvegdugós, vékony nyakú, 10, 25, 100 és 500 ml-es mérőlombikok
4.2.3. 1 000 ml-es, kúpos választótölcsérek, csiszolt üvegdugóval
4.2.4. 250 ml-es, körte alakú lombikok, csiszolt üvegnyakkal
4.3. Allihn-féle hűtő, köpeny hossza 300 mm, csiszolt üvegcsatlakozóval, adapteres gázadagoló csővel
4.4. Redős szűrőpapír a fázisok szétválasztásához, 185 mm átmérőjű (pl. Schleicher & Schuell 597 HY 1/2)
4.5. HPLC-berendezés, befecskendező rendszerrel
4.5.1. Folyadékkromatográfiás oszlop, 250 mm × 4 mm, C18, 5 vagy 10 μm töltet vagy azzal egyenértékű berendezés (teljesítménykritérium: az alkalmazott HPLC paraméterek mellett az összes retinol-izomer csak egyetlen csúcsot adjon)
4.5.2. Állítható hullámhosszúságú UV- vagy fluoreszcens detektor,
4.6. Spektrofotométer 10 mm-es kvarcküvettákkal
4.7. Vízfürdő, mágneses keverővel
4.8. Extrahálókészülék (lásd az 1. ábrát), amely az alábbiakból áll:
4.8.1. 1 l térfogatú üveghenger, csiszolt üvegnyakkal és -dugóval
4.8.2. Oldalirányú bevezetőcsővel és állítható, a feltét közepén áthaladó csővel ellátott, csiszolt üvegből készült feltét. Az állítható csőnek U alakú alsó véggel, az ellenkező oldalon pedig fúvókával kell rendelkeznie, hogy a hengerben levő felső folyadékréteg átvihető legyen a választótölcsérbe.
5. A vizsgálat módja
Megjegyzés: Az A-vitamin (UV-) fényre és oxidációra érzékeny. Minden műveletet fénytől elzárva (barna üvegedényekben vagy alumíniumfóliába tekert üvegedényekben), és oxigén kizárásával (nitrogénáramban) kell végezni. Az extrahálás során a folyadék feletti levegőt nitrogénnel kell eltávolítani (a dugó időszakonkénti meglazításával kerüljük el a túlzottan nagy nyomás kialakulását).
5.1. A minta előkészítése
Ügyelve a hőképződés elkerülésére, őröljük a mintát annyira, hogy az átjusson egy 1 mm-es szemnagyságú szitán. Az őrlést közvetlenül a súlymérés és a szappanosítás előtt kell végezni, különben A-vitamin-veszteség léphet fel. Ne őröljük a mintá(ka)t, ha megfelelő a szemcsenagyság eloszlása (pl. előkeverékek és takarmány-adalékanyagok).
5.2. Szappanosítás
Az A-vitamin-tartalomtól függően mérjünk be 1 mg pontossággal 2-25 g mintát egy 500 ml-es, lapos fenekű vagy Erlenmeyer-lombikba (4.2.1. pont). Alacsony koncentrációk esetében a minta súlya növelhető annak érdekében, hogy elegendő szemcse legyen a vizsgálati adagban. Keverés közben adjunk hozzá egymást követően 130 ml etanolt (3.1. pont), körülbelül 100 mg BHT-t (3,13. pont), 2 ml nátrium-aszkorbát-oldatot (3.5. pont) és 2 ml nátrium-szulfid-oldatot (3.6. pont). Illesszünk a lombikhoz hűtőt (4.3. pont), és merítsük mágneses keverővel felszerelt vízfürdőbe (4.7. pont). Hevítsük forrásig a lombik tartalmát, majd hagyjuk visszacsepegni öt percig. Ezután a hűtőn keresztül (4.3. pont) adjunk hozzá 25 ml kálium-hidroxid-oldatot (3.4. pont), és hagyjuk visszacsepegni újabb 25 percig, lassú nitrogénáramban történő keverés közben. Ezt követően kb. 20 ml vízzel öblítsük a hűtőt, és hűtsük le a lombik tartalmát szobahőmérsékletűre.
5.3. Extrahálás
250 ml vízzel történő átöblítéssel dekantáljuk a szappanosítási oldatot teljes mennyiségében egy 1 000 ml-es választótölcsérbe (4.2.3. pont) vagy az extrahálókészülékbe (4.8. pont). Öblítsük a szappanosításhoz használt lombikot egymást követően 25 ml etanollal (3.1. pont) és 100 ml petroléterrel (3.2. pont), majd az öblítő oldószereket töltsük a választótölcsérbe vagy az extrahálókészülékbe. Az egyesített oldatokban a víz és az etanol aránya körülbelül 2:1 legyen. Rázassuk erőteljesen két percig, majd két percen át hagyjuk ülepedni.
5.3.1. Extrahálás választótölcsérrel (4.2.3. pont)
Amikor a rétegek már elkülönültek (lásd az Észrevételek 7.3. pontját), vigyük át a petroléterréteget egy másik választótölcsérbe (4.2.3. pont). Az extrahálást ismételjük meg kétszer 100 ml petroléterrel (3.2. pont), kétszer pedig 50 ml petroléterrel (3.2. pont).
Az egyesített kivonatokat a választótölcsérben mossuk kétszer 100-100 ml vízadaggal, lassú keverés közben (az emulzióképződés megakadályozása céljából), majd többszöri rázatás közben további 100 ml-es vízadagokkal egészen addig, amíg a víz, fenolftaleinoldat (3.7. pont) hozzáadását követően színtelen nem marad (négyszeri mosás rendszerint elegendő). A szuszpendált víz eltávolítása céljából, a mosott kivonatot szűrjük át a fázisok elkülönítésére szolgáló száraz, redős szűrőpapíron (4.4. pont) egy 500 ml-es mérőlombikba (4.2.2. pont). A választótölcsért és a szűrőt öblítsük 50 ml petroléterrel (3.2. pont), töltsük fel jelig petroléterrel (3.2. pont), és a tartalmát alaposan keverjük össze.
5.3.2. Extrahálás extrahálókészülékkel (4.8. pont)
Amikor a rétegek szétváltak (lásd az Észrevételek 7.3. pontját), helyezzük az üveghenger (4.8.1. pont) dugója helyére a csiszolt üvegfeltétet (4.8.2. pont), és helyezzük az állítható cső U alakú, alsó végét úgy, hogy az éppen a rétegek találkozási felülete felett legyen. Egy nitrogénvezetékből nyomást gyakorolva az oldalcsőre, vigyük át a felső petroléterréteget egy 1 000 ml-es választótölcsérbe (4.2.3. pont). Mérjünk 100 ml petrolétert (3.2. pont) az üveghengerbe, helyezzük vissza dugót, és alaposan rázzuk össze. Hagyjuk szétválni a rétegeket, majd a felső réteget vigyük át a választótölcsérbe az előbbiekben ismertetett módon. Ismételjük meg az extrahálási műveletet további 100 ml petroléterrel (3.2. pont), majd kétszer 50 ml petroléterrel (3.2. pont), és vigyük át a petroléterrétegeket a választótölcsérbe.
Mossuk az egyesített petroléterkivonatokat az 5.3.1. pontban leírt módon, majd folytassuk a műveletet az említett pontban leírtak szerint.
5.4. A mintaoldat elkészítése a HPLC-hez
Pipettázzuk a petroléteroldat aliquot részét (5.3.1. vagy 5.3.2. pont) egy 250 ml-es, körte alakú lombikba (4.2.4. pont). Az oldószert pároljuk még éppen nedves állapotúra a rotációs bepárlókészüléken (4.1. pont), csökkentett nyomáson, legfeljebb 40 °C-os vízfürdőben. Nitrogén (3.10. pont) bejuttatásával állítsuk vissza az atmoszferikus nyomást, és vegyük le a lombikot a rotációs bepárlókészülékről. Nitrogén beáramoltatásával (3.10. pont) távolítsuk el a maradék oldószert, és a maradékot azonnal oldjuk fel ismert térfogatú (10-100 ml) metil-alkoholban (3.3. pont) (az A-vitamin koncentrációjának 5 NE/ml és 30 NE/ml között kell lennie).
5.5. Meghatározás HPLC-vel
Az A-vitamin elválasztása fordított fázisú C18 oszlopon (4.5.1. pont) történik, koncentrációját pedig UV-detektor (325 nm) vagy fluoreszcens detektor (gerjesztés: 325 nm, emisszió: 475 nm) (4.5.2. pont) segítségével mérjük.
Fecskendezzünk be az 5.4. pont szerint nyert metil-alkoholos oldatból egy aliquot részt (pl. 20 μl), és eluáljuk a mozgófázissal (3.9. pont). Ugyanabból a mintaoldatból többszöri befecskendezés után számítsuk ki a mintaoldathoz tartozó átlagos csúcsmagasságot (területet), valamint a kalibrálóoldatból (5.6.2. pont) többszöri befecskendezés után számítsuk ki a kalibrálóoldathoz tartozó átlagos csúcsmagasságokat (területeket).
HPLC paraméterek
Az alábbi vizsgálati feltételek útmutatásként szolgálnak, ettől eltérő feltételeket is lehet használni, feltéve, hogy ezzel egyenértékű eredményt adnak.
| Folyadékkromatográfiás oszlop (4.5.1. pont): | C18, 250 mm × 4 mm, 5 vagy 10 μm-es töltettel, vagy ezzel egyenértékű berendezés. |
| Mozgófázis (3.9. pont): | metil-alkohol (3.3. pont) és víz keveréke, pl. 980 + 20 (v + v). |
| Átáramlási sebesség: | 1–2 ml/perc |
| Detektor (4.5.2. pont): | UV-detektor (325 nm) vagy fluoreszcens detektor (gerjesztés: 325 nm/emisszió: 475 nm) |
5.6. Kalibrálás
5.6.1. A standard munkaoldatok elkészítése
Pipettázzunk az A-vitamin-acetát törzsoldatból (3.11.1. pont) 20 ml-t vagy az A-vitamin-palmitát törzsoldatból (3.12.1. pont) 20 ml-t egy 500 ml-es, lapos fenekű vagy Erlenmeyer-lombikba (4.2.1. pont), és hidrolizáljuk az 5.2. pontban leírt módon, de BHT hozzáadása nélkül. Ezt követően extraháljuk petroléterrel (3.2. pont) az 5.3. pont szerint, és töltsük fel 500 ml-re petroléterrel (3.2. pont). Ebből a kivonatból 100 ml-t pároljunk még éppen nedves állapotig a rotációs bepárlókészüléken (lásd 5.4. pont), nitrogénáramoltatással távolítsuk el a maradék oldószert (3.10. pont), majd oldjuk a maradékot 10,0 ml metil-alkoholban (3.3. pont). Ennek az oldatnak a névleges koncentrációja 560 NE A-vitamin/ml. A pontos A-vitamin-tartalmat az 5.6.3.3. pont szerint kell meghatározni. A standard munkaoldatot felhasználás előtt frissen kell elkészíteni.
Ebből a standard munkaoldatból 2,0 ml-t pipettázzunk egy 20 ml-es mérőlombikba, majd töltsük azt fel jelig metil-alkohollal (3.3. pont), és keverjük össze. E hígított standard munkaoldat névleges koncentrációja 56 NE A-vitamin/ml.
5.6.2. A kalibrálóoldatok és a kalibrációs görbe elkészítése
A hígított standard munkaoldatból vigyünk át 1,0, 2,0, 5,0 és 10,0 ml-t 20 ml-es mérőlombikokba, töltsük fel a lombikokat jelig metil-alkohollal (3.3. pont), és keverjük össze a tartalmukat. Ezeknek az oldatoknak a névleges koncentrációja 2,8, 5,6, 14,0 és 28,0 NE A-vitamin/ml.
Minden kalibrálóoldatból fecskendezzünk be 20 μl-t több alkalommal, és határozzuk meg az átlagos csúcsmagasságokat (területeket). Figyelembe véve az UV-ellenőrzés (5.6.3.3. pont) eredményeit, az átlagos csúcsmagasságok (területek) alapján szerkesszük meg a kalibrációs görbét.
5.6.3. A standardoldatok UV hitelesítése
5.6.3.1. A-vitamin-acetát törzsoldat
Pipettázzunk az A-vitamin-acetát törzsoldatból (3.11.1. pont) 2,0 ml-t egy 50 ml-es mérőlombikba (4.2.2. pont), és a lombikot töltsük fel jelig 2-propanollal (3.8. pont). Ennek az oldatnak a névleges koncentrációja 56 NE A-vitamin/ml. Ebből a hígított A-vitamin-acetát oldatból pipettázzunk 3,0 ml-t egy 25 ml-es mérőlombikba, és a lombikot töltsük fel jelig 2-propanollal (3.8. pont). Ennek az oldatnak a névleges koncentrációja 6,72 NE A-vitamin/ml. Mérjük meg ennek az oldatnak az UV-spektrumát spektrofotométeren (4.6. pont) 2-propanollal (3.8. pont) szemben, 300 nm és 400 nm között. Az extinkció maximális értékének 325 nm és 327 nm között kell lennie.
Az A-vitamin-tartalom kiszámítása:
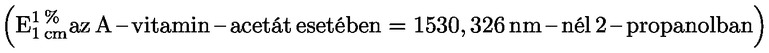
5.6.3.2. A-vitamin-palmitát törzsoldat
Pipettázzunk az A-vitamin-palmitát törzsoldatból (3.12.1. pont) 2,0 ml-t egy 50 ml-es mérőlombikba (4.2.2. pont), és a lombikot töltsük fel jelig 2-propanollal (3.8. pont). Ennek az oldatnak a névleges koncentrációja 56 NE A-vitamin/ml. Ebből a hígított A-vitamin-palmitát oldatból pipettázzunk 3,0 ml-t egy 25 ml-es mérőlombikba, és a lombikot töltsük fel jelig 2-propanollal (3.8. pont). Ennek az oldatnak a névleges koncentrációja 6,72 NE A-vitamin/ml. Mérjük meg ennek az oldatnak az UV-spektrumát spektrofotométeren (4.6. pont) 2-propanollal (3.8. pont) szemben, 300 nm és 400 nm között. Az extinkció maximális értékének 325 nm és 327 nm között kell lennie.
Az A-vitamin-tartalom kiszámítása:
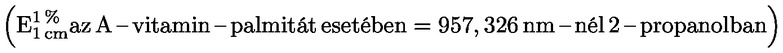
5.6.3.3. A-vitamin standard munkaoldat
Pipettázzunk az 5.6.1. pont szerint elkészített, hígítatlan A-vitamin standard munkaoldatból 3,0 ml-t egy 50 ml-es mérőlombikba (4.2.2. pont), és a lombikot töltsük fel jelig 2-propanollal (3.8. pont). Ebből az oldatból pipettázzunk 5,0 ml-t egy 25 ml-es mérőlombikba, és a lombikot töltsük fel jelig 2-propanollal (3.8. pont). Ennek az oldatnak a névleges koncentrációja 6,72 NE A-vitamin/ml. Mérjük meg ennek az oldatnak az UV-spektrumát spektrofotométeren (4.6. pont) 2-propanollal (3.8. pont) szemben, 300 nm és 400 nm között. Az extinkció maximális értékének 325 nm és 327 nm között kell lennie.
Az A-vitamin-tartalom kiszámítása:
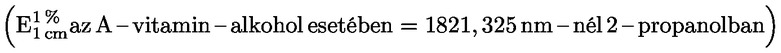
6. Az eredmények kiszámítása
A mintaoldat A-vitamin csúcsainak átlagos magasságából (területéből) határozzuk meg a mintaoldat koncentrációját NE/ml-ben, a kalibrációs görbét (5.6.2. pont) használva.
Az A-vitamin-tartalmat (w) NE/kg mintában a következő képlet adja:
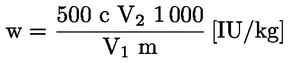
ahol:
c = va mintaoldat (5.4. pont) A-vitamin-koncentrációja, NE/ml-ben
V1 = a mintaoldat (5.4. pont) térfogata, ml-ben
V2 = az 5.4. pont szerint vett aliquot rész térfogata, ml-ben
m = a vizsgálati adag tömege g-ban
7. Észrevételek
7.1. Alacsony A-vitamin-koncentrációjú minták esetében a HPLC-meghatározáshoz hasznos lehet két szappanosítás (kimért mennyiség: 25 g) petroléterkivonatait egy mintaoldatban egyesíteni.
7.2. Az analizáláshoz bemért minta nem tartalmazhat 2 g-nál több zsírt.
7.3. Ha a fázisok nem válnak szét, adjunk az elegyhez körülbelül 10 ml etanolt (3.1. pont) az emulzió megbontása céljából.
7.4. Tőkehalmájolaj és más tiszta zsírok esetében a szappanosítási időt 45-60 percre kell meghosszabbítani.
7.5. BHT helyett hidrokinon is használható.
7.6. Normálfázisú oszlop használata esetén a retinol-izomerek szétválaszthatók. De ebben az esetben valamennyi cisz- és transz-izomer csúcsmagasságát (területét) összesíteni kell a számításokhoz.
7.7. Nátrium-aszkorbát-oldat helyett mintegy 150 mg aszkorbinsav is használható.
7.8. Nátrium-szulfid-oldat helyett mintegy 50 mg EDTA is használható.
7.9. A tejpótlók A-vitamin tartalmának analízise esetében kiemelt figyelmet kell fordítani a következőkre:
- szappanosításkor (5.2. pont): a mintában jelen lévő zsír mennyisége miatt szükséges lehet a kálium-hidroxid oldat (3.4. pont) mennyiségének növelése,
- extraháláskor (5.3. pont): az emulziók jelenléte miatt szükséges lehet a víz/etanol 2:1 arány módosítása.
Annak ellenőrzése érdekében, hogy a használt analitikai módszer megbízható eredményt ad-e az adott mátrixon (tejpótló), visszanyerési próbát kell végezni egy további vizsgálati adagon. Ha a visszanyerés 80 % alatti, az analízis eredményét korrigálni kell a visszanyeréssel.
8. Ismételhetőség
Ugyanazon a mintán végzett két párhuzamos meghatározás eredményének különbsége nem haladhatja meg a magasabb érték 15 %-át.
9. A körvizsgálat eredményei ( 20 )
| Előkeverékek | Előkeverék-takarmány | Ásványi koncentrátumok | Fehérjetakarmány | Malactáp | |
| L | 13 | 12 | 13 | 12 | 13 |
| n | 48 | 45 | 47 | 46 | 49 |
| átlag [NE/kg] | 17,02 × 106 | 1,21 × 106 | 537 100 | 151 800 | 18 070 |
| sr [NE/kg] | 0,51 × 106 | 0,039 × 106 | 22 080 | 12 280 | 682 |
| r [NE/kg] | 1,43 × 106 | 0,109 × 106 | 61 824 | 34 384 | 1 910 |
| CVr [%] | 3,0 | 3,5 | 4,1 | 8,1 | 3,8 |
| sR [NE/kg] | 1,36 × 106 | 0,069 × 106 | 46 300 | 23 060 | 3 614 |
| R [NE/kg] | 3,81 × 106 | 0,193 × 106 | 129 640 | 64 568 | 10 119 |
| CVR [%] | 8,0 | 6,2 | 8,6 | 15 | 20 |
| L: a laboratóriumok száma n: az egyedi értékek száma sr: az ismételhetőség szórása sR: a reprodukálhatóság szórása r: az ismételhetőség R: a reprodukálhatóság CVr: az ismételhetőség relatív szórása CVR: a reprodukálhatóság relatív szórása | |||||
1. ábra
Extrahálókészülék (4.8)
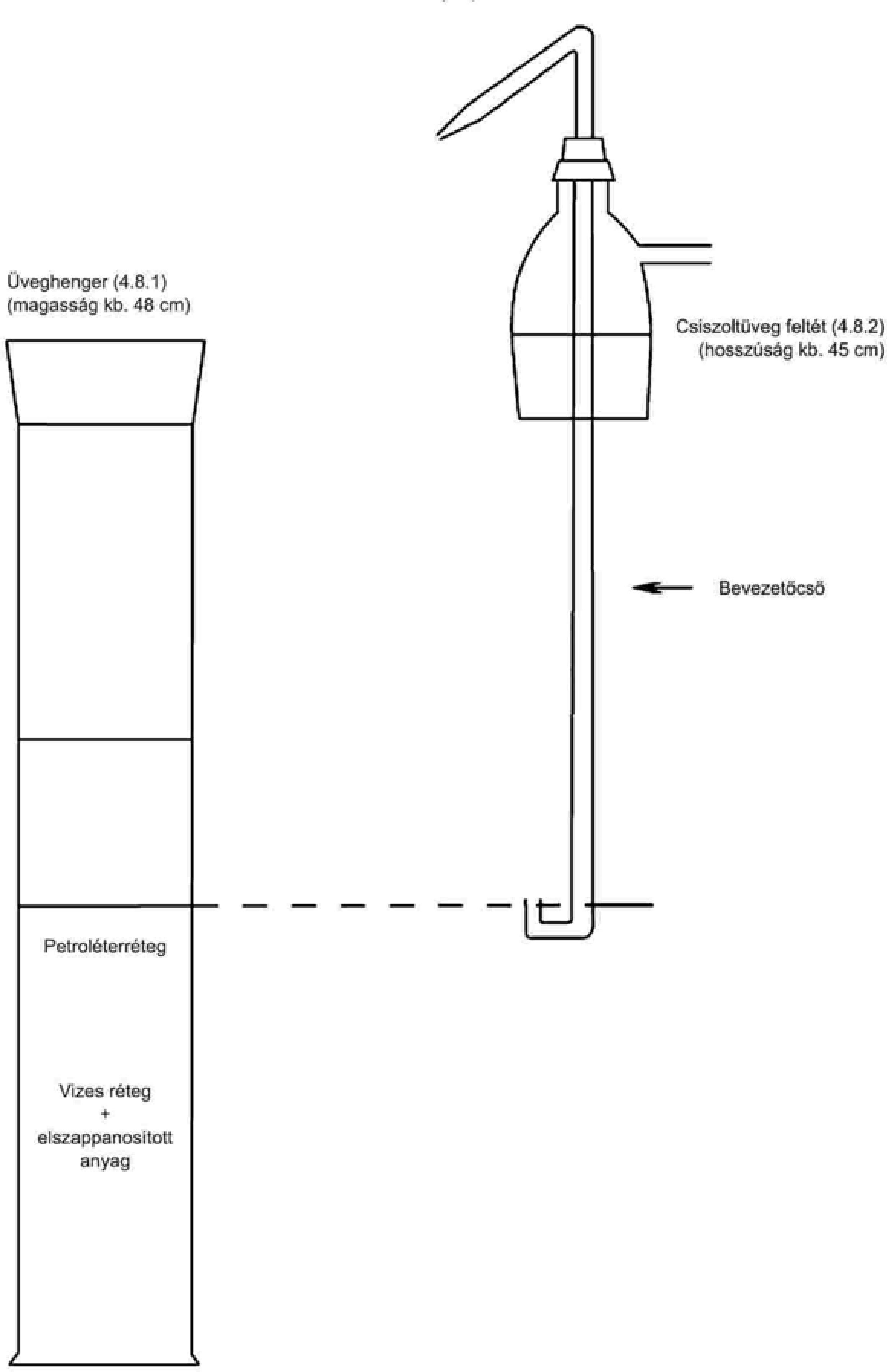
B. Az E-vitamin meghatározása
Az E-vitamint az alábbiak szerint kell meghatározni:
- a "Takarmányok. Mintavételi és elemzési módszerek. Az A-, E- és D-vitamin-tartalom meghatározása ( 21 ). Szilárd fázisú extrakciós (SPE-) tisztítás és nagy hatékonyságú folyadékkromatográfiás (HPLC-) módszer" című EN 17547 szabványban előírt analitikai módszerrel, vagy
- fordított fázisú, nagy teljesítményű folyadékkromatográfiával (RP-HPLC) UV vagy fluoreszcens detektor használatával, az alábbi 1-9. pontban leírtak szerint.
1. Cél és alkalmazási terület
Ez a módszer lehetővé teszi a takarmányok E-vitamin- (retinol-) tartalmának meghatározását. Az E-vitamin-tartalmat mg DL-α-tokoferol-acetát/kg-ban fejezzük ki. Egy mg DL-α-tokoferol-acetát 0,91 mg DL-α-tokoferolnak (E-vitaminnak) felel meg.
A meghatározási határ 2 mg E-vitamin/kg. Ez a meghatározási határ csak fluoreszcens detektorral érhető el. UV-detektorral a meghatározási határ 10 mg/kg.
2. Vizsgálati alapelv
A mintát etanolos kálium-hidroxid oldattal hidrolizáljuk, és az E-vitamint petroléterrel extraháljuk. Az oldószert bepárlással eltávolítjuk, a maradékot metanolban oldjuk, és - szükség esetén - a kívánt koncentrációra hígítjuk. Az E-vitamin-tartalmat fordított fázisú, nagy teljesítményű folyadék-kromatográfiával (RP-HPLC) határozzuk meg, fluoreszcens vagy UV-detektor alkalmazásával.
3. Reagensek
3.1. Etanol, σ = 96 %
3.2. Petroléter, forrásponttartomány: 40-60 °C
3.3. Metil-alkohol
3.4. Kálium-hidroxid-oldat, c = 50 g/100 ml
3.5. Nátrium-aszkorbát-oldat, c = 10 g/100 ml (lásd az Észrevételek 7.7. pontját)
3.6. Nátrium-szulfid, Na2S · x H2O (x = 7 - 9)
3.6.1. Nátrium-szulfid-oldat, c = 0,5 mol/l glicerinben, β = 120 g/l (x = 9-nél) (lásd az Észrevételek 7.8. pontját)
3.7. Fenolftaleinoldat, c = 2 g/100 ml etanolban (3.1. pont)
3.8. Mozgófázis a HPLC-hez: metil-alkohol (3.3. pont) és víz keveréke, pl. 980 + 20 (v + v). A pontos arányt az alkalmazott oszlop tulajdonságai határozzák meg.
3.9. Nitrogén, oxigénmentes
3.10. DL-α-tokoferol-acetát, fokozottan tiszta, igazolt aktivitású
3.10.1. DL-α-tokoferol-acetát törzsoldat: mérjünk be 0,1 mg-os pontossággal 100 mg DL-α-tokoferol-acetátot (3.10. pont) egy 100 ml-es mérőlombikba. Oldjuk fel etanolban (3.1. pont), majd töltsük fel jelig ugyanezzel az oldószerrel. Ebből az oldatból 1 ml 1 mg DL-α-tokoferol-acetátot tartalmaz. (Az UV-ellenőrzést lásd az 5.6.1.3. pontban; a stabilizálást lásd az Észrevételek 7.4. pontjában.)
3.11. DL-α-tokoferol, fokozottan tiszta, igazolt aktivitású
3.11.1. DL-α-tocopherol törzsoldat: mérjünk be 0,1 mg-os pontossággal 100 mg DL-α-tokoferolt (3.11. pont) egy 100 ml-es mérőlombikba. Oldjuk fel etanolban (3.1. pont), majd töltsük fel jelig ugyanezzel az oldószerrel. Ebből az oldatból 1 ml 1 mg DL-α-tokoferolt tartalmaz. (Az UV-ellenőrzést lásd az 5.6.2.3. pontban; (Az UV-ellenőrzést lásd az 5.6.1.3. pontban;
3.12. 2,6-di-terc-butil-4-metilfenol (BHT) (lásd az Észrevételek 7.5. pontját)
4. Eszközök
4.1. Rotációs, vékonyfilmes bepárló
4.2. Barna üvegedények
4.2.1. 500 ml-es, lapos fenekű vagy Erlenmeyer-lombikok, csiszolt üvegnyakkal
4.2.2. Csiszolt üvegdugós, vékony nyakú, 10, 25, 100 és 500 ml-es mérőlombikok
4.2.3. 1 000 ml-es, kúpos választótölcsérek, csiszolt üvegdugóval
4.2.4. 250 ml-es, körte alakú lombikok, csiszolt üvegnyakkal
4.3. Allihn-féle hűtő, köpeny hossza 300 mm, csiszolt üvegcsatlakozóval, adapteres gázadagoló csővel
4.4. Redős szűrőpapír a fázisok szétválasztásához, 185 mm átmérőjű (pl. Schleicher & Schuell 597 HY 1/2)
4.5. HPLC-berendezés, befecskendező rendszerrel
4.5.1. Folyadék-kromatográfiás oszlop, 250 mm × 4 mm, C18, 5 vagy 10 μm töltet vagy azzal egyenértékű berendezés
4.5.2. Állítható hullámhosszúságú, fluoreszcens- vagy UV-detektor
4.6. Spektrofotométer 10 mm-es kvarcküvettákkal
4.7. Vízfürdő, mágneses keverővel
4.8. Extrahálókészülék (lásd az 1. ábrát), amely az alábbiakból áll:
4.8.1. 1 l térfogatú üveghenger, csiszolt üvegnyakkal és -dugóval
4.8.2. Oldalirányú bevezetőcsővel és állítható, a feltét közepén áthaladó csővel ellátott, csiszolt üvegből készült feltét. Az állítható csőnek U alakú alsó véggel, az ellenkező oldalon pedig fúvókával kell rendelkeznie, hogy a hengerben levő felső folyadékréteg átvihető legyen a választótölcsérbe.
5. A vizsgálat módja
Megjegyzés: Az E-vitamin (UV-) fényre és oxidációra érzékeny. Minden műveletet fénytől elzárva (barna üvegedényekben vagy alumíniumfóliába tekert üvegedényekben), és oxigén kizárásával (nitrogénáramban) kell végezni. Az extrahálás során a folyadék feletti levegőt nitrogénnel kell eltávolítani (a dugó időszakonkénti meglazításával kerüljük el a túlzottan nagy nyomás kialakulását).
5.1. A minta előkészítése
Ügyelve a hőképződés elkerülésére, őröljük a mintát annyira, hogy az átjusson egy 1 mm-es szemnagyságú szitán. Az őrlést közvetlenül a súlymérés és a szappanosítás előtt kell végezni, különben E-vitamin-veszteség léphet fel.
5.2. Szappanosítás
Az E-vitamin-tartalomtól függően mérjünk be 0,01 g pontossággal 2-25 g mintát egy 500 ml-es, lapos fenekű vagy Erlenmeyer-lombikba (4.2.1. pont). Keverés közben adjunk hozzá egymást követően 130 ml etanolt (3.1. pont), körülbelül 100 mg BHT-t (3.12. pont), 2 ml nátrium-aszkorbát-oldatot (3.5. pont) és 2 ml nátrium-szulfid-oldatot (3.6. pont). Illesszük a lombikhoz a hűtőt (4.3. pont), és merítsük a mágneses keverővel felszerelt vízfürdőbe (4.7. pont). Hevítsük forrásig a lombik tartalmát, majd hagyjuk visszacsepegni öt percig. Ezután a hűtőn keresztül (4.3. pont) adjunk hozzá 25 ml kálium-hidroxid-oldatot (3.4. pont), és hagyjuk visszacsepegni újabb 25 percig, lassú nitrogénáramban történő keverés közben. Ezt követően kb. 20 ml vízzel öblítsük a hűtőt, és hűtsük le a lombik tartalmát szobahőmérsékletűre.
5.3. Extrahálás
250 ml vízzel történő átöblítéssel dekantáljuk a szappanosítási oldatot teljes mennyiségében egy 1 000 ml-es választótölcsérbe (4.2.3. pont) vagy az extrahálókészülékbe (4.8. pont). Öblítsük a szappanosításhoz használt lombikot egymást követően 25 ml etanollal (3.1. pont) és 100 ml petroléterrel (3.2. pont), majd az öblítő oldószereket töltsük a választótölcsérbe vagy az extrahálókészülékbe. Az egyesített oldatokban a víz és az etanol aránya körülbelül 2:1 legyen. Rázassuk erőteljesen két percig, majd két percen át hagyjuk ülepedni.
5.3.1. Extrahálás választótölcsérrel (4.2.3. pont)
Amikor a rétegek már elkülönültek (lásd az Észrevételek 7.3. pontját), vigyük át a petroléterréteget egy másik választótölcsérbe (4.2.3. pont). Az extrahálást ismételjük meg kétszer 100 ml petroléterrel (3.2. pont), kétszer pedig 50 ml petroléterrel (3.2. pont).
Az egyesített kivonatokat a választótölcsérben mossuk kétszer 100-100 ml vízadaggal, lassú keverés közben (az emulzióképződés megakadályozása céljából), majd többszöri rázatás közben további 100 ml-es vízadagokkal egészen addig, amíg a víz, fenolftaleinoldat (3.7. pont) hozzáadását követően színtelen nem marad (négyszeri mosás rendszerint elegendő). A szuszpendált víz eltávolítása céljából, a mosott kivonatot szűrjük át a fázisok elkülönítésére szolgáló száraz, redős szűrőpapíron (4.4. pont) egy 500 ml-es mérőlombikba (4.2.2. pont). A választótölcsért és a szűrőt öblítsük 50 ml petroléterrel (3.2. pont), töltsük fel jelig petroléterrel (3.2. pont), és a tartalmát alaposan keverjük össze.
5.3.2. Extrahálás extrahálókészülékkel (4.8. pont)
Amikor a rétegek szétváltak (lásd az Észrevételek 7.3. pontját), helyezzük az üveghenger (4.8.1. pont) dugója helyére a csiszolt üvegfeltétet (4.8.2. pont), és helyezzük az állítható cső U alakú, alsó végét úgy, hogy az éppen a rétegek találkozási felülete felett legyen. Egy nitrogénvezetékből nyomást gyakorolva az oldalcsőre, vigyük át a felső petroléterréteget egy 1 000 ml-es választótölcsérbe (4.2.3. pont). Mérjünk 100 ml petrolétert (3.2. pont) az üveghengerbe, helyezzük vissza dugót, és alaposan rázzuk össze. Hagyjuk szétválni a rétegeket, majd a felső réteget vigyük át a választótölcsérbe az előbbiekben ismertetett módon. Ismételjük meg az extrahálási műveletet további 100 ml petroléterrel (3.2. pont), majd kétszer 50 ml petroléterrel (3.2. pont), és vigyük át a petroléterrétegeket a választótölcsérbe.
Mossuk az egyesített petroléterkivonatokat az 5.3.1. pontban leírt módon, majd folytassuk a műveletet az ott leírtak szerint.
5.4. A mintaoldat elkészítése a HPLC-hez
Pipettázzuk a petroléteroldat aliquot részét (5.3.1. vagy 5.3.2. pont) egy 250 ml-es, körte alakú lombikba (4.2.4. pont). Az oldószert pároljuk még éppen nedves állapotúra a rotációs bepárlókészüléken (4.1. pont), csökkentett nyomáson, legfeljebb 40 °C-os vízfürdőben. Nitrogén (3.9. pont) bejuttatásával állítsuk vissza az atmoszferikus nyomást, és vegyük le a lombikot a rotációs bepárlókészülékről. Nitrogénáram (3.9. pont) segítségével távolítsuk el a maradék oldószert, és a maradékot azonnal oldjuk fel ismert térfogatú (10-100 ml) metil-alkoholban (3.3. pont) (a DL-α-tokoferol koncentrációjának 5 μg/ml és 30 μg/ml között kell lennie).
5.5. Meghatározás HPLC-vel
Az E-vitamin elválasztása fordított fázisú C18 oszlopon (4.5.1. pont) történik, koncentrációját fluoreszcens detektor (gerjesztés: 295 nm, emisszió: 330 nm) vagy UV-detektor (292 nm) (4.5.2. pont) segítségével mérjük.
Fecskendezzünk be az 5.4. pont szerint nyert metil-alkoholos oldatból egy aliquot részt (pl. 20 μl), és eluáljuk a mozgófázissal (3.8. pont). Ugyanabból a mintaoldatból többször befecskendezve számítsuk ki a mintaoldathoz tartozó átlagos csúcsmagasságokat (területeket), valamint a kalibrálóoldatokból (5.6.2. pont) többször befecskendezve a kalibrálóoldatokhoz tartozó átlagos csúcsmagasságokat (területeket).
HPLC paraméterek
Az alábbi vizsgálati feltételek útmutatásként szolgálnak, ettől eltérő feltételeket is lehet használni, feltéve, hogy ezzel egyenértékű eredményt adnak.
| Folyadékkromatográfiás oszlop (4.5.1. pont): | C18, 250 mm × 4 mm, 5 vagy 10 μm-es töltettel, vagy ezzel egyenértékű berendezés. |
| Mozgófázis (3,8. pont): | metil-alkohol (3.3. pont) és víz keveréke, pl. 980 + 20 (v + v). |
| Átáramlási sebesség: | 1–2 ml/perc |
| Detektor (4.5.2. pont) | UV-detektor (gerjesztés: 295 nm/ emisszió: 330 nm) vagy UV-detektor (292 nm) |
5.6. Kalibrálás (DL-α-tokoferol-acetát vagy DL-α-tokoferol)
5.6.1. DL-α-tokoferol-acetát standard
5.6.1.1. A standard munkaoldat elkészítése
Pipettázzunk a DL-α-tokoferol-acetát törzsoldatból (3.10.1. pont) 25 ml-t egy 500 ml-es, lapos fenekű vagy Erlenmeyer-lombikba (4.2.1. pont), és hidrolizáljuk az 5.2. pontban leírt módon. Ezt követően extraháljuk petroléterrel (3.2. pont) az 5.3. pont szerint, és töltsük fel 500 ml-re petroléterrel. Ebből a kivonatból 25 ml-t pároljunk még éppen nedves állapotig a rotációs bepárlókészüléken (lásd 5.4. pont), nitrogénáramoltatással távolítsuk el a maradék oldószert (3.9. pont), majd oldjuk a maradékot 25,0 ml metil-alkoholban (3.3. pont). Ennek az oldatnak a névleges koncentrációja 45,5 μg DL-α-tokoferol/ml, ami egyenértékű 50 μg DL-α-tokoferol-acetát/ml-rel. A standard munkaoldatot felhasználás előtt frissen kell elkészíteni.
5.6.1.2. A kalibrálóoldatok és a kalibrációs görbe elkészítése
A standard munkaoldatból vigyünk át 1,0, 2,0, 4,0 és 10,0 ml-t 20 ml-es mérőlombikokba, töltsük fel a lombikokat jelig metil-alkohollal (3.3. pont), és keverjük össze a tartalmukat. Ezeknek az oldatoknak a névleges koncentrációja 2,5, 5,0, 10,0 és 25,0 μg/ml DL-α-tokoferol-acetát, azaz 2,28, 4,55, 9,10 μg/ml és 22,8 μg/ml DL-α-tokoferol.
Minden kalibrálóoldatból fecskendezzünk be 20 μl-t több alkalommal, és határozzuk meg az átlagos csúcsmagasságokat (területeket). Az átlagos csúcsmagasságok (területek) alapján szerkesszük meg a kalibrációs görbét.
5.6.1.3. A DL-α-tokoferol-acetát törzsoldat (3.10.1. pont) UV hitelesítése
Etanollal hígítsuk a DL-α-tokoferol-acetát törzsoldat (3.10.1. pont) 5,0 ml-ét 25,0 ml-re, majd mérjük meg ennek az oldatnak az UV-spektrumát spektrofotométeren (4.6. pont) etanollal (3.1. pont) szemben, 250 nm és 320 nm között.
Az abszorbancia maximális értéke 284 nm kell, hogy legyen:
Ennél a hígításnál 0,84 és 0,88 közötti extinkciós értéket kell kapni.
5.6.2. DL-α-tokoferol standard
5.6.2.1. A standard munkaoldat elkészítése
Pipettázzunk a DL-α-tokoferol törzsoldatból (3.11.1. pont) 2 ml-t egy 50 ml-es mérőlombikba, oldjuk fel metil-alkoholban (3.3. pont), és töltsük fel jelig metil-alkohollal. Ennek az oldatnak a névleges koncentrációja 40 μg DL-α-tokoferol/ml, ami egyenértékű 44,0 μg DL-α-tokoferol-acetát/ml-rel. A standard munkaoldatot felhasználás előtt frissen kell elkészíteni.
5.6.2.2. A kalibrálóoldatok és a kalibrációs görbe elkészítése
A standard munkaoldatból vigyünk át 1,0, 2,0, 4,0 és 10,0 ml-t 20 ml-es mérőlombikokba, töltsük fel a lombikokat jelig metil-alkohollal (3.3. pont), és keverjük össze a tartalmukat. Ezeknek az oldatoknak a névleges koncentrációja 2,0, 4,0, 8,0 és 20,0 μg/ml DL-α-tokoferol, azaz 2,20, 4,40, 8,79 μg/ml és 22,0 μg/ml DL-α-tokoferol-acetát.
Minden kalibrálóoldatból fecskendezzünk be 20 μl-t több alkalommal, és határozzuk meg az átlagos csúcsmagasságokat (területeket). Az átlagos csúcsmagasságok (területek) alapján szerkesszük meg a kalibrációs görbét.
5.6.2.3. A DL-α-tokoferol törzsoldat (3.11.1. pont) UV hitelesítése
Etanollal hígítsuk a DL-α-tokoferol törzsoldat (3.11.1. pont) 2,0 ml-ét 25,0 ml-re, majd mérjük meg ennek az oldatnak az UV-spektrumát spektrofotométeren (4.6. pont) etanollal (3.1. pont) szemben, 250 nm és 320 nm között. Az abszorbancia maximális értéke 292 nm kell, hogy legyen:
Ennél a hígításnál 0,6-os extinkciós értéket kell kapni.
6. Az eredmények kiszámítása
A mintaoldat E-vitamin-csúcsainak átlagos magasságából (területéből) a kalibrációs görbét (5.6.1.2. vagy 5.6.2.2. pont) használva határozzuk meg a mintaoldat koncentrációját μg/ml-ben (DL-α-tokoferol-acetátban számolva).
Az E-vitamin-tartalmat (w) mg/kg mintában kifejezve a következő képlet adja:
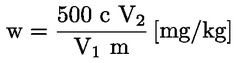
ahol:
c = a mintaoldat (5.4. pont) E-vitamin-koncentrációja (DL-α-tokoferol-acetátként), μg/ml-ben
V1 = a mintaoldat (5.4. pont) térfogata, ml-ben
V2 = az 5.4. pont szerint vett aliquot rész térfogata, ml-ben
m = a vizsgálati adag tömege g-ban
7. Észrevételek
7.1. Alacsony E-vitamin-koncentrációjú minták esetében a HPLC-meghatározáshoz hasznos lehet a két szappanosítás petroléterkivonatait (kimért mennyiség: 25 g) petroléterkivonatait egy mintaoldatban egyesíteni.
7.2. Az analizáláshoz bemért minta nem tartalmazhat 2 g-nál több zsírt.
7.3. Ha a fázisok nem válnak szét, adjunk az elegyhez körülbelül 10 ml etanolt (3.1. pont) az emulzió megbontása céljából.
7.4. A DL-α-tokoferol-acetát- vagy DL-α-tokoferol-oldat 5.6.1.3., illetve 5.6.2.3. szerinti spektrofotometriás mérése után adjunk kb. 10 mg BHT-t (3.12. pont) az oldathoz (3.10.1. vagy 3.10.2. pont), és tartsuk az oldatot hűtőszekrényben (a tárolási időtartam legfeljebb négy hét).
7.5. BHT helyett hidrokinon is használható.
7.6. Normálfázisú oszloppal lehetséges az α-, β-, γ- és δ-tokoferol elválasztása.
7.7. Nátrium-aszkorbát-oldat helyett mintegy 150 mg aszkorbinsav is használható.
7.8. Nátrium-szulfid-oldat helyett mintegy 50 mg EDTA is használható.
7.9. Az E-vitamin-acetát lúgos közegben igen gyorsan hidrolizálódik, ezért igen érzékeny az oxidációra, különösen olyan nyomelemek jelenlétében, mint a vas vagy a réz. E-vitamin előkeverékekben 5 000 mg/kg szint felett történő meghatározásakor ennek következménye az E-vitamin bomlása lehet. Ezért megerősítésre egy, az E-vitamin lúgos szappanosításának lépését nem tartalmazó, enzimes bontással járó HPLC-módszer ajánlott.
8. Ismételhetőség
Ugyanazon a mintán végzett két párhuzamos meghatározás eredményének különbsége nem haladhatja meg a magasabb érték 15 %-át.
9. A körvizsgálat eredményei ( 22 )
| Előkeverékek | Előkeverék-takarmány | Ásványi koncentrátumok | Fehérjetakarmány | Malactáp | |
| L | 12 | 12 | 12 | 12 | 12 |
| n | 48 | 48 | 48 | 48 | 48 |
| középérték [mg/kg] | 17 380 | 1 187 | 926 | 315 | 61,3 |
| sr [mg/kg] | 384 | 45,3 | 25,2 | 13,0 | 2,3 |
| r [mg/kg] | 1 075 | 126,8 | 70,6 | 36,4 | 6,4 |
| CVr [%] | 2,2 | 3,8 | 2,7 | 4,1 | 3,8 |
| sR mg/kg] | 830 | 65,0 | 55,5 | 18,9 | 7,8 |
| R [mg/kg] | 2 324 | 182,0 | 155,4 | 52,9 | 21,8 |
| CVR [%] | 4,8 | 5,5 | 6,0 | 6,0 | 12,7 |
| L: a laboratóriumok száma n: az egyedi értékek száma sr: az ismételhetőség szórása SR: a reprodukálhatóság szórása r: az ismételhetőség R: a reprodukálhatóság CVr: az ismételhetőség relatív szórása CVR: a reprodukálhatóság relatív szórása | |||||
1. ábra
Extrahálókészülék (4.8)
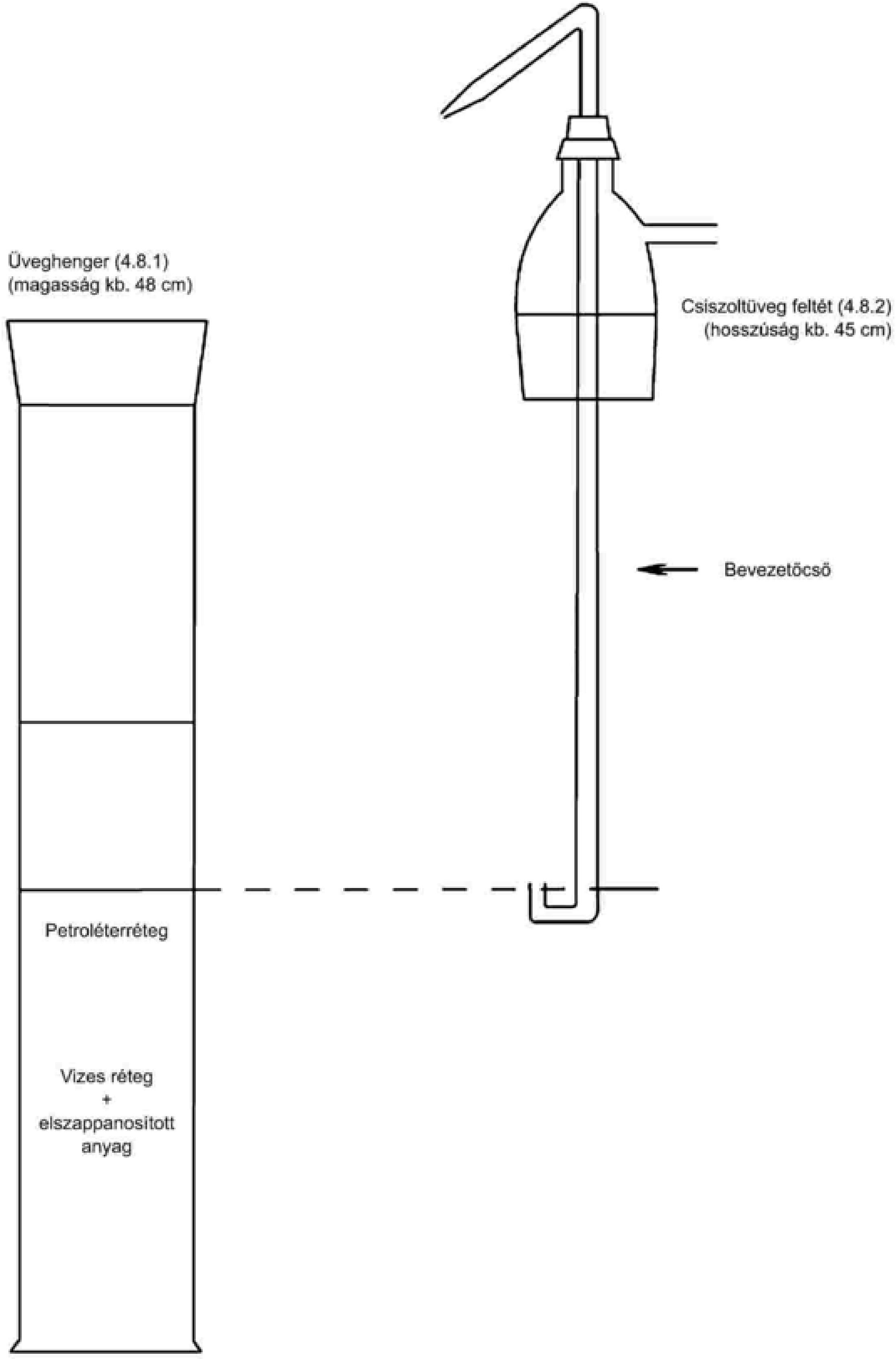
C. A VAS, RÉZ, MANGÁN ÉS CINK NYOMELEMEK MEGHATÁROZÁSA
A vas-, réz-, mangán- és cinktartalmat az alábbiak szerint kell meghatározni:
- a "Takarmányok. Mintavételi és elemzési módszerek. A kalcium-, nátrium-, foszfor-, magnézium-, kálium-, vas-, cink-, réz-, mangán-, kobalt-, molibdén- és ólomtartalom meghatározása ICP-AES-sel" című EN 15510 szabványban előírt analitikai módszerrel, vagy
- a "Takarmányok. Mintavételi és elemzési módszerek. A kalcium-, nátrium-, foszfor-, magnézium-, kálium-, kén-, vas-, cink-, réz-, mangán- és kobalttartalom meghatározása ICP-AES-sel, nyomás alatti feltárás után" című EN 15621 szabványban előírt analitikai módszerrel, vagy
- a "Takarmányok. Mintavételi és elemzési módszerek. A nyomelemek, a nehézfémek és más elemek meghatározása takarmányban ICP-MS-sel (multimódszer) " című EN 17053 szabványban előírt analitikai módszerrel, vagy
- a "Takarmányok. A kalcium-, réz-, vas-, magnézium-, mangán-, kálium-, nátrium- és cinktartalom meghatározása. Atomabszorpciós spektrometriás módszer" című EN ISO 6869 szabványban előírt analitikai módszerrel, vagy
- láng-atomabszorpciós spektrometriás (FAAS) módszerrel, az alábbi 1-8. pontban leírtak szerint.
1. Cél és alkalmazási terület
A módszer lehetővé teszi a vas, réz, mangán és cink nyomelemek takarmányokban történő meghatározását ( 23 ). A meghatározás méréshatárai a következők:
- vas (Fe): 20 mg/kg
- réz (Cu): 10 mg/kg
- mangán (Mn): 20 mg/kg
- cink (Zn): 20 mg/kg
2. Vizsgálati alapelv
Az esetleg jelen lévő szerves anyag roncsolása után a mintát sósavban oldjuk. A vas, réz, mangán és cink nyomelemeket megfelelő hígítás után atomabszorpciós spektrometriával határozzuk meg.
3. Reagensek
Előzetes megjegyzések
A reagensek és az analitikai oldatok elkészítéséhez használt víznek a meghatározás alá eső kationoktól mentesnek kell lennie, amelyet vagy boroszilikát üveg, illetve kvarc lepárlókészülékben történő kétszeres desztillálással, vagy ioncserélő gyantával végzett kétszeres kezeléssel érhetünk el.
A reagenseknek legalább analitikai tisztaságúaknak kell lenniük. A meghatározandó elemektől való mentességet vakpróbával kell ellenőrizni. Szükség esetén a reagenseket tovább kell deríteni.
Az alább leírásra kerülő standardoldatok helyett használhatunk olyan kereskedelmi forgalomban lévő standardoldatokat is, amelyek garantált minőségűek, és ezt felhasználásuk előtt le is ellenőrizték.
3.1. Sósav (d: 1,19 g/ml).
3.2. Sósav (6 mol/liter).
3.3. Sósav (0,5 mol/liter).
3.4. 38-40 %-os (v/v) fluorhidrogénsav, amelynek vastartalma kevesebb mint 1 mg Fe/liter és a bepárlás utáni maradék kevesebb mint 10 mg (mint szulfát)/liter.
3.5. Kénsav (d: 1,84 g/ml).
3.6. Hidrogén-peroxid (megközelítőleg 100 térfogat oxigén [30 tömegszázalék]).
3.7. Standard vasoldat (1 000 μg Fe/ml) a következőképpen elkészítve vagy ezzel egyenértékű, kereskedelmi forgalomban kapható oldat: oldjunk fel 1 g vashuzalt 200 ml 6 mol/liter koncentrációjú sósavban (3.2. pont), adjunk hozzá 16 ml hidrogén-peroxidot (3.6. pont), és töltsük fel vízzel 1 literre.
3.7.1. Standard vas-munkaoldat (100 μg Fe/ml), amelyet a standardoldat (3.7. pont) egy részének 9 rész vízzel történő hígításával készítünk el.
3.8. Standard rézoldat (1 000 μg Cu/ml) a következőképpen elkészítve vagy ezzel egyenértékű, kereskedelmi forgalomban kapható oldat:
- oldjunk fel 1 g porított rezet 25 ml 6 mol/liter koncentrációjú sósavban (3.2. pont), adjunk hozzá 5 ml hidrogén-peroxidot (3.6. pont), és töltsük fel vízzel 1 literre.
3.8.1. Standard réz-munkaoldat (10 μg Cu/ml), amelyet a standardoldat (3.8. pont) egy részének 9 rész vízzel történő hígításával, majd az így kapott oldat egy részének 9 rész vízzel történő hígításával készítünk el.
3.9. Standard mangánoldat (1 000 μg Mn/ml) a következőképpen elkészítve vagy ezzel egyenértékű, kereskedelmi forgalomban kapható oldat:
- oldjunk fel 1 g porított mangánt 25 ml 6 mol/liter koncentrációjú sósavban (3.2. pont), és töltsük fel vízzel 1 literre.
3.9.1. Standard mangán-munkaoldat (10 μg Mn/ml), amelyet a standardoldat (3.9. pont) egy részének 9 rész vízzel történő hígításával, majd az így kapott oldat egy részének 9 rész vízzel történő hígításával készítünk el.
3.10. Standard cinkoldat (1 000 μg Zn/ml) a következőképpen elkészítve vagy ezzel egyenértékű kereskedelmi forgalomban kapható oldat:
- oldjunk fel 1 g cinkszalagot vagy -fóliát 25 ml 6 mol/liter koncentrációjú sósavban (3.2. pont), és töltsük fel vízzel 1 literre.
3.10.1. Standard cink-munkaoldat (10 μg Zn/ml), amelyet a standardoldat (3.10. pont) egy részének 9 rész vízzel történő hígításával, majd az így kapott oldat egy részének 9 rész vízzel történő hígításával készítünk el.
3.11. Lantán-klorid-oldat: oldjunk fel 12 g lantán-oxidot 150 ml vízben, adjunk hozzá 100 ml 6 mol/liter koncentrációjú sósavat (3.2. pont), és töltsük fel vízzel 1 literre.
4. Eszközök
4.1. Hőmérséklet-szabályozóval és lehetőleg regisztrálókészülékkel ellátott tokos kemence.
4.2. Magas rezisztenciájú boroszilikát üveg eszközök. Ajánlatos kimondottan nyomelemek meghatározására szolgáló eszközöket használni.
4.3. Atomabszorpciós spektrofotométer, amely érzékenység és pontosság vonatkozásában a kívánt tartományban megfelel a módszer követelményeinek.
5. Az eljárás ( 24 )
5.1. Szerves anyagot tartalmazó minták
5.1.1. Hamvasztás és az oldatok előkészítése az analízisre ( 25 )
5.1.1.1. Helyezzünk egy 0,2 mg pontossággal kimért, 5-10 g tömegű mintát kvarc- vagy platinatégelybe (lásd a b) megjegyzést), szárítsuk ki szárítószekrényben 105 °C-on, majd rakjuk be a tégelyt a hideg tokos kemencébe (4.1. pont). Zárjuk a kemencét (lásd a c) megjegyzést) és fokozatosan emeljük a hőmérsékletet kb. 90 perc alatt 450-475 °C-ra. Tartsuk ezt a hőmérsékletet 4-16 óráig (pl. egész éjszaka), hogy eltávozzanak a széntartalmú anyagok, majd nyissuk ki a kemencét, és hagyjuk kihűlni (lásd a d) megjegyzést).
Vízzel nedvesítsük meg a hamut, és tegyük egy 250 ml-es főzőpohárba. Mossuk a tégelyt kb. 5 ml sósavval (3.1. pont), amit aztán lassan és óvatosan öntsünk a főzőpohárba (a CO2-képződés miatt heves reakció léphet fel). Adjuk hozzá cseppenként a sósavat (3.1. pont) addig kevergetve, amíg a pezsgés meg nem szűnik. Párologtassuk el belőle a nedvességet, időnként üvegbottal megkeverve.
Ezután adjunk a maradékanyaghoz 15 ml, 6 mol/liter koncentrációjú sósavat (3.2. pont), majd kb. 120 ml vizet. Keverjük meg az üvegbottal, amit a főzőpohárban hagyunk, és fedjük be a főzőpoharat egy kémlelőüveggel. Lassan forraljuk és tartsuk forrásponton addig, amíg a hamu feloldódása befejeződik. Szűrjük le hamumentes szűrőpapíron, és vegyük fel a szűrletet egy 250 ml-es mérőlombikban. Mossuk a főzőpoharat és a szűrőt 5 ml, forró, 6 mol/liter koncentrációjú sósavval (3.2. pont) és kétszer forrásban lévő vízzel. Töltsük fel a mérőlombikot jelig vízzel (a HCl-koncentráció kb. 0,5 mol/liter).
5.1.1.2. Ha a szűrőben lévő maradékanyag feketének látszik (szén), tegyük vissza a kemencébe, és hamvasszuk újra 450-475 °C-on. Ez a hamvasztás, amely csak pár órát vesz igénybe (kb. három-öt órát), akkor fejeződik be, amikor a hamu fehérnek vagy majdnem fehérnek tűnik. Oldjuk fel a maradékanyagot kb. 2 ml sósavval (3.1. pont), párologtassuk el belőle a nedvességet és adjunk hozzá 5 ml, 6 mol/liter koncentrációjú sósavat (3.2. pont). Hevítsük, szűrjük az oldatot mérőlombikba, és töltsük fel a mérőlombikot jelig vízzel (a HCl-koncentráció kb. 0,5 mol/liter).
Megjegyzések:
a) A nyomelemek meghatározásakor fontos odafigyelni a - főleg a cink, réz és vas képezte - szennyeződés jelentette kockázatokra. Ezért a minták előkészítésére használt eszközöknek mentesnek kell lennie ezen fémektől.
A szennyeződés általános kockázatának csökkentése érdekében dolgozzunk pormentes környezetben, gondosan tisztított eszközökkel és alaposan elmosott üvegedényekkel. Különösen a cink meghatározása érzékeny a szennyeződések számos típusára, pl. az üvegkészítményektől, a reagensektől, a portól stb. származókra.
b) Az elhamvasztandó minta tömege a takarmány hozzávetőleges nyomelemtartalma alapján számítható ki, az alkalmazott spektrofotométer érzékenységéhez viszonyítva. Egyes, kevés nyomelemet tartalmazó takarmányok esetén 10-20 g-os mintát kell venni, és a végső oldatot csak 100 ml-re kell feltölteni.
c) A hamvasztást zárt kemencében kell elvégezni, levegő vagy oxigén befúvatása nélkül.
d) A pirométer által jelzett hőmérséklet nem lépheti túl a 475 °C-ot.
5.1.2. Spektrofotometriás meghatározás
5.1.2.1. A kalibrálóoldatok elkészítése
Minden egyes meghatározandó elemhez készítsünk a 3.7.1., 3.8.1., 3.9.1. és 3.10.1. pontban megadott standard munkaoldatokból egy kalibrálóoldat-sorozatot, úgy, hogy minden egyes kalibrálóoldatnak a HCl-koncentrációja kb. 0,5 mol/liter és (vas, mangán és cink esetében) a lantán-klorid koncentrációja 0,1 %-os (w/v) lantánnal egyenértékű legyen.
A kiválasztott nyomelem-koncentrációknak az alkalmazott spektrofotométer érzékenységi tartományán belül kell lenniük. Az alábbi táblázatok példákon keresztül mutatják a kalibrálóoldat-sorozatok egy mintasorozatát; az alkalmazott spektrofotométer típusától és érzékenységi tartományától függően, adott esetben más koncentrációkat kell kiválasztani.
Vas
| μg Fe/ml | 0 | 0,5 | 1 | 2 | 3 | 4 | 5 |
| standard munkaoldat, ml-ben (3.7.1. pont) (1 ml = 100 μg Fe) | 0 | 0,5 | 1 | 2 | 3 | 4 | 5 |
| ml HCl (3.2. pont) | 7 | 7 | 7 | 7 | 7 | 7 | 7 |
| + 10 ml lantán-klorid oldat (3.11. pont), töltsük fel vízzel 100 ml-re. | |||||||
Réz
| μg Cu/ml | 0 | 0,1 | 0,2 | 0,4 | 0,6 | 0,8 | 1,0 |
| standard munkaoldat, ml-ben (3.8.1. pont) (1 ml = 10 μg Cu) | 0 | 1 | 2 | 4 | 6 | 8 | 10 |
| ml HCl (3.2. pont) | 8 | 8 | 8 | 8 | 8 | 8 | 8 |
Mangán
| μg Mn/ml | 0 | 0,1 | 0,2 | 0,4 | 0,6 | 0,8 | 1,0 |
| standard munkaoldat, ml-ben (3.9.1. pont) (1 ml = 10 μg Mn) | 0 | 1 | 2 | 4 | 6 | 8 | 10 |
| ml HCl (3.2. pont) | 7 | 7 | 7 | 7 | 7 | 7 | 7 |
| + 10 ml lantán-klorid oldat (3.11. pont), töltsük fel vízzel 100 ml-re. | |||||||
Cink
| μg Zn/ml | 0 | 0,05 | 0,1 | 0,2 | 0,4 | 0,6 | 0,8 |
| standard munkaoldat, ml-ben (3.10.1. pont) (1 ml = 10 μg Zn) | 0 | 0,5 | 1 | 2 | 4 | 6 | 8 |
| ml HCl (3.2. pont) | 7 | 7 | 7 | 7 | 7 | 7 | 7 |
| + 10 ml lantán-klorid oldat (3.11. pont), töltsük fel vízzel 100 ml-re. | |||||||
5.1.2.2. Az oldat előkészítése az analízisre
A réz meghatározásához az 5.1.1. pont szerint elkészített oldatot általában közvetlenül fel lehet használni. Ha mégis korrigálni kell koncentrációját a kalibrálóoldat-sorozaton belül, egy aliquot részt pipettázzunk egy 100 ml-es mérőlombikba, és töltsük fel jelig 0,5 mol/liter koncentrációjú sósavval (3.3. pont).
A vas, mangán és cink meghatározásához pipettázzunk egy aliquot részt az 5.1.1. pont szerint elkészített oldatból egy 100 ml-es mérőlombikba, adjunk hozzá 10 ml lantán-klorid oldatot (3.11. pont), és töltsük fel jelig 0,5 mol/liter koncentrációjú sósavval (3.3. pont) (lásd még az Észrevételek 8. pontját).
5.1.2.3. Vakpróba
A vakpróba során a módszer összes meghatározott lépését el kell végezni, azzal a különbséggel, hogy a mintaanyagot elhagyjuk. A "0" kalibrálóoldatot nem szabad vakoldatként használni.
5.1.2.4. Az atomabszorpció mérése
Mérjük meg a kalibrálóoldatok és az analizálandó oldat atomabszorpcióját oxidáló, levegő-acetilén láng alkalmazásával, a következő hullámhosszokon:
Fe: 248,3 nm
Cu: 324,8 nm
Mn: 279,5 nm
Zn: 213,8 nm
Minden mérést négyszer végezzünk el.
5.2. Ásványi takarmányok
Ha a minta nem tartalmaz szerves anyagot, az előzetes hamvasztás szükségtelen. Folytassuk a műveletet az 5.1.1.1. pont második bekezdésétől. A fluorhidrogénsavval végzett bepárlás elhagyható.
6. Az eredmények kiszámítása
Egy kalibrációs görbe segítségével számoljuk ki az analizálandó oldat nyomelem-koncentrációját, az eredményt a minta kilogrammonkénti nyomelemtartalmára vonatkoztatva, milligrammban adjuk meg (ppm).
7. Ismételhetőség
Ugyanazon a mintán, ugyanazon analitikus által végzett két párhuzamos meghatározás eredménye közötti különbség nem haladhatja meg a:
- a vizsgált nyomelemtartalom legfeljebb 50 mg/kg értéke esetén abszolút értékben az 5 mg/kg-ot,
- a vizsgált nyomelemtartalom 50 és 100 mg/kg koncentráció közötti értéke esetén a magasabb érték 10 %-át,
- a vizsgált nyomelemtartalom 100 és 200 mg/kg közötti értéke esetén abszolút értékben a 10 mg/kg-ot,
- a vizsgált nyomelemtartalom 200 mg/kg feletti értéke esetén a magasabb érték 5 %-át.
8. Észrevételek
Nagy mennyiségű foszfát jelenléte interferenciát okozhat a vassal, mangánnal és cinkkel. Az intereferenciát lantán-klorid oldat (3.11. pont) hozzáadásával kell korrigálni. Ha azonban a mintában a Ca + Mg/P súlyaránya > 2, elhagyható a lantán-klorid oldatnak (3.11. pont) a vizsgálandó oldathoz és a kalibrálóoldatokhoz való hozzáadása.
D. A HALOFUGINON MEGHATÁROZÁSA
DL-transz-7-bromo-6-kloro-3-[3-(3-hidroxi-2-piperidil)acetonil]-quinazolin-4-(3H)-egy hidrobromid
A halofuginontartalmat az alábbiak szerint kell meghatározni:
- a "Takarmányok. Mintavételi és elemzési módszerek. Az engedélyezett kokcidiosztatikumok szűrése és meghatározása additív, illetve 1 %-os és 3 %-os keresztszennyeződési szinten, valamint a nem regisztrált kokcidiosztatikumok és egy antibiotikum szűrése és meghatározása szubadditív szinteken, összetett takarmányban, nagy hatékonyságú folyadékkromatográfiával. Tandem-tömegspektrometriás kimutatás (LC-MS/MS)" című EN 17299 szabványban előírt analitikai módszerrel, vagy
- fordított fázisú, nagy teljesítményű folyadékkromatográfiával (HPLC), UV detektor használatával, az alábbi 1-8. pontban leírtak szerint.
1. Cél és alkalmazási terület
A módszer a halofuginon takarmányokban történő meghatározására szolgál. A meghatározási határ 1 mg/kg.
2. Vizsgálati alapelv
Forró vízzel történt kezelés után a halofuginont szabad bázis formában etilén-acetátba extraháljuk, ezt követően pedig hidroklorid formában vizes savoldatba partícionáljuk. A kivonatot ioncserélő kromatográfiával tisztítjuk. A halofuginontartalom meghatározását fordított fázisú, nagy teljesítményű folyadékkromatográfiás (HPLC) módszerrel és UV-detektor alkalmazásával végezzük.
3. Reagensek
3.1. Acetonitril, HPLC-minőségű.
3.2. Amberlite XAD-2 műgyanta
3.3. Ammónium-acetát
3.4. Etil-acetát
3.5. Jégecet
3.6. Halofuginon standard anyag (DL-transz-7-bromo-6-kloro-3-[3-(3-hidroxi-2-piperidil)acetonil] quinazolin-4-(3H)-egy hidrobromid, E 764)
3.6.1. Halofuginon standard törzsoldat, 100 μg/ml
Mérjünk be egy 500 ml-es mérőlombikba 0,1 mg pontossággal 50 mg halofuginont (3.6. pont), oldjuk fel ammónium-acetát pufferoldatban (3.18. pont), töltsük fel a lombikot jelig a pufferoldattal, majd keverjük el. Ezen oldat 5 °C hőmérsékleten és sötétben tárolva három héten át stabil marad.
3.6.2. Kalibrálóoldatok
100 ml-es mérőlombikokba adagoljunk 1,0, 2,0, 3,0, 4,0 és 6,0 ml standard törzsoldatot (3.6.1. pont). A mozgófázissal (3.21. pont) töltsük fel jelig, és keverjük el. Ezen oldatok halofuginon-koncentrációja 1,0, 2,0, 3,0, 4,0, illetve 6,0 μg/ml lesz. Az oldatokat használat előtt frissen kell elkészíteni.
3.7. Sósav (ρ20 megközelítőleg 1,16 g/ml)
3.8. Metil-alkohol
3.9. Ezüst-nitrát
3.10. Nátrium-aszkorbát
3.11. Nátrium-karbonát
3.12. Nátrium-klorid
3.13. EDTA (etilén-diamin-tetraecetsav, dinátrium só)
3.14. Víz, HPLC-minőségű
3.15. Nátrium-karbonát-oldat, c = 10 g/100 ml
3.16. Nátrium-kloriddal telített nátrium-karbonát-oldat, c = 5 g/100 ml
Oldjunk fel 50 g nátrium-karbonátot (3.11. pont) vízben, hígítsuk fel 1 literre, és adjunk hozzá nátrium-kloridot (3.12. pont), amíg az oldat telített nem lesz.
3.17. Sósav, megközelítőleg 0,1 mol/l
10 ml HCl-t (3.7. pont) hígítsunk vízzel egy literre.
3.18. Ammónium-acetát pufferoldat, megközelítőleg 0,25 mol/l koncentrációjú
Oldjunk fel 19,3 g ammónium-acetátot (3.3. pont) és 30 ml ecetsavat (3.5. pont) vízben (3.14. pont), és hígítsuk 1 literre.
3.19. Amberlite XAD-2 műgyanta előkészítése
Megfelelő mennyiségű Amberlite-et (3.2. pont) mossunk addig vízzel, amíg az összes kloridiont el nem távolítottuk, mindezt ezüst-nitrátnak (3.20. pont) az elöntött vizes fázishoz való hozzáadásával ellenőrizhetjük. Ezután mossuk ki a gyantát 50 ml metil-alkoholban (3.8. pont), öntsük le a metil-alkoholt, és a gyantát friss metil-alkohol alatt tároljuk.
3.20. Ezüst-nitrát oldat, megközelítőleg 0,1 mol/l
Oldjunk fel 0,17 g ezüst-nitrátot (3.9. pont) 10 ml vízben.
3.21. HPLC mozgófázis
Keverjünk el 500 ml acetonitrilt (3.1. pont) 300 ml ammónium-acetát pufferoldattal (3.18. pont) és 1 200 ml vízzel (3.14. pont). Az oldat pH-ját ecetsavval (3.5. pont) állítsuk be 4,3-ra. Szűrjük át egy 0,22 μm-es szűrőn (4.8. pont) és gázmentesítsük az oldatot (pl. 10 percig tartó ultrahangos kezeléssel). Az oldat egy hónapon át stabil marad, ha zárt edényben, sötétben tároljuk.
4. Eszközök
4.1. Ultrahangos fürdő
4.2. Rotációs, vékonyfilmes bepárló
4.3. Centrifuga
4.4. HPLC-készülék, változtatható hullámhosszú UV-detektorral vagy diódasoros detektorral
4.4.1. Folyadékkromatográfiás oszlop, 300 mm × 4 mm, C18, 10 μm-es töltet vagy azzal egyenértékű berendezés
4.5. Zsugorított üvegszűrővel és elzárócsappal ellátott üvegoszlop (300 mm × 10 mm)
4.6. Üvegszálas szűrők, 150 mm-es átmérővel
4.7. Membránszűrők, 0,45 μm
4.8. Membránszűrők, 0,22 μm
5. A vizsgálat módja
Megjegyzés: A halofuginon szabad bázis formájában nem stabil a lúgos és etil-acetát oldatokban. Harminc percnél tovább nem maradhat etil-acetátban.
5.1. Általános szabályok
5.1.1. Vizsgáljunk meg egy takarmányvakmintát annak ellenőrzésére, hogy sem halofuginon, sem interferáló anyagok nincsenek jelen.
5.1.2. A mintában találhatóval közel azonos mennyiségű halofuginonnal dúsított takarmányvakminta analízisével végezzünk visszanyerési próbát. A 3 mg/kg-os szintre történő dúsításhoz adjunk 300 μl standard törzsoldatot (3.6.1. pont) 10 g takarmányvakmintához, keverjük össze, majd várjunk 10 percet, mielőtt folytatnánk a műveletet az extrahálással (5.2. pont).
Megjegyzés: Ennél a módszernél a takarmányvakminta a mintával azonos fajtájú takarmány legyen, amelyben az analízis során halofuginon nem mutatható ki.
5.2. Extrahálás
Egy 200 ml-es centrifugacsőbe 0,1 g-os pontossággal mérjünk ki 10 g-ot az előkészített mintából, adjunk hozzá 0,5 g nátrium-aszkorbátot (3.10. pont), 0,5 g EDTA-oldatot (3.13. pont) és 20 ml vizet, majd keverjük össze. A csövet helyezzük öt percre egy 80 °C-os vízfürdőbe. Miután visszahűlt szobahőmérsékletre, adjunk hozzá 20 ml nátrium-karbonát oldatot (3.15. pont), és keverjük össze. Közvetlenül ezután adjunk hozzá 100 ml etil-acetátot (3.4. pont), és kézzel erősen rázzuk 15 másodpercen keresztül. Ezután tegyük a csövet három percig az ultrahangos fürdőbe (4.1. pont), és lazítsuk meg a dugóját. Centrifugáljuk két percen át, majd dekantáljuk az etil-acetát fázist egy üvegszálas szűrőn (4.6. pont) keresztül egy 500 ml-es választótölcsérbe. Ismételjük meg az extrahálást a mintán egy második 100 ml-es etil-acetát adaggal. Mossuk az elegyített kivonatot 50 ml nátrium-kloriddal telített nátrium-karbonát oldattal (3.16. pont) egy percen keresztül, és a vizes fázist öntsük el.
A szerves réteget 1 percen keresztül 50 ml sósavval (3.17. pont) extraháljuk. Az alsó savas réteget eresszük le egy 250 ml-es választótölcsérbe. A szerves réteget ismételten extraháljuk másfél percen keresztül további 50 ml sósavval, és elegyítsük az első kivonattal. Mossuk az elegyített savas kivonatokat úgy, hogy megközelítőleg tíz másodpercig locsoljuk 10 ml etil-acetáttal (3.4. pont).
A vizes réteget teljes mennyiségben vigyük át egy 250 ml-es gömblombikba, és öntsük el a szerves fázist. A rotációs, vékonyfilmes bepárlókészülékkel (4.2. pont) párologtassuk el az összes visszamaradó etil-acetátot a savas oldatból. A vízfürdő hőmérséklete nem haladhatja meg a 40 °C-ot. Körülbelül 25 mbar vákuumban 38 °C-on az összes visszamaradt etil-acetát öt perc alatt elpárolog.
5.3. Tisztítás
5.3.1. Az Amberlite-oszlop előkészítése
Minden mintakivonat számára külön XAD-2 oszlopot készítünk. Vigyünk át metil-alkohollal (3.8. pont) 10 g előkészített Amberlite-et (3.19. pont) egy üvegoszlopba (4.5. pont). A gyantaágy tetejére tegyünk egy kis üvegvatta dugót. Eresszük le az oszlopról a metil-alkoholt, és 100 ml vízzel mossuk el a gyantát, de állítsuk meg az átfolyást, amikor a folyadék elérte a gyantaágy tetejét. Használat előtt, 10 percig engedjük, hogy az oszlop egyensúlyba kerüljön. Soha ne hagyjuk az oszlopot kiszáradni.
5.3.2. A minta tisztítása
A kivonatot (5.2. pont) teljes mennyiségben vigyük át az előkészített Amberlite-oszlop (5.3.1. pont) tetejére, eluáljuk, majd az eluátumot öntsük el. Az eluálás sebessége nem lehet több, mint 20 ml/perc. Öblítsük el a gömblombikot 20 ml sósavval (3.17. pont), és ezt használjuk a gyantaoszlop elmosásához is. Légbefúvással távolítsunk el minden savoldat-maradékot. A mosóoldatot öntsük el. Adjunk 100 ml metil-alkoholt (3.8. pont) az oszlopra, és engedjünk 5-10 ml-t eluálódni, miközben az eluátumot egy 250 ml-es gömblombikban fogjuk fel. Tíz percig hagyjuk, hogy a maradék metil-alkohol egyensúlyba kerüljön a műgyantával, majd folytassuk az eluálást legfeljebb 20 ml/perc sebességen, miközben az eluátumot ugyanabban a gömblombikban fogjuk fel. A metil-alkoholt a rotációs, vékonyfilmes bepárlókészüléken párologtassuk el (4.2. pont), a vízfürdő hőmérséklete nem haladhatja meg a 40 °C-ot. A maradékot a mozgófázis (3.21. pont) segítségével teljes mennyiségben vigyük át egy 10 ml-es mérőlombikba. Mozgófázissal töltsük fel jelig, és keverjük el. Egy aliquot részt szűrjünk át membránszűrőn (4.7. pont) Ezt az oldatot tegyük el a HPLC-meghatározáshoz (5.4. pont).
5.4. HPLC-meghatározás
5.4.1. Paraméterek
A következő vizsgálati feltételek útmutatásként szolgálnak, egyéb feltételek is használhatók, feltéve, hogy ezzel egyenértékű eredményeket adnak.
Folyadékkromatográfiás oszlop (4.4.1. pont)
HPLC mozgófázis (3.21. pont)
Átáramlási sebesség: 1,5-2 ml/perc
Kimutatási hullámhossz: 243 nm
Befecskendezett mennyiség: 40-100 μl
Ellenőrizzük a kromatográfiás rendszer stabilitását oly módon, hogy 3,0 μg/ml koncentrációjú kalibrálóoldatot (3.6.2. pont) fecskendezünk be többször, amíg a csúcsmagasságok (vagy területek) és a retenciós idők állandóvá nem válnak.
5.4.2. Kalibrációs görbe
Fecskendezzük be többször egymás után az egyes kalibrálóoldatokat (3.6.2. pont), és mérjük meg az egyes koncentrációkhoz tartozó csúcsok magasságát (területét). A kalibrálóoldatok átlagos csúcsmagassága vagy területe legyen az ordinátatengelyen, a megfelelő koncentrációk pedig μg/ml-ben az abszcisszán, és ennek alapján szerkesszünk kalibrációs görbét.
5.4.3. Mintaoldat
Ugyanolyan mennyiségű oldatot használva, mint amennyit a kalibrálóoldatoknál használtunk, fecskendezzük be a mintakivonatot (5.3.2. pont) egymás után többször, és határozzuk meg a halofuginoncsúcsok átlagos magasságát (területét).
6. Az eredmények kiszámítása
A mintaoldat halofuginoncsúcsainak átlagos magasságát (területét) a kalibrációs görbével (5.4.2. pont) egybevetve határozzuk meg a mintaoldat koncentrációját μg/ml-ben.
A minta (mg/kg-ban kifejezett) w halofuginontartalmát az alábbi képlet segítségével kapjuk meg:
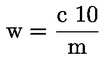
ahol:
c : a mintaoldat halofuginon-koncentrációja, μg/ml-ben,
m : a vizsgálati adag tömege grammban.
7. Az eredmények validálása
7.1. Azonosítás
Az analizált anyag azonosítása párhuzamos kromatográfiával vagy diódasoros detektor használatával erősíthető meg, amely során a mintakivonat és a 6,0 μg/ml-es kalibrálóoldat (3.6.2. pont) spektrumát hasonlítjuk össze.
7.1.1. Párhuzamos kromatográfia
A mintakivonatot megfelelő mennyiségű kalibrálóoldattal (3.6.2. pont) dúsítjuk. A hozzáadott halofuginon mennyiségének a mintakivonatban található halofuginon becsült mennyiségével közel azonosnak kell lennie.
A hozzáadott mennyiséget és a kivonat hígítását figyelembe véve, csak a halofuginoncsúcs emelkedhet. A csúcs szélességének a legnagyobb magasság felénél az eredeti szélesség ± 10 %-on belül kell lennie.
7.1.2. Diódasoros detektálás
Az eredményeket a következő kritériumok szerint értékeljük:
a) a minta és a standard spektrumában a kromatogram csúcsmaximumán rögzített legnagyobb elnyeléshez tartozó hullámhossznak a detektor felbontóképessége által meghatározott tűréshatáron belül azonosnak kell lennie. A diódasoros detektoroknál ez a tűréshatár jellemzően ± 2 nm;
b) 225 és 300 nm között a spektrumnak a relatív abszorbancia 10 és 100 %-a közötti tartományba eső részein a mintának és a standardnak a kromatogram csúcsmaximumán rögzített spektruma nem térhet el egymástól. Ez a kritérium akkor teljesül, ha ugyanazok a maximális értékek vannak jelen, és a két spektrum egyetlen vizsgált ponton sem tér el a standard abszorbanciájának 15 %-ánál nagyobb mértékben;
c) 225 és 300 nm között a mintakivonat által kiváltott csúcs felszálló ágának, csúcsmaximumának és leszálló ágának spektrumai nem térhetnek el egymástól a spektrumnak a relatív abszorbancia 10 és 100 %-a közötti tartományba eső részein. Ez a kritérium akkor teljesül, ha ugyanazok a maximális értékek vannak jelen, és a spektrumok egyetlen vizsgált ponton sem térnek el a csúcsmaximum spektruma abszorbanciájának 15 %-ánál nagyobb mértékben.
Amennyiben ezen kritériumok valamelyike nem teljesül, az analizált anyag jelenléte nem bizonyított.
7.2. Ismételhetőség
Két azonos mintán végzett párhuzamos meghatározás eredménye közötti különbség 3 mg/kg halofuginontartalomig nem lehet több, mint 0,5 mg/kg.
7.3. Visszanyerés
A dúsított takarmányvakminta esetében legalább 80 %-os visszanyerési arányt kell elérni.
8. A körvizsgálat eredményei
Egy körvizsgálat ( 26 ) során három mintát analizáltak nyolc laboratóriumban.
Eredmények
| A. minta (vak) átvételkor | B. minta (takarmányliszt) | C. minta (granulátum) | |||
| átvételkor | két hónap után | átvételkor | két hónap után | ||
| Középérték [mg/kg] | ND | 2,80 | 2,42 | 2,89 | 2,45 |
| SR [mg/kg] | -- | 0,45 | 0,43 | 0,40 | 0,42 |
| CVR [%] | -- | 16 | 18 | 14 | 17 |
| vissza. [%] | 86 | 74 | 88 | 75 | |
| ND= nem kimutatható SR= a reprodukálhatóság szórása CVR= a reprodukálhatóság relatív szórása (%) vissza.= visszanyerés (%) | |||||
E. A ROBENIDIN MEGHATÁROZÁSA
1,3-bisz[(4-klorobenzilidén)amino]guanidin-hidroklorid
A robenidintartalmat az alábbiak szerint kell meghatározni:
- a "Takarmányok. Mintavételi és elemzési módszerek. Az engedélyezett kokcidiosztatikumok szűrése és meghatározása additív, illetve 1 %-os és 3 %-os keresztszennyeződési szinten, valamint a nem regisztrált kokcidiosztatikumok és egy antibiotikum szűrése és meghatározása szubadditív szinteken, összetett takarmányban, nagy hatékonyságú folyadékkromatográfiával. Tandem-tömegspektrometriás kimutatás (LC-MS/MS)" című EN 17299 szabványban előírt analitikai módszerrel, vagy
- fordított fázisú, nagy teljesítményű folyadékkromatográfiával (HPLC), UV detektor használatával, az alábbi 1-8. pontban leírtak szerint.
1. Cél és alkalmazási terület
Ez a módszer lehetővé teszi a takarmányokban lévő robenidin szintjének meghatározását. A meghatározási határ 5 mg/kg.
2. Vizsgálati alapelv
A mintát savas metil-alkohollal extraháljuk. A kivonatot szárítjuk, és egy aliquot részét alumínium-oxid oszlopon tisztítjuk. A robenidint az oszlopról metil-alkohollal eluáljuk, koncentráljuk, majd a mozgófázissal megfelelő térfogatra töltjük fel. A robenidintartalom meghatározását fordított fázisú, nagy teljesítményű folyadékkromatográfiával (HPLC), UV-detektor alkalmazásával végezzük.
3. Reagensek
3.1. Metil-alkohol
3.2. Savas metil-alkohol
Öntsünk át 4,0 ml sósavat (ρ20 = 1,18 g/ml) egy 500 ml-es mérőlombikba, töltsük fel jelig metil-alkohollal (3.1. pont), majd keverjük el. Ezt az oldatot használat előtt frissen kell elkészíteni.
3.3. Acetonitril, HPLC-minőségű.
3.4. Molekuláris szűrő
3A típusú, 8-12 szitaszemcse (1,6-2,5 mm-es szemcsék, kristályos alumínium-szilikát, 0,3 mm pórusátmérő).
3.5. Alumínium-oxid: I. savas aktivitási fokozatú anyag oszlopkromatográfiás célra
Öntsünk át 100 g alumínium-oxidot egy megfelelő tárolóedénybe, és adjunk hozzá 2,0 ml vizet. Zárjuk le, és rázzuk kb. 20 percen keresztül. Jól lezárt tárolóedényben tartsuk.
3.6. Kálium-dihidrogén-foszfát-oldat, c = 0,025 mol/l
Oldjunk fel vízzel 3,40 g kálium-dihidrogén-foszfátot (HPLC-minőségű) egy 1 000 ml-es mérőlombikban, töltsük fel jelig, és keverjük el.
3.7. Dinátrium-hidrogén-foszfát-oldat, c = 0,025 mol/l
Oldjunk fel 3,55 g vízmentes (vagy 4,45 g dehidrát, illetve 8,95 g dodekahidrát) dinátrium-hidrogén-foszfátot vízben (HPLC-minőségű) egy 1 literes mérőlombikban, töltsük fel jelig, és keverjük el.
3.8. HPLC mozgófázis
Az alábbi reagenseket keverjük össze:
650 ml acetonitril (3.3. pont),
250 ml víz (HPLC minőségű),
50 ml kálium-dihidrogén-foszfát oldat (3.6. pont),
50 ml dinátrium-hidrogén-foszfát oldat (3.7. pont).
Szűrjük át egy 0,22 μm-es szűrőn (4.6. pont) és gázmentesítsük az oldatot (pl. 10 percig tartó ultrahangos kezeléssel).
3.9. Standard anyag
Tiszta robenidin: 1,3-bisz[(4-klorobenzilidén)amino]guanidin-hidroklorid.
3.9.1. Robenidin standard törzsoldat: 300 μg/ml
Mérjünk ki 0,1 mg-os pontossággal 30 mg robenidin standard anyagot (3.9. pont). Oldjuk fel savas metil-alkohollal (3.2. pont) egy 100 ml-es mérőlombikban, töltsük fel jelig ugyanezzel az oldószerrel, és keverjük el. A lombikot tekerjük alumíniumfóliába, és tároljuk sötét helyen.
3.9.2. Robenidin közbenső standardoldat: 12 μg/ml
A standard törzsoldatból (3.9.1. pont) öntsünk át 10,0 ml-t egy 250 ml-es mérőlombikba, töltsük fel jelig a mozgófázissal (3.8. pont), és keverjük el. A lombikot tekerjük alumíniumfóliába, és tároljuk sötét helyen.
3.9.3. Kalibrálóoldatok
50 ml-es mérőlombikokba adagoljunk 5,0, 10,0, 15,0, 20,0 és 25,0 ml közbenső standardoldatot (3.9.2. pont). A mozgófázissal (3.8. pont) töltsük fel jelig, és keverjük el. Ezen oldatok 1,2, 2,4, 3,6, 4,8, illetve 6,0 μg/ml robenidin-koncentrációnak felelnek meg. Az oldatokat használat előtt frissen kell elkészíteni.
3.10. Víz, HPLC-minőségű
4. Eszközök
4.1. Üvegoszlop
Barna üvegből készült, elzáró csappal és egy megközelítőleg 150 ml-es tartállyal ellátott, 10-15 mm belső átmérőjű és 250 mm hosszú oszlop.
4.2. Mechanikus rázógép vagy mágneses keverő
4.3. Rotációs, vékonyfilmes bepárló
4.4. 250-400 mm-es érzékenységi tartományú HPLC-berendezés, változtatható hullámhosszú UV-detektorral vagy diódasoros detektorral
4.4.1. Folyadékkromatográfiás oszlop: C18, 300 mm × 4 mm, 10 μm-es töltettel, vagy ezzel egyenértékű berendezés
4.5. Üvegszálas szűrőpapír (Whatman GF/A vagy azzal egyenértékű más papír)
4.6. Membránszűrők, 0,22 μm
4.7. Membránszűrők, 0,45 μm
5. A vizsgálat módja
Megjegyzés: A robenidin fényérzékeny. Minden műveletnél barna üvegeszközöket kell használni.
5.1. Általános szabályok
5.1.1. Visgáljunk meg egy takarmányvakmintát annak ellenőrzésére, hogy sem robenidin, sem interferáló anyagok nincsenek jelen.
5.1.2. A mintában találhatóval közel azonos mennyiségű robenidinnel dúsított takarmányvakminta (5.1.1. pont) analízisével végezzünk visszanyerési próbát. A 60 mg/kg-os szintre történő dúsításhoz öntsünk 3,0 ml standard törzsoldatot (3.9.1. pont) egy 250 ml-es Erlenmeyer-lombikba. Az oldatot nitrogénáramban kb. 0,5 ml-re pároljuk be. Adjunk hozzá 15 g takarmányvakmintát, keverjük össze, és hagyjuk állni 10 percig, mielőtt folytatjuk a műveletet az extrahálással (5.2. pont).
Megjegyzés: Ehhez a módszerhez a takarmányvakminta a mintával azonos fajtájú takarmány legyen, amelyben az analízis során robenidin nem mutatható ki.
5.2. Extrahálás
Az előkészített mintából mérjünk ki 15 g-t, 0,01 g-os pontossággal. Tegyük egy 250 ml-es Erlenmeyer-lombikba, adjunk hozzá 100,0 ml savas metil-alkoholt (3.2. pont), zárjuk le, és rázassuk egy órán keresztül a rázógépen (4.2. pont). Az oldatot szűrjük át üvegszálas szűrőpapíron (4.5. pont), majd az egész szűrletet vegyük fel egy 150 ml-es Erlenmeyer-lombikban. Adjunk hozzá 7,5 g molekuláris szűrőt (3.4. pont), zárjuk le, és öt percig rázassuk. Azonnal szűrjük át egy üvegszálas szűrőpapíron. Ezt az oldatot használjuk a derítésnél (5.3. pont).
5.3. Derítés
5.3.1. Az alumínium-oxid oszlop előkészítése
Az üvegoszlop (4.1. pont) alsó végébe illesszünk egy kis üvegvatta dugót, és üvegbottal tömködjük le egészen az aljára. Mérjünk ki az előkészített alumínium-oxidból (3.5. pont) 11,0 g-ot, és öntsük át az oszlopba. Ügyeljünk arra, hogy az oszlop tartalma, a vizsgálat ezen szakaszában minél kevésbé legyen a levegő hatásának kitéve. Gyengéden ütögessük a megtöltött oszlop alsó végét, hogy a benne lévő alumínium-oxid leülepedjen.
5.3.2. A minta derítése
Az (5.2. pont) pontban előkészített mintakivonatból 5,0 ml-t pipettázzunk az oszlop tetejére. A pipetta csúcsát az oszlop falának közelébe fektessük, és hagyjuk, hogy az oldatot az alumínium-oxid elnyelje. 100 ml metil-alkohol (3.1. pont) segítségével, 2-3 ml/perc átfolyási sebességgel eluáljuk a robenidint az oszlopról, és vegyük fel az eluátumot egy 250 ml-es gömblombikban. Csökkentett nyomáson, 40 °C hőmérsékleten egy rotációs, vékonyfilmes bepárló (4.3. pont) segítségével teljes száradásig párologjuk be a metil-alkoholt. A maradékot újra oldjuk fel 3-4 ml mozgófázisban (3.8. pont), és öntsük át teljes mennyiségben egy 10 ml-es mérőlombikba. Többször öblítsük el a lombikot 1-2 ml mozgófázissal, és az öblítő folyadékot is öntsük bele a mérőlombikba. Töltsük fel jelig ugyanezzel az oldószerrel, majd keverjük el. Egy aliquot részt szűrjünk át egy 0,45 μm-es membránszűrőn (4.7. pont). Ezt az oldatot tegyük el a HPLC-meghatározáshoz (5.4. pont).
5.4. HPLC-meghatározás
5.4.1. Paraméterek
Az alábbi vizsgálati feltételek csak útmutatásként szolgálnak, ettől eltérő feltételeket is lehet használni, feltéve, hogy ezzel egyenértékű eredményt adnak:
Folyadékkromatográfiás oszlop (4.4.1. pont),
HPLC mozgófázis (3.8. pont),
Átáramlási sebesség: 1,5-2 ml/perc,
Kimutatási hullámhossz: 317 nm,
Befecskendezett mennyiség: 20-50 μl.
Ellenőrizzük a kromatográfiás rendszer stabilitását oly módon, hogy 3,6 μg/ml anyagot tartalmazó kalibrálóoldatot (3.9.3. pont) fecskendezünk be többször, míg a csúcsmagasságok és a retenciós idők állandóvá nem válnak.
5.4.2. Kalibrációs görbe
Fecskendezzük be többször egymás után az egyes kalibrálóoldatokat (3.9.3. pont), és mérjük meg az egyes koncentrációkhoz tartozó csúcsok magasságát (területét). A kalibrálóoldatok átlagos csúcsmagassága vagy területe legyen az ordinátatengelyen, a megfelelő koncentrációk μg/ml-ben az abszcisszán, és ennek alapján szerkesszünk kalibrációs görbét.
5.4.3. Mintaoldat
Ugyanolyan mennyiségű oldatot használva, mint amennyit a kalibrálóoldatoknál használtunk, fecskendezzük be a mintakivonatot (5.3.2. pont) egymás után többször, és határozzuk meg a robenidincsúcsok átlagos magasságát (területét).
6. Az eredmények kiszámítása
A mintaoldat robenidincsúcsainak átlagmagasságából (területéből) határozzuk meg a mintaoldat koncentrációját μg/ml-ben a kalibrációs görbe (5.4.2. pont) alapján.
A minta, (mg/kg-ban kifejezett) w robenidintartalmát az alábbi képlet segítségével kapjuk meg:
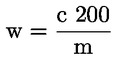
ahol:
c = a mintaoldat robenidin-koncentrációja, μg/ml-ben,
m = a vizsgálati adag tömege grammban.
7. Az eredmények validálása
7.1. Azonosítás
Az analizált anyag azonosítása párhuzamos kromatográfiával vagy diódasoros detektor használatával erősíthető meg, amely során a mintakivonat és a 6 μg/ml-es kalibrálóoldat (3.9.3. pont) spektrumát hasonlítjuk össze.
7.1.1. Párhuzamos kromatográfia
A mintakivonatot megfelelő mennyiségű kalibrálóoldattal (3.9.3. pont) dúsítjuk. A hozzáadott robenidin mennyisége és a mintakivonatban található robenidin becsült mennyisége közel azonos legyen.
A hozzáadott mennyiséget és a kivonat hígítását figyelembe véve, csak a robenidincsúcs magassága emelkedhet. A csúcs szélességének a legnagyobb magasság felénél az eredeti szélesség ± 10 %-on belül kell lennie.
7.1.2. Diódasoros detektálás
Az eredményeket a következő kritériumok szerint értékeljük:
a) a minta és a standard spektrumában a kromatogram csúcsmaximumán rögzített legnagyobb elnyeléshez tartozó hullámhossznak a detektor felbontóképessége által meghatározott tűréshatáron belül azonosnak kell lennie. A diódasoros detektoroknál ez a tűréshatár jellemzően ± 2 nm;
b) 250 és 400 nm között a spektrumnak a relatív abszorbancia 10 és 100 %-a közötti tartományba eső részein a mintának és a standardnak a kromatogram csúcsmaximumán rögzített spektruma nem térhet el egymástól. Ez a kritérium akkor teljesül, ha ugyanazok a maximális értékek vannak jelen, és a két spektrum egyetlen vizsgált ponton sem tér el a standard abszorbanciájának 15 %-ánál nagyobb mértékben;
c) 250 és 400 nm között a mintakivonat által kiváltott csúcs felszálló ágának, csúcsmaximumának és leszálló ágának spektrumai nem térhetnek el egymástól a spektrumnak a relatív abszorbancia 10 és 100 %-a közötti tartományba eső részein. Ez a kritérium akkor teljesül, ha ugyanazok a maximális értékek vannak jelen, és a spektrumok egyetlen vizsgált ponton sem térnek el a csúcsmaximum spektruma abszorbanciájának 15 %-ánál nagyobb mértékben.
Amennyiben ezen kritériumok valamelyike nem teljesül, az analizált anyag jelenléte nem bizonyított.
7.2. Ismételhetőség
Két azonos mintán végzett párhuzamos meghatározás eredménye közötti különbség, 15 mg/kg-nál magasabb robenidintartalom esetén, nem haladhatja meg a magasabb érték 10 %-át.
7.3. Visszanyerés
A dúsított vakminta esetében legalább 85 %-os visszanyerési arányt kell elérni.
8. A körvizsgálat eredményei
Uniós körvizsgálatot szerveztek, amely során őrlemény, illetve pellet formájában négy baromfi- és négy nyúltakarmánymintát elemeztek 12 laboratóriumban. Minden mintán kettős analízist végeztek. Az eredmények a következő táblázatban szerepelnek.
| Baromfi | Nyúl | |||
| Őrlemény | Pellet | Őrlemény | Pellet | |
| Középérték [mg/kg] | 27,00 | 27,99 | 43,6 | 40,1 |
| sr [mg/kg] | 1,46 | 1,26 | 1,44 | 1,66 |
| CVr [%] | 5,4 | 4,5 | 3,3 | 4,1 |
| SR [mg/kg] | 4,36 | 3,36 | 4,61 | 3,91 |
| CVR [%] | 16,1 | 12,0 | 10,6 | 9,7 |
| Visszanyerés [%] | 90,0 | 93,3 | 87,2 | 80,2 |
| sr= az ismételhetőség szórása CVr= az ismételhetőség relatív szórása, % SR= a reprodukálhatóság szórása CVR= a reprodukálhatóság relatív szórása, % | ||||
F. A DIKLAZURIL MEGHATÁROZÁSA
(+)-4-klórfenil [2,6-diklór-4-(2,3,4,5-tetrahidro-3,5-dioxo-1,2,4-triazin-2-il) fenil] acetonitril
A diklazuriltartalmat az alábbiak szerint kell meghatározni:
- a "Takarmányok. Mintavételi és elemzési módszerek. Az engedélyezett kokcidiosztatikumok szűrése és meghatározása additív, illetve 1 %-os és 3 %-os keresztszennyeződési szinten, valamint a nem regisztrált kokcidiosztatikumok és egy antibiotikum szűrése és meghatározása szubadditív szinteken, összetett takarmányban, nagy hatékonyságú folyadékkromatográfiával. Tandem-tömegspektrometriás kimutatás (LC-MS/MS)" című EN 17299 szabványban előírt analitikai módszerrel, vagy
- háromkomponensű, fordított fázisú, nagy teljesítményű folyadékkromatográfiával (HPLC), UV detektor használatával, az alábbi 1-9. pontban leírtak szerint.
1. Cél és alkalmazási terület
Ez a módszer lehetővé teszi a takarmánykeverékekben és előkeverékekben lévő diklazuril meghatározását ( 27 ). A kimutatási határérték 0,1 mg/kg, a meghatározás határértéke 0,5 mg/kg. Alacsonyabb meghatározási határok is elérhetők, de ezt a felhasználónak validálnia kell.
2. Vizsgálati alapelv
Egy belső standard hozzáadását követően a mintát savas metil-alkohollal extraháljuk. A takarmányok esetében a kivonat egy aliquot részét C18 szilárdfázisú extrahálótöltetben derítjük. A diklazurilt savas metil-alkohol és víz keverékével eluáljuk a töltetből. Bepárlás után a maradékot DMF/vízben feloldjuk. Előkeverékek esetében a kivonatot bepároljuk, és a maradékot DMF/vízben feloldjuk. A diklazuriltartalmat háromkomponensű, fordított fázisú, nagy teljesítményű folyadékkromatográfiával (HPLC) határozzuk meg, UV-detektor használatával.
3. Reagensek
3.1. Víz, HPLC minőségű
3.2. Ammónium-acetát
3.3. Tetrabutil-ammónium-hidrogén-szulfát (TBHS)
3.4. Acetonitril, HPLC-minőségű
3.5. Metil-alkohol, HPLC-minőségű
3.6. N, N-dimetilformamid (DMF)
3.7. Sósav, ρ20 = 1,19 g/ml
3.8. Standard anyag: diklazuril: (+)-4-klórfenil [2,6-diklór-4-(2,3,4,5-tetrahidro3,5 -dioxo-1,2,4-triazin-2-il) fenil] acetonitril, garantált tisztaságú
3.8.1. Diklazuril standard törzsoldat, 500 μg/ml
Mérjünk ki 0,1 mg pontossággal 25 mg diklazuril standard anyagot (3.8. pont) egy 50 ml-es mérőlombikba. Oldjuk fel DMF-ben (3.6. pont), töltsük fel jelig DMF-fel (3.6. pont), és keverjük össze. Tekerjük a lombikot alumíniumfóliába, vagy használjunk barna üveglombikot, és tároljuk hűtőben. 4 °C-on vagy ennél alacsonyabb hőmérsékleten az oldat egy hónapig stabil ( 28 ).
3.8.2. Diklazuril standardoldat, 50 μg/ml
Öntsünk át 5,00 ml standard törzsoldatot (3.8.1. pont) egy 50 ml-es mérőlombikba, töltsük fel jelig DMF-fel (3.6. pont), és keverjük össze. Tekerjük a lombikot alumíniumfóliába, vagy használjunk barna üveglombikot, és tároljuk hűtőben. 4 °C-on vagy ennél alacsonyabb hőmérsékleten az oldat egy hónapig stabil.
3.9. Belső standard anyag: 2,6-diklór-α-(4-klórfenil)-4-(4,5 dihidro3,5 -dioxo-1,2,4-triazin-2 (3H)-il) α-metilbenzol-acetonitril (metil-diklazuril)
3.9.1. Belső standard törzsoldat, 500 μg/ml
Mérjünk be 0,1 mg pontossággal 25 mg belső standard anyagot (3.9. pont) egy 50 ml-es mérőlombikba. Oldjuk fel DMF-ben (3.6. pont), töltsük fel jelig DMF-fel (3.6. pont), és keverjük össze. Tekerjük a lombikot alumíniumfóliába, vagy használjunk barna üveglombikot, és tároljuk hűtőben. 4 °C-on vagy ennél alacsonyabb hőmérsékleten az oldat egy hónapig stabil.
3.9.2. Belső standardoldat, 50 μg/ml
Öntsünk át 5,00 ml belső standard törzsoldatot (3.9.1. pont) egy 50 ml-es mérőlombikba, töltsük fel jelig DMF-fel (3.6. pont), és keverjük össze. Tekerjük a lombikot alumíniumfóliába, vagy használjunk barna üveglombikot, és tároljuk hűtőben. 4 °C-on vagy ennél alacsonyabb hőmérsékleten az oldat egy hónapig stabil.
3.9.3. Belső standardoldat előkeverékekhez, p/1 000 mg/ml (p = az előkeverék mg/kg-ban kifejezett névleges diklazuriltartalma)
Mérjünk be 0,1 mg-os pontossággal p/10 mg belső standardoldatot egy 100 ml-es mérőlombikba, oldjuk fel DMF-ben (3.6. pont) egy ultrahangos fürdőben (4.7. pont), töltsük fel jelig DMF-fel, és keverjük össze. Tekerjük a lombikot alumíniumfóliába, vagy használjunk barna üveglombikot, és tároljuk hűtőben. 4 °C-on vagy ennél alacsonyabb hőmérsékleten az oldat egy hónapig stabil.
3.10. Kalibrálóoldatok
3.10.1. Kalibrálóoldat, 1 μg/ml (diklazuril)
Pipettázzunk 1,00 ml diklazuril standardoldatot (3.8.2. pont) és 2,00 ml belső standardoldatot (3.9.2. pont) egy 50 ml-es mérőlombikba. Adjunk hozzá 17 ml DMF-t (3.6. pont), töltsük fel jelig vízzel (3.1. pont), és keverjük össze. Ezt az oldatot közvetlenül a felhasználás előtt kell elkészíteni.
3.10.2. Kalibrálóoldat, 2 μg/ml (diklazuril)
Pipettázzunk 2,00 ml diklazuril standardoldatot (3.8.2. pont) és 2,00 ml belső standardoldatot (3.9.2. pont) egy 50 ml-es mérőlombikba. Adjunk hozzá 16 ml DMF-t (3.6. pont), töltsük fel jelig vízzel (3.1. pont), és keverjük össze. Ezt az oldatot közvetlenül a felhasználás előtt kell elkészíteni.
3.10.3. Kalibrálóoldat, 3 μg/ml (diklazuril)
Pipettázzunk 3,00 ml diklazuril standardoldatot (3.8.2. pont) és 2,00 ml belső standardoldatot (3.9.2. pont) egy 50 ml-es mérőlombikba. Adjunk hozzá 15 ml DMF-t (3.6. pont), töltsük fel jelig vízzel (3.1. pont), és keverjük össze. Ezt az oldatot közvetlenül a felhasználás előtt kell elkészíteni.
3.10.4. Kalibrálóoldat, 4 μg/ml (diklazuril)
Pipettázzunk 4,00 ml diklazuril standardoldatot (3.8.2. pont) és 2,00 ml belső standardoldatot (3.9.2. pont) egy 50 ml-es mérőlombikba. Adjunk hozzá 14 ml DMF-t (3.6. pont), töltsük fel jelig vízzel (3.1. pont), és keverjük össze. Ezt az oldatot közvetlenül a felhasználás előtt kell elkészíteni.
3.10.5. Kalibrálóoldat, 5 μg/ml (diklazuril)
Pipettázzunk 5,00 ml diklazuril standardoldatot (3.8.2. pont) és 2,00 ml belső standardoldatot (3.9.2. pont) egy 50 ml-es mérőlombikba. Adjunk hozzá 13 ml DMF-t (3.6. pont), töltsük fel jelig vízzel (3.1. pont), és keverjük össze. Ezt az oldatot közvetlenül a felhasználás előtt kell elkészíteni.
MEGJEGYZÉS: a kalibráló oldatok (3.10.1., 3.10.2., 3.10.3., 3.10.4. és 3.10.5. pont) a takarmányban lévő diklazuril 0,5-2,5 mg/kg közötti koncentrációtartományát fedik le a jelen eljárás alkalmazása esetén.
3.11. C18 szilárdfázisú extrahálótöltet, pl. Mega Bond Elut, méret: 20 cm3, szorbens tömege: 5 000 mg (előkondicionálás a beszállítói iránymutatások szerint).
3.12. Extraháló oldószer: savas metil-alkohol.
Pipettázzunk 5,0 ml sósavat (3.7. pont) 1 000 ml metil-alkoholba (3.5. pont), és keverjük össze.
3.13. Mozgófázis a HPLC-hez:
3.13.1. A. eluens: ammónium-acetát - tetrabutil-ammónium-hidrogénszulfát oldat.
Oldjunk fel 5 g ammónium-acetátot (3.2. pont) és 3,4 g TBHS-t (3.3. pont) 1 000 ml vízben (3.1. pont), és keverjük össze.
3.13.2. B. eluens: acetonitril (3.4. pont).
3.13.3. C. eluens: metil-alkohol (3.5. pont).
4. Eszközök
4.1. Mechanikus rázógép
4.2. Felszerelés a háromkomponensű HPLC-hez:
4.2.1. Hypersil ODS folyadékkromatográfiás oszlop, 100 mm × 4,6 mm, 3 μm-es töltettel, vagy ezzel egyenértékű berendezés.
4.2.2. Változtatható hullámhosszon mérő UV-detektor vagy diódasoros detektor
4.3. Rotációs, vékonyfilmes bepárló
4.4. Membránszűrő (pl. vegyi anyagokkal szemben ellenálló nejlon), 0,45 μm-es
4.5. Egyszer használatos fecskendő, 5 ml-es
4.6. Vákuumos elosztóvezeték.
4.7. Ultrahangos fürdő
5. A vizsgálat módja
5.1. Általános szabályok
5.1.1. Takarmányvakminta
Vizsgáljunk meg egy takarmányvakmintát annak ellenőrzésére, hogy sem diklazuril, sem interferáló anyagok nincsenek jelen. A takarmányvakminta a mintával azonos típusú takarmány legyen, amelyben az analízis során sem diklazuril, sem interferáló anyagok nem mutathatók ki.
5.1.2. Visszanyerési próba
Végezzünk visszanyerési próbát a mintában meglévővel közel azonos mennyiségű diklazurillel dúsított takarmányvakminta analízisével. Az 1 mg/kg szintre való dúsításhoz adjunk 0,1 ml standard törzsoldatot (3.8.1. pont) 50 g takarmányvakmintához, keverjük össze alaposan, és hagyjuk állni 10 percig, keverjük össze néhányszor, mielőtt folytatjuk a műveletet (5.2. pont).
Amennyiben a mintával megegyező takarmányfajtából vett takarmányvakminta nem áll rendelkezésre (lásd az 5.1.1. pontot), a visszanyerési próbát a standard hozzáadásos módszerrel lehet elvégezni. Ebben az esetben a vizsgált mintát az abban már jelen lévővel azonos mennyiségű diklazurillal dúsítjuk. Ezt a mintát a nem dúsított mintával együtt elemezzük, és a visszanyerést kivonással számítjuk ki.
5.2. Extrahálás
5.2.1. Takarmánykeverékek
0,01 g pontossággal mérjünk ki 50 g mintát. Öntsük át egy 500 ml-es Erlenmeyer-lombikba, adjunk hozzá 1,00 ml belső standardoldatot (3.9.2. pont), 200 ml extraháló oldószert (3.12. pont), és zárjuk le a lombikot. Rázassuk a keveréket a rázógépen (4.1. pont) egy éjszakán át. Hagyjuk ülepedni 10 percig. A felülúszó anyag 20 ml-es aliquot részét öntsük át egy üvegedénybe, és hígítsuk fel 20 ml vízzel (3.1. pont). Öntsük át ezt az oldatot egy extrahálótöltetbe (3.11. pont), és vákuum segítségével engedjük át rajta (4.6. pont). Mossuk a töltetet 25 ml extraháló oldószer (3.12. pont) és víz (3.1. pont) 65 + 35 (V + V) arányú keverékével. Öntsük el a felvett frakciókat, és eluáljuk az alkotóelemeket 25 ml extraháló oldószer (3.12. pont) és víz 80 + 20 (V + V) arányú keverékével. Pároljuk be ezt a frakciót a rotációs bepárló (4.3. pont) segítségével 60 °C-on, amíg éppen beszárad. A maradékot oldjuk fel 1,0 ml DMF-ben (3.6. pont), adjunk hozzá 1,5 ml vizet (3.1. pont), és keverjük össze. Szűrjük át egy egyszer használatos fecskendőre (4.5. pont) illesztett membránszűrőn (4.4. pont). Folytassuk a műveletet a HPLC-meghatározással (5.3. pont).
5.2.2. Előkeverékek
0,001 g pontossággal mérjünk ki 1 g mintát. Öntsük át egy 500 ml-es Erlenmeyer-lombikba, adjunk hozzá 1,00 ml belső standardoldatot (3.9.3. pont), 200 ml extraháló oldószert (3.12. pont), és zárjuk le a lombikot. Rázassuk a keveréket a rázógépen (4.1. pont) egy éjszakán át. Hagyjuk ülepedni 10 percig. A felülúszó anyag egy 10 000 /p ml (p = az előkeverék mg/kg-ban kifejezett névleges diklazuriltartalma) aliquot részét öntsük át egy megfelelő méretű gömblombikba. Pároljuk be ezt a frakciót a rotációs bepárló (4.3. pont) segítségével 60 °C-on, csökkentett nyomáson, amíg éppen beszárad. A maradékot oldjuk fel 10,0 ml DMF-ben (3.6. pont), adjunk hozzá 15,0 ml vizet (3.1. pont), és keverjük össze. Folytassuk a műveletet a HPLC-meghatározással (5.3. pont).
5.3. HPLC-meghatározás
5.3.1. Paraméterek
Az alábbi vizsgálati feltételek útmutatásként szolgálnak, ettől eltérő feltételeket is lehet használni, feltéve, hogy ezzel egyenértékű vagy jobb eredményeket adnak.
- Folyadékkromatográfiás oszlop (4.2.1. pont): 100 mm × 4,6 mm méretű, Hypersil ODS 3 μm-es töltettel, vagy ezzel egyenértékű berendezés.
- Mozgófázis
- A. eluens (3.13.1. pont): ammónium-acetát-tetrabutil-ammónium-hidrogénszulfát vizes oldata.
- B. eluens (3.13.2. pont): acetonitril.
- C. eluens (3.13.3. pont): metil-alkohol.
- Eluálás módja: - lineáris gradiens
- kiindulási feltételek: A + B + C = 60 + 20 + 20 (V + V + V);
- 10 perces gradiens eluálás után 30 percig: A + B + C = 45 + 20 + 35 (V + V + V);
- majd öblítés B-eluenssel 10 percig.
- Átáramlási sebesség: 1,5-2 ml/perc
- Befecskendezett mennyiség: 20 μl.
- Kimutatási hullámhossz: 280 nm
Ellenőrizzük a kromatográfiás rendszer stabilitását oly módon, hogy 2 μg/ml diklazurilt és belső standardot tartalmazó kalibrálóoldatot (3.10.2. pont) fecskendezünk be többször, amíg a csúcsok magassága és a retenciós idők állandóvá nem válnak.
5.3.2. A kalibrálóoldatok kromatográfiás vizsgálata
Fecskendezzünk be 20 μl-t minden kalibrálóoldatból (3.10.1., 3.10.2., 3.10.3., 3.10.4. és 3.10.5. pont) kétszer, azonosítsuk és integráljuk a diklazurilcsúcsot és a belső standard csúcsot, majd szerkesszünk kalibrációs görbét a diklazurilcsúcsok átlagmagasságát vagy átlagterületét a belső standard csúcsok átlagmagasságához vagy átlagterületéhez viszonyított arányszámok és a kalibrálóoldatokban lévő diklazuril-koncentrációk (μg/ml) alapján.
5.3.3. A mintaoldatok kromatográfiás vizsgálata
Fecskendezzünk be 20 μl mintaoldatot (5.2.1. vagy 5.2.2. pont) kétszer, és határozzuk meg a diklazurilcsúcsok és a belső standard csúcsok átlagmagasságát vagy átlagterületét.
6. Az eredmények kiszámítása
6.1. Takarmánykeverékek
A minta mg/kg-ban kifejezett w diklazuriltartalmát a következő képlet segítségével kapjuk meg:
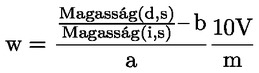
vagy
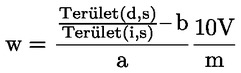
ahol:
- Magasság(d,s): a diklazuril csúcsmagassága a mintaoldatban (5.2.1. pont)
- Terület(d,s): a diklazuril csúcsterülete a mintaoldatban (5.2.1. pont)
- Magasság(i,s): a belső standard csúcsmagassága a mintaoldatban (5.2.1. pont)
- Terület(i,s): a belső standard csúcsterülete a mintaoldatban (5.2.1. pont)
- b: a kalibráló oldatokból (3.10.1., 3.10.2., 3.10.3., 3.10.4. és 3.10.5. pont) az 5.3.2. pont szerint szerkesztett kalibrációs görbe tengelymetszete
- a: a kalibráló oldatokból (3.10.1., 3.10.2., 3.10.3., 3.10.4. és 3.10.5. pont) az 5.3.2. pont szerint szerkesztett kalibrációs görbe meredeksége
- m: a vizsgált mennyiség tömege grammban
- V: a mintakivonat végső térfogata milliliterben az 5.2.1. pont szerinti újrafeloldás után (azaz 2,5 ml)
6.2. Előkeverékek
A minta mg/kg-ban kifejezett w diklazuriltartalmát a következő képlet segítségével kapjuk meg:
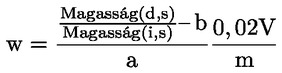
p vagy
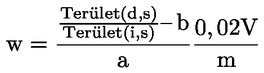
p
ahol:
- Magasság(d,s): a diklazuril csúcsmagassága a mintaoldatban (5.2.2. pont)
- Terület(d,s): a diklazuril csúcsterülete a mintaoldatban (5.2.2. pont)
- Magasság(i,s): a belső standard csúcsmagassága a mintaoldatban (5.2.2. pont)
- Terület(i,s): a belső standard csúcsterülete a mintaoldatban (5.2.2. pont)
- b: a kalibráló oldatokból (3.10.1., 3.10.2., 3.10.3., 3.10.4. és 3.10.5. pont) az 5.3.2. pont szerint szerkesztett kalibrációs görbe metszete
- a: a kalibráló oldatokból (3.10.1., 3.10.2., 3.10.3., 3.10.4. és 3.10.5. pont) az 5.3.2. pont szerint szerkesztett kalibrációs görbe meredeksége
- m: a vizsgált mennyiség tömege grammban
- V: a mintakivonat végső térfogata milliliterben az 5.2.2. pont szerinti újrafeloldás után (azaz 25 ml)
- p: az előkeverék névleges diklazuriltartalma, mg/kg-ban kifejezve
7. Az eredmények validálása
7.1. Azonosítás
Az analizált anyag azonosítása párhuzamos kromatográfiával vagy diódasoros detektor használatával erősíthető meg, amely során a mintakivonat (5.2.1. vagy 5.2.2. pont) és a kalibrálóoldat (3.10.2. pont) spektrumát hasonlítjuk össze.
7.1.1. Párhuzamos kromatográfia
A mintakivonatot (5.2.1. vagy 5.2.2. pont) megfelelő mennyiségű kalibrálóoldattal (3.10.2. pont) dúsítjuk. A hozzáadott diklazuril mennyisége közel azonos legyen a mintakivonatban meglévő diklazuril mennyiségével.
A hozzáadott mennyiséget és a kivonat hígítását figyelembe véve, csak a diklazuril csúcs és a belső standard csúcs magassága emelkedhet. A csúcs szélességének a legnagyobb magasság felénél a dúsítás előtti mintakivonat diklazuril csúcsa vagy belső standard csúcsa eredeti szélességének ± 10 %-án belül kell lennie.
7.1.2. Diódasoros detektálás
Az eredményeket a következő kritériumok szerint értékeljük:
a) a minta és a standard spektrumában a kromatogram csúcsmaximumán rögzített legnagyobb elnyeléshez tartozó hullámhossznak a detektor felbontóképessége által meghatározott tűréshatáron belül azonosnak kell lennie. A diódasoros detektoroknál ez a tűréshatár jellemzően ± 2 nm.
b) 230 és 320 nm között a spektrumnak a relatív abszorbancia 10 és 100 %-a közötti tartományba eső részein a mintának és a standardnak a kromatogram csúcsmaximumán rögzített spektruma nem térhet el egymástól. Ez a kritérium akkor teljesül, ha ugyanazok a maximális értékek vannak jelen, és a két spektrum egyetlen vizsgált ponton sem tér el a standard abszorbanciájának 15 %-ánál nagyobb mértékben.
c) 230 és 320 nm között a mintakivonat által kiváltott csúcs felszálló ágának, csúcsmaximumának és leszálló ágának spektrumai nem térhetnek el egymástól a spektrumnak a relatív abszorbancia 10 és 100 %-a közötti tartományba eső részein. Ez a kritérium akkor teljesül, ha ugyanazok a maximális értékek vannak jelen, és a spektrumok egyetlen vizsgált ponton sem térnek el a csúcsmaximum spektruma abszorbanciájának 15 %-ánál nagyobb mértékben.
Amennyiben ezen kritériumok valamelyike nem teljesül, az analizált anyag jelenléte nem bizonyított.
7.2. Ismételhetőség
A két részmintán végzett két független mérés eredményei közötti különbség nem haladhatja meg:
- 0,5 mg/kg és 2,5 mg/kg közötti diklazuriltartalom esetén a magasabb érték 30 %-át,
- 2,5 mg/kg és 5 mg/kg közötti diklazuriltartalom esetén a 0,75 mg/kg értéket,
- 5 mg/kg feletti diklazuriltartalom esetén a magasabb érték 15 %-át.
7.3. Visszanyerés
Dúsított (vak-)minta esetében a visszanyerésnek legalább 80 %-osnak kell lennie.
8. A körvizsgálat eredményei
Két körvizsgálatot szerveztek. Az elsőben, amelyet egy másik laboratóriumcsoport 1994-ben szervezett, az elemzett minták két előkeverék (O 100, A 100) és három, baromfinak szánt kiegészítő takarmány (L1, Z1, K1) voltak. Az egyik előkeverék-mintát egy szerves mátrixszal (O 100), a másikat pedig egy szervetlen mátrixszal (A 100) keverték össze. Az elméleti diklazuriltartalom 100 mg/kg volt. A laboratóriumokat utasították, hogy a vizsgálatokat egyszer vagy kétszeres ismétléssel vizsgálják. (Az első kör vizsgálatra vonatkozóan részletesebb információk találhatók a következő helyen: Journal of AOAC International, 77. évfolyam, 6. szám, 1994, 1359-1361. o.).
A második körvizsgálatban három, baromfi takarmányozására szánt takarmánykeveréket vizsgáltak meg, amelyek 0,9 mg/kg (1. ANYAG), 1,5 mg/kg (2. ANYAG) koncentrációban diklazurilt, illetve és takarmányvakmintát (3. ANYAG) tartalmaztak. A második körvizsgálatra vonatkozó részletes információk a JRC technikai jelentésében (JRC Technical report, 2016) és a Journal of AOAC Internationalben (102. évfolyam, 2. szám, 2019, 646-652. o.) találhatók. A két körvizsgálat eredményeit az alábbi táblázat tartalmazza.
| 1. minta A 100 | 2. minta O 100 | 3. minta L1 | 4. minta Z1 | 5. minta K1 | 6. minta 1. ANYAG | 7. minta 2. ANYAG | 8. minta 3. ANYAG | |
| L | 11 | 11 | 11 | 11 | 6 | 10 | 9 | 10 |
| n | 19 | 18 | 19 | 19 | 12 | 20 | 18 | 10 |
| Átlag (mg/kg) | 100,8 | 103,5 | 0,89 | 1,15 | 0,89 | 1,0 | 1,5 | < LOQ |
| Sr (mg/kg) | 5,88 | 7,64 | 0,15 | 0,02 | 0,03 | 0,11 | 0,07 | — |
| CVr (%) | 5,83 | 7,38 | 17,32 | 1,92 | 3,34 | 11,2 | 4,5 | — |
| SR (mg/kg) | 7,59 | 7,64 | 0,17 | 0,11 | 0,12 | 0,18 | 0,21 | — |
| CVR (%) | 7,53 | 7,38 | 18,61 | 9,67 | 13,65 | 18,1 | 14,3 | — |
| Névleges tartalom (mg/kg) | 100 | 100 | 1,0 | 1,0 | 1,0 | 0,9 | 1,5 | — |
| Hivatkozás (*1) | Az 1 994 -es első vizsgálat | Az 1 994 -es első vizsgálat | Az 1 994 -es első vizsgálat | Az 1 994 -es első vizsgálat | Az 1 994 -es első vizsgálat | A 2 015 -ös második vizsgálat | A 2 015 -ös második vizsgálat | A 2 015 -ös második vizsgálat |
| (*1) Az 1994-es első vizsgálat: Journal of AOAC International, 77. kötet, 6. szám, 1994., 1359–1361. o.; A 2015-ös második vizsgálat: A JRC technikai jelentése: „Re-validation of a method for the determination of diclazuril by collaborative study” (Egy a diklazuril körvizsgálattal történő meghatározására szolgáló módszer újravalidálása) (2016). L = a laboratóriumok száma n = az egyedi értékek száma Sr = az ismételhetőség szórása CVr = az ismételhetőség relatív szórása SR = a reprodukálhatóság szórása CVR = a reprodukálhatóság relatív szórása LOQ = a meghatározási határ | ||||||||
9. Általános megjegyzések
A diklazuril-reakciót korábban lineárisnak találták a mért koncentrációk sávja felett.
Legalább a magas zsírtartalmú (e célból a 12 %-ot meghaladó zsírtartalmú) takarmánykeverékben lévő diklazuril vizsgálatánál a vizsgálati módszer helyettesíthető más HPLC-alapú módszerekkel, például nagy teljesítményű folyadék-kromatográffal kapcsolt tömegspektrometrián (HPLC-MS) alapuló módszerrel, feltéve, hogy az alternatív módszer egyenértékű teljesítményjellemzőkkel rendelkezik (visszanyerési arány, precizitás az ismételhetőségi és reprodukálhatósági feltételek melletti).
G. A LAZALOCID-NÁTRIUM MEGHATÁROZÁSA
A Streptomyces lasaliensis által termelt poliéter-monokarboxilsav nátriumsója
A lazalocid-nátrium-tartalmat az alábbiak szerint kell meghatározni:
- a "Takarmányok. Mintavételi és elemzési módszerek. Az engedélyezett kokcidiosztatikumok szűrése és meghatározása additív, illetve 1 %-os és 3 %-os keresztszennyeződési szinten, valamint a nem regisztrált kokcidiosztatikumok és egy antibiotikum szűrése és meghatározása szubadditív szinteken, összetett takarmányban, nagy hatékonyságú folyadékkromatográfiával. Tandem-tömegspektrometriás kimutatás (LC-MS/MS)" című EN 17299 szabványban előírt analitikai módszerrel, vagy
- fordított fázisú, nagy teljesítményű folyadékkromatográfiával (HPLC), spektrofotometriás (fluoreszcens) detektor használatával, az alábbi 1-8. pontban leírtak szerint.
1. Cél és alkalmazási terület
A módszer a takarmányok lazalocid-nátrium-tartalmának meghatározására szolgál. A kimutatási határérték 5 mg/kg, a meghatározás határértéke 10 mg/kg.
2. Vizsgálati alapelv
A lazalocid-nátriumot a mintából savas metil-alkohollal extraháljuk, majd fordított fázisú, nagy teljesítményű folyadékkromatográfiával (HPLC), spektrofotometriás (fluoreszcens) detektor segítségével határozzuk meg.
3. Reagensek
3.1. Kálium-dihidrogén-foszfát (KH2PO4)
3.2. Ortofoszforsav, w (w/w) = 85 %
3.3. Ortofoszforsav-oldat, c = 20 %
Hígítsunk fel vízzel 23,5 ml ortofoszforsavat (3.2. pont) 100 ml-re.
3.4. 6-metil-2-heptilamin (1,5-dimetil-hexilamin), w (w/w) = 99 %
3.5. Metil-alkohol, HPLC-minőségű
3.6. Sósav, sűrűség = 1,19 g/ml
3.7. Foszfátpuffer-oldat, c = 0,01 mol/l
Oldjunk fel 1,36 g KH2PO4-ot (3.1. pont) 500 ml vízben (3.11. pont), adjunk hozzá 3,5 ml ortofoszforsavat (3.2. pont) és 10,0 ml 6-metil-2-heptilamint (3.4. pont). Állítsuk be a pH-t 4,0-re ortofoszforsav-oldattal (3.3. pont), és hígítsuk fel vízzel 1 000 ml-re (3.11. pont).
3.8. Savas metil-alkohol
Öntsünk 5,0 ml sósavat (3.6. pont) egy 1 000 ml-es mérőlombikba, töltsük fel jelig metil-alkohollal (3.5. pont), és keverjük össze. Ezt az oldatot közvetlenül a felhasználás előtt kell elkészíteni.
3.9. HPLC mozgófázis, foszfátpuffer-metil-alkohol-oldat, 5 + 95 (V + V)
Keverjünk össze 5 ml foszfátpuffer-oldatot (3.7. pont) 95 ml metil-alkohollal (3.5. pont).
3.10. Lazalocid-nátrium garantált tisztaságú standard anyag, C34H53O8Na (a poliéter-monokarboxilsav nátriumsója, amelyet a Streptomyces lasaliensis termel), E763
3.10.1. Lazalocid-nátrium standard törzsoldat, 500 μg/ml
Mérjünk be 0,1 mg-os pontossággal 50 mg lazalocid-nátriumot (3.10. pont) egy 100 ml-es mérőlombikba, oldjuk fel savas metil-alkoholban (3.8. pont), töltsük fel jelig ugyanezzel az oldószerrel, és keverjük össze. Ezt az oldatot használat előtt frissen kell elkészíteni.
3.10.2. Lazalocid-nátrium közbenső standardoldat, 50 μg/ml
Pipettázzunk 10,0 ml standard törzsoldatot (3.10.1. pont) egy 100 ml-es mérőlombikba, töltsük fel jelig savas metil-alkohollal (3.8. pont), és keverjük össze. Ezt az oldatot közvetlenül a felhasználás előtt kell elkészíteni.
3.10.3. Kalibrálóoldatok
50 ml-es mérőlombikokba öntsünk 1,0, 2,0, 4,0, 5,0, illetve 10,0 ml közbenső standardoldatot (3.10.2. pont). Öntsük fel jelig savas metil-alkohollal (3.8. pont), és keverjük össze. Ezek az oldatok egyenként 1,0, 2,0, 4,0, 5,0 és 10,0 μg/ml lazalocid-nátriumnak felelnek meg. Ezeket az oldatokat közvetlenül a felhasználás előtt kell elkészíteni.
3.11. Víz, HPLC-minőségű
4. Eszközök
4.1. Ultrahangos fürdő (vagy vízfürdős rázógép), hőmérséklet-szabályozóval
4.2. Membránszűrők, 0,45 μm
4.3. HPLC-berendezés 20 μl mennyiség befecskendezésére alkalmas befecskendező rendszerrel
4.3.1. Folyadékkromatográfiás oszlop, 125 mm × 4 mm, fordított fázisú C18, 5 μm töltet vagy azzal egyenértékű berendezés
4.3.2. Spektrofluoriméter, változtatható gerjesztési és emissziós hullámhossz-szabályozással
5. A vizsgálat módja
5.1. Általános szabályok
5.1.1. Takarmányvakminta
A visszanyerési próba (5.1.2. pont) elvégzése érdekében elemezzünk egy takarmányvakmintát annak ellenőrzésére, hogy sem lazalocid-nátrium, sem egyéb interferáló anyag nincs jelen. A takarmányvakminta a mintához hasonló típusú legyen, és benne lazalocid-nátrium vagy más interferáló anyag ne legyen kimutatható.
5.1.2. Visszanyerési próba
Végezzünk visszanyerési próbát egy olyan takarmányvakminta analizálásával, amelyet a mintában jelen lévőhöz hasonló mennyiségű lazalocid-nátriummal dúsítottak. 100 mg/kg-os szintre történő dúsításhoz öntsünk 10,0 ml standard törzsoldatot (3.10.1. pont) egy 250 ml-es Erlenmeyer-lombikba, és pároljuk be az oldatot kb. 0,5 ml-re. Adjunk hozzá 50 g takarmányvakmintát, keverjük össze alaposan, és hagyjuk állni 10 percig, majd keverjük össze újra néhányszor, mielőtt az extrahálással (5.2. pont) folytatjuk a műveletet.
Amennyiben a mintával megegyező takarmányfajtából vett takarmányvakminta nem áll rendelkezésre (lásd az 5.1.1. pontot), a visszanyerési próbát a standard hozzáadásos módszerrel lehet elvégezni. Ebben az esetben a vizsgálandó mintát az abban már jelen lévővel közel azonos mennyiségű lazalocid-nátriummal dúsítjuk. Ezt a mintát a nem dúsított mintával együtt analizáljuk, a visszanyerést kivonással számítjuk ki.
5.2. Extrahálás
5.2.1. Takarmány
Mérjünk be 0,01 g-os pontossággal 5-10 g mintát egy dugós, 250 ml-es Erlenmeyer-lombikba. Pipettával adjunk hozzá 100,0 ml savas metil-alkoholt (3.8. pont). Dugaszoljuk le lazán, és keverjük, míg diszperziót nem kapunk. Helyezzük a lombikot körülbelül 40 °C-os ultrahangos fürdőbe (4.1. pont) 20 percre, majd vegyük ki, és hűtsük le szobahőmérsékletre. Hagyjuk állni kb. 1 óráig, amíg a szuszpendált anyag leülepszik, majd egy 0,45 μm-es membránszűrőn (4.2. pont) szűrjünk egy aliquot részt egy megfelelő edénybe. Folytassuk a műveletet a HPLC-meghatározással (5.3. pont).
5.2.2. Előkeverékek
Mérjünk be 0,001 g-os pontossággal 2 g őröletlen előkeveréket egy 250 ml-es mérőlombikba. Adjunk hozzá 100,0 ml savas metil-alkoholt (3.8. pont), és keverjük, amíg diszperziót nem kapunk. Helyezzük a mérőlombikot tartalmával együtt kb. 40 °C-os ultrahangos fürdőbe (4.1. pont) 20 percre, majd vegyük ki, és hűtsük le szobahőmérsékletre. Hígítsuk a jelig savas metil-alkohollal (3.8. pont), és alaposan keverjük össze. Hagyjuk állni 1 óráig, amíg a szuszpendált anyag leülepszik, majd egy 0,45 μm-es membránszűrőn (4.2. pont) szűrjünk át egy aliquot részt. Hígítsuk a tiszta filtrátum megfelelő mennyiségét savas metil-alkohollal (3.8. pont), hogy így kb. 4 μg/ml lazalocid-nátriumot tartalmazó végső vizsgálati oldatot kapjunk. Folytassuk a műveletet a HPLC-meghatározással (5.3. pont).
5.3. HPLC-meghatározás
5.3.1. Paraméterek
Az alábbi vizsgálati feltételek csak útmutatásként szolgálnak, ettől eltérő feltételeket is lehet használni, feltéve, hogy ezzel egyenértékű eredményt adnak:
| Folyadékkromatográfiás oszlop, (4.3.1. pont) | 125 mm × 4 mm, fordított fázisú C18, 5 μm töltet vagy azzal egyenértékű berendezés |
| Mozgófázis (3.9. pont): | Foszfátpuffer-oldat (3.7. pont) és metil-alkohol (3.5. pont) keveréke, 5 + 95 (V + V) |
| Átáramlási sebesség: | 1,2 ml/perc |
| Kimutatási hullámhossz: | gerjesztés: 310 nm |
| emisszió: 419 nm | |
| Befecskendezett mennyiség: | 20 μl |
Ellenőrizzük a kromatográfiás rendszer stabilitását oly módon, hogy 4,0 μg/ml-es koncentrációjú kalibrálóoldatot (3.10.3. pont) fecskendezünk be többször, amíg a csúcsmagasságok (vagy területek) és retenciós idők állandóvá nem válnak.
5.3.2. Kalibrációs görbe
Fecskendezzük be többször egymás után az egyes kalibrálóoldatokat (3.10.3. pont), és határozzuk meg az egyes koncentrációk átlagos csúcsmagasságait (területeit). Készítsük el a kalibrációs görbét úgy, hogy az átlagos csúcsmagasságokat (területeket) az ordinátán, a μg/ml-ben kifejezett megfelelő koncentrációkat az abszcisszán ábrázoljuk.
5.3.3. Mintaoldat
Ugyanolyan mennyiségű oldatot használva, mint amennyit a kalibrálóoldatoknál használtunk, fecskendezzük be az 5.2.1. vagy 5.2.2. pont szerint nyert mintakivonatokat egymás után többször, és határozzuk meg a lazalocid-nátrium-csúcsok átlagos csúcsmagasságait (területeit).
6. Az eredmények kiszámítása
A mintaoldat (5.3.3. pont) befecskendezése után kapott átlagos csúcsmagasságokból (területekből), a kalibrációs görbe alapján határozzuk meg a lazalocid-nátrium koncentrációját μg/ml-ben.
6.1. Takarmány
A minta w lazalocid-nátrium-tartalma (mg/kg) a következő képlet szerint adható meg:
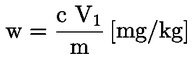
ahol:
c = a mintaoldat (5.2.1. pont) μg/ml-ben kifejezett lazalocid-nátrium koncentrációja
V1 = a mintakivonat 5.2.1. pont szerint ml-ben megadott (= 100) térfogata
m = a vizsgálati adag tömege g-ban
6.2. Előkeverékek
A minta w lazalocid-nátrium-tartalma (mg/kg) a következő képlet szerint adható meg:
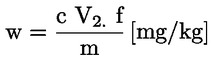
ahol:
c = a mintaoldat (5.2.2. pont) μg/ml-ben kifejezett lazalocid-nátrium koncentrációja
V2 = a mintakivonat 5.2.2. pont szerint ml-ben megadott (= 250) térfogata
f = az 5.2.2. pont szerinti hígítási tényező
m = a vizsgálati adag tömege g-ban
7. Az eredmények validálása
7.1. Azonosítás
A spektro-fluorimetrián alapuló módszerek kevésbé érzékenyek az interferenciára, mint azok az eljárások, amelyek során UV-detektort használnak. Az analizált anyag azonossága párhuzamos kromatografiával ellenőrizhető.
7.1.1. Párhuzamos kromatográfia
A mintakivonatot (5.2.1. vagy 5.2.2. pont) megfelelő mennyiségű kalibrálóoldattal (3.10.3. pont) dúsítjuk. A hozzáadott lazalocid-nátrium mennyisége közel azonos legyen a mintaoldatban található lazalocid-nátrium mennyiségével. A hozzáadott lazalocid-nátrium mennyiségét és a kivonat hígulását figyelembe véve csak a lazalocid-nátrium csúcsmagassága emelkedhet. A csúcs szélességének a legnagyobb magasság felénél a nem dúsított mintakivonat eredeti csúcsszélessége ± 10 %-on belül kell lennie.
7.2. Ismételhetőség
Az ugyanazon mintán végzett két párhuzamos meghatározás eredménye közötti különbség nem haladhatja meg:
- 30-100 mg/kg közötti lazalocid-nátrium-tartalom esetén a magasabb érték 15 %-át,
- 100-200 mg/kg közötti lazalocid-nátrium-tartalom esetén a 15 mg/kg értéket,
- 200 mg/kg-nál nagyobb lazalocid-nátrium-tartalom esetén a magasabb érték 7,5 %-át.
7.3. Visszanyerés
A dúsított (vakpróba) takarmányminta esetében a visszanyerés foka legalább 80 %-os legyen. A dúsított előkeverék-minta esetében legalább 90 %-os visszanyerési arányt kell elérni.
8. A körvizsgálat eredményei
Egy körvizsgálat () keretében 12 laboratóriumban 2 előkeveréket (1. és 2. minta) és 5 takarmányt (3-7. minta) analizáltak. Minden mintán kettős analízist végeztek. Az eredmények a következő táblázatban szerepelnek.
| 1. minta Csirke-előkeverék | 2. minta Pulyka-előkeverék | 3. minta Pulykapellet | 4. minta Csirkederce | 5. minta Pulykatakarmány | 6. minta Baromfi A. takarmány | 7. minta Baromfi B. takarmány | |
| L | 12 | 12 | 12 | 12 | 12 | 12 | 12 |
| n | 23 | 23 | 23 | 23 | 23 | 23 | 23 |
| Középérték [mg/kg] | 5 050 | 16 200 | 76,5 | 78,4 | 92,9 | 48,3 | 32,6 |
| sr [mg/kg] | 107 | 408 | 1,71 | 2,23 | 2,27 | 1,93 | 1,75 |
| CVr [%] | 2,12 | 2,52 | 2,24 | 2,84 | 2,44 | 4,00 | 5,37 |
| sR [mg/kg] | 286 | 883 | 3,85 | 7,32 | 5,29 | 3,47 | 3,49 |
| CVR [%] | 5,66 | 5,45 | 5,03 | 9,34 | 5,69 | 7,18 | 10,70 |
| Névleges tartalom [mg/kg] | 5 000 (*2) | 16 000 (*2) | 80 (*2) | 105 (*2) | 120 (*2) | 50 () | 35 () |
| (*1) Analyst, 1995, 120., 2175–2180. o. (*2) Gyártó által feltüntetett tartalom. (1) Laboratóriumban készített takarmány. L= a laboratóriumok száma n= az egyedi értékek száma sr= az ismételhetőség szórása SR= a reprodukálhatóság szórása CVr= az ismételhetőség relatív szórása, % CVR= a reprodukálhatóság relatív szórása, % | |||||||
H. AZ AMPRÓLIUM-HIDROKLORID MEGHATÁROZÁSA - 1-[(4-amino-2-propil-5-pirimidinil)metil]-2-metilpiridinium klorid monohidroklorid
1. Cél és alkalmazási terület
Ez a módszer lehetővé teszi a takarmányokban lévő amprólium szintjének meghatározását. A kimutatási határ 1 mg/kg, a meghatározási határ 5 mg/kg.
2. Vizsgálati alapelv
A mintát metil-alkohol és víz keverékével extraháljuk. A mozgófázissal történő hígítás és membránszűrés után az ampróliumtartalmat kationcserélő, nagy teljesítményű folyadékkromatográfiával (HPLC), UV-detektor használatával határozzuk meg.
3. Reagensek
3.1. Metil-alkohol
3.2. Acetonitril, HPLC-minőségű.
3.3. Víz, HPLC-minőségű
3.4. Nátrium-dihidrogén-foszfát oldat, c = 0,1 mol/l
Oldjunk fel 13,80 g nátrium-dihidrogén-foszfát-monohidrátot vízben (3.3. pont), egy 1 000 ml-es mérőlombikban, töltsük fel vízzel (3.3. pont) jelig és keverjük össze.
3.5. Nátrium-perklorát oldat, c = 1,6 mol/l
Oldjunk fel 224,74 g nátrium-perklorát-monohidrátot vízben (3.3. pont) egy 1 000 ml-es mérőlombikban, töltsük fel vízzel (3.3. pont) jelig, és keverjük össze.
3.6. Mozgófázis a HPLC-hez (lásd az Észrevételek 9.1. pontját).
Acetonitril (3.2. pont), nátrium-dihidrogén- foszfát oldat (3.4. pont) és nátrium-perklorát oldat (3.5. pont) 450 + 450 + 100 (v + v + v) arányú keveréke. Használat előtt szűrjük át egy 0,22 μm-es membránszűrőn (4.3. pont) és gázmentesítsük az oldatot (pl. ultrahangos fürdőben (4.4. pont) legalább 15 percig).
3.7. Standard anyag: tiszta amprólium, 1-[(4-amino-2-propil-5-pirimidinil)metil]-2-metilpiridinium klorid, E 750 (lásd a 9.2. pontot)
3.7.1. Amprólium standard törzsoldat, 500 μg/ml
Mérjünk ki 0,1 mg-os pontossággal 50 mg ampróliumot (3.7. pont) egy 100 ml-es mérőlombikba, oldjuk fel 80 ml metil-alkoholban (3.1. pont), és helyezzük a lombikot 10 percre egy ultrahangos fürdőbe (4.4. pont). Az ultrahangos kezelés után állítsuk be az oldatot szobahőmérsékletűre, töltsük fel jelig vízzel, és keverjük össze. ≤ 4 °C hőmérsékleten az oldat egy hónapig stabil.
3.7.2. Amprólium közbenső standardoldat, 50 μg/ml
Pipettázzunk 5,0 ml standard törzsoldatot (3.7.1. pont) egy 50 ml-es mérőlombikba, töltsük fel jelig az extraháló oldószerrel (3.8. pont), és keverjük össze. ≤ 4 °C hőmérsékleten az oldat egy hónapig stabil.
3.7.3. Kalibrálóoldatok
Adagoljunk 0,5, 1,0 és 2,0 ml közbenső standardoldatot (3.7.2. pont) 50 ml-es mérőlombikokba. Töltsük fel a lombikokat jelig a mozgófázissal (3.6. pont), és keverjük össze. Ezek az oldatok 0,5, 1,0, illetve 2,0 μg/ml ampróliumnak felelnek meg. Ezeket az oldatokat közvetlenül a felhasználás előtt kell elkészíteni.
3.8. Extraháló oldószer
Metil-alkohol (3.1. pont) és víz 2 + 1 (v + v) arányú keveréke.
4. Eszközök
4.1. HPLC-berendezés 100 μl mennyiség befecskendezésére alkalmas befecskendező rendszerrel
4.1.1. 125 mm × 4 mm méretű, kationcserélő Nucleosil 10 SA folyadékkromatográfiás oszlop, 5 vagy 10 μm-es töltettel, vagy ezzel egyenértékű berendezés
4.1.2. Változtatható hullámhosszon mérő UV-detektor vagy diódasoros detektor.
4.2. Membránszűrő, PTFE anyag, 0,45 μm
4.3. Membránszűrő, 0,22 μm
4.4. Ultrahangos fürdő
4.5. Mechanikus rázógép vagy mágneses keverő
5. A vizsgálat módja
5.1. Általános szabályok
5.1.1. Takarmányvakminta
A visszanyerési próba (5.1.2. pont) előtt vizsgáljunk meg egy takarmányvakmintát annak ellenőrzésére, hogy sem amprólium, sem interferáló anyagok nincsenek jelen. A takarmányvakminta a mintával azonos fajtájú takarmány legyen, és ampróliumot vagy interferáló anyagokat ne tartalmazzon.
5.1.2. Visszanyerési próba
A mintában találhatóval közel azonos mennyiségű ampróliummal dúsított takarmányvakminta analízisével végezzünk visszanyerési próbát. A 100 mg/kg szintre való dúsításhoz öntsünk át a standard törzsoldatból (3.7.1. pont) 10,0 ml-t egy 250 ml-es Erlenmeyer-lombikba, és párologtassuk az oldatot körülbelül 0,5 ml-ig. Adjunk hozzá 50 g takarmányvakmintát, alaposan keverjük össze, és hagyjuk 10 percig állni, majd többször keverjük meg újra, mielőtt folytatjuk a műveletet az extrahálással (5.2. pont).
Amennyiben a mintával megegyező takarmányfajtából vett takarmányvakminta nem áll rendelkezésre (lásd az 5.1.1. pontot), a visszanyerési próbát a standard hozzáadásos módszerrel lehet elvégezni. Ebben az esetben a vizsgálandó mintát az abban már jelen lévővel közel azonos mennyiségű ampróliummal dúsítjuk. Ezt a mintát a nem dúsított mintával együtt elemezzük, és a visszanyerést kivonással számítjuk ki.
5.2. Extrahálás
5.2.1. Előkeverékek (ampróliumtartalom < 1 %) és takarmányok
Mérjünk ki 0,01 g pontossággal, az ampróliumtartalomtól függően, 5-40 g mintát egy 500 ml-es Erlenmeyer-lombikba, és adjunk hozzá 200 ml extraháló oldószert (3.8. pont). Helyezzük a lombikot az ultrahangos fürdőbe (4.4. pont), és hagyjuk benne 15 percig. Vegyük ki a lombikot az ultrahangos fürdőből, és egy óráig rázassuk a rázógépen, vagy keverjük a mágneses keverővel (4.5. pont). Oldjuk a kivonat egy aliquot részét a mozgófázissal (3.6. pont) 0,5-2 μg/ml ampróliumtartalomig és keverjük össze (lásd az Észrevételek 9.3. pontját). Szűrjük át e hígított oldat 5-10 ml-ét egy membránszűrőn (4.2. pont). Folytassuk a műveletet a HPLC-meghatározással (5.3. pont).
5.2.2. Előkeverékek (amprómiumtartalom ≥ 1 %)
Mérjünk ki 0,001 g pontossággal, az ampróliumtartalomtól függően, 1-4 g előkeveréket egy 500 ml-es Erlenmeyer-lombikba, és adjunk hozzá 200 ml extraháló oldószert (3.8. pont). Helyezzük a lombikot az ultrahangos fürdőbe (4.4. pont), és hagyjuk benne 15 percig. Vegyük ki a lombikot az ultrahangos fürdőből, és egy óráig rázassuk a rázógépen, vagy keverjük a mágneses keverővel (4.5. pont). Oldjuk a kivonat egy aliquot részét a mozgófázissal (3.6. pont) 0,5-2 μg/ml ampróliumtartalomig, és keverjük össze. Szűrjük át e hígított oldat 5-10 ml-ét egy membránszűrőn (4.2. pont). Folytassuk a műveletet a HPLC-meghatározással (5.3. pont).
5.3. HPLC-meghatározás
5.3.1. Paraméterek:
Az alábbi vizsgálati feltételek útmutatásként szolgálnak, ettől eltérő feltételeket is lehet használni, feltéve, hogy ezzel egyenértékű eredményt adnak.
| Folyadékkromatográfiás oszlop (4.1.1. pont): | 125 mm × 4 mm méretű, kationcserélő Nucleosil 10 SA, 5 vagy 10 μm-es töltettel, vagy ezzel egyenértékű berendezés. |
| Mozgófázis (3.6. pont): | Acetonitril (3.2. pont), nátrium-dihidrogén- foszfát oldat (3.4. pont) és nátrium-perklorát oldat (3.5. pont) 450 + 450 + 100 (v + v + v) arányú keveréke. |
| Átáramlási sebesség: | 0,7 –1 ml/perc |
| Kimutatási hullámhossz: | 264 nm |
| Befecskendezett mennyiség: | 100 μl |
Ellenőrizzük a kromatográfiás rendszer stabilitását 1,0 μg/ml-t tartalmazó kalibrálóoldat (3.7.3. pont) többszöri befecskendezésével addig, amíg a csúcsmagasságok és retenciós idők stabilakká nem válnak.
5.3.2. Kalibrációs görbe
Fecskendezzünk be minden kalibrálóoldatot (3.7.3. pont) többször egymás után, és határozzuk meg az egyes koncentrációkhoz tartozó csúcsmagasságok (területének) átlagát. A kalibrálóoldatok átlagos csúcsmagassága (területe) legyen az ordinátatengelyen, a megfelelő koncentrációk pedig μg/ml-ben az abszcisszán, és ennek alapján szerkesszünk kalibrációs görbét.
5.3.3. Mintaoldat
Ugyanolyan mennyiségú oldatot használva, mint amennyit a kalibrálóoldatoknál használtunk, fecskendezzük be a mintakivonatot (5.2. pont) egymás után többször, és határozzuk meg az ampróliumcsúcsok átlagos csúcsmagasságát (területét).
6. Az eredmények kiszámítása
A mintaoldat ampróliumcsúcsainak átlagmagasságából (területéből) határozzuk meg a mintaoldat μg/ml-ben kifejezett koncentrációját a kalibrációs görbe (5.3.2. pont) alapján.
A minta mg/kg-ban kifejezett w ampróliumtartalmát a következő képlettel kapjuk meg:
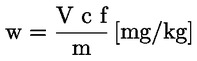
ahol:
V = az extraháló oldószer (3.8. pont) ml-ben kifejezett térfogata, az 5.2. pont szerint (azaz 200 ml)
c = a mintakivonat (5.2. pont) μg/ml-ben kifejezett ampróliumkoncentrációja
f = az 5.2. pont szerinti hígítási tényező
m = a vizsgálati adag tömege g-ban
7. Az eredmények validálása
7.1. Azonosítás
Az analizált anyag azonosítását párhuzamos kromatográfiával vagy diódasoros detektor alkalmazásával lehet megerősíteni, amelyek segítségével a mintakivonat (5.2. pont) spektruma és a 2,0 μg/ml koncentrációjú kalibrálóoldat (3.7.3. pont) spektruma összehasonlítható.
7.1.1. Párhuzamos kromatográfia
A mintakivonatot (5.2. pont) megfelelő mennyiségű kalibrálóoldattal (3.7.3. pont) dúsítjuk. A hozzáadott amprólium mennyisége közel azonos legyen a mintakivonatban található amprólium mennyiségével.
A hozzáadott mennyiséget és a kivonat hígítását figyelembe véve, csak az ampróliumcsúcs magassága emelkedhet. A csúcs szélességének a legnagyobb magasság felénél a dúsítás előtti mintakivonat ampróliumcsúcsa eredeti szélessége ± 10 %-on belül kell lennie.
7.1.2. Diódasoros detektálás
Az eredményeket a következő kritériumok szerint értékeljük:
a) a minta és a standard spektrumában a kromatogram csúcsmaximumán rögzített legnagyobb elnyeléshez tartozó hullámhossznak a detektor felbontóképessége által meghatározott tűréshatáron belül azonosnak kell lennie. A diódasoros detektoroknál ez a tűréshatár jellemzően ± 2 nm.
b) 210 és 320 nm között a spektrumnak a relatív abszorbancia 10 és 100 %-a közötti tartományba eső részein a mintának és a standardnak a kromatogram csúcsmaximumán rögzített spektruma nem térhet el egymástól. Ez a kritérium akkor teljesül, ha ugyanazok a maximális értékek vannak jelen, és a két spektrum egyetlen vizsgált ponton sem tér el a standard abszorbanciájának 15 %-ánál nagyobb mértékben.
c) 210 és 320 nm között a mintakivonat által kiváltott csúcs felszálló ágának, csúcsmaximumának és leszálló ágának spektrumai nem térhetnek el egymástól a spektrumnak a relatív abszorbancia 10 és 100 %-a közötti tartományba eső részein. Ez a kritérium akkor teljesül, ha ugyanazok a maximális értékek vannak jelen, és a spektrumok egyetlen vizsgált ponton sem térnek el a csúcsmaximum spektruma abszorbanciájának 15 %-ánál nagyobb mértékben.
Amennyiben ezen kritériumok valamelyike nem teljesül, az analizált anyag jelenléte nem bizonyított.
7.2. Ismételhetőség
Az ugyanazon mintán végzett két párhuzamos meghatározás eredménye közötti különbség nem haladhatja meg:
- 25 mg/kg és 500 mg/kg közötti ampróliumtartalom esetén a magasabb érték 15 %-át
- 500 mg/kg és 1 000 mg/kg közötti ampróliumtartalom esetén a 75 mg/kg értéket
- 1 000 mg/kg feletti ampróliumtartalom esetén a magasabb érték 7,5 %-át
7.3. Visszanyerés
Dúsított (vak)minta esetében a visszanyerésnek legalább 90 %-osnak kell lennie.
8. A körvizsgálat eredményei
Egy körvizsgálatot szerveztek, amely során három baromfitakarmányt (1-3. minta) egy ásványi takarmányt (4. minta) és egy előkeveréket (5. minta) analizáltak. Az eredmények a következő táblázatban szerepelnek.
| 1. minta (takarmányvakminta) | 2. minta | 3. minta | 4. minta | 5. minta | |
| L | 14 | 14 | 14 | 14 | 15 |
| n | 56 | 56 | 56 | 56 | 60 |
| középérték [mg/kg] | -- | 45,5 | 188 | 5 129 | 25 140 |
| sr [mg/kg] | -- | 2,26 | 3,57 | 178 | 550 |
| CVr [%] | -- | 4,95 | 1,90 | 3,46 | 2,20 |
| sR [mg/kg] | -- | 2,95 | 11,8 | 266 | 760 |
| CVR [%] | -- | 6,47 | 6,27 | 5,19 | 3,00 |
| névleges tartalom [mg/kg] | -- | 50 | 200 | 5 000 | 25 000 |
| L: a laboratóriumok száma n: az egyedi értékek száma sr: az ismételhetőség szórása CVr: az ismételhetőség relatív szórása SR: a reprodukálhatóság szórása CVR: a reprodukálhatóság relatív szórása | |||||
9. Észrevételek
9.1. Amennyiben a minta tiamint tartalmaz, a tiamincsúcs valamivel az ampróliumcsúcs előtt jelenik meg a kromatogramon. Ezt a módszert követve az ampróliumot és a tiamint el kell választani. Amennyiben az ebben a módszerben használt oszlopon (4.1.1. pont) nem sikerül szétválasztani az ampróliumot és a tiamint, akkor a mozgófázis (3.6. pont) acetonitril részének legfeljebb 50 %-át helyettesítsük metil-alkohollal.
9.2. A British Pharmacopoeia szerint az ampróliumoldat (c = 0,02 mol/l) spektruma sósavban (c = 0,1 mol/l) 246 nm és 262 nm között ad maximumértékeket. Az abszorbancia értéke 246 nm-nél 0,84, 262 nm-nél 0,80.
9.3. A kivonatot mindig fel kell hígítani a mozgófázissal, mert egyébként az ampróliumcsúcs retenciós ideje jelentősen eltolódhat az ionerőben történő változásoknak köszönhetően.
I. A NARAZIN MEGHATÁROZÁSA
A narazintartalmat az alábbiak szerint kell meghatározni:
- a "Takarmányok. Mintavételi és elemzési módszerek. Az engedélyezett kokcidiosztatikumok szűrése és meghatározása additív, illetve 1 %-os és 3 %-os keresztszennyeződési szinten, valamint a nem regisztrált kokcidiosztatikumok és egy antibiotikum szűrése és meghatározása szubadditív szinteken, összetett takarmányban, nagy hatékonyságú folyadékkromatográfiával. Tandem-tömegspektrometriás kimutatás (LC-MS/MS)" című EN 17299 szabványban előírt analitikai módszerrel, vagy
- a "Takarmányok. A monenzin-, a narizin- és a szalinomicin-tartalom meghatározása. Oszlop utáni származékképzést alkalmazó folyadékkromatográfiás módszer" című EN ISO 14183 szabványban előírt módszerrel.
J. A NIKARBAZIN MEGHATÁROZÁSA
A nikarbazintartalmat az alábbiak szerint kell meghatározni:
- a "Takarmányok. Mintavételi és elemzési módszerek. Az engedélyezett kokcidiosztatikumok szűrése és meghatározása additív, illetve 1 %-os és 3 %-os keresztszennyeződési szinten, valamint a nem regisztrált kokcidiosztatikumok és egy antibiotikum szűrése és meghatározása szubadditív szinteken, összetett takarmányban, nagy hatékonyságú folyadékkromatográfiával. Tandem-tömegspektrometriás kimutatás (LC-MS/MS)" című EN 17299 szabványban előírt analitikai módszerrel, vagy
- a »Takarmányok. A nikarbazin meghatározása. Nagy hatékonyságú folyadékkromatográfiás módszer" című EN 15782 szabványban előírt módszerrel.
K. A DEKOKINÁT MEGHATÁROZÁSA
A dekokináttartalmat az alábbiak szerint kell meghatározni:
- a "Takarmányok. Mintavételi és elemzési módszerek. Az engedélyezett kokcidiosztatikumok szűrése és meghatározása additív, illetve 1 %-os és 3 %-os keresztszennyeződési szinten, valamint a nem regisztrált kokcidiosztatikumok és egy antibiotikum szűrése és meghatározása szubadditív szinteken, összetett takarmányban, nagy hatékonyságú folyadékkromatográfiával. Tandem-tömegspektrometriás kimutatás (LC-MS/MS)" című EN 17299 szabványban előírt analitikai módszerrel, vagy
- a »Takarmányok. A dekokinát meghatározása HPLC fluoreszcenciás detektálással" című EN 16162 szabványban előírt módszerrel.
L. A MONENZIN MEGHATÁROZÁSA
A monenzintartalmat az alábbiak szerint kell meghatározni:
- a "Takarmányok. Mintavételi és elemzési módszerek. Az engedélyezett kokcidiosztatikumok szűrése és meghatározása additív, illetve 1 %-os és 3 %-os keresztszennyeződési szinten, valamint a nem regisztrált kokcidiosztatikumok és egy antibiotikum szűrése és meghatározása szubadditív szinteken, összetett takarmányban, nagy hatékonyságú folyadékkromatográfiával. Tandem-tömegspektrometriás kimutatás (LC-MS/MS)" című EN 17299 szabványban előírt analitikai módszerrel, vagy
- a "Takarmányok. A monenzin-, a narizin- és a szalinomicin-tartalom meghatározása. Oszlop utáni származékképzést alkalmazó folyadékkromatográfiás módszer" című EN ISO 14183 szabványban előírt módszerrel.
M. A SZALINOMICIN MEGHATÁROZÁSA
A szalinomicintartalmat az alábbiak szerint kell meghatározni:
- a "Takarmányok. Mintavételi és elemzési módszerek. Az engedélyezett kokcidiosztatikumok szűrése és meghatározása additív, illetve 1 %-os és 3 %-os keresztszennyeződési szinten, valamint a nem regisztrált kokcidiosztatikumok és egy antibiotikum szűrése és meghatározása szubadditív szinteken, összetett takarmányban, nagy hatékonyságú folyadékkromatográfiával. Tandem-tömegspektrometriás kimutatás (LC-MS/MS)" című EN 17299 szabványban előírt analitikai módszerrel, vagy
- a "Takarmányok. A monenzin-, a narizin- és a szalinomicin-tartalom meghatározása. Oszlop utáni származékképzést alkalmazó folyadékkromatográfiás módszer" című EN ISO 14183 szabványban előírt módszerrel.
N. A SZEMDURAMICIN MEGHATÁROZÁSA
A szemduramicintartalmat az alábbiak szerint kell meghatározni:
- a "Takarmányok. Mintavételi és elemzési módszerek. Az engedélyezett kokcidiosztatikumok szűrése és meghatározása additív, illetve 1 %-os és 3 %-os keresztszennyeződési szinten, valamint a nem regisztrált kokcidiosztatikumok és egy antibiotikum szűrése és meghatározása szubadditív szinteken, összetett takarmányban, nagy hatékonyságú folyadékkromatográfiával. Tandem-tömegspektrometriás kimutatás (LC-MS/MS) ó" című EN 17299 szabványban előírt analitikai módszerrel, vagy
- a "Takarmányok. A szemduramicintartalom meghatározása. Folyadékkromatográfiás módszer 'faszerkezetű' analitikai megközelítéssel" című EN 16158 szabványban előírt módszerrel kell meghatározni.
O. EN SZABVÁNYOK
Az (EU) 2017/625 rendelet 34. cikke (2) bekezdése a) pontjának alkalmazásában a következő EN szabványok relevánsak:
EN ISO 30024 Takarmányok. A fitázaktivitás meghatározása
EN 17050 Takarmányok. Mintavételi és vizsgálai módszerek. A jód meghatározása takarmányban ICP-MS-sel
EN 17550 Takarmányok. A karotinoidok meghatározása összetett takarmányokban és előkeverékekben nagy hatékonyságú folyadékkromatográfiával. UV-detektálás (HPLC-UV)
EN 15784 Takarmányok. Mintavételi és elemzési módszerek. A Bacillus spp. elkülönítése és megszámlálása
EN 15785 Takarmányok. A Bifidobacterium spp. elkülönítése és megszámlálása
EN 15786 Takarmányok. Mintavételi és elemzési módszerek. A Pediococcus spp. kimutatása és számlálása
EN 15787 Takarmányok. Takarmányok. Mintavételi és elemzési módszerek. A takarmány-adalékanyagként használt Lactobacillus spp. kimutatása és számlálása
EN 15788 Takarmányok. Mintavételi és elemzési módszerek. A takarmány-adalékanyagként használt Enterococcus (E. faecium) spp. kimutatása és számlálása
EN 15789 Takarmányok. Mintavételi és elemzési módszerek. A takarmány-adalékanyagként használt Saccharomyces cerevisiae kimutatása és számlálása
EN 15510 Takarmányok. Mintavételi és elemzési módszerek. A kalcium-, nátrium-, foszfor-, magnézium-, kálium-, vas-, cink-, réz-, mangán-, kobalt-, molibdén- és ólomtartalom meghatározása ICP-AES-sel (a kobalt és a molibdén takarmány-adalékanyagok vizsgálata céljából)
EN 15621 Takarmányok. Mintavételi és elemzési módszerek. A kalcium-, nátrium-, foszfor-, magnézium-, kálium-, kén-, vas-, cink-, réz-, mangán- és kobalttartalom meghatározása ICP-AES-sel, nyomás alatti feltárás után (a kobalt takarmány-adalékanyag vizsgálata céljából)
EN 16159 Takarmányok. A szelén meghatározása hidridfejlesztéses atomabszorpciós spektrometriával (HGAAS), mikrohullámú feltárás után (feltárás 65 %-os salétromsavval és 30 %-os hidrogén-peroxiddal)
EN 17053: Takarmányok. Mintavételi és elemzési módszerek. A nyomelemek, a nehézfémek és más elemek meghatározása takarmányban ICP-MS-sel (multimódszer) (a kobalt, a molibdén és a szelén takarmány-adalékanyagok vizsgálata céljából)
V. MELLÉKLET
ANALITIKAI MÓDSZEREK A TAKARMÁNYOKBAN LÉVŐ NEMKÍVÁNATOS ANYAGOK ELLENŐRZÉSÉRE
A. DIOXINOK (PCDD/PCDF) ÉS PCB-K KONCENTRÁCIÓJÁNAK MEGHATÁROZÁSA
I. FEJEZET
MINTAVÉTELI MÓDSZEREK ÉS AZ ANALITIKAI EREDMÉNYEK ÉRTELMEZÉSE
1. Cél és alkalmazási kör
A takarmányokban előforduló poliklórozott dibenzo-p-dioxinok (PCDD-k), poliklórozott dibenzofuránok (PCDF-ek), dioxin jellegű poliklórozott bifenilek (PCB-k) ( 29 ) és nem dioxin jellegű PCB-k koncentrációjának hatósági ellenőrzésére az I. mellékletnek megfelelően kell mintát venni. Alkalmazni kell az I. melléklet 5.1. pontjában a takarmányban egyenletesen eloszló anyagok vagy termékek ellenőrzésére vonatkozóan előírt mennyiségi követelményeket. Az így nyert egyesített minták reprezentatívnak tekintendők azon tételekre és altételekre, amelyekből a mintavétel történt. A 2002/32/EK irányelvben előírt felső határértékeknek való megfelelést a laboratóriumi minták elemzésével kapott koncentrációk alapján kell megállapítani.
E rész alkalmazásakor az (EU) 2021/808 bizottsági végrehajtási rendelet ( 30 ) I. mellékletében meghatározott fogalommeghatározások alkalmazandók.
Az említett fogalommeghatározásokon kívül e rész alkalmazásában a következő fogalommeghatározásokat kell alkalmazni:
"Szűrőmódszerek": azoknak a mintáknak a kiválasztására használt módszerek, melyekben a PCDD/F-ek és a dioxin jellegű PCB-k koncentrációja meghaladja a felső határértéket vagy a beavatkozási küszöbértéket. Segítségükkel költséghatékonyan lehet nagy mennyiségű mintát feldolgozni, és így nagyobb az esély az olyan új szennyeződési esetek felismerésére, amelyek nagy fogyasztói expozíciót és egészségi kockázatot jelenthetnek. A szűrőmódszerek bioanalitikai módszereken vagy gázkromatográfiás/tömegspektrometriás (GC-MS) módszereken alapszanak. A felső határértékeknek való megfelelés ellenőrzéséhez használt cut-off értéket meghaladó mintákból kapott eredményeket az eredeti minta megerősítő módszerrel történő teljes újbóli vizsgálata által kell ellenőrizni.
"Megerősítő módszerek" olyan teljes vagy kiegészítő információkkal szolgálnak, amelyek lehetővé teszik a PCDD/F-ek és a dioxin jellegű PCB-k maximális koncentrációban vagy szükség esetén a beavatkozási küszöbértéken történő azonosítását és egyértelmű mennyiségi meghatározását. E módszerek gázkromatográfiát/nagyfelbontású tömegspektrometriát (GC-HRMS) vagy gázkromatográfiát/tandem tömegspektrometriát (GC-MS/MS) alkalmaznak.
2. A tétel vagy altétel megfelelése a felső határértéknek
2.1. A nem dioxin jellegű PCB-k tekintetében
A tétel vagy altétel akkor felel meg a felső határértéknek, ha a PCB 28, PCB 52, PCB 101, PCB 138, PCB 153 és PCB 180 (a továbbiakban: nem dioxin jellegű PCB-k) összege tekintetében az analitikai eredmény - a kiterjesztett mérési bizonytalanságot is figyelembe véve - nem haladja meg a 2002/32/EK irányelvben megállapított felső határértéket ( 31 ). A tétel vagy altétel nem felel meg a 2002/32/EK irányelvben megállapított felső határértéknek, ha két párhuzamos elemzéssel ( 32 ), nyert két felfelé kerekített ( 33 ) analitikai eredmény átlaga - figyelembe véve a kiterjesztett mérési bizonytalanságot is - az észszerűség határain belül kétséget kizáróan meghaladja a felső határértéket; azaz a megfelelés megállapítására az elemzett koncentrációt alkalmazzák a kiterjesztett mérési bizonytalanság levonása után.
A kiterjesztett mérési bizonytalanság kb. 95 %-os konfidenciaszintnek megfelelő 2-es kiterjesztési tényezővel számítandó ki. A tétel vagy altétel nem megfelelő, ha az átlagos mért értékekből az átlag kiterjesztett bizonytalansági értékét kivonva a megállapított felső határérték feletti értéket kapunk.
Az e pont alatt, a fenti bekezdésekben említett szabályok a hatósági ellenőrzés céljából vett mintákkal kapott analitikai eredményekre alkalmazandók. A második szakértői vélemény vagy referencia céljából végzett elemzések esetében a nemzeti szabályok alkalmazandók.
2.2. A PCDD/F-ek és a dioxin jellegű PCB-k tekintetében
A tétel vagy altétel megfelel a felső határértékeknek, ha
- 5 % alatti "hamis megfelelő" arányt biztosító szűrőmódszerrel végzett egyetlen elemzés eredménye azt mutatja, hogy a koncentráció nem haladja meg a 2002/32/EK irányelvben a PCDD/F-ekre, illetve a PCDD/F-ek és a dioxin jellegű PCB-k összegére megállapított felső határértéket,
- a megerősítő módszerrel végzett egyetlen elemzés eredménye - a kiterjesztett mérési bizonytalanságot is figyelembe véve - nem haladja meg a 2002/32/EK irányelvben a PCDD/F-ekre, illetve a PCDD/F-ek és a dioxin jellegű PCB-k összegére megállapított felső határértéket.
A szűrőtesztekhez cut-off értéket kell megállapítani annak meghatározásához, hogy a minta megfelel-e a PCDD/F-ekre, illetve a PCDD/F-ek és a dioxin jellegű PCB-k összegére megállapított határértéknek.
A tétel vagy altétel nem felel meg a 2002/32/EK irányelvben megállapított felső határértéknek, ha megerősítő módszerrel végzett két párhuzamos elemzéssel, ( 34 ) nyert két felfelé kerekített ( 35 ) analitikai eredmény átlaga - figyelembe véve a kiterjesztett mérési bizonytalanságot is - az észszerűség határain belül kétséget kizáróan meghaladja a felső határértéket; azaz a megfelelés megállapítására az elemzett koncentrációt alkalmazzák a kiterjesztett mérési bizonytalanság levonása után.
A kiterjesztett mérési bizonytalanság kb. 95 %-os konfidenciaszintnek megfelelő 2-es kiterjesztési tényezővel számítandó ki. A tétel vagy altétel nem megfelelő, ha az átlagos mért értékekből az átlag kiterjesztett bizonytalansági értékét kivonva a megállapított felső határérték feletti értéket kapunk.
A PCDD/F-ek és a dioxin jellegű PCB-k külön-külön kapott analitikai eredményei becsült kiterjesztett bizonytalanságainak összegét kell használni a PCDD/F-ek és a dioxin jellegű PCB-k összegének kiszámításához.
Az e pont alatt, a fenti bekezdésekben említett szabályok a hatósági ellenőrzés céljából vett mintákkal kapott analitikai eredményekre alkalmazandók. Az érdekvédelmi vagy szakértői célból végzett elemzések esetében a nemzeti szabályok alkalmazandók.
3. A 2002/32/EK irányelv II. mellékletében megállapított beavatkozási küszöbértékeket meghaladó eredmények
A beavatkozási küszöbértékek a minták kiválasztásának eszközei azokban az esetekben, amelyekben azonosítani kell a szennyeződés forrását, és intézkedni kell a csökkentéséről vagy megszüntetéséről. A szűrőmódszereknél meg kell határozni a megfelelő cut-off értékeket az ilyen minták kiválasztásához. Ha jelentős erőfeszítések szükségesek a szennyeződés forrásának beazonosításához és a szennyeződés csökkentéséhez vagy megszüntetéséhez, célszerű eljárás két párhuzamos, megerősítő módszert alkalmazó elemzést végezni - a kiterjesztett mérési bizonytalanságot is figyelembe véve - a beavatkozási küszöbérték túllépésének megerősítésére ( 36 ).
II. FEJEZET
A TAKARMÁNYOKBAN ELŐFORDULÓ DIOXINOK (PCDD/F-EK) ÉS DIOXIN JELLEGŰ PCB-K KONCENTRÁCIÓJÁNAK HATÓSÁGI ELLENŐRZÉSÉHEZ HASZNÁLT MINTA-ELŐKÉSZÍTÉS ÉS AZ ELEMZÉSI MÓDSZEREKRE VONATKOZÓ KÖVETELMÉNYEK
1. Alkalmazási kör
Az e fejezetben előírt követelményeket kell alkalmazni a takarmányoknak a 2,3,7,8-as szénatomokon helyettesített PCDD/F-ek és a dioxin jellegű PCB-k koncentrációja tekintetében történő hatósági ellenőrzési célú elemzéséhez, továbbá az egyéb szabályozási célokat szolgáló minta-előkészítés és analitikai követelmények tekintetében, ideértve 183/2005/EK európai parlamenti és tanácsi rendelet ( 37 ) rendelkezéseinek való megfelelés biztosítása érdekében a takarmányipari vállalkozó által végzett ellenőrzéseket.
A takarmányokban előforduló PCDD/F-ek és dioxin jellegű PCB-k jelenlétének figyelemmel kísérése két különböző analitikai módszerrel történhet:
a) Szűrőmódszerek
A szűrőmódszerek célja azoknak a mintáknak a kiválasztása, melyekben a PCDD/F-ek és a dioxin jellegű PCB-k koncentrációja meghaladja a felső határértéket vagy a beavatkozási küszöbértéket. A szűrőmódszerek segítségével költséghatékonyan lehet nagy mennyiségű mintát feldolgozni, és így nagyobb az esély az olyan új szennyeződési esetek felismerésére, amelyek nagy fogyasztói expozíciót és egészségi kockázatot jelenthetnek. Alkalmazásuknak arra kell irányulnia, hogy ne forduljanak elő "hamis megfelelő" eredmények. A szűrőmódszerek közé tartozhatnak bioanalitikai módszerek és gázkromatográfiás/tömegspektrometriás (GC-MS) módszerek.
A szűrőmódszerek az analitikai eredményt összehasonlítják egy cut-off értékkel, és igen/nem választ adnak a felső határérték vagy a beavatkozási küszöbérték lehetséges túllépéséről. Az olyan mintákban, amelyek esetében feltételezhető a felső határértéket meghaladó eredmény, a PCDD/F-ek koncentrációját, valamint a PCDD/F-ek és a dioxin jellegű PCB-k összkoncentrációját megerősítő módszerrel kell meghatározni vagy megerősíteni.
Emellett a szűrőmódszerek jelezhetik a mintában lévő PCDD/F-ek és dioxin jellegű PCB-k koncentrációját. Bioanalitikai szűrőmódszerek alkalmazása esetén az eredmény bioanalitikai egyenértékként (BEQ) fejezhető ki, míg fizikai-kémiai GC-MS módszerek alkalmazása esetén toxicitási egyenértékként (TEQ). A szűrőmódszerek számadattal megadott eredményei megfelelően mutatják a megfelelést, a feltételezett meg nem felelést vagy a beavatkozási küszöbérték túllépését, és amennyiben megerősítő módszerekkel történő nyomon követés szükséges, jelzik a koncentrációtartományt. Nem alkalmasak olyan célokra, mint például a háttér-koncentrációk értékelése, a bevitel becslése, a koncentráció időbeli tendenciáinak nyomon követése vagy a beavatkozási küszöbérték, illetve a felső határértékek felülvizsgálata.
b) Megerősítő módszerek
A megerősítő módszerek lehetővé teszik a mintában jelen lévő PCDD/F-ek és dioxin jellegű PCB-k egyértelmű azonosítását és mennyiségi meghatározását, és teljeskörű információt adnak az egyes kongénerek szintjéről. Ezért e módszerek lehetővé teszik a felső határértékek és a beavatkozási küszöbértékek ellenőrzését, ideértve a szűrőmódszerekkel kapott eredmények megerősítését. Ezen túlmenően az eredmények egyéb célokra - például a takarmányellenőrzésben az alacsony háttér-koncentrációk meghatározására, az időbeli tendenciák követésére, az expozíció felmérésére, valamint a beavatkozási küszöbérték és a felső határértékek esetleges felülvizsgálatát szolgáló adatbázis létrehozására - is alkalmazhatók. Fontosak továbbá a kongénerkombinációk megállapításához, hogy azonosítani lehessen egy szennyeződés lehetséges forrását. Az ilyen módszerek GC-HRMS-t alkalmaznak. A felső határértéknek való megfelelés/meg nem felelés megerősítésére GC-MS/MS is alkalmazható.
2. Háttér
A toxicitási egyenértékben (TEQ) kifejezett koncentráció kiszámításához az egyes anyagok adott mintában mért koncentrációját meg kell szorozni a hozzájuk tartozó toxicitási egyenérték-tényezővel (TEF) (lásd az I. fejezet 1. lábjegyzetét), majd össze kell adni őket, és ez adja a dioxin jellegű vegyületek toxicitási egyenértékben (TEQ) kifejezett összkoncentrációját.
Az ezen A. rész alkalmazásában egy adott kongéner specifikus meghatározási határa a mintában az a legkisebb analittartalom, amely elfogadható statisztikai bizonyossággal mérhető, teljesítve a nemzetközileg elfogadott szabványokban, így például az EN 16215:2012 szabványban (Takarmányok. A dioxinok és a dioxionhoz hasonló PCB-k meghatározása GC/HRMS-sel és ezek indikátor-PCB-inek meghatározása GC/HRMS-sel) és/vagy a felülvizsgált EPA 1613 és 1668 módszerben leírt azonosítási kritériumokat.
Egy adott kongéner meghatározási határa a következőképpen azonosítható:
a) a mintakivonatban mért azon analitkoncentráció, amelynél két különböző vizsgált ion olyan analitikai jelet ad, amelynél a jel/zaj (S/N) arány a kisebb intenzitású jelre 3:1; vagy
b) amennyiben technikai okokból a jel-zaj viszony számítása nem ad megbízható eredményt, egy kalibrációs görbe legkisebb koncentrációs pontja, amely elfogadható (≤ 30 %) és következetes (a minták analitikai sorozatának legalább a kezdetén és a végén mért) eltérést mutat az egyes mintasorozatokban a kalibrációs görbe valamennyi pontjára kiszámított átlagos relatív válaszfaktoroktól. A meghatározási határt a legkisebb koncentrációs pontból kell kiszámítani, figyelembe véve a belső standardok visszanyerését és a beviteli mintamennyiséget.
A bioanalitikai szűrőmódszerek az eredményeket nem a kongénerek szintjén adják meg, hanem csupán bioanalitikai egyenértékként (BEQ) jelzik ( 38 ) a TEQ-értéket, ami azt jelenti, hogy a méréskor analitikai jelet adó, a mintakivonatban jelen lévő vegyületek közül esetleg nem felel meg mindegyik a TEQ-elv összes követelményének.
Egy adott mátrix ellenőrzésére csak akkor alkalmazhatók szűrési és megerősítő módszerek, ha azok kellően érzékenyek ahhoz, hogy megbízhatóan ki tudják mutatni a beavatkozási küszöbértéket vagy a felső határértéket.
3. Minőségbiztosítási előírások
3.1. Intézkedéseket kell hozni, hogy a mintavétel és az analitikai eljárás szakaszaiban ne fordulhasson elő a minták keresztszennyeződése.
3.2. A mintákat olyan üveg, alumínium, polipropilén vagy polietilén edényekben kell tárolni és szállítani, amelyek alkalmasak a minták olyan tárolására, hogy az azokban lévő PCDD/F-ek és dioxin jellegű PCB-k koncentrációját semmilyen hatás ne érhesse. A papírpormaradékokat el kell távolítani a mintatartóból.
3.3. A minták tárolása és szállítása során gondoskodni kell a takarmányminta épségének megőrzéséről.
3.4. Adott esetben az egyes laboratóriumi mintákat finomra kell őrölni és alaposan össze kell keverni bizonyítottan teljes homogenizációt eredményező eljárással (például őrlés olyan finomságúra, hogy 1 mm-es szitán áthulljon). A túl nagy nedvességtartalmú mintákat őrlés előtt szárítani kell.
3.5. Ellenőrizni kell, hogy a reagensek, a laboratóriumi üvegeszközök és berendezések esetleg nem befolyásolják-e a TEQ-értékre vagy BEQ-értékre alapozott eredményeket.
3.6. Vakpróbát kell végezni, azaz végre kell hajtani a teljes analitikai eljárást, a mintát kihagyva belőle.
3.7. Bioanalitikai módszereknél az elemzéshez használt összes laboratóriumi üvegeszközt és oldószert ellenőrizni kell, hogy mentesek legyenek olyan vegyületektől, melyek a munkatartományban zavarhatnák a keresett vegyületek kimutatását. Az üvegeszközöket ki kell öblíteni oldószerrel, vagy fel kell melegíteni olyan hőmérsékletre, amelyen a felületükről eltávoznak a nyomokban esetleg ott lévő PCDD/F-ek, dioxin jellegű vegyületek és zavaró vegyületek.
3.8. Az extrakcióhoz használt minta mennyiségének elegendőnek kell lennie ahhoz, hogy teljesüljenek a felső határérték vagy a beavatkozási küszöbérték koncentrációit magában foglaló, kellően alacsony munkatartományra vonatkozó követelmények.
3.9. A vizsgált termékekre alkalmazott adott minta-előkészítési eljárásoknak nemzetközileg elfogadott iránymutatásokat - pl. EN ISO 6498 - kell követniük.
4. A laboratóriumokra vonatkozó követelmények
4.1. Az (EU) 2017/625 rendelet rendelkezéseinek megfelelően az analitikai minőségbiztosítás alkalmazásának biztosítása érdekében a laboratóriumokat az ISO/IEC 58 útmutató szerint működő elismert testületnek kell akkreditálnia. A laboratóriumokat az EN ISO/IEC 17025 szabvány szerint kell akkreditálni. A "Technical Guidelines for the estimation of measurement uncertainty and limits of quantification for PCDD/F and PCB analysis" (Műszaki iránymutatás a mérési bizonytalanság és meghatározási határ becsléséhez PCDD/F és PCB vizsgálata tekintetében) dokumentumban foglalt elvek követendők ( 39 ).
4.2. A laboratóriumok szakmai felkészültségét olyan laboratóriumközi körvizsgálatokban való folyamatos, sikeres részvétellel kell bizonyítani, amelyek a PCDD/F-ek és a dioxin jellegű PCB-k meghatározására irányulnak a megfelelő takarmánymátrixokban és koncentrációtartományokban.
4.3. A minták rutinszerű ellenőrzésére szűrőmódszereket alkalmazó laboratóriumoknak szoros együttműködést kell kialakítaniuk a megerősítő módszert alkalmazó laboratóriumokkal mind a minőség-ellenőrzés, mind a feltehetően nem megfelelő minták analitikai eredményének megerősítése terén.
5. A dioxinok (PCDD/F-ek) és dioxin jellegű PCB-k tekintetében alkalmazott analitikai eljárásokra vonatkozó alapkövetelmények
5.1. Alacsony munkatartomány és a mennyiségi meghatározás határértéke
A PCDD/F-ek esetében a kimutatható mennyiségnek a femtogrammos (10-15 g) tartomány felső részébe kell esnie, mivel az említett vegyületek közül több rendkívül mérgező. A legtöbb PCB-kongéner esetében már az is elegendő, ha a meghatározási határ értéke a nanogrammos (10-9 g) tartományban van. A mérgezőbb dioxin jellegű PCB-kongénerek (és különösen a nem orto szubsztituált kongénerek) méréséhez a munkatartomány aljának el kell érnie a pikogrammos (10-12 g) tartomány alját. Az összes többi PCB-kongéner esetében elegendő, ha a meghatározási határ értéke a nanogrammos (10-9 g) tartományba esik.
5.2. Nagy szelektivitás (specifikusság)
5.2.1. Meg kell tudni különböztetni a PCDD/F-eket és a dioxin jellegű PCB-ket azoktól a sokféle, együtt extrahálódó és a mérést esetleg zavaró más vegyületektől, amelyek több nagyságrenddel nagyobb koncentrációban lehetnek jelen, mint a keresett analitok. A GC-MS módszerekkel meg kell tudni különböztetni egymástól a különböző kongénereket, így például a mérgező kongénereket (például a 2,3,7,8-as szénatomokon helyettesített tizenhét PCDD/F-et és a tizenkét dioxin jellegű PCB-t) a többi kongénertől.
5.2.2. A bioanalitikai módszerekkel ki kell tudni mutatni a keresett vegyületeket a PCDD/F-ek, és/vagy a dioxin jellegű PCB-k összegeként. A mintatisztítás célja az olyan vegyületek eltávolítása, amelyek "hamis nem megfelelő" eredményeket okoznak, vagy az olyanokét, amelyek a reakció gyengítésével "hamis megfelelő" eredményt okoznak.
5.3. Nagy adatpontosság (valódiság és módszerpontosság, biológiai próba során észlelt visszanyerés)
5.3.1. A GC-MS módszerekkel a meghatározásnak hiteles becslést kell adnia a minta valódi koncentrációjáról. Nagy adatpontosság szükséges ahhoz, hogy a mintaelemzés eredményét ne kelljen elutasítani a kapott TEQ-érték gyenge megbízhatósága miatt. Az adatpontosság a valódiság (egy hitelesített anyagban az adott analitra mért átlagérték és a hitelesített érték közötti különbség, ez utóbbi százalékában kifejezve) és a módszerpontosság (a reprodukálhatósági körülmények között kapott eredményekből kiszámított relatív szórás [RSDR] összege).
5.3.2. A bioanalitikai módszereknél a biológiai próba során észlelt visszanyerést kell meghatározni. A biológiai próba során észlelt visszanyerés a TCDD vagy a PCB-126 kalibrációs görbéjéből számított, a vakminta eredményeivel korrigált BEQ-érték osztva a megerősítő módszerrel meghatározott TEQ-értékkel. Célja az olyan hatások korrigálása, mint például a PCDD/F-ek és a dioxin jellegű vegyületek vesztesége az extrakció és a mintatisztítás során, az extrakcióval átmenő más vegyületek reakciót erősítő vagy gyengítő (agonisztikus és antagonisztikus) hatása, a görbeilleszkedés minősége vagy a toxikológiai egyenérték-tényező (TEF) és a relatív hatóképesség (REP) értékei közötti különbségek. A biológiai próba során észlelt visszanyerés kiszámítása reprezentatív kongénerkombinációkat a keresett koncentráció körüli koncentrációkban tartalmazó, alkalmas referenciamintákból történik.
5.4. Hitelesítés a felső határérték tartományában és általános minőség-ellenőrző intézkedések
5.4.1. A laboratóriumoknak a hitelesítés és a rutinszerű elemzés során igazolniuk kell a módszerek teljesítőképességét a felső határérték esetében - például a felső határérték felénél, egyszeresénél és kétszeresénél - azzal, hogy az ismételt elemzések variációs együtthatója elfogadható.
5.4.2. Belső minőség-ellenőrzési intézkedésként rendszeresen vakpróbákat, adalékolási kísérleteket vagy kontrollminta-elemzést (lehetőleg hitelesített referenciaanyaggal) kell végezni. A vakpróbákról, az adalékolási kísérletekről vagy a kontrollminta-elemzésekről minőség-ellenőrzési nyilvántartást kell vezetni, és ellenőrizni kell, hogy az analitikai teljesítőképesség valóban megfelel-e a követelményeknek.
5.5. A meghatározási határ
5.5.1. A bioanalitikai szűrőmódszereknél nem feltétlenül szükséges, hogy meg legyen határozva a meghatározási határ (LOQ), de a módszerről bizonyítani kell, hogy meg tudja különböztetni a vakmintával kapott eredményeket a cut-off értéktől. BEQ-érték megadásakor az e koncentráció alatti reakciót mutató minták kezelése céljából meg kell határozni egy jelentési határértéket. A jelentési határértéknek igazoltan az elemzési vakmintákkal kapott eredmény legalább háromszorosának kell lennie úgy, hogy az érték a munkatartomány alá esik. Ezért ezt a keresett vegyületet az előírt minimumkoncentráció körüli koncentrációban tartalmazó mintákból, nem pedig a jel/zaj arányból vagy mérési vakmintából kell kiszámítani.
5.5.2. Megerősítő módszer esetében a meghatározási határ értékének a felső határérték körülbelül egyötödénél kell lennie.
5.6. Analitikai kritériumok
Ahhoz, hogy a megerősítő vagy a szűrőmódszerek megbízható eredményeket adjanak, a következő kritériumoknak kell teljesülniük a felső határérték vagy a beavatkozási küszöbérték tartományában a TEQ-értékre, illetve a BEQ-értékre, függetlenül attól, hogy a meghatározás TEQ-összértékként (a PCDD/F-ek és a dioxin jellegű PCB-k összegéként) vagy külön a PCDD/F-ekre és külön a dioxin jellegű PCB-kre történik:
| Szűrés bioanalitikai vagy fizikai-kémiai módszerekkel | Megerősítő módszerek | |
| „Hamis megfelelő” arány (*1) | < 5 % | |
| Valódiság | -20 % és 20 % között | |
| Megismételhetőség (RSDr) | < 20 % | |
| Közbenső precizitás (RSDR) | < 25 % | < 15 % |
| (*1) A felső határértékhez képest. | ||
5.7. A szűrőmódszerekre vonatkozó egyedi követelmények
5.7.1. A szűréshez mind GC-MS módszerek, mind bioanalitikai módszerek használhatók. A GC-MS módszerekre a 6. pontban előírt követelményeket kell alkalmazni. A sejtalapú bioanalitikai módszerekre vonatkozó egyedi követelményeket a 7. pont írja le.
5.7.2. A minták rutinszerű ellenőrzéséhez szűrőmódszereket alkalmazó laboratóriumoknak szoros együttműködést kell kialakítaniuk a megerősítő módszert alkalmazó laboratóriumokkal.
5.7.3. A szűrőmódszerek teljesítőképességét a rutinszerű elemzések során analitikai minőség-ellenőrzéssel és a módszer folyamatos hitelesítésével ellenőrizni kell. A "megfelelő" eredmények ellenőrzésére folyamatos programot kell alkalmazni.
5.7.4. A sejtreakció és a citotoxicitás esetleges elnyomásának ellenőrzése:
A mintakivonatok 20 %-át kell mérni rutinszerű szűréssel, a felső határértéknek vagy a beavatkozási küszöbértéknek megfelelően hozzáadott 2,3,7,8-TCDD-vel és anélkül, ellenőrizendő, hogy előfordulhat-e reakcióelnyomás a mintakivonatban jelen lévő zavaró anyagok miatt. Az adalékolt minta mért koncentrációját össze kell hasonlítani az adalékolás nélküli kivonat koncentrációjának és az adalékolási koncentrációnak az összegével. Ha a mért koncentráció több mint 25 %-kal kisebb a számított (össz)koncentrációnál, akkor ez azt mutatja, hogy fennáll a reakcióelnyomás lehetősége, és az illető mintát meg kell vizsgálni GC-HRMS megerősítő elemzéssel. Az eredményeket figyelemmel kell kísérni a minőség-ellenőrző nyilvántartásban.
5.7.5. A "megfelelő" minták minőség-ellenőrzése:
A mintamátrixtól és a laboratóriumi gyakorlattól függően a "megfelelő" minták megközelítőleg 2-10 %-ával el kell végezni a megerősítést GC-HRMS módszerrel.
5.7.6. A "hamis megfelelő" arányok meghatározása a minőség-ellenőrzési adatokból:
A felső határérték vagy a beavatkozási küszöbérték alatti és feletti minták szűrése alapján meg kell határozni a "hamis megfelelő" eredmények arányát. A tényleges "hamis megfelelő" aránynak 5 % alatt kell lennie. Ha mátrixonként vagy mátrixcsoportonként van legalább 20 megerősített eredmény a "megfelelő" minták minőség-ellenőrzéséből, akkor a "hamis megfelelő" arányt ebből az adatbázisból kell megállapítani. A körvizsgálatokban vagy a szennyeződési esetek során elemzett minták eredményei, amelyek például akár a felső határérték (ML) kétszereséig terjedő koncentrációtartományokba is eshetnek, szintén felvehetők a "hamis megfelelő" arány kiszámításához minimumként használandó 20 eredmény közé. A mintáknak le kell fedniük a különféle forrásokat reprezentáló, leggyakoribb kongénerkombinációkat.
Noha a szűrőteszteknek lehetőleg azt kell célozniuk, hogy kimutassák a beavatkozási küszöbértéket meghaladó mintákat, a "hamis megfelelő" arányok meghatározásának kritériuma a felső határérték, figyelembe véve a megerősítő módszer mérési bizonytalanságát.
5.7.7. A szűrés alapján "feltehetően nem megfelelő" mintákat az eredeti mintának a teljeskörű újraelemzése útján megerősítő módszerrel kell mindig ellenőrizni. Ezek a minták a "hamis nem megfelelő" eredmények arányának kiszámításához is használhatók. A szűrőmódszereknél a "hamis nem megfelelő" eredmények aránya a korábbi szűréskor "feltehetően nem megfelelő"-nek minősített minták megerősítő elemzéssel "megfelelő"-ként megerősített eredmények hányada. A szűrőmódszer előnyösségének értékelése a "hamis nem megfelelő" mintáknak az összes ellenőrzött mintával való összehasonlításán alapul. Ennek az aránynak elég kicsinek kell lennie ahhoz, hogy a szűrővizsgálati módszer alkalmazása előnyös legyen.
5.7.8. A bioanalitikai módszereknek a hitelesítési körülmények között, hitelesen kell jelezniük - a BEQ-érték kiszámításával és megadásával - a TEQ-értéket.
A megismételt körülmények között alkalmazott bioanalitikai módszerekre is igaz, hogy a laboratóriumon belüli RSDr jellemzően kisebb, mint reprodukálhatósági körülmények között (RSDR).
6. A szűrés és megerősítés céljából alkalmazott GC/MS módszerekre vonatkozó különleges követelmények
6.1. A felfelé kerekített és a lefelé kerekített WHO-TEQ-koncentrációk közötti elfogadható különbség
A megerősített felső és alsó határ közötti különbség nem haladhatja meg a 20 %-ot a felső határérték vagy szükség esetén a beavatkozási küszöbérték túllépésének megerősítése céljából.
6.2. A visszanyerések ellenőrzése
6.2.1. Az elemzési eljárás hitelesítéséhez a 2,3,7,8-as szénatomokon klórral helyettesített PCDD/F-ekből készített, 13C-jelölésű belső standardokat és a dioxin jellegű PCB-kből készített, 13C-jelölésű belső standardokat az analitikai eljárás legelején, például az extrakció előtt hozzá kell adni a mintákhoz. A tetraklórozottól az oktaklórozottig terjedő PCDD/F homológ csoportok mindegyikéből legalább egy kongénert, továbbá a dioxin jellegű PCB homológ csoportok mindegyikéből legalább egy kongénert kell hozzáadni a mintához (más megoldásként legalább egy-egy kongénert a PCDD/F-ek és dioxin jellegű PCB-k ellenőrzéséhez használt tömegspektrometriás szelektív ionkövetéses funkciókhoz). A megerősítő módszerek esetében a 2,3,7,8-as szénatomokon helyettesített mind a tizenhét PCDD/F-ből készített, 17 13C-jelölésű belső standardot és mind a tizenkét dioxin jellegű PCB-ből készített, 12 13C-jelölésű belső standardot használni kell.
6.2.2. Ezenkívül megfelelő kalibráló oldatok alkalmazásával relatív válaszfaktorokat kell meghatározni azon kongénerekre, amelyek 13C-jelölésű analógja nem lett hozzáadva a mintához.
6.2.3. A növényi eredetű takarmányok és a 10 %-nál kevesebb zsírt tartalmazó állati eredetű takarmányok esetében a belső standardokat kötelezően az extrakció előtt kell a mintához adni. A 10 %-nál nagyobb zsírtartalmú állati eredetű takarmányok esetében a belső standardokat vagy a zsírextrakció előtt, vagy azt követően kell a mintához adni. Az extrakció hatékonyságát megfelelően hitelesíteni kell, figyelembe véve, hogy a belső standardok hozzáadása melyik szakaszban történt.
6.2.4. GC-MS elemzés előtt a visszanyerés mérésére egy vagy két (kísérő) standardot is hozzá kell adni a mintához.
6.2.5. A visszanyerést ellenőrizni kell. A megerősítő módszerek esetében az egyes belső standardoknál a visszanyerésnek a 60-120 %-os tartományban kell lennie. Egyes kongénereknél, különösen néhány 7, illetve 8 klóratomot tartalmazó dibenzo-p-dioxin és dibenzo-furán esetében ennél kisebb vagy nagyobb visszanyerés is elfogadható, feltéve, hogy TEQ-értékük részaránya a (PCDD/F-ek és dioxin jellegű PCB-k összegén alapuló) TEQ-összértéken belül legfeljebb 10 %. GC-MS szűrőmódszerek esetében a visszanyerésnek a 30-140 %-os tartományban kell lennie.
6.3. A zavaró anyagok eltávolítása
- A PCDD/F-eket megfelelő kromatográfiás eljárással (lehetőleg florisil-, alumínium-oxid- és/vagy szénoszlopon) el kell választani a zavaró klórozott vegyületektől, így például a nem dioxin jellegű PCB-ktől és a klórozott difenil-éterektől.
- Izomerek gázkromatográfiás elválasztásánál az 1,2,3,4,7,8-HxCDF és az 1,2,3,6,7,8-HxCDF esetében a csúcsok közötti eltérésnek 25 %-nál kisebbnek kell lennie.
6.4. Kalibrálás standard görbével
A kalibrációs görbe tartományának le kell fednie a felső határérték vagy a beavatkozási küszöbérték megfelelő tartományát.
6.5. A megerősítő módszerekre vonatkozó egyedi kritériumok
- A GC-HRMS esetében:
A HRMS esetében a felbontás a teljes tömegtartományra nézve jellemzően legalább 10 000 vagy annál nagyobb, 10 %-os mélypont mellett.
A nemzetközileg elismert szabványokban - például az EN 16215:2012 szabványban (Takarmányok. A dioxinok és a dioxionhoz hasonló PCB-k meghatározása GC/HRMS-sel és ezek indikátor-PCB-inek meghatározása GC/HRMS-sel) és/vagy a felülvizsgált EPA 1613 és 1668 módszerekben - ismertetett további azonosítási és megerősítési kritériumok teljesülése.
- A GC-MS/MS esetében:
legalább két specifikus anyaion nyomon követése, melyek a vizsgálat keretében minden jelölt és nem jelölt analit tekintetében rendelkeznek egy specifikus, megfelelő ionátmenettel.
A relatív ionintenzitásokhoz engedélyezett felső tűréshatár a kiválasztott ionátmenetek esetében a kiszámított vagy mért értékekhez viszonyítva ± 15 % (a kalibrációs standardokból származó átlag), az analit minden egyes átmenete esetében azonos MS/MS-feltételek alkalmazása mellett (mindenekelőtt ütközési energia és ütközési gáznyomás).
Az egyes quadrupolok felbontását úgy kell beállítani, hogy - a vizsgálat szempontjából fontos analitokkal kapcsolatos esetleges interferencia minimalizálása érdekében - megegyezzen az egységnyi tömegfelbontással, vagy nagyobb legyen annál (egységnyi tömegfelbontás: ahhoz elegendő felbontás, hogy egy tömegegységet két csúcsra bontson).
A nemzetközileg elismert szabványokban - például az EN 16215:2012 szabványban (Takarmányok. A dioxinok és a dioxionhoz hasonló PCB-k meghatározása GC/HRMS-sel és ezek indikátor-PCB-inek meghatározása GC/HRMS-sel) és/vagy a felülvizsgált EPA 1613 és 1668 módszerekben - ismertetett további kritériumok teljesülése, a GC-HRMS alkalmazására vonatkozó kötelezettséget leszámítva.
7. A BIOANALITIKAI MÓDSZEREKRE VONATKOZÓ EGYEDI KÖVETELMÉNYEK
A bioanalitikai módszerek biológiai alapelveken alapuló módszerek, mint például sejtalapú tesztek, receptortesztek vagy immuntesztek. E pont általánosságban leírja a bioanalitikai módszerekre vonatkozó követelményeket.
Egy szűrőmódszer alapvetően "megfelelő"-nek vagy "feltehetően nem megfelelő"-nek minősít egy mintát. Ehhez a számított BEQ-értéket össze kell hasonlítani a cut-off értékkel (lásd a 7.3. pontot). A cut-off érték alatti minták "megfelelő"-nek minősülnek, az azzal egyenlő vagy a feletti értékű minták pedig "feltehetően nem megfelelő"-nek, és ezeket megerősítő módszerrel elemezni kell. A gyakorlatban a felső határérték 2/3-ának megfelelő BEQ-érték alkalmazható cut-off értékként, amennyiben biztosított, hogy a "hamis megfelelő" arány 5 % alatt van, és a "hamis nem megfelelő" eredmények aránya is elfogadható. Mivel külön felső határértékek vonatkoznak a PCDD/F-ekre, illetve a PCDD/F-ek és a dioxin jellegű PCB-k összegére, a minták frakcionálás nélküli megfelelőség-ellenőrzéséhez a biológiai próbáknál megfelelő cut-off értékek szükségesek a PCDD/F-ek vonatkozásában. A beavatkozási küszöbértékeket meghaladó minták ellenőrzéséhez az adott beavatkozási küszöbértékek megfelelő százalékaránya az alkalmas cut-off érték.
Amennyiben tájékoztató BEQ-érték van megadva, a mintából származó eredményeknek a munkatartományon belül kell lenniük, és meg kell haladniuk a jelentési határértéket (lásd a 7.1.1. és a 7.1.6. pontot).
7.1. A mérési reakciók kiértékelése
7.1.1. Általános követelmények
- Ha a koncentrációk kiszámítása TCDD-s kalibrációs görbével történik, akkor a görbe felső végénél lévő értékek nagy szórást (nagy variációs együtthatót [CV]) mutatnak. A munkatartomány az a terület, ahol ez a CV kisebb, mint 15 %. A munkatartomány alját (jelentési határérték) úgy kell meghatározni, hogy az legalább háromszorosa legyen az elemzési vakmintákkal kapott eredményeknek. A munkatartomány tetejét általában az EC70 érték jelenti (a legnagyobb hatásos koncentráció 70 %-a), de ennél kisebbnek kell lennie, ha ebben a tartományban a CV nagyobb, mint 15 %. A munkatartományt a hitelesítés során kell meghatározni. A cut-off értékeknek (lásd a 7.3. pontot) a munkatartomány belsejében kell lenniük.
- A standardoldatokat és a mintakivonatokat legalább három vagy legalább két párhuzamos elemzéssel kell mérni. Párhuzamos vizsgálatkor a lemezen elosztva 4-6 cellában mért standard oldatnak vagy kontrollkivonatnak olyan reakciókat vagy koncentrációkat kell adnia (ez csak a munkatartományban lehetséges), amelyek variációs együtthatója CV < 15 %.
7.1.2. Kalibrálás
7.1.2.1. Kalibrálás standard görbével
- A mintában lévő mennyiségeket a mérési reakció és a TCDD-vel (vagy a PCB-126-tal vagy pedig PCDD-ből/PCDF-ből/dioxin jellegű PCB-ből álló standard keverékkel) készített kalibrációs görbe összehasonlításával kell megbecsülni, és ez alapján lehet kiszámítani a kivonatra és ezt követően a mintára jellemző BEQ-értéket.
- A kalibrációs görbéknek 8-12 (legalább két párhuzamos elemzéssel kapott) koncentrációt kell tartalmazniuk úgy, hogy elegendő koncentráció legyen a görbe alsó részén (a munkatartományban). Külön figyelmet kell fordítani a munkatartományban a görbeilleszkedés minőségére. Ennél az R2 érték kevéssé vagy egyáltalán nem használható az illeszkedés jóságának becslésére a nem lineáris regresszió esetében. Jobb illeszkedést kell elérni a görbe munkatartományában számított és mért koncentrációk közötti legkisebb különbségek, például a legkisebb maradék négyzetösszeg alkalmazásával.
- A mintakivonat becsült koncentrációját ezután korrigálni kell egy vakmátrixra vagy vakoldószerre számított BEQ-értékkel (az alkalmazott oldószerek és vegyszerek szennyeződéseinek figyelembevétele céljából) és a látszólagos visszanyeréssel (ennek kiszámítása a reprezentatív kongénerkombinációkat a felső határérték vagy a beavatkozási küszöbérték körül tartalmazó, alkalmas referenciaminták BEQ-értékéből történik). A visszanyeréssel való korrigáláshoz a látszólagos visszanyerésnek az előírt tartományba kell esnie (lásd a 7.1.4. pontot). A visszanyeréssel való korrigáláshoz használt referenciamintáknak meg kell felelniük a 7.2. pontban ismertetett követelményeknek.
7.1.2.2. Kalibrálás referenciamintákkal
Más megoldásként használható legalább négy, a felső határérték vagy a beavatkozási szint körüli koncentrációjú referenciamintával (lásd a 7.2.4. pontot): egy vakmátrix és három, a felső határérték vagy a cselekvési küszöbérték 0,5-szörösét, 1-szeresét, illetve 2-szeresét képviselő referenciaminta - felhasználásával készített kalibrációs görbe; így nincs szükség a vakminta eredményeivel és a visszanyeréssel történő korrigálásra, ha a referenciaminták mátrixtulajdonságai megfelelnek az ismeretlen minták tulajdonságainak. Ebben az esetben a felső határérték 2/3-ának megfelelő mérési reakció (lásd a 7.3. pontot) közvetlenül kiszámítható ezekből a mintákból és cut-off értékként használható. A beavatkozási küszöbértékeket meghaladó minták ellenőrzéséhez a beavatkozási küszöbértékek megfelelő százalékaránya az alkalmas cut-off érték.
7.1.3. A PCDD/F-ek és a dioxin jellegű PCB-k meghatározása külön-külön
A kivonatok szétválaszthatók olyan frakciókra, melyek PCDD/F-eket, illetve dioxin jellegű PCB-ket tartalmaznak, így külön-külön meghatározható a PCDD/F-ek, illetve a dioxin jellegű PCB-k TEQ-értéke (BEQ-értékként kifejezve). A dioxin jellegű PCB-ket tartalmazó frakció eredményeinek kiszámításához lehetőleg a PCB-126-tal készített standarddal felvett kalibrációs görbét kell használni.
7.1.4. Biológiai próbák során észlelt visszanyerések
A "biológiai próbák során észlelt visszanyerést" a felső határérték vagy a beavatkozási küszöbérték körüli reprezentatív kongénerkombinációkat tartalmazó alkalmas referenciamintákból kell kiszámítani és a BEQ-értéknek a TEQ-értékhez képesti százalékában kell megadni. A teszt típusától és az alkalmazott TEF-értékektől ( 40 ) függően a dioxin jellegű PCB-kre vonatkozó TEF- és REP-tényezők közötti különbségek a dioxin jellegű PCB-k esetében a PCDD/F-ekhez képest kis látszólagos visszanyerést okozhatnak. Ezért ha a PCDD/F-ek és a dioxin jellegű PCB-k meghatározása külön-külön történik, akkor a biológiai próba során észlelt visszanyerésnek a következőnek kell lennie: dioxin jellegű PCB-knél 20-60 %, PCDD/F-eknél pedig 50-130 % (a tartományok a TCDD-s kalibrációs görbére vonatkoznak). Mivel a dioxin jellegű PCB-k hozzájárulása a PCDD/F-ek és a dioxin jellegű PCB-k összegéhez a mátrixoktól és a mintáktól függően változhat, a PCDD/F-ek és a dioxin jellegű PCB-k összegére vonatkozóan a biológiai próba során észlelt visszanyerések tükrözik ezeket a tartományokat, és ezeknek 30 % és 130 % között kell lenniük. A PCDD/F-ekre és a dioxin jellegű PCB-kre vonatkozó uniós szabályozásban a TEF-értékek lényeges módosítása esetén ezeket a tartományokat is módosítani kell.
7.1.5. A visszanyerések ellenőrzése mintatisztításnál
A mintatisztítás során előforduló vegyületvesztést a hitelesítéskor ellenőrizni kell. A különböző kongénerek keverékével adalékolt vakmintán el kell végezni a mintatisztítást (n = legalább 3), és a visszanyerést és a variabilitást megerősítő módszerrel kell ellenőrizni. A visszanyerésnek 60 % és 120 % között kell lennie, főként olyan kongénereknél, amelyek 10 %-nál nagyobb mértékben járulnak hozzá a TEQ-értékhez a különféle keverékekben.
7.1.6. Jelentési határérték
A BEQ-értékeknek a jelentésben történő megadásához jelentési határértéket kell meghatározni jellemző kongénerkombinációkat tartalmazó adott mátrixmintákból, de nem a standard kalibrációs görbéből, mivel a görbe alján kicsi a módszerpontosság. Figyelembe kell venni az extrakció és a mintatisztítás hatásait. A jelentési határértéket az elemzési vakmintákkal kapott érték legalább háromszorosaként kell megállapítani.
7.2. Referenciaminták használata
7.2.1. A referenciamintáknak reprezentálniuk kell a mintamátrixot, a kongénerkombinációkat és a PCDD/F-eknek és a dioxin jellegű PCB-knek a felső határérték vagy a beavatkozási küszöbérték körüli koncentrációtartományait.
7.2.2. Mindegyik méréssorozatban lennie kell vakmátrixnak vagy, ha ez nem lehetséges, elemzési vakmintának, valamint a felső határérték vagy a beavatkozási küszöbérték körüli koncentrációjú referenciamintának. Ezeket a mintákat egy időben és azonos körülmények között kell extrahálni és elemezni. A referenciamintának egyértelműen nagyobb reakciót kell adnia, mint a vakmintának, így biztosítva az elemzés alkalmasságát. Ezek a minták használhatók a vakminta eredményeivel és a visszanyeréssel való korrigáláshoz.
7.2.3. A visszanyeréssel való korrigáláshoz választott referenciamintáknak reprezentatívaknak kell lenniük az elemzett mintákra, ami azt jelenti, hogy a kongénerkombinációk nem okozhatják a koncentrációk alábecslését.
7.2.4. A felső határérték vagy a beavatkozási küszöbérték ellenőrzésére például a felső határérték vagy a beavatkozási küszöbérték felének és kétszeresének megfelelő további referenciamintákat is alkalmazni lehet annak igazolására, hogy az elemzés alkalmassága a keresett tartományban megfelelő. Ezek a minták együttesen használhatók az elemzett minták BEQ-értékeinek kiszámítására (lásd a 7.1.2.2. pontot).
7.3. A cut-off értékek meghatározása
Meg kell határozni a BEQ-értékként kapott bioanalitikai eredmények és a megerősítő módszerrel kapott TEQ-értékek közötti összefüggést, például egyeztetett mátrixokkal végzett kalibrálási kísérletekkel, a felső határérték (ML) 0-szorosával, 0,5-szeresével, 1-szeresével és 2-szeresével egyenlő koncentrációkra adalékolt referenciamintákat használva, mindegyik koncentrációnál hat-hat ismétléssel (n = 24). Ebből az összefüggésből meg lehet becsülni a korrigálási tényezőket (vakminta és visszanyerés), de azokat a 7.2.2. pont szerint ellenőrizni kell.
Cut-off értékeket kell megállapítani annak eldöntéséhez, hogy a minta megfelel-e a PCDD/F-ekre és a dioxin jellegű PCB-kre külön-külön, vagy pedig a PCDD/F-ek és a dioxin jellegű PCB-k összegére meghatározott felső határértéknek, illetve adott esetben a beavatkozási küszöbértéknek. Ezeket a (vakminta eredményeivel és a visszanyeréssel korrigált) bioanalitikai eredmények eloszlásának alsó végpontja jelenti, amely megfelel a megerősítő módszernél használt, 5 %-nál kisebb "hamis megfelelő" arányt és 25 %-nál kisebb RSDR-t feltételező 95 %-os megbízhatósági szinten alapuló döntési határértéknek. A megerősítő módszernél használt döntési határérték a felső határérték, figyelembe véve a kiterjesztett mérési bizonytalanságot.
A (BEQ-értékként kifejezett) cut-off érték a 7.3.1., a 7.3.2. és a 7.3.3. pontban ismertetett módszerek egyikével számítható ki (lásd: 1. ábra).
7.3.1. A 95 %-os predikciós intervallum alsó sávja a megerősítő módszernél használt döntési határértéknél
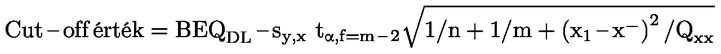
ahol:
BEQDL A megerősítő módszer döntési határértékének - amely a kiterjesztett mérési bizonytalanságot is figyelembe vevő felső határérték - megfelelő BEQ
sy,x maradék szórás
t α,f = m-2 Student-tényező (α = 5 %, f = szabadságfok, egyoldali)
m a kalibrációs pontok összesen (j index)
n n az ismétlések száma az egyes koncentrációknál
xi az i. kalibrációs pontnál a minta megerősítő módszerrel mért koncentrációja (TEQ-értékként)
[Kép #3] az összes kalibrálási minta koncentrációjának (TEQ-érték) átlaga
Kép #3
[Kép #4] négyzetösszeg-paraméter, i = az i. kalibrálási pont indexe
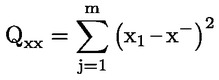
Kép #4
7.3.2. A megerősítő módszer döntési határértékének megfelelő szennyezettségű minták párhuzamos elemzéseinek (n ≥ 6) (a vakminta eredményeivel és a visszanyeréssel korrigált) bioanalitikai eredményeiből, az eloszlási görbének a megfelelő BEQ-átlagértéknél lévő alsó végpontjaként:
ahol:
SDR a biológiai tesztek BEQDL-értékre kapott eredményeinek szórása, a laboratóriumon belüli reprodukálhatóság körülményei között mérve.
7.3.3. A felső határérték vagy a beavatkozási küszöbérték 2/3-ának megfelelő szennyezettségű minták párhuzamos elemzéseivel (n ≥ 6) kapott (BEQ-értékként kifejezett, a vakminta eredményeivel és a visszanyeréssel korrigált) bioanalitikai eredmények átlaga, annak a megfigyelésnek az alapján, hogy ez az érték a 7.3.1. vagy 7.3.2. pont szerint meghatározott cut-off érték körül lesz:
5 %-nál kisebb "hamis megfelelő" arányt és 25 %-nál kisebb RSDR-t feltételező 95 %-os megbízhatósági szinten alapuló cut-off értékek kiszámítása:
1. a 95 %-os predikciós intervallum alsó sávjából a megerősítő módszernél használt döntési határértéknél;
2. a megerősítő módszer döntési határértékének megfelelő szennyezettségű minták párhuzamos elemzéseiből (n ≥ 6), az eloszlási görbének (az ábrán a harang alakú görbe) a megfelelő BEQ-átlagértéknél lévő alsó végpontjaként.
1. ábra
7.3.4. A cut-off értékek korlátozásai
A korlátozott számú, különböző mátrixú/kongénerkombinációjú mintákkal végzett hitelesítés során kapott RSDR-ből kiszámított, BEQ-értéken alapuló cut-off értékek nagyobbak lehetnek a TEQ értéken alapuló felső határértéknél vagy beavatkozási küszöbértéknél, mivel nagyobb módszerpontosság érhető el, mint a rutinszerű elemzéseknél, ahol a lehetséges kongénerkombinációk ismeretlen skáláját kell ellenőrizni. Ilyen esetekben a cut-off értékek kiszámításához RSDR = 25 %-ot kell venni, vagy pedig a felső határérték vagy a beavatkozási küszöbérték kétharmadát célszerű cut-off értéknek tekinteni.
7.4. Alkalmasságjellemzők
7.4.1. Mivel a bioanalitikai módszereknél nem alkalmazható belső standard, a méréssorozatokon belüli és a sorozatok közötti szórás megállapításához vizsgálni kell a bioanalitikai módszerek megismételhetőségét. A megismételhetőségnek 20 % alatt, a laboratóriumon belüli reprodukálhatóságnak pedig 25 % alatt kell lennie. Ehhez a vakminta eredményeivel és a visszanyeréssel korrigált BEQ-értékként számított koncentrációkat kell használni.
7.4.2. A hitelesítési eljárás részeként a tesztről ki kell mutatni, hogy különbséget tesz a vakminta eredménye és a cut-off alsó küszöbérték között, és így lehetővé teszi a megfelelő cut-off érték felett lévő minták azonosítását (lásd a 7.1.2. pontot).
7.4.3. Meg kell határozni a keresett vegyületeket, az esetleges interferenciákat és a vakminták elfogadható legnagyobb értékeit.
7.4.4. Egy mintakivonat három párhuzamos meghatározásakor kapott reakciók vagy az ilyen reakcióból számított koncentrációk (csak a munkatartományban lehetséges) százalékos szórása nem lehet nagyobb 15 %-nál.
7.4.5. A referenciaminta/referenciaminták BEQ-értékként kifejezett, korrigálás (vakminta és felső határérték vagy beavatkozási küszöbérték) nélküli eredményeit kell használni a bioanalitikai módszer egy meghatározott időszak alatti alkalmasságának értékelésére.
7.4.6. Az elemzési vakmintákról és a referenciaminták egyes típusairól minőség-ellenőrzési nyilvántartást kell vezetni, és ellenőrizni kell, hogy az analitikai alkalmasság biztosan megfelel-e a követelményeknek, az elemzési vakminták esetében főként a munkatartomány aljához képest előírt legkisebb különbség, a referenciaminták esetében pedig főként a laboratóriumon belüli reprodukálhatóság tekintetében. Az elemzési vakmintáknál ügyelni kell arra, hogy a kivonás miatt ne keletkezhessenek "hamis megfelelő" eredmények.
7.4.7. A szűrőmódszer alkalmasságának, valamint a BEQ és a TEQ közötti összefüggés értékeléséhez a feltehetően nem megfelelő mintáknak és a megfelelő minták 2-10 %-ának (mátrixonként legalább 20 minta) megerősítő módszerrel végzett elemzéséből származó eredményeket kell összegyűjteni és felhasználni. Ez az adatbázis használható a hitelesített mátrixokkal végzett rutinszerű elemzésekre vonatkozó cut-off értékek újraértékeléséhez.
7.4.8. A módszerek alkalmassága igazolható körvizsgálatokban való részvétellel is. A körvizsgálatokban elemzett minták eredményei, a felső határérték kétszereséig terjedő koncentrációtartományokban, felhasználhatók a "hamis megfelelő" arány kiszámításához, ha a laboratórium igazolni tudja az alkalmasságot. A mintáknak le kell fedniük a különféle forrásokat reprezentáló, leggyakoribb kongénerkombinációkat.
7.4.9. Szennyeződési eseteknél a cut-off értékek átértékelhetők, hogy tükrözzék az adott külön esemény egyedi mátrix- és kongénerkombinációit.
8. AZ EREDMÉNYEK JELENTÉSE
8.1. Megerősítő módszerek
8.1.1. Amennyire az alkalmazott analitikai eljárás lehetővé teszi, az analitikai eredmények között fel kell tüntetni az egyes PCDD/F- és dioxin jellegű PCB-kongénerek koncentrációit, és ezeket felfelé, lefelé és középre kerekített értékként kell a jelentésben megadni, hogy az az eredményekről a lehető legtöbb információt tartalmazza, lehetővé téve az eredményeknek a vonatkozó követelmények szerinti értelmezését.
8.1.2. A jelentésnek tartalmaznia kell a PCDD/F-ek és a dioxin jellegű PCB-k extrakciójához használt módszert.
8.1.3. Meg kell adni az egyes belső standardok visszanyeréseit abban az esetben, ha a visszanyerések kívül esnek a 6.2.5. pontban megadott tartományon, ha az eredmények meghaladják a felső határértéket (ebben az esetben meg kell adni a két párhuzamos elemzés egyikének visszanyeréseit) vagy kérésre más esetekben.
8.1.4. Mivel a minta megfelelőségéről való döntésnél figyelembe kell venni a kiterjesztett mérési bizonytalanságot, ezt a paramétert is meg kell adni. Az analitikai eredményeket tehát x +/- U formában kell megadni a jelentésben, ahol x az analitikai eredmény, U a kiterjesztett mérési bizonytalanság, a kiterjesztési tényező 2, amely körülbelül 95 %-os megbízhatósági szintet eredményez. A PCDD/F-ek és a dioxin jellegű PCB-k külön meghatározása esetén a PCDD/F-ek és a dioxin jellegű PCB-k külön-külön kapott analitikai eredményeinél becsült kiterjesztett bizonytalanságok összegét kell használni a PCDD/F-ek és a dioxin jellegű PCB-k összegének kiszámításához.
8.1.5. Az eredményeket ugyanabban a mértékegységben és legalább ugyanannyi tizedesjeggyel kell megadni, ahogy a 2002/32/EK irányelv a felső határértékeket megadja.
8.2. Bioanalitikai szűrőmódszerek
8.2.1. A szűrés eredményét "megfelelő" vagy "feltehetően nem megfelelő" megjelöléssel kell megadni.
8.2.2. Ezenkívül megadható egy indikatív eredmény a PCDD/F-ekre és/vagy a dioxin jellegű PCB-kre - TEQ érték helyett - BEQ-értékként.
8.2.3. A jelentési határérték alatti reakciókat adó mintákat "jelentési határérték alatt" megjelöléssel kell feltüntetni. A munkatartomány feletti reakciókat mutató mintákat a "munkatartományt meghaladókként" kell jelenteni, és a munkatartomány tetejének megfelelő szintet BEQ-értékként kell megadni.
8.2.4. A jelentésnek minden mintamátrix-típusnál tartalmaznia kell az értékelés alapját képező felső határértéket vagy beavatkozási küszöbértéket.
8.2.5. A jelentésnek tartalmaznia kell az alkalmazott teszt típusát, a teszt alapelvét és a kalibráció típusát.
8.2.6. A jelentésnek tartalmaznia kell a PCDD/F-ek és a dioxin jellegű PCB-k extrakciójához használt módszert.
8.2.7. Ha valamely mintával kapcsolatban felmerül, hogy feltehetően nem felel meg, a jelentésben szerepelnie kell a szükséges intézkedésre vonatkozó megjegyzésnek. A jelentős koncentrációt tartalmazó mintákban a PCDD/F-ek koncentrációját, valamint a PCDD/F-ek és a dioxin jellegű PCB-k összkoncentrációját megerősítő módszerrel kell meghatározni/megerősíteni.
8.2.8. A nem megfelelő eredményeket csak megerősítő elemzés után kell jelenteni.
8.3. Fizikai-kémiai szűrőmódszerek
8.3.1. A szűrés eredményét "megfelelő" vagy "feltehetően nem megfelelő" megjelöléssel kell megadni.
8.3.2. A jelentésnek minden mintamátrix-típusnál tartalmaznia kell az értékelés alapját képező felső határértéket vagy beavatkozási küszöbértéket.
8.3.3. Emellett jelenteni kell az egyes PCDD/F- és/vagy dioxin jellegű PCB-kongénerek koncentrációit és a TEQ-értékeket, és ezeket felfelé, lefelé és középre kerekített értékként kell a jelentésben megadni. Az eredményeket ugyanabban a mértékegységben és legalább ugyanannyi tizedesjeggyel kell megadni, ahogy a 2002/32/EK irányelv a felső határértékeket megadja.
8.3.4. Meg kell adni az egyes belső standardok visszanyeréseit abban az esetben, ha a visszanyerések kívül esnek a 6.2.5. pontban megadott tartományon, ha az eredmények meghaladják a felső határértéket (ebben az esetben meg kell adni a két párhuzamos elemzés egyikének visszanyeréseit) vagy kérésre más esetekben.
8.3.5. A jelentésnek tartalmaznia kell az alkalmazott GC-MS módszert.
8.3.6. A jelentésnek tartalmaznia kell a PCDD/F-ek és a dioxin jellegű PCB-k extrakciójához használt módszert.
8.3.7. Ha valamely mintával kapcsolatban felmerül, hogy feltehetően nem felel meg, a jelentésben szerepelnie kell a szükséges intézkedésre vonatkozó megjegyzésnek. A jelentős koncentrációt tartalmazó mintákban a PCDD/F-ek koncentrációját, valamint a PCDD/F-ek és a dioxin jellegű PCB-k összkoncentrációját megerősítő módszerrel kell meghatározni/megerősíteni.
8.3.8. A nem megfelelésről csak megerősítő elemzés után lehet határozni.
III. FEJEZET
A takarmányokban előforduló nem dioxin jellegű PCB-k koncentrációjának hatósági ellenőrzéséhez használt minta-előkészítés és az elemzési módszerekre vonatkozó követelmények
1. Alkalmazási kör
Az e fejezetben előírt követelményeket kell alkalmazni a takarmányoknak a nem dioxin jellegű PCB-k koncentrációja tekintetében történő hatósági ellenőrzési célú elemzéséhez, továbbá az egyéb szabályozási célokat szolgáló minta-előkészítés és analitikai követelmények tekintetében, ideértve a 183/2005/EK rendelet rendelkezéseinek való megfelelés biztosítása érdekében a takarmányipari vállalkozó által végzett ellenőrzéseket.
2. Az alkalmazandó kimutatási módszerek
Gázkromatográfia/elektronbefogásos kimutatás (GC-ECD), GC-LRMS, GC-MS/MS, GC-HRMS vagy egyenértékű módszerek.
3. A keresett analitok beazonosítása és megerősítése
3.1. A belső standardokhoz vagy referenciastandardokhoz viszonyított relatív retenciós idő (elfogadható eltérés: +/- 0,25 %).
3.2. A nem dioxin jellegű PCB-k gázkromatográfiás elválasztása a zavaró anyagoktól, főként az együtt eluálódó PCB-ktől, különösen, ha a minták koncentrációi a jogszabályi határértékek tartományában vagy afölött vannak, és a meg nem felelést meg kell erősíteni ( 41 ).
3.3. A GC-MS módszerekre vonatkozó követelmények
Legalább az alábbi számú molekuláris ion vagy a molekuláris klaszterben lévő jellemző ionok nyomon követése:
a) a HRMS-nél két specifikus ion;
b) az LRMS-nél három specifikus ion;
c) két specifikus anyaion; mindegyik esetében egy megfelelő ionátmenettel az MS-MS tekintetében.
Kiválasztott tömegfragmentumok gyakoriságára megengedett tűrések:
Kiválasztott tömegfragmentumok gyakoriságának relatív eltérése az elméleti gyakorisághoz vagy a keresett ion (a vizsgált ionok közül a leggyakoribb) és a minősítő ion(ok) kalibrációs standardjához képest: ± 15 %.
3.4. A GC-ECD módszerekre vonatkozó követelmények
A felső határértéket meghaladó eredményeket különböző polaritású állófázist használó két gázkromatográfiás oszloppal meg kell erősíteni.
4. A módszer alkalmasságának igazolása
A módszer alkalmasságát a felső határérték tartományában (a felső határérték felétől a kétszereséig) kell hitelesíteni úgy, hogy az ismételt elemzések elfogadható variációs együtthatót adnak (a közbenső precizitásra vonatkozó előírásokat lásd a 9. pontban).
5. A meghatározási határ
A nem dioxin jellegű PCB-k meghatározási határának ( 42 ) összege nem lehet magasabb a felső határérték egyharmadánál ( 43 ).
6. Minőség-ellenőrzés
Rendszeres vakkontrollmérések, adalékolt minták elemzése, minőség-ellenőrzési minták, részvétel a vonatkozó mátrixokkal végzett laboratóriumközi körvizsgálatokban.
7. A visszanyerések ellenőrzése
7.1. A keresett analitokhoz hasonló fizikai-kémiai tulajdonságokkal rendelkező alkalmas belső standardokat kell használni.
7.2. Belső standardok hozzáadása:
Hozzáadás a termékekhez (extrakció és mintatisztítás előtt).
7.3. A mind a hat izotópos jelölésű nem dioxin jellegű PCB-kongénert használó módszerekre vonatkozó előírások:
a) az eredményeket korrigálni kell a belső standardok visszanyeréseivel;
b) az izotópjelölésű belső standardok visszanyerésének 60 és 120 % között kell lennie;
c) anem dioxin jellegű PCB-k összegéhez 10 %-nál kisebb mértékben hozzájáruló egyes kongénereknél elfogadható ennél kisebb vagy nagyobb visszanyerés.
7.4. A nem mind a hat izotópos jelölésű belső standardot vagy más belső standardokat használó módszerekre vonatkozó előírások:
a) a belső standard(ok) visszanyerését minden minta esetében ellenőrizni kell;
b) a belső standard(ok) visszanyeréseinek 60 % és 120 % között kell lenniük;
c) az eredményeket korrigálni kell a belső standardok visszanyeréseivel.
7.5. Az izotóppal nem jelölt kongénerek visszanyerését olyan adalékolt mintákkal vagy minőség-ellenőrzési mintákkal kell ellenőrizni, amelyek koncentrációja a felső határérték tartományába esik. Ezen kongénereknél a visszanyerést elfogadhatónak kell tekinteni, ha az 60 és 120 % között van.
8. A laboratóriumokra vonatkozó követelmények
Az (EU) 2017/625 rendelet rendelkezéseinek megfelelően az analitikai minőségbiztosítás alkalmazásának biztosítása érdekében a laboratóriumokat az ISO/IEC 58 útmutató szerint működő elismert testületnek kell akkreditálnia. A laboratóriumokat az EN ISO/IEC 17025 szabvány szerint kell akkreditálni. Emellett a "Technical Guidelines for the estimation of measurement uncertainty and limits of quantification for PCB analysis" (Műszaki iránymutatás a mérési bizonytalanság és meghatározási határ becsléséhez a PCB vizsgálata tekintetében) dokumentumban foglalt elvek követendők ( 44 ).
9. Alkalmasságjellemzők: a felső határértékben jelen lévő hat indikátor PCB összegére vonatkozó kritériumok
| Izotóphígításos tömegspekometria (*1) | Egyéb technikák | |
| Valódiság | -20 % és 20 % között | -30 % és 30 % között |
| Közbenső precizitás (RSD%) | ≤ 15 % | ≤ 20 % |
| A felfelé és a lefelé kerekítés közötti különbség | ≤ 20 % | ≤ 20 % |
| (*1) Belső standardként mind a hat, 13C-jelölésű analógot használni kell. | ||
10. Az eredmények jelentése
10.1. Az analitikai eredmények között fel kell tüntetni az egyes nem dioxin jellegű PCB-k koncentrációit és az ilyen PCB-kongénerek összegét, és ezeket felfelé, lefelé és középre kerekített értékként kell a jelentésben megadni, hogy az az eredményekről a lehető legtöbb információt tartalmazza, lehetővé téve az eredményeknek a vonatkozó követelmények szerinti értelmezését.
10.2. A jelentésnek tartalmaznia kell a PCB-k extrakciójához használt módszert.
10.3. Meg kell adni az egyes belső standardok visszanyeréseit abban az esetben, ha a visszanyerések kívül esnek a 7. pontban megadott tartományon, ha az eredmények meghaladják a felső határértéket vagy kérésre más esetekben.
10.4. Mivel a minta megfelelőségéről való döntésnél figyelembe kell venni a kiterjesztett mérési bizonytalanságot, ezt a paramétert is meg kell adni. Az analitikai eredményeket tehát x +/- U formában kell megadni a jelentésben, ahol x az analitikai eredmény, U a kiterjesztett mérési bizonytalanság, a kiterjesztési tényező 2, amely körülbelül 95 %-os megbízhatósági szintet eredményez.
10.5. Az eredményeket ugyanabban a mértékegységben és legalább ugyanannyi tizedesjeggyel kell megadni, ahogy a 2002/32/EK irányelv a felső határértékeket megadja.
B. EN SZABVÁNYOK
Az (EU) 2017/625 rendelet 34. cikke (2) bekezdése a) pontjának alkalmazásában a következő EN szabványok a relevánsak:
EN 17194: Takarmányok. Mintavétel és vizsgálati módszerek. Deoxinivalenol, aflatoxin B1, fumonizin B1 és B2, T-2 és HT-2 toxinok, zearalenon és ochratoxin A meghatározása takarmány-alapanyagokban és összetett takarmányokban, LC-MS/MS-sel
EN 17270: Takarmányok. Mintavételi és elemzési módszerek. Teobromin meghatározása takarmány-alapanyagokban és összetett takarmányokban, beleértve a kakaóból származó összetevőket, folyadékkromatográfiával
EN 17504 Takarmányok. Mintavételi és elemzési módszerek. A gosszipol meghatározása gyapotmagban és takarmányokban, LC-MS/MS-sel
EN 17362 Takarmányok. Mintavételi és elemzési módszerek. A pentaklór-fenol (PCP) meghatározása takarmány-alapanyagokban és összetett takarmányokban, LC-MS/MS-sel
EN 16279: Takarmányok. A fluoridtartalom meghatározása sósavas kezelés után, ionszelektív elektródos módszerrel (ISE)
EN 17053: Takarmányok. Mintavételi és elemzési módszerek. A nyomelemek, a nehézfémek és más elemek meghatározása takarmányban ICP-MS-sel (multimódszer)
EN 15550 Takarmányok. Mintavételi és elemzési módszerek. A kadmium- és ólomtartalom meghatározása grafitkemencés atomabszorpciós spektrometriával (GF-AAS), nyomás alatti roncsolás után
EN 16206 Takarmányok. Az arzén meghatározása hidridfejlesztéses atomabszorpciós spektrometriával (HGAAS), mikrohullámú nyomás alatti feltárás után (feltárás 65 %-os salétromsavval és 30 %-os hidrogén-peroxiddal)
EN 16277 Takarmányok. A higany meghatározása hideg gőzös atomabszorpciós spektrometriával (CVAAS), mikrohullámú nyomás alatti feltárás után (extrakció 65 %-os salétromsavval és 30 %-os hidrogén-peroxiddal)
EN 16278 Takarmányok. A szervetlen arzén meghatározása hidridfejlesztéses atomabszorpciós spektrometriával (HG-AAS), mikrohullámú extrakció és szilárd fázisú extrakciós (SPE) elválasztás után
EN 17374 Takarmányok. Takarmányok. Mintavétel és vizsgálati módszerek. Szervetlen arzén meghatározása takarmányban anioncserés HPLC-ICP-MS-sel
VI. MELLÉKLET
A TAKARMÁNYOK HATÓSÁGI ELLENŐRZÉSE SORÁN AZ ÁLLATI EREDETŰ ALKOTÓELEMEK MEGHATÁROZÁSÁRA SZOLGÁLÓ ANALITIKAI MÓDSZEREK
1. CÉL ÉS ALKALMAZÁSI TERÜLET
E melléklet rendelkezéseinek értelmében a takarmányban lévő állati eredetű alkotóelemeket fénymikroszkóppal vagy polimeráz láncreakcióval (PCR) kell meghatározni.
E két módszer lehetővé teszi az állati eredetű alkotóelemek kimutatását az előkeverékekben, a takarmány-alapanyagokban és a takarmánykeverékekben. Azonban ezekkel a módszerekkel az említett alkotóelemek mennyiségét az előkeverékekben, a takarmány-alapanyagokban és a takarmánykeverékekben nem lehet meghatározni. A kimutatási határérték mindkét módszer esetén 0,1 m/m %.
A PCR-módszerrel lehetséges az előkeverékekben, a takarmány-alapanyagokban és a takarmánykeverékekben jelen lévő állati fehérje rendszertani csoportjának meghatározása.
Ezeket a módszereket kell alkalmazni a 999/2001/EK európai parlamenti és tanácsi rendelet ( 45 ) 7. cikkének (1) bekezdésében, az említett rendelet IV. mellékletében és az 1069/2009/EK európai parlamenti és tanácsi rendelet ( 46 ) 11. cikkének (1) bekezdésében megállapított tilalmak alkalmazásának ellenőrzésére.
A vizsgált takarmány típusától függően a takarmányokban előforduló állati fehérjéket vizsgáló uniós referencialaboratórium (EURL-AP) által meghatározott és a laboratórium honlapján közzétett szabványműveleti eljárásoknak (SOP) megfelelően ezek a módszerek külön-külön vagy egymással kombinálva egy műveleti eljáráson belül is alkalmazhatók ( 47 ).
2. MÓDSZEREK
2.1. Fénymikroszkóp
2.1.1. A módszer alapelve
Az elemzésre küldött előkeverékekben, takarmány-alapanyagokban és takarmánykeverékekben jelen lévő állati eredetű alkotóelemek a típusos, mikroszkóppal felismerhető jellegzetességek alapján azonosíthatók (pl. izomrostok és egyéb húsrészek, porc, csontok, szaru, szőr, sörte, gerinctelenek kutikulatöredékei, rovarok tracheális részei, vértermékek, tejcseppecskék, laktózkristályok, tollak, tojáshéjak, halcsontok, pikkelyek).
A minták ülepítéssel történő előkészítése után mikroszkópos vizsgálatokat kell végezni.
A mintákat ülepítési lépésnek kell alávetni az alábbiak szerint:
a) a szárazföldi gerinctelenektől eltérő állatokból származó alkotóelemek kimutatása érdekében egyszeri tetraklór-etilén (TCE) ülepítési lépés a 2.1.3.4.3. pontban leírtak szerint;
b) a szárazföldi gerinctelenekből származó alkotóelemek kimutatása érdekében kétlépcsős petroléter/tetraklóretilén (PE/TCE) ülepítési lépés a 2.1.3.4.4. pontban leírtak szerint.
2.1.2. Reagensek és eszközök
2.1.2.1. Reagensek
2.1.2.1.1. Koncentráló anyagok
- Tetraklór-etilén (fajsúly: 1,62).
- Petroléter (PE) forráspont 40-60 °C (fajsúly 0,65).
2.1.2.1.2. Színezékek
- Alizarinvörös-oldat (fel kell oldani 2,5 ml 1 M-os sósavat 100 ml vízben, majd hozzá kell adni ehhez az oldathoz 200 mg alizarinvöröset)
2.1.2.1.3. Beágyazó közegek
- Lúg (NaOH 2,5 % w/v vagy KOH 2,5 %w/v)
- Glicerin (tömény, viszkozitás: 1 490 cP) vagy ezzel egyenértékű tulajdonságokkal rendelkező, nem tartós tárgylemez előkészítésére alkalmas beágyazó közeg.
- Norland ® Optical Adhesive 65 (viszkozitás: 1 200 cP) vagy ezzel egyenértékű tulajdonságokkal rendelkező, tartós tárgylemez-előkészítésre alkalmas gyanta.
2.1.2.1.4. Színező tulajdonsággal rendelkező beágyazó közegek.
- Lúgoldat (fel kell oldani 2 g kálium-jodidot 100 ml vízben, majd hozzáadni 1 g jódot gyakori rázogatás közben)
- Cisztin reagens (2 g ólom-acetát, 10 g NaOH/100 ml víz)
- Fehling reagens (felhasználás előtt az A és B törzsoldatból vett egyenlő egységekből (1/1) készül. A oldat: fel kell oldani 6,9 g réz(II)-szulfát-pentahidrátot 100 ml vízben. B oldat: fel kell oldani 34,6 g kálium-nátrium-tartarát-tetrahidrátot és 12 g NaOH-t 100 ml vízben)
- Tetrametilbenzidin/Hidrogén-peroxid. (fel kell oldani 1 g 3,3',5,5'-tetrametilbenzidint (TMB) 100 ml jégecetben és 150 ml vízben. Felhasználás előtt össze kell keverni négy rész TMB-oldatot és egy rész 3 %-os hidrogén-peroxidot.
2.1.2.1.5. Öblítő reagensek
- Etanol ≥ 96 % (technikai minőségű)
- Aceton (technikai minőségű)
2.1.2.1.6. Fehérítő reagens
- Kereskedelmi nátriumhipoklorit-oldat (9-14 % aktív klór)
2.1.2.2. Felszerelés
- 0,001 g pontosságú analitikai mérleg.
- Örlőeszközök: késes vagy rotoros malom. Rotoros malom esetén ≤ 0,5 mm-es szembőségű szita használata tilos.
- 0,25 mm-es és 1 mm-es lyukátmérőjű négyzethálós szita. Az anyagveszteség elkerülése érdekében a minta előszitálásának kivételével a szita átmérője nem haladhatja meg a 10 cm-t. A sziták kalibrálása nem követelmény.
- 250 ml űrtartalmú kúpos üveg elválasztó tölcsér, a tölcsér kúpos végén teflon vagy csiszolt üveg elzárócsappal. Az elzárócsap nyitási átmérője ≥ 4 mm. Alternatív megoldásként - csak az egyszeri TCE-ülepítés esetében - kúpos aljú főzőpohár is használható, amennyiben a laboratórium bizonyítja, hogy a kimutatási szintek megegyeznek a kúpos üveg elválasztó tölcsér alkalmazásával elért kimutatási szintekkel.
Elválasztó tölcsér
- Sztereomikroszkóp, legalább 6,5-40-szeresig terjedő nagyítású.
- Összetett mikroszkóp legalább 100-400-szorosig terjedő nagyítású, áteső fényű világos látótérrel. Kiegészítésként polarizált fény és differenciál-interferenciakontraszt is alkalmazható.
- Szabványos laboratóriumi üvegedények
- Felszerelés tárgylemez előkészítéséhez: klasszikus mikroszkóp-tárgylemezek, vésett tárgylemezek, fedőlemezek (20 × 20 mm), csipeszek, finom spatula.
- Laboratóriumi szárítószekrény.
- Centrifuga.
- Szűrőpapír: kvalitatív cellulózszűrő (pórusméret 4-11 μm).
2.1.3. Mintavételezés és minta-előkészítés
2.1.3.1. Mintavétel
Az I. mellékletben foglaltak szerint vett reprezentatív mintát kell használni.
2.1.3.1.1. Mintaszárítás:
a 14 %-nál magasabb nedvességtartalmú mintákat kezelés előtt a III. mellékletben meghatározottak szerint ki kell szárítani.
2.1.3.1.2. Minta előszitálása:
a takarmány lehetséges környezeti szennyeződésére vonatkozó információk összegyűjtése érdekében a pelletált takarmányokat és magvakat ajánlatos 1 mm-es lyukátmérőjű szitán előszitálni, majd a külön mintáknak tekintendő két kapott frakciót külön-külön előkészíteni, elemezni, és azokról külön jelentést írni.
2.1.3.2. Óvintézkedések
A laboratóriumban a keresztszennyeződés elkerülése érdekében a többször használatos eszközöket használat előtt gondosan meg kell tisztítani. Az elválasztó tölcsért tisztítás előtt részekre kell szedni. Az elválasztó tölcsért és az üvegedényeket először kézzel el kell mosni, és ezt követően mosogatógépben tisztítani. A szitákat erősszálú műszálas kefével kell megtisztítani. Zsíros anyag (például halliszt) átszitálását követően acetonnal és sűrített levegővel javasolt a szitákon végső tisztítást végezni.
2.1.3.3. Zsírt vagy olajat tartalmazó minták előkészítése
A következő módszer alkalmazható a zsírt tartalmazó minták előkészítéséhez:
- Ha a zsír szilárd, addig kell melegíteni szárítószekrényben, amíg folyékonnyá nem válik,
- 40 ml zsírt át kell pipettázni a minta aljáról egy centrifugacsőbe.
- A mintát 10 percig 4 000 ford./perc fordulatszámon centrifugálni kell.
- Ha a zsír a centrifugálás után szilárd, addig kell melegíteni szárítószekrényben, amíg folyékonnyá nem válik,
- A centrifugálást 5 percig 4 000 ford./perc fordulatszámon meg kell ismételni.
- A leülepedett szennyeződések felét egy kiskanállal vagy egy spatulával tegyük át egy mikroszkóp-tárgylemezre vizsgálat céljából. Beágyazószerként a glicerin használata javasolt.
- A fennmaradó szennyeződéseket a 2.1.3.4.3. pont első francia bekezdésének megfelelően az üledék előkészítésére kell használni.
Az olajat tartalmazó minták előkészítésére az első és a negyedik francia bekezdés kivételével ugyanezt az eljárást kell alkalmazni.
2.1.3.4. Zsírtól vagy olajtól eltérő minták előkészítése
2.1.3.4.1. Részminta-vételezés és őrlés: a mintából legalább 50 g részmintát kell venni elemzés céljára, majd leőrölni.
2.1.3.4.2. A nyersanyag előkészítése: az őrölt részmintából legalább 5 g-ot kell előkészíteni. 0,25 mm lyukátmérőjű szitán át kell szitálni és az így kapott két frakciót meg kell vizsgálni.
2.1.3.4.3. Egyszeri TCE-ülepítés a szárazföldi gerincesektől eltérő állatokból származó alkotóelemek kimutatására.
- Üledék kivonása és előkészítése:
A 10 g tömegű (0,01 g pontossággal bemért) őrölt részmintát elválasztó tölcsérbe vagy kúpos aljú főzőpohárba kell helyezni, és legalább 50 ml tetraklór-etilént hozzáadni. Hallisztből vagy egyéb tiszta állati termékből, ásványi alkotóelemből vagy olyan előkeverékekből, amelyek 10 %-nál nagyobb mennyiségű üledéket képeznek, 3 g-nál nagyobb mennyiség nem kerülhet a tölcsérbe. A keveréket legalább 30 mp-ig erőteljesen kell rázni, majd óvatosan hozzáadni legalább 50 ml tetraklór-etilént a tölcsér belső falának leöblítésével, a rátapadt részecskék eltávolítása érdekében. A kapott keveréket legalább öt percig állni kell hagyni, mielőtt az elzárócsap megnyitásával az üledéket leválasztjuk.
Kúpos aljú főzőpohár használatakor a keveréket legalább 15 mp-ig erőteljesen fel kell keverni, majd legalább 10 ml tiszta tetraklór-etilénnel óvatosan leöblíteni a pohár belső faláról az odatapadt részecskéket. A keveréket legalább 3 percig állni kell hagyni, majd további 15 mp-ig keverni és legalább 10 ml tiszta tetraklór-etilénnel óvatosan leöblíteni a pohár belső faláról az odatapadt részecskéket. Az így kapott keveréket legalább 5 percig állni kell hagyni, majd a folyadékfrakciót eltávolítani és óvatos dekantálással kiönteni úgy, hogy az összes üledék megmaradjon.
Az üledéket tölcsérbe helyezett szűrőpapíron kell összegyűjteni, ami lehetővé teszi a maradék tetraklór-etilén leválasztását, és ugyanakkor annak elkerülését, hogy zsír rakódjon le az üledékbe. Az üledéket ki kell szárítani. Az ülepítés ellenőrzése céljából javasolt ezután az üledék lemérése (0001 g pontossággal). Végül - hacsak a szitálás nem tűnik szükségtelennek - az üledéket 0,25 mm lyukátmérőjű szitán át kell szitálni, és az így kapott két frakciót kell vizsgálni.
- A flotátum kivonása és előkészítése:
Az üledék fentiek szerinti módszerrel történő kinyerése után két fázis marad vissza az elválasztó tölcsérben: a tetraklór-etilénből álló folyadék és a felülúszó anyagból álló szilárd halmazállapotú anyag. A szilárd fázist (a flotátumot) a tetraklór-etilénnek az elzárócsapon keresztüli kiengedésével kell kinyerni a tölcsérből. Az elválasztó tölcsér megfordításával a flotátumot egy nagy petri-csészébe kell önteni, majd füstelszívó süvegben levegős szárítással megszárítani. 0,25 mm lyukátmérőjű szitán át kell szitálni és az így kapott két frakciót meg kell vizsgálni.
- Színezőanyagok használata:
Az állati eredetű alkotóelemek helyes meghatározásának megkönnyítése érdekében, az EURL-AP által kiadott és a honlapján is elérhető útmutatókkal összhangban a laboratóriumi munkatárs használhat színezékeket a minta előkészítése során.
Az üledék alizarinvörös-oldattal történő festésekor az alábbi eljárás alkalmazandó:
- A szárított üledéket üvegkémcsőbe kell tölteni, és kétszer kb. 5 ml etanollal átöblíteni (minden alkalommal 30 mp-ig Vortex keverőt kell használni, az oldószert másfél percig hagyni leülepedni, majd leönteni),
- Az üledéket legalább 1 ml nátrium-hipoklorit oldat hozzáadásával fehéríteni kell. A folyamatnak 10 perc reakcióidőt kell biztosítani. A kémcsövet megtöltjük vízzel, az üledéket 2-3 percig hagyjuk leülepedni, és a vizet és a szuszpendált részecskéket óvatosan leöntjük,
- Az üledéket még kétszer kb. 10 ml vízzel le kell öblíteni (30 mp-ig Vortex keverőt kell használni, hagyni leülepedni, és a vizet minden alkalommal leönteni),
- hozzá kell adni 2-10 csepp alizarinvörös-oldatot, majd Vortex keverővel összekeverni. A reakció 30 mp-ig tarthat, és a színezett üledéket kétszer kb. 5 ml etanollal, majd egyszer acetonnal át kell öblíteni (minden alkalommal 30 mp-ig Vortex keverőt kell használni, az oldószert kb. egy percig hagyni leülepedni, majd leönteni).
- A színezett üledéket ki kell szárítani.
2.1.3.4.4. Kétlépcsős PE/TCE-ülepítés a szárazföldi gerinctelenekből származó alkotóelemek kimutatására.
Minden lépést 250 ml űrtartalmú kúpos üveg elválasztó tölcsérben kell végrehajtani a 2.1.2.2. pont negyedik francia bekezdésében leírtak szerint.
- Az őrölt részmintából egy 10 g-os (0,01 g-os) részt át kell helyezni az elválasztó tölcsérbe, és először egyszeri TCE-ülepítésnek kell alávetni a 2.1.3.4.3. pontban leírtak szerint, beleértve az üledék kinyerését egy tölcsérre helyezett papírszűrőn. Ezt az üledéket úgy lehet felhasználni, mint a 2.1.3.4.3. pont szerint kapott üledéket.
- Az üledékkel együtt leürített kis mennyiségű tetraklór-etilént egy beosztásos mérőhengerbe kell átvezetni. Az elválasztó tölcsér elzáró csapjának felnyitásával a beosztásos mérőhengert tovább kell tölteni 30 ml TCE eléréséig. E térfogat elérése után a csapot le kell zárni.
- Ezt az összegyűjtött TCE-mennyiséget 30 ml 40-60 °C-os forráspontú petroléter elválasztó tölcsérbe történő hozzáadásával kell helyettesíteni. Az elválasztó tölcsér tartalmát alaposan össze kell keverni, hogy 30 % PE/70 % TCE keveréket kapjunk (kb. 1,26 g.cm-3 sűrűséggel). Engedjük, hogy az anyag 10 percig leülepedjen. Két új frakció fog elkülönülni: egy második üledék és egy végső flotátum (< 1,26 g.cm-3). A második üledéket a petri-csészében (vagy a tölcsérre helyezett papírszűrőn) kell visszanyerni az elzáró csap felnyitásával, amíg csak kevés oldószerkeverék és a végső flotátum marad az elválasztó tölcsérben. A maradék folyadékot és a végső flotátumot külön kell összegyűjteni egy tölcsérre helyezett papírszűrőn. Az elválasztó tölcsér falát petroléterrel le kell öblíteni, hogy a végső flotátumból minden anyagot összegyűjtsünk. A végső flotátumot hagyni kell száradni. Az üledéket 0,25 mm lyukátmérőjű szitán át kell szitálni, és az így kapott két frakciót kell vizsgálni a szárazföldi gerinctelenekből származó alkotóelemek kimutatására, hacsak a szitálás nem tűnik szükségtelennek.
2.1.4. Mikroszkópos vizsgálat
2.1.4.1. Tárgylemez-előkészítés
Az üledékből és a laboratóriumi munkatárs választásától függően a flotátumból vagy a nyersanyagból tárgylemezen preparátumot készítünk. Adott esetben, kizárólag a szárazföldi gerinctelenekből származó alkotóelemek kimutatása céljából a 2.1.3.4.4. pont szerint nyert végső flotátumból is elő kell készíteni tárgylemezeket. Az így kapott két frakciót (a finomat és a durvát) elő kell készíteni. A frakcióból a tárgylemezre terített vizsgálati mennyiségeknek az egész frakcióra nézve reprezentatívaknak kell lenniük.
Elegendő mennyiségű tárgylemezt kell előkészíteni annak érdekében, hogy a 2.1.4.2. pontban ismertetett vizsgálati eljárást teljes egészében végre lehessen hajtani.
A tárgylemezeket az EURL-AP által meghatározott és a honlapján közzétett szabványműveleti eljárásokat követve a megfelelő beágyazószerrel kell beágyazni. A tárgylemezeket fedőlemezekkel le kell fedni.
2.1.4.2. Megfigyelési folyamatábra állati részecskék kimutatására takarmánykeverékekben, takarmány-alapanyagokban és előkeverékekben
Az előkészített tárgylemezeket az 1. és 2. ábrában meghatározott megfigyelési folyamatábra szerint kell megfigyelni.
Az üledék és a laboratóriumi munkatárs döntésétől függően a flotátum vagy a nyersanyag mikroszkópos megfigyelését összetett mikroszkóppal kell elvégezni. Ezenkívül a szárazföldi gerinctelenekből származó alkotóelemek kimutatása céljából a megfigyeléseket a 2.1.3.4.4. pont szerint nyert végső flotátumon is el kell végezni a 3. ábra szerint. A durva frakciók megfigyelésekor az összetett mikroszkóp mellett sztereomikroszkóp is használható. Minden tárgylemezt többféle nagyítással kell teljes egészében megvizsgálni. A folyamatábrák használatára vonatkozó pontos magyarázatot az EURL-AP által meghatározott és a saját honlapján közzétett SOP részletesen tartalmazza.
A megfigyelési folyamatábra minden szakaszában szigorúan be kell tartani a tárgylemezek megfigyelésre előírt minimális számát, kivéve, ha a frakció teljes mérete nem teszi lehetővé a megadott tárgylemezszám elérését, például akkor, ha nem keletkezett üledék. Meghatározásonként legfeljebb 6 tárgylemezt kell használni a részecskeszám rögzítésére.
Amikor specifikusabb, a 2.1.2.1.4. pontban leírtak szerinti színező tulajdonsággal rendelkező beágyazó közeg használatával további tárgylemezek készülnek a flotátum vagy a nyersanyag vizsgálatára, a 2.1.2.1.3. pontban meghatározott, más beágyazóközeggel készített tárgylemezeken kimutatott szerkezetek (pl. tollak, szőrök, izom- vagy vérrészecskék) további jellemzése céljából, a részecskék számát meghatározásonként legfeljebb 6 tárgylemez alapján kell meghatározni, a specifikusabb beágyazó közeggel készült további tárgylemezeket is beleértve. A szárazföldi gerinctelenekből származó alkotóelemek kimutatására szolgáló, a 2.1.3.4.4. pontban leírtak szerint a végső flotátumból készített további tárgylemezeket nem kell figyelembe venni más kategóriák (szárazföldi gerincesek és halak) azonosítása céljából.
A részecskék tulajdonságainak és eredetének meghatározását megkönnyítendő a laboratóriumi munkatárs alkalmazhat segédeszközöket, például döntéstámogató rendszereket, képkönyvtárakat és referenciamintákat.
1. ábra
Megfigyelési folyamatábra egyszeri TCE-ülepítés után a szárazföldi gerinctelenekből származó állati részecskék kimutatására takarmánykeverékekben, takarmány-alapanyagokban és előkeverékekben az első meghatározáshoz
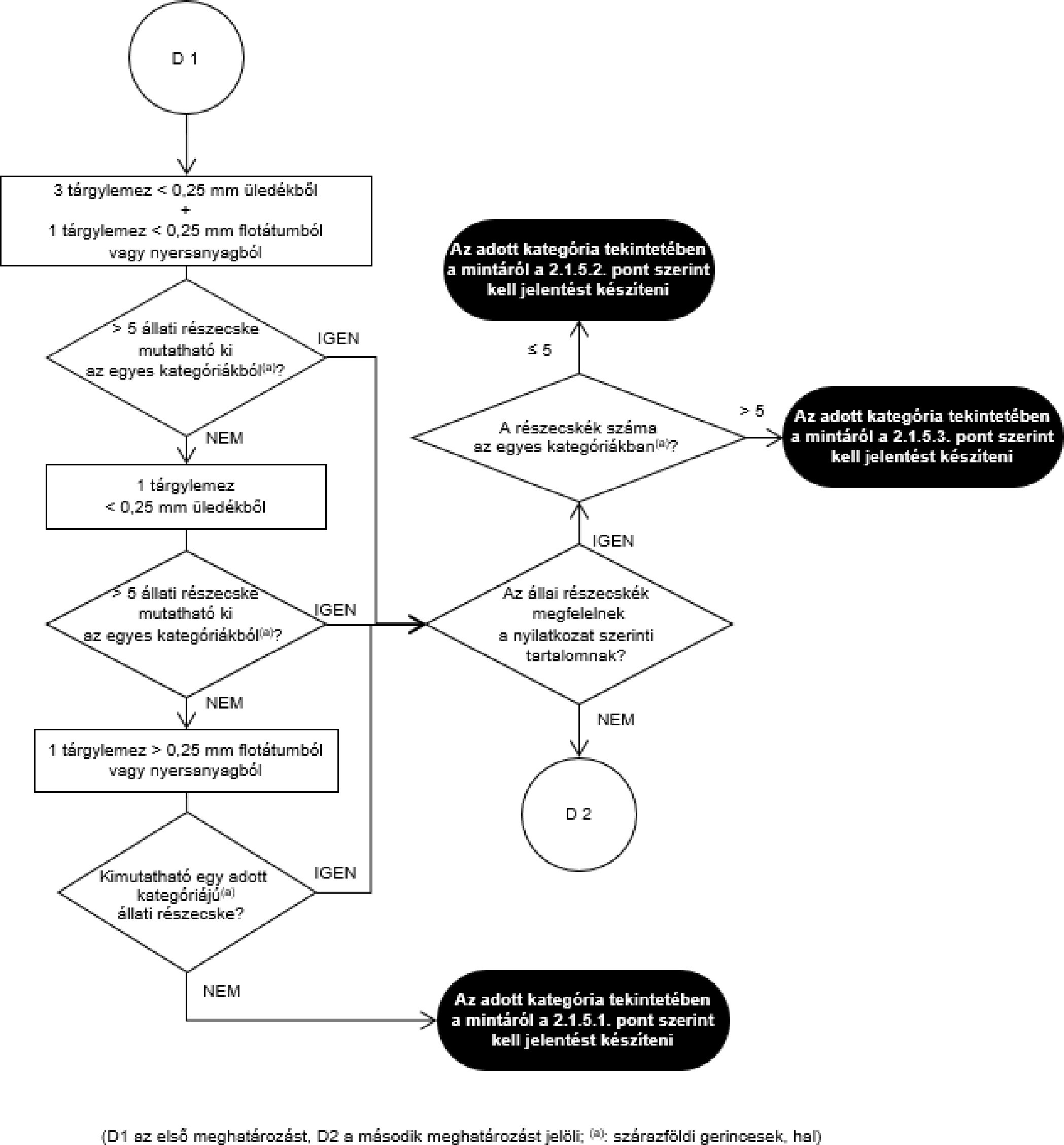
2. ábra
Megfigyelési folyamatábra egyszeri TCE-ülepítés után a szárazföldi gerinctelenekből származó állati részecskék kimutatására takarmánykeverékekben, takarmány-alapanyagokban és előkeverékekben a második meghatározáshoz
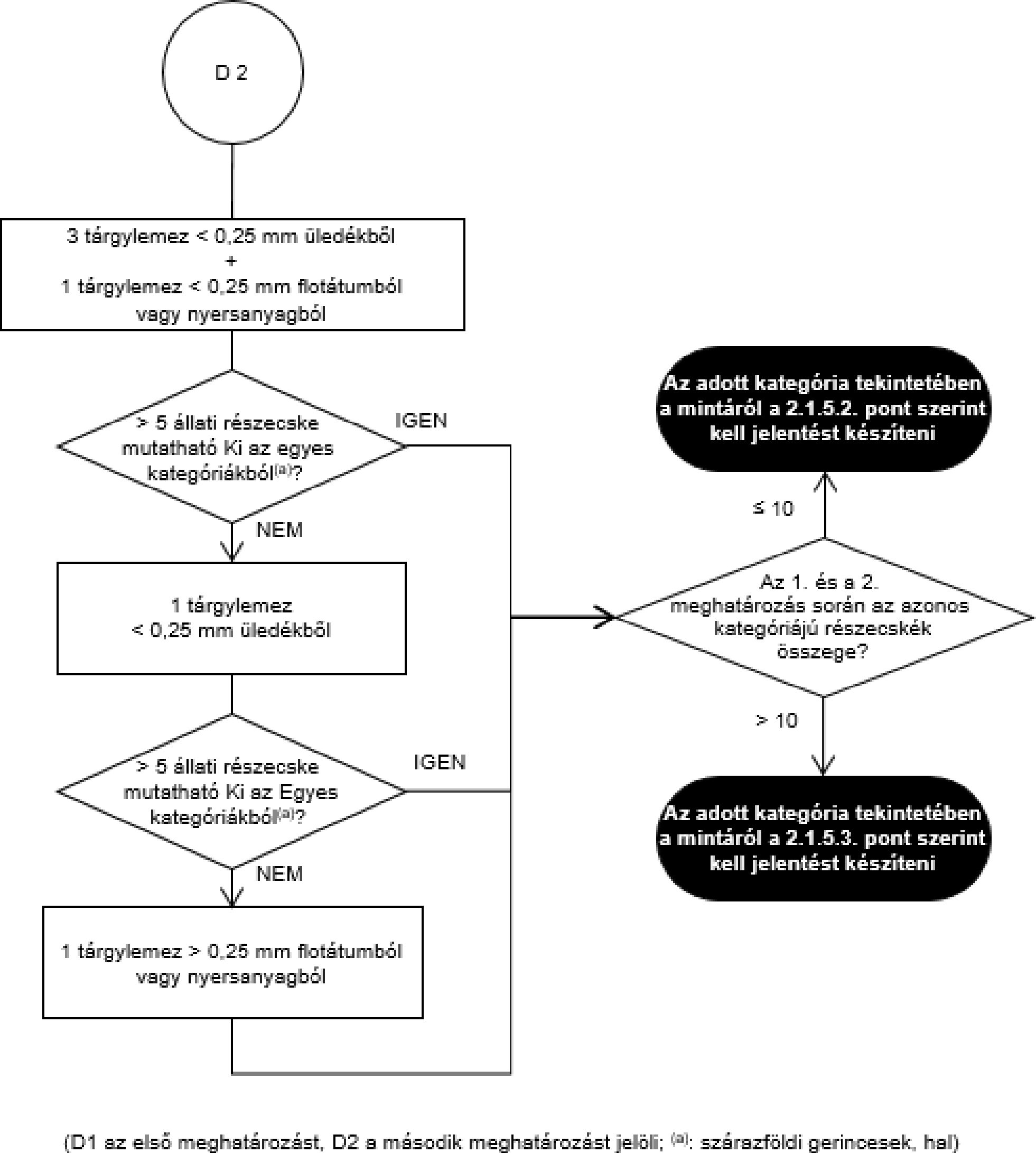
3. ábra
Megfigyelési folyamatábra kétlépcsős PE/TCE-ülepítés után a szárazföldi gerinctelenekből származó alkotóelemek kimutatására takarmánykeverékekben, takarmány-alapanyagokban és előkeverékekben
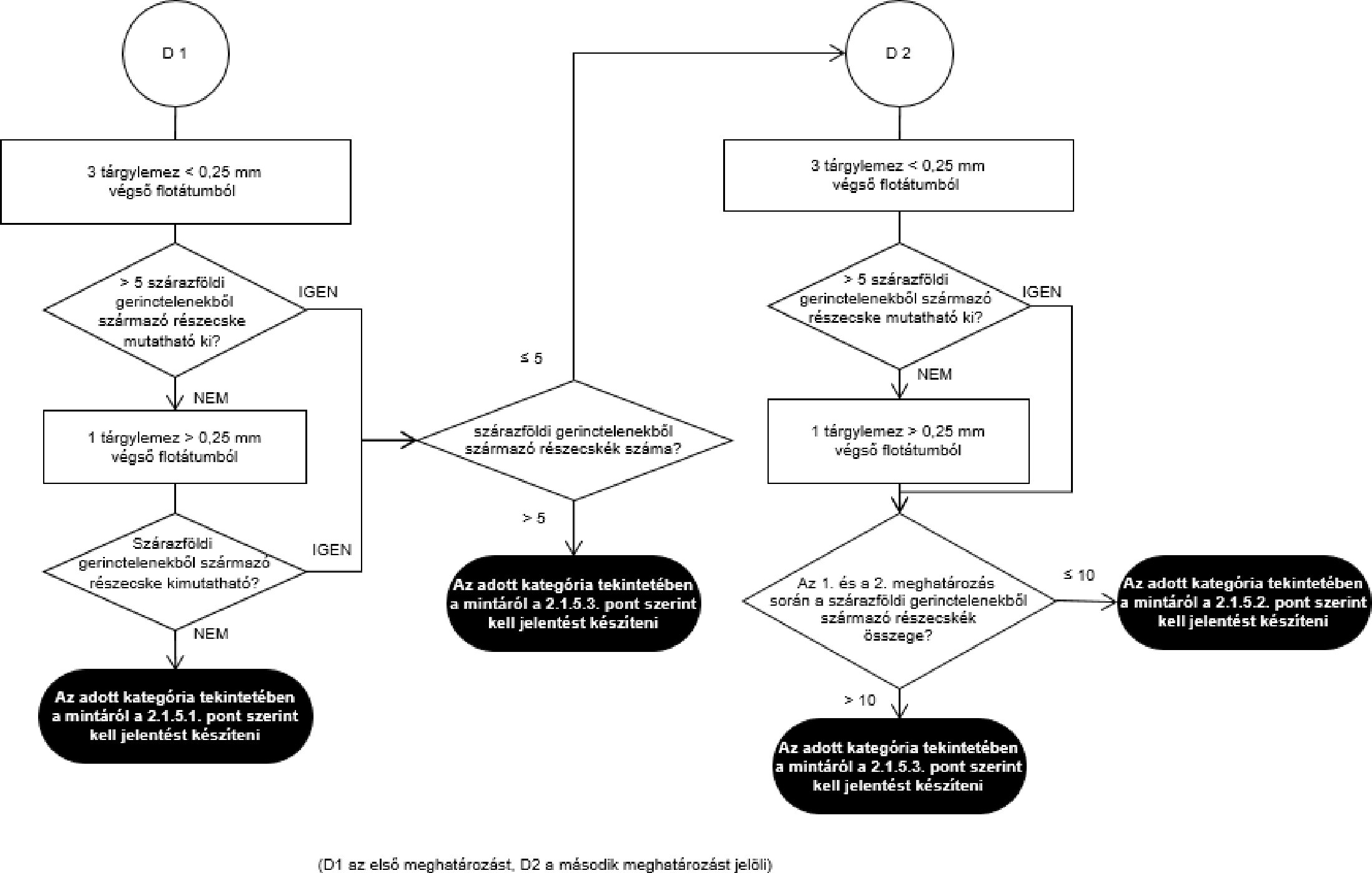
2.1.4.3. Meghatározások száma
A meghatározásokat különböző, egyenként 50 g-os almintákon kell elvégezni.
Ha az 1. ábrán vagy adott esetben a 3. ábrán szereplő megfigyelési folyamatábra szerint végzett első meghatározás nem mutatta ki állati részecske jelenlétét, nincs szükség további meghatározásra, és az elemzés eredményéről a 2.1.5.1. pontban foglalt megszövegezés alkalmazásával kell jelentést készíteni.
Ha az 1. ábrában meghatározott megfigyelési folyamatábra szerint végzett első meghatározás kimutatta egy vagy több, egy adott kategóriához tartozó (azaz szárazföldi gerinces vagy hal) állati részecske jelenlétét, és a talált részecskék kategóriája megfelel a minta nyilatkozat szerinti tartalmának, nincs szükség második meghatározásra. Ha az első meghatározás során kimutatott, egy adott kategóriához tartozó állati részecskék száma 5-nél több, az elemzés eredményéről állatkategóriánként a 2.1.5.3. pontban foglalt megfogalmazás alkalmazásával kell jelentést készíteni. Különben az elemzés eredményéről állatkategóriánként a 2.1.5.2. pontban foglalt megfogalmazás alkalmazásával kell jelentést készíteni.
Ha a 3. ábrán szereplő megfigyelési folyamatábra szerint végzett első meghatározás 5-nél több, szárazföldi gerinctelenkből származó részecske jelenlétét mutatta ki, nincs szükség második meghatározásra, és az elemzés eredményéről a 2.1.5.3. pontban foglalt megfogalmazás alkalmazásával kell jelentést készíteni.
Minden más esetben, így akkor is, ha a laboratórium nem kapott a tartalomra vonatkozó nyilatkozatot, egy újabb almintán második meghatározást kell végezni. Ha a 2. ábrán vagy adott esetben a 3. ábrán szereplő megfigyelési folyamatábra szerint végzett második meghatározás után a két meghatározás során kimutatott adott kategóriájú állati részecskék számának összege nagyobb 10-nél, az elemzés eredményéről a 2.1.5.3. pontban foglalt megfogalmazás alkalmazásával kell jelentést készíteni. Különben az elemzés eredményéről állatkategóriánként a 2.1.5.2. pontban foglalt megfogalmazás alkalmazásával kell jelentést készíteni.
2.1.5. Az eredmények közlése
A laboratórium az eredmények jelentésekor megadja a vizsgált anyagok típusát (üledék, flotátum, végső flotátum, nyersanyag). A jelentésben egyértelműen fel kell tüntetni, hogy hány meghatározást végeztek el, és hogy a tárgylemezek előkészítése előtt sor került-e a frakciók 2.1.3.4.3. pont első franciabekezdésének harmadik bekezdése vagy a 2.1.3.4.4. pont harmadik franciabekezdése szerinti átszitálására.
A laboratóriumi jelentésnek tartalmaznia kell legalább a szárazföldi gerincesekből és a halból származó alkotóelemek jelenlétére vonatkozó információt.
A különböző eseteket a következő módon kell jelenteni:
2.1.5.1. Nem kimutatható adott kategóriájú állati részecske:
- "A beküldött mintában fénymikroszkópos vizsgálat alapján, szárazföldi gerincesekből származó részecske nem volt kimutatható."
- "A beküldött mintában fénymikroszkópos vizsgálat alapján, halból származó részecske nem volt kimutatható."
- "A beküldött mintában fénymikroszkópos vizsgálat alapján, szárazföldi gerinctelenekből származó részecske nem volt kimutatható."
2.1.5.2. Amennyiben egy adott kategóriájú állati részecskéből egyetlen meghatározás elvégzése esetén 1-5, illetve két meghatározás esetén 1-10 került kimutatásra (a kimutatott részecskék száma nem éri el a takarmányokban előforduló állati fehérjéket vizsgáló EURL-AP szabványműveleti eljárásaiban (SOP) megállapított és a honlapján közzétett döntési határétéket):
Egyetlen meghatározás elvégzése esetén:
- "A beküldött mintában fénymikroszkópos vizsgálat alapján legfeljebb 5, szárazföldi gerincesekből származó részecske volt kimutatható. A részecskék a következőkként azonosíthatók: ... [csont, porc, izom, szőr, szaru, egyéb (kérjük, adja meg a megfelelőt)]. Ez a kis mértékű jelenlét az e mikroszkópos módszer tekintetében megállapított döntési határérték alatt van."
- "A beküldött mintában fénymikroszkópos vizsgálat alapján legfeljebb 5, halból származó részecske volt kimutatható. A részecskék a következőkként azonosíthatók: ... [halcsont, halpikkely, porc, izom, hallókő (otolith), kopoltyú, egyéb (kérjük, adja meg a megfelelőt)]. Ez a kis mértékű jelenlét az e mikroszkópos módszer tekintetében megállapított döntési határérték alatt van."
Két meghatározás elvégzése esetén:
- "A beküldött mintában fénymikroszkópos vizsgálat alapján a két meghatározás során összesen legfeljebb 10, szárazföldi gerincesekből származó részecske volt kimutatható. A részecskék a következőkként azonosíthatók: ... [csont, porc, izom, szőr, szaru, egyéb (kérjük, adja meg a megfelelőt)]. Ez a kis mértékű jelenlét az e mikroszkópos módszer tekintetében megállapított döntési határérték alatt van."
- "A beküldött mintában fénymikroszkópos vizsgálat alapján a két meghatározás során összesen legfeljebb 10, halból származó részecske volt kimutatható. A részecskék a következőkként azonosíthatók: ... [halcsont, halpikkely, porc, izom, hallókő (otolith), kopoltyú, egyéb (kérjük, adja meg a megfelelőt)]. Ez a kis mértékű jelenlét az e mikroszkópos módszer tekintetében megállapított döntési határérték alatt van."
- "A beküldött mintában fénymikroszkópos vizsgálat alapján a két meghatározás során összesen legfeljebb 10, szárazföldi gerinctelenekből származó részecske volt kimutatható. A részecskék a következőkként azonosíthatók: ... [kutikula, szájrészek, izmok, tracheális szerkezetek, egyéb (kérjük, adja meg a megfelelőt)]. Ez a kis mértékű jelenlét az e mikroszkópos módszer tekintetében megállapított döntési határérték alatt van."
Továbbá:
- A minta előszitálása esetén a laboratóriumi jelentés tartalmazza, hogy melyik frakcióban (átszitált frakció, pelletált frakció vagy magvak) mutattak ki állati részecskéket, mivel a kizárólag az átszitált frakcióban kimutatott állati részecskék jelenléte környezeti szennyezésre utalhat.
- Ha csak olyan állati részecskék mutathatók ki (pl. izomrostok), amelyek nem kategorizálhatók szárazföldi gerincesből vagy halból származóként, a jelentésben meg kell említeni, hogy csak ilyen állati részecskéket mutattak ki, és nem zárható ki, hogy azok szárazföldi gerincesekből származnak.
2.1.5.3. Egy adott kategóriájú állati részecskéből egyetlen meghatározás elvégzése esetén több mint 5-öt vagy két meghatározás esetén több mint 10-et mutattak ki:
Egyetlen meghatározás elvégzése esetén:
- "A beküldött mintában fénymikroszkópos vizsgálat alapján 5-nél több, szárazföldi gerincesekből származó részecske volt kimutatható. A részecskék a következőkként azonosíthatók: ... [csont, porc, izom, szőr, szaru, egyéb (kérjük, adja meg a megfelelőt)]."
- "A beküldött mintában fénymikroszkópos vizsgálat alapján 5-nél több, halból származó részecske volt kimutatható. A részecskék a következőkként azonosíthatók: ... [halcsont, halpikkely, porc, izom, hallókő (otolith), kopoltyú, egyéb (kérjük, adja meg a megfelelőt)]."
- "A beküldött mintában fénymikroszkópos vizsgálat alapján 5-nél több, szárazföldi gerinctelenekből származó részecske volt kimutatható. A részecskék a következőkként azonosíthatók: ... [kutikula, szájrészek, izmok, tracheális szerkezetek, egyéb (kérjük, adja meg a megfelelőt)]."
Két meghatározás elvégzése esetén:
- "A beküldött mintában fénymikroszkópos vizsgálat alapján a két meghatározás során összesen 10-nél több, szárazföldi gerincesekből származó részecske volt kimutatható. A részecskék a következőkként azonosíthatók: ... [csont, porc, izom, szőr, szaru, egyéb (kérjük, adja meg a megfelelőt)]."
- "A beküldött mintában fénymikroszkópos vizsgálat alapján a két meghatározás során összesen 10-nél több, halból származó részecske volt kimutatható. A részecskék a következőkként azonosíthatók: ... [halcsont, halpikkely, porc, izom, hallókő (otolith), kopoltyú, egyéb (kérjük, adja meg a megfelelőt)]."
- "A beküldött mintában fénymikroszkópos vizsgálat alapján a két meghatározás során összesen 10-nél több, szárazföldi gerinctelenekből származó részecske volt kimutatható. A részecskék a következőkként azonosíthatók: ... [kutikula, szájrészek, izmok, tracheális szerkezetek, egyéb (kérjük, adja meg a megfelelőt)]."
Továbbá:
- A minta előszitálása esetén a laboratóriumi jelentés tartalmazza, hogy melyik frakcióban (átszitált frakció, pelletált frakció vagy magvak) mutattak ki állati részecskéket, mivel a kizárólag az átszitált frakcióban kimutatott állati részecskék jelenléte környezeti szennyezésre utalhat.
- Ha csak olyan állati részecskék mutathatók ki (pl. izomrostok), amelyek nem kategorizálhatók szárazföldi gerincesből vagy halból származóként, a jelentésben meg kell említeni, hogy csak ilyen állati részecskéket mutattak ki, és nem zárható ki, hogy azok szárazföldi gerincesekből származnak.
2.2. PCR
2.2.1. A módszer alapelve
A takarmány-alapanyagokban vagy az összetett takarmányokban előforduló állati eredetű dezoxiribonukleinsav- (DNS-) fragmentumok jelenlétét a PCR-módszerrel végzett genetikai amplifikációs technika révén mutatják ki, meghatározott fajspecifikus DNS-szakaszok kiválasztásával
A PCR-módszer első lépése a DNS-extrakció. Az így kapott DNS-kivonat egy amplifikációs eljárással alkalmassá válik a vizsgálandó állatfaj jelenlétének kimutatására.
2.2.2. Reagensek és eszközök
2.2.2.1. Reagensek
2.2.2.1.1. Reagensek a DNS-extrakció szakaszában
Kizárólag az EURL-AP által jóváhagyott és a honlapján közzétett reagensek használhatók.
2.2.2.1.2. Reagensek a génamplifikáció szakaszában
2.2.2.1.2.1. Primerek és próbák
Kizárólag az EURL-AP által validált oligonukleotid szekvenciákat tartalmazó primerek és próbák használhatók ( 48 ).
2.2.2.1.2.2. Master Mix
Kizárólag olyan Master Mix oldatok használhatóak, amelyek nem tartalmaznak állati eredetű DNS komponenseket, melyek hibás mérési eredményhez vezetnek ( 49 ).
2.2.2.1.2.3. Dekontaminációs reagens
2.2.2.1.2.3.1. Sósavoldat (0,1 N)
2.2.2.1.2.3.2. Nátriumhipoklorit-oldat, 0,15 % aktív klór
2.2.2.1.2.3.3. Nem korrozív reagensek nagy értékű eszközök, például analitikai mérlegek fertőtlenítéséhez (pl. az MP Biomedicals által gyártott DNA EraseTM)
2.2.2.2. Felszerelés
2.2.2.2.1. 0,001 g pontosságú analitikai mérleg
2.2.2.2.2. Örlőeszközök
2.2.2.2.3. Valós idejű PCR-t lehetővé tevő termocykler
2.2.2.2.4. Mikrocentrifuga mikrofugacsövekhez
2.2.2.2.5. 1 μl-1 000 μl pipettázására alkalmas mikropipetta-készlet
2.2.2.2.6. Standard molekuláris biológiai műanyag edények, mikrofugacsövek, szűrős műanyag hegy mikropipettához, lemezek a termocyklerhez
2.2.2.2.7. Fagyasztószekrény a minták és reagensek tárolására
2.2.3. Mintavételezés és minta-előkészítés
2.2.3.1. Mintavétel
Az azonosításra az I. mellékletben megállapított rendelkezések szerint vett reprezentatív mintát kell használni.
2.2.3.2. Minta-előkészítés
A DNS-extrakcióra szolgáló laboratóriumi minták készítése során figyelembe kell venni a II. mellékletben meghatározott követelményeket. A mintából elemzés céljára legalább 50 g részmintát kell venni, majd megőrölni.
Az ISO 24276 szabványban meghatározottak értelmében a minta-előkészítést más helyiségben kell elvégezni, mint a DNS-extrakciót és a génamplifikációt.
Két, egyenként legalább 100 mg vizsgálati mennyiséget kell előkészíteni.
2.2.4. DNS-extrakció
A DNS-extrakciót minden egyes elkészített vizsgálati mintán az EURL-AP által meghatározott és a honlapján közzétett szabványműveleti eljárásokat (SOP) alkalmazva kell elvégezni.
Az ISO 24276 szabvány értelmében minden extrakciós sorozathoz két extrakciós kontrollt kell készíteni:
- egy extrakciós vakpróbát,
- egy pozitív DNS-extrakciós kontrollt.
2.2.5. Genetikai amplifikáció
A genetikai amplifikációt a meghatározni kívánt egyes fajokra vonatkozóan validált módszerek alkalmazásával kell végezni. Ezeket a módszereketaz EURL-AP által meghatározott és a honlapján közzétett szabványműveleti eljárások állapítják meg. Az inhibíció felmérése érdekében a DNS-kivonatokat legalább két különböző hígításban szükséges vizsgálni.
Az ISO 24276 szabvány értelmében minden célállatfajra vonatkozóan két amplifikációs kontrollt kell előkészíteni:
- egy pozitív DNS-kontrollt minden lemezhez vagy PCR vizsgálati sorozathoz,
- egy nontemplát kontrollt (más néven templátmentes kontrollt) minden lemezhez vagy PCR vizsgálati sorozathoz
2.2.6. Az eredmények értékelése és közlése
- A laboratóriumnak az eredmények közlésekor meg kell jelölnie legalább a felhasznált vizsgálati minta súlyát, az alkalmazott kivonási technikát, az elvégzett meghatározások számát és a módszer kimutatási határértékét.
Ha negatív templátmentes kontroll mellett a pozitív DNS-extrakciós kontroll és a pozitív DNS-kontroll nem ad pozitív eredményt a vizsgált célra vonatkozóan, az eredményeket nem kell értékelni és közölni.
Ha a két párhuzamos vizsgálat eredménye nem egyezik, legalább a génamplifikációs lépést meg kell ismételni. Amennyiben a laboratórium azt feltételezi, hogy az ellentmondások a DNS-kivonatokból adódnak, az eredmények értékelését megelőzően újabb DNS-extrakciót, majd ezt követően génamplifikációt kell végezni.
Az EURL-AP által meghatározott és a honlapján közzétett szabványműveleti eljárások (SOP) értelmében az eredmények végső közlésének két páthuzamos mérés együttes értékelésén kell alapulnia.
2.2.6.1. Negatív eredmény
A negatív eredményt a következők szerint kell közölni:
A vizsgálatra beküldött mintában nem volt kimutatható X-ből származó DNS (X alatt a vizsgálat által megcélzott állatfaj vagy fajcsoport értendő).
2.2.6.2. Pozitív eredmény
A pozitív eredményt a következők szerint kell közölni:
A vizsgálatra beküldött mintában X-ből származó DNS volt kimutatható (X alatt a vizsgálat által megcélzott állatfaj vagy fajcsoport értendő).
VII. MELLÉKLET
A BAROMFITAKARMÁNYOK ENERGIAÉRTÉKÉNEK SZÁMÍTÁSI MÓDSZERE
1. Az energiaérték számításának és kifejezésének módszere
A baromfitakarmány-keverékek energiaértékét az alábbi képletnek megfelelően kell kiszámítani a takarmány bizonyos analitikai alkotóelemeinek százalékos aránya alapján. Ezt az értéket a takarmánykeverék kilogrammonkénti, nitrogénnel korrigált, metabolizálható energiájával (ME) kell kifejezni, megajoule (MJ) mértékegységben:
ME (MJ/kg) = 0,1551 × nyersfehérje % + 0,3431 × nyers zsír % + 0,1669 × keményítő % + 0,1301 × összes cukor % (szacharózban kifejezve)
2. A feltüntetett értékekre alkalmazandó tűréshatárok
Amennyiben a hatósági ellenőrzés eltérést (a takarmány megemelkedett vagy lecsökkent energiaértéke) mutat ki az ellenőrzés eredménye és a feltüntetett energiaérték között, az ME 0,4 MJ/kg-os tűréshatárát engedélyezik.
3. Az eredmény kifejezése
A fenti képlet alkalmazásával kapott eredményt egy tizedesjegyre kerekítve kell megadni.
4. A mintavétel és analitikai módszerek
A takarmánykeverék mintavételét és a számítási módszerben jelzett analitikai alkotóelemek tartalmának meghatározását a takarmányok hatósági ellenőrzésére szolgáló uniós mintavételi és analitikai módszerekkel összhangban kell elvégezni.
A következőket kell alkalmazni:
- a nyerszsírtartalom meghatározására: a III. melléklet G. részében a nyersolajok és zsírok meghatározására szolgáló B. eljárás,
- a keményítőtartalom meghatározására: a III. melléklet K. részében megállapított polarimetriás módszer.
A MACSKÁKNAK ÉS KUTYÁKNAK SZÁNT TAKARMÁNY-ALAPANYAGOK ÉS ÖSSZETETT TAKARMÁNYOK ENERGIAÉRTÉKÉNEK SZÁMÍTÁSI MÓDSZERE
A macskáknak és kutyáknak szánt takarmány-alapanyagok és összetett takarmányok energiaértékét a "Takarmányok. Mintavételi és elemzési módszerek. A kutyáknak és macskáknak szánt takarmány-alapanyagok és összetett takarmányok (kedvtelésből tartott állatok eledelei) metabolizálható energiaszintjének prediktív egyenletei, beleértve a diétás eledeleket" című EN 16967 szabványnak megfelelően kell kiszámítani.
IX. MELLÉKLET
A 6. CIKKBEN EMLÍTETT MEGFELELÉSI TÁBLÁZATOK
| 71/250/EGK irányelv | Ez a rendelet |
| 1. cikk, első albekezdés | 3. cikk |
| 1. cikk, második albekezdés | 2. cikk |
| 2. cikk | — |
| 3. cikk | — |
| Melléklet, 1. rész | II. melléklet |
| Melléklet, 2. rész | — |
| Melléklet, 3. rész | — |
| Melléklet, 4. rész | III. melléklet, O. rész |
| Melléklet, 5. rész | III. melléklet, M. rész |
| Melléklet, 6. rész | III. melléklet, N. rész |
| Melléklet, 7. rész | III. melléklet, Q. rész |
| Melléklet, 9. rész | III. melléklet, K. rész |
| Melléklet, 10. rész | — |
| Melléklet, 11. rész | — |
| Melléklet, 12. rész | III. melléklet, J. rész |
| Melléklet, 14. rész | III. melléklet, D. rész |
| Melléklet, 16. rész | — |
| 71/393/EGK irányelv | Ez a rendelet |
| 1. cikk | 3. cikk |
| 2. cikk | — |
| 3. cikk | — |
| Melléklet, I. rész | III. melléklet, A. rész |
| Melléklet, II. rész | III. melléklet, E. rész |
| Melléklet, III. rész | III. melléklet, P. rész |
| Melléklet, IV. rész | III. melléklet, H. rész |
| 72/199/EGK irányelv | Ez a rendelet |
| 1. cikk | 3. cikk |
| 2. cikk | — |
| 3. cikk | — |
| 4. cikk | — |
| I. melléklet, 1. rész | III. melléklet, L. rész |
| I. melléklet, 2. rész | III. melléklet, C. rész |
| I. melléklet, 3. rész | — |
| I. melléklet, 4. rész | — |
| I. melléklet, 5. rész | V. melléklet, A. rész |
| II. melléklet | — |
| 73/46/EGK irányelv | Ez a rendelet |
| 1. cikk | 3. cikk |
| 3. cikk | — |
| 4. cikk | — |
| I. melléklet, 1. rész | III. melléklet, B. rész |
| I. melléklet, 2. rész | — |
| I. melléklet, 3. rész | III. melléklet, I. rész |
| 76/371/EGK irányelv | Ez a rendelet |
| 1. cikk | 1. cikk |
| 2. cikk | — |
| 3. cikk | — |
| Melléklet | I. melléklet |
| 76/372/EGK irányelv | Ez a rendelet |
| 1. cikk | — |
| 2. cikk | — |
| 3. cikk | — |
| Melléklet | — |
| 78/633/EGK irányelv | Ez a rendelet |
| 1. cikk | 3. cikk |
| 2. cikk | — |
| 3. cikk | — |
| Melléklet, 1. rész | — |
| Melléklet, 2. rész | — |
| Melléklet, 3. rész | IV. melléklet, C. rész |
| 81/715/EGK irányelv | Ez a rendelet |
| 1. cikk | — |
| 2. cikk | — |
| 3. cikk | — |
| Melléklet | — |
| 84/425/EGK irányelv | Ez a rendelet |
| 1. cikk | — |
| 2. cikk | — |
| 3. cikk | — |
| Melléklet | — |
| 86/174/EGK irányelv | Ez a rendelet |
| 1. cikk | 4. cikk |
| 2. cikk | — |
| 3. cikk | — |
| Melléklet | VII. melléklet |
| 93/70/EGK irányelv | Ez a rendelet |
| 1. cikk | 3. cikk |
| 2. cikk | — |
| 3. cikk | — |
| Melléklet | IV. melléklet, D. rész |
| 93/117/EK irányelv | Ez a rendelet |
| 1. cikk | 3. és 5. cikk |
| 2. cikk | — |
| 3. cikk | — |
| Melléklet, 1. rész | IV. melléklet, E. rész |
| Melléklet, 2. rész | VIII. melléklet, A. rész |
| 98/64/EK irányelv | Ez a rendelet |
| 1. cikk | 3. és 5. cikk |
| 2. cikk | — |
| 3. cikk | — |
| 4. cikk | — |
| Melléklet, A. rész | III. melléklet, F. rész |
| Melléklet, C. rész | VIII. melléklet, B. rész |
| 1999/27/EK irányelv | Ez a rendelet |
| 1. cikk | 3. és 5. cikk |
| 2. cikk | — |
| 3. cikk | — |
| 4. cikk | — |
| 5. cikk | — |
| 6. cikk | — |
| 7. cikk | — |
| Melléklet, A. rész | VIII. melléklet, C. rész |
| Melléklet, B. rész | IV. melléklet, F. rész |
| Melléklet, C. rész | VIII. melléklet, D. rész |
| 1999/76/EK irányelv | Ez a rendelet |
| 1. cikk | 3. cikk |
| 2. cikk | — |
| 3. cikk | — |
| 4. cikk | — |
| Melléklet | IV. melléklet, G. rész |
| 2000/45/EK irányelv | Ez a rendelet |
| 1. cikk | 3. cikk |
| 2. cikk | — |
| 3. cikk | — |
| 4. cikk | — |
| Melléklet, A. rész | IV. melléklet, A. rész |
| Melléklet, B. rész | IV. melléklet, B. rész |
| Melléklet, C. rész | III. melléklet, G. rész |
| 2002/70/EK irányelv | Ez a rendelet |
| 1. cikk | 1. cikk |
| 2. cikk | 2. és 3. cikk |
| 3. cikk | — |
| 4. cikk | — |
| 5. cikk | — |
| I. melléklet | I. melléklet és V. melléklet, B. (I.) rész |
| II. melléklet | II. melléklet és V. melléklet, B. (II.) rész |
| 2003/126/EK irányelv | Ez a rendelet |
| 1. cikk | 3. cikk |
| 2. cikk | — |
| 3. cikk | — |
| 4. cikk | — |
| 5. cikk | — |
| 6. cikk | — |
| Melléklet | VI. melléklet |
( 1 ) Az Európai Parlament és a Tanács 1831/2003/EK rendelete (2003. szeptember 22.) a takarmányozási célra felhasznált adalékanyagokról (HL L 268., 2003.10.18., 29. o.).
( 2 ) Az Európai Parlament és a Tanács 2002/32/EK irányelve (2002. május 7.) a takarmányban előforduló nemkívánatos anyagokról (HL L 140., 2002.5.30., 10. o.).
( 3 ) HL L 70., 2005.3.16., 1. o.
( 4 ) Az Európai Parlament és a Tanács 767/2009/EK rendelete (2009. július 13.) a takarmányok forgalomba hozataláról és felhasználásáról, az 1831/2003/EK európai parlamenti és tanácsi rendelet módosításáról, valamint a 79/373/EGK tanácsi irányelv, a 80/511/EGK bizottsági irányelv, a 82/471/EGK, 83/228/EGK, 93/74/EGK, 93/113/EK és 96/25/EK tanácsi irányelv és a 2004/217/EK bizottsági határozat hatályon kívül helyezéséről (HL L 229., 2009.9.1., 1. o.).
( 5 ) Az esetlegesen előforduló csomókat össze kell törni (a csomókat szükség esetén ki is lehet venni, porlasztás után pedig visszatenni a mintába).
( 6 ) Kivéve a kis fajlagos tömegű szálastakarmányokat/zöldtakarmányokat.
( 7 ) Kivéve a kis fajlagos tömegű szálastakarmányokat/zöldtakarmányokat.
( 8 ) Az Európai Parlament és a Tanács 178/2002/EK rendelete (2002. január 28.) az élelmiszerjog általános elveiről és követelményeiről, az Európai Élelmiszerbiztonsági Hatóság létrehozásáról és az élelmiszerbiztonságra vonatkozó eljárások megállapításáról (HL L 31., 2002.2.1., 1. o.).
( 9 ) Az Európai Parlament és a Tanács (EU) 2019/4 rendelete (2018. december 11.) a gyógyszeres takarmányok előállításáról, forgalomba hozataláról és felhasználásáról, a 183/2005/EK európai parlamenti és tanácsi rendelet módosításáról, valamint a 90/167/EGK tanácsi irányelv hatályon kívül helyezéséről (HL L 4., 2019.1.7., 1. o.).
( 10 ) A 95 %-os konfidencia-intervallumot egy másik tényező, például a Student-féle (t) együttható felhasználásával is el lehet érni.
( 11 ) A 95 %-os konfidencia-intervallumot egy másik tényező, például a t-tényező felhasználásával is el lehet érni.
( 12 ) A gabonafélék, lisztek, darafélék és derce szárítására szolgáló szárítószekrény olyan hőkapacitású legyen, hogy 131 °C-ra beállítva kevesebb mint 45 perc alatt visszaálljon a beállított hőfokra, azt követően, hogy a maximális számú vizsgálati mintát elhelyezték benne egyidejű szárítás céljából. A szellőztetése olyan legyen, hogy amikor a maximális befogadóképességnek megfelelő számú közönséges búzamintát elhelyezzük benne és két órán át szárítjuk, akkor az így kapott eredmény mindössze kevesebb mint 0,15 %-kal térjen el attól az eredménytől, amelyet a négy órán át folytatott szárítással kapunk.
( 13 ) A számítással kapcsolatos további részletekért lásd az EN ISO 6498 - Takarmányok - A minta-előkészítés irányelvei című szabványt.
( 14 ) A nitrogéntartalom minden takarmányban meghatározható, de előfordulhat, hogy a nyersfehérje-tartalom kiszámításához használt 6,25 értékű átváltási együttható nem alkalmazható a rovaralapú takarmány-alapanyagokra (alacsonyabb átváltási együttható) és egyes hobbiállat-eledelekre és vérplazmafehérjékre (magasabb átváltási együttható)
( 15 ) Az EN 17180 szabványban előírt analitikai módszerre a 10 %-nál több aminosavat tartalmazó takarmányokban található aminosavak meghatározására szolgáló, hatósági ellenőrzés során alkalmazott alternatív módszerként hivatkoznak.
( 16 ) Ezt a módszert nem validálták a kollaboratív vizsgálat során a 10 %-nál több takarmány-adalékanyagot tartalmazó előkeverékek esetében. A módszer azonban ezekre a mátrixokra is alkalmazható, megfelelő kisebb módosításokkal, feltéve, hogy a módszert ezt követően a laboratóriumon belül validálják. További információk a https://ec.europa.eu/jrc/en/eurl/feed-additives/authorisation weboldalon érhetők el.
( 17 ) Amennyiben az olajon vagy a zsíron ezt követően minőség-ellenőrzést végeznek, a horzsakődarabokat üveggyöngyökkel helyettesítsük.
( 18 ) A Bizottság 121/2008/EK rendelete (2008. február 11.) az állatok etetésére szolgáló készítmények (KN-kód: 2309 ) keményítőtartalmának meghatározására szolgáló analitikai módszer megállapításáról (HL L 37., 2008.2.12., 3. o.).
( 19 ) Az e melléklet A. részében az A-vitamin meghatározására és az e melléklet B. részében az E-vitamin meghatározására leírt módszer helyett az EN 17547 szabványban előírt analitikai módszer a hatósági ellenőrzés céljára alkalmazható alternatív módszernek minősül.
( 20 ) A vizsgálatot a Verband Deutscher Landwirtschaftlicher Untersuchungs- und Forschungsanstalten (VDLUFA) takarmány-munkacsoportja végezte.
( 21 ) Az e melléklet A. részében az A-vitamin meghatározására és az e melléklet B. részében az E-vitamin meghatározására leírt módszer helyett az EN 17547 szabványban előírt analitikai módszer a hatósági ellenőrzés céljára alkalmazható alternatív módszernek minősül.
( 22 ) A vizsgálatot a Verband Deutscher Landwirtschaftlicher Untersuchungs- und Forschungsanstalten (VDLUFA) takarmány-munkacsoportja végezte.
( 23 ) Ezt a módszert különböző takarmánymátrixokra vonatkozó kollaboratív vizsgálattal validálták. További információk a https://ec.europa.eu/jrc/en/eurl/feed-additives/authorisation weboldalon érhetők el.
( 24 ) Más roncsolási módszerek is használhatók, ha azok igazoltan hasonló eredményt adnak (mint például a nyomás alatti mikrohullámú roncsolás).
( 25 ) A (friss vagy szárított) zöldtakarmányok nagy mennyiségű növényi eredetű kovasavat tartalmazhatnak, amely nyomelemeket tarthat vissza, ezért el kell távolítani. Ilyen takarmányokból vett minták esetén ezért a következő, módosított módszert kell követni. Végezzük el az 5.1.1.1. pontban ismertetett műveletet egészen a szűrésig. Az oldhatatlan maradékot tartalmazó szűrőpapírt mossuk át kétszer forrásban lévő vízzel, és helyezzük egy kvarc- vagy platinatégelybe.
Égessük el a tokos kemencében (4.1. pont) 550 °C alatti hőmérsékleten, amíg az összes széntartalmú anyag teljesen el nem tűnik. Hagyjuk kihűlni, adjunk hozzá néhány csepp vizet, ezt követően pedig 10-15 ml fluorhidrogénsavat (3.4. pont), és párologtassuk el belőle a nedvességet kb. 150 °C-on. Ha maradt kovasav a maradékanyagban, oldjuk fel újra néhány milliliter fluorhidrogénsavban (3.4. pont), és párologtassuk el belőle a nedvességet. Adjunk hozzá öt csepp kénsavat (3.5. pont), és melegítsük addig, amíg megszűnik a fehér füst képződése. Miután hozzáadtunk 5 ml 6 mol/liter koncentrációjú sósavat (3.2. pont) és kb. 30 ml vizet, melegítsük meg, szűrjük le az oldatot egy 250 ml-es mérőlombikba, és töltsük fel vízzel a jelig (a HCl-koncentráció kb. 0,5 mol/liter). Folytassuk a meghatározást az 5.1.2. ponttól.
( 26 ) The Analyst 108., 1983, 1252-1256. o.
( 27 ) A módszer a takarmány-alapanyagokban található diklazuril meghatározására is alkalmazható.
( 28 ) Hosszabb (legfeljebb 1 éves) stabilitás lehetséges, de azt az egyes laboratóriumoknak igazolniuk kell.
( 29 ) A PCDD-k, PCDF-ek és dioxin jellegű PCB-k toxicitási egyenérték-tényezőit (TEF) tartalmazó táblázat: Az Egészségügyi Világszervezet (WHO) Nemzetközi Kémiai Biztonsági Programja (IPCS) keretében 2005 júniusában Genfben tartott szakértői konferencia megállapításai alapján számított, a humán kockázatok felméréséhez használt WHO-TEF-értékek (Martin van den Berg és mtsai, The 2005 World Health Organization Re-evaluation of Human and Mammalian Toxic Equivalency Factors for Dioxins and Dioxin-like Compounds. [Dioxinok és dioxin jellegű vegyületek emberekre és emlősökre vonatkozó toxicitási egyenérték-tényezőinek az Egészségügyi Világszervezet által 2005-ben végzett újraértékelése] Toxicological Sciences 93(2), 223-241. oldal (2006).
| Kongéner | TEF-érték | Kongéner | TEF-érték |
| Dibenzo-p-dioxinok (PCDD-k) és dibenzo-p-furánok (PCDF-ek) | Dioxin jellegű PCB-k: nem orto-PCB-k + mono-orto-PCB-k | ||
| 2,3,7,8-TCDD | 1 | ||
| 1,2,3,7,8-PeCDD | 1 | Non-ortho PCBs | |
| 1,2,3,4,7,8-HxCDD | 0,1 | PCB 77 | 0,0001 |
| 1,2,3,6,7,8-HxCDD | 0,1 | PCB 81 | 0,0003 |
| 1,2,3,7,8,9-HxCDD | 0,1 | PCB 126 | 0,1 |
| 1,2,3,4,6,7,8-HpCDD | 0,01 | PCB 169 | 0,03 |
| OCDD | 0,0003 | Mono-ortho PCBs | |
| 2,3,7,8-TCDF | 0,1 | PCB 105 | 0,00003 |
| 1,2,3,7,8-PeCDF | 0,03 | PCB 114 | 0,00003 |
| 2,3,4,7,8-PeCDF | 0,3 | PCB 118 | 0,00003 |
| 1,2,3,4,7,8-HxCDF | 0,1 | PCB 123 | 0,00003 |
| 1,2,3,6,7,8-HxCDF | 0,1 | PCB 156 | 0,00003 |
| 1,2,3,7,8,9-HxCDF | 0,1 | PCB 157 | 0,00003 |
| 2,3,4,6,7,8-HxCDF | 0,1 | PCB 167 | 0,00003 |
| 1,2,3,4,6,7,8-HpCDF | 0,01 | PCB 189 | 0,00003 |
| 1,2,3,4,7,8,9-HpCDF | 0,01 | ||
| OCDF | 0,0003 | ||
Alkalmazott rövidítések: "T" = tetra; "Pe" = penta; "Hx" = hexa; "Hp" = hepta; "O" = okta; "CDD" = klór-dibenzo-dioxin; "CDF" = klór-dibenzo-furán; "CB" = klór-bifenil.
( 30 ) A Bizottság (EU) 2021/808 végrehajtási rendelete (2021. március 22.) az élelmiszer-termelő állatokon alkalmazott gyógyszerhatóanyagok maradékanyagaira vonatkozó analitikai módszerek elvégzéséről és az eredmények értelmezéséről, valamint az alkalmazandó mintavételi módszerekről és a 2002/657/EK és 98/179/EK határozatok hatályon kívül helyezéséről (HL L 180., 2021.5.21., 84. o.).
( 31 ) Adott esetben a "Guidance Document on Measurement Uncertainty for Laboratories performing PCDD/F and PCB Analysis using Isotope Dilution Mass Spectrometry" (Iránymutatás a mérési bizonytalanság tekintetében PCDD/F és PCB vizsgálatát végző, izotóphígításos tömegspektrometriát alkalmazó laboratóriumok számára) (https://food.ec.europa.eu/system/files/2017-05/animal-feed-guidance_document_pcdd-f_pcb_en.pdf) című dokumentumban meghatározott elvek követendők.
( 32 ) Párhuzamos elemzés: a keresett analitok külön vizsgálata ugyanazon homogenizált minta második alikvotjának használatával.Általában a II. melléklet C. fejezetének 3. pontjában előírt követelmények alkalmazandók a párhuzamos elemzésre. Ugyanakkor az adott analitoknál a 13C-jelölésű belső standardot alkalmazó módszerek esetében a két párhuzamos elemzésre csak akkor van szükség, ha az ilyen első megerősítés eredménye nem mutat megfelelést. A két párhuzamos elemzésre azért van szükség, hogy ki lehessen zárni a minták belső keresztszennyeződésének vagy véletlen összecserélődésének lehetőségét. Ha szennyeződési eset miatt történik az elemzés, el lehet tekinteni a két párhuzamos elemzéssel történő megerősítéstől abban az esetben, ha az elemzésre kiválasztott minták nyomon követhetően a szennyeződéshez kapcsolódnak, a megállapított koncentráció pedig jelentős mértékben meghaladja a felső határértéket.
( 33 ) A felfelé kerekítés fogalma szerint a mennyiségileg nem meghatározott egyes kongénerek hozzájárulásának értéke a meghatározási határ értékével egyenlő. A lefelé kerekítés fogalma szerint a mennyiségileg nem meghatározott egyes kongénerek hozzájárulásának értéke nullával egyenlő. A középre kerekítés fogalma szerint a mennyiségileg nem meghatározott egyes kongénerek hozzájárulásának értéke a meghatározási határ értékének felével egyenlő.
( 34 ) Általában a II. melléklet C. fejezetének 3. pontjában előírt követelmények alkalmazandók a párhuzamos elemzésre. Ugyanakkor az adott analitoknál a 13C-jelölésű belső standardot alkalmazó megerősítő módszerek esetében a két párhuzamos elemzésre csak akkor van szükség, ha az ilyen első megerősítés eredménye nem mutat megfelelést. A két párhuzamos elemzésre azért van szükség, hogy ki lehessen zárni a minták belső keresztszennyeződésének vagy véletlen összecserélődésének lehetőségét. Ha szennyeződési eset miatt történik az elemzés, el lehet tekinteni a két párhuzamos elemzéssel történő megerősítéstől abban az esetben, ha az elemzésre kiválasztott minták nyomon követhetően a szennyeződéshez kapcsolódnak, a megállapított koncentráció pedig jelentős mértékben meghaladja a felső határértéket.
( 35 ) A felfelé kerekítés fogalma szerint a mennyiségileg nem meghatározott egyes kongénereknek a toxicitási egyenértékhez (TEQ) való hozzájárulása a meghatározási határ értékével egyenlő. A lefelé kerekítés fogalma szerint a mennyiségileg nem meghatározott egyes kongénereknek a toxicitási egyenértékhez (TEQ) való hozzájárulása nullával egyenlő. A középre kerekítés fogalma szerint a mennyiségileg nem meghatározott egyes kongénereknek a toxicitási egyenértékhez (TEQ) való hozzájárulása a meghatározási határ értékének felével egyenlő.
( 36 ) A beavatkozási küszöbértékek ellenőrzésére szolgáló párhuzamos elemzés indokolása és követelményei azonosak a felső határértékre az 5. lábjegyzetben leírtakkal.
( 37 ) Az Európai Parlament és a Tanács 183/2005/EK rendelete (2005. január 12.) a takarmányhigiénia követelményeinek meghatározásáról (HL L 35., 2005.2.8., 1. o.).
( 38 ) A bioanalitikai módszerek nem specifikusak a TEF-rendszerben szereplő kongénerekre. A mintakivonatban lehetnek szerkezetileg hasonló más AhR-aktív vegyületek, amelyek hozzájárulnak az összreakcióhoz. A bioanalitikai eredményeket ezért nem lehet a minta TEQ-értéke becslésének tekinteni, hanem csupán jelzik azt.
( 39 ) Guidance Document on Measurement Uncertainty for Laboratories performing PCDD/F and PCB Analysis using Isotope Dilution Mass Spectrometry (Iránymutatás a mérési bizonytalanság tekintetében PCDD/F és PCB vizsgálatát végző, izotóphígításos tömegspektrometriát alkalmazó laboratóriumok számára) (https://food.ec.europa.eu/system/files/2017-05/animal-feed-guidance_document_pcdd-f_pcb_en.pdf), Guidance Document on the Estimation of LOD and LOQ for Measurements in the Field of Contaminants in Feed and Food (Iránymutatás az érzékelési határ és a meghatározási határ méréséhez a takarmányban és élelmiszerekben előforduló szennyező anyagok tekintetében) (https://food.ec.europa.eu/system/files/2016-10/cs_contaminants_sampling_analysis-report_2004_en.pdf).
( 40 ) A jelenlegi követelmények az alábbi kiadványban közzétett TEF-értékeken alapulnak: M. Van den Berg és mtsai., Toxicol Sci 93 (2), 223-241 (2006).
( 41 ) A tapasztalatok szerint a következő kongénereknél például gyakran előfordul az együttes eluáció: PCB-28/31, PCB-52/69 és PCB-138/163/164. A GC-MS módszernél figyelembe kell venni az esetleg a nagyobb mértékben klórozott kongénerek fragmentumai által okozott esetleges zavarást is.
( 42 ) Adott esteben a "Guidance Document on the Estimation of LOD and LOQ for Measurements in the Field of Contaminants in Feed and Food" (Iránymutatás az érzékelési határ és a meghatározási határ méréséhez a takarmányban és élelmiszerekben előforduló szennyező anyagok tekintetében) (https://data.europa.eu/doi/10.2787/8931) dokumentumban meghatározott elvek követendők.
( 43 ) Határozottan ajánlott, hogy a reagenssel készített vakminta koncentrációjának hozzájárulása a mintában lévő szennyeződés koncentrációjához ennél kisebb legyen. A laboratórium felelőssége a vakminták koncentrációeltéréseinek ellenőrzés alatt tartása, különösen akkor, ha a vakmintákkal kapott eredményeket le kell vonni az analitikai eredményekből.
( 44 ) Lásd a 9. lábjegyzetet.
( 45 ) Az Európai Parlament és a Tanács 999/2001/EK rendelete (2001. május 22.) egyes fertőző szivacsos agyvelőbántalmak megelőzésére, az ellenük való védekezésre és a felszámolásukra vonatkozó szabályok megállapításáról (HL L 147., 2001.5.31., 1. o.).
( 46 ) Az Európai Parlament és a Tanács 1069/2009/EK rendelete (2009. október 21.) a nem emberi fogyasztásra szánt állati melléktermékekre és a belőlük származó termékekre vonatkozó egészségügyi szabályok megállapításáról és az 1774/2002/EK rendelet hatályon kívül helyezéséről (állati melléktermékekre vonatkozó rendelet) (HL L 300., 2009.11.14., 1. o.).
( 47 ) https://www.eurl.craw.eu/legal-sources-and-sops/method-of-reference-and-sops/.
( 48 ) Az EURL-AP honlapján megtalálható a vizsgálat által megcélzott állatfajokhoz használandó primerek és próbák jegyzéke.
( 49 ) A célnak megfelelő Master Mix oldatok megtalálhatók az EURL-AP honlapján.
Lábjegyzetek:
[1] A dokumentum eredetije megtekinthető CELEX: 32009R0152 - https://eur-lex.europa.eu/legal-content/HU/ALL/?uri=CELEX:32009R0152&locale=hu Utolsó elérhető, magyar nyelvű konszolidált változat CELEX: 02009R0152-20250514 - https://eur-lex.europa.eu/legal-content/HU/ALL/?uri=CELEX:02009R0152-20250514&locale=hu